
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scalable electrocatalyzed formation of C–O bonds using flow reactor technology†
Michael
Prieschl
a,
David
Cantillo
 ab,
C. Oliver
Kappe
ac and
Gabriele
Laudadio
*ac
ab,
C. Oliver
Kappe
ac and
Gabriele
Laudadio
*ac
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: gabriele.laudadio@uni-graz.at
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland 4072, Australia
cCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
First published on 21st October 2024
Abstract
The development of modular and robust synthetic routes that can serve both in medicinal and process chemistry settings is rare. Generally, highly modular medicinal chemistry routes are too hazardous and expensive to be translated into a process chemistry environment. Taking the case study of delamanid, a pharmaceutical compound used for multidrug-resistant tuberculosis treatment, the development of a sustainable and modular but scalable formation of C–O bonds via an electrocatalytic method is presented. In this work, the electrochemical batch reaction was studied, addressing critical reproducibility issues related with the process. Furthermore, the reaction was successfully translated to a flow electrochemical reactor design, which allowed the use of carbon felt electrodes. The high modularity of the protocol was demonstrated by the synthesis of 11 different examples, while the scalability of the reaction was proven by a gram scale preparation of a key intermediate for the synthesis of delamanid.
The ideation of scalable and reliable synthetic routes for the preparation of active pharmaceutical ingredients (APIs) is an essential step in the workflow towards the approval of a new drug candidate.1,2 Ideally, this synthetic plan should guarantee sufficient robustness across different scales, from mg to decagram in the initial phases, providing access to multi-kilogram processes when the commercialization of the bioactive compound is approaching.3 However, the reality behind the thinking process does not adhere with this principle, due to the conceptual differences between drug discovery and process development route requirements. In the discovery phase, the candidates are generally synthesized in the most modular and versatile way to enable the variation of key molecular fragments.4 When the first part of the campaign is complete, the route is usually not amenable to be performed on a large scale, due to the toxic/hazardous reagents involved or the prohibitive costs associated with the reactants.1 Hence, process chemists have to redesign the synthetic sequence in order to obtain scalable protocols (Fig. 1).
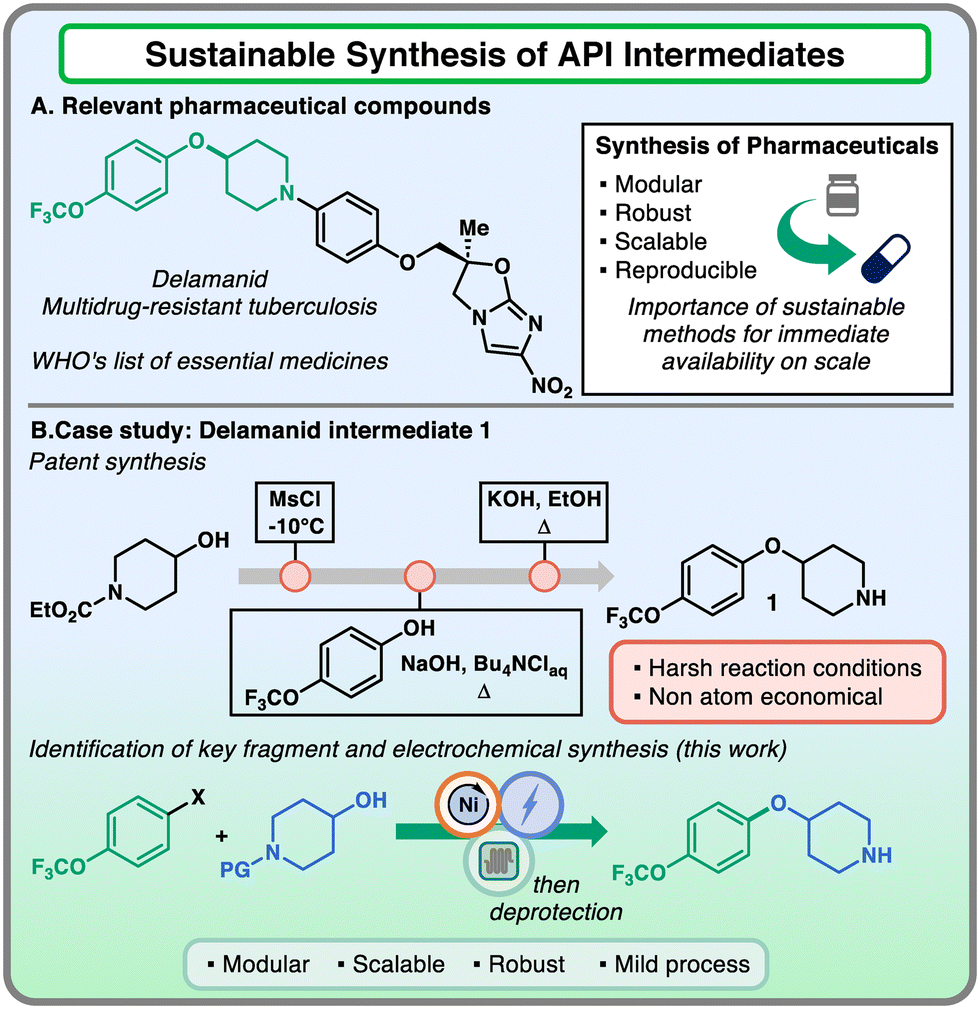 | ||
| Fig. 1 Sustainable synthesis of API intermediates. (A) Delamanid, a relevant pharmaceutical compound for multidrug-resistant tuberculosis treatment. (B) Process route towards the synthesis of intermediate 1 and the new sustainable approach. | ||
With the advent of new technologies in organic chemistry, innovative synthetic methodologies have been developed across the years, providing sustainable alternatives to the traditional transformations.5–7 In particular, the employment of sustainable electrochemical reactions have impacted the way of conceiving redox processes, dramatically improving the atom-economy and the safety of the reactions by avoiding the involvement of dangerous oxidants or reductants.8,9 Furthermore, electrochemical and photochemical transformations express their scalable potential in combination with flow technology, indicating the feasibility of these methods across a broad scale regime.10
Intrigued by the possibility of providing versatile tools to the pharmaceutical industry that combine modularity and scalability features, we identified relevant APIs that would benefit from this exploration. Among these compounds, delamanid was found to be of interest both for its application and molecular characteristics.11 This API has been prescribed for multidrug-resistant tuberculosis treatment and appeared in the World Health Organization's List of Essential Medicines in 2014.11,12 From a synthetic point of view, the aryl ether piperidine core attracted our attention as a critical fragment of the molecule for its functional group handles, which allowed the linkage of different portions of the scaffold. The patent synthetic protocol reported that the protected 4-hydroxy piperidine was subjected to mesylation under cryogenic conditions, followed by nucleophilic substitution on 4-trifluoromethoxy phenol under basic conditions at high temperatures and final deprotection under reflux conditions.13 This three-step sequence is considered a non-atom economical and harsh way to access the key intermediate 1. In this work, the robust, scalable, and sustainable synthesis of this system employing Ni-electrocatalyzed etherification is presented, providing a valuable example of conciliation between high versatility and robustness across different scale regimes thanks to an electrochemical flow reactor.
We commenced our investigation by establishing the appropriate coupling conditions for our designated substrates, 4-trifluoromethoxy bromobenzene (2a) and 4-hydroxy N-Boc piperidine (3a, Fig. 2). The conditions reported by Baran and collaborators for nickel electrocatalyzed C–O bond formation were identified as a valid starting point for the synthesis of compound 4a.14 Indeed, as the replication of the electrochemical protocol led to the formation of the product in good yield (entry 1, 48%), we sought to further explore this reaction in order to facilitate the experimental protocol, bearing in mind the importance of the late-stage scale-up phase. The first important experimental detail that emerged from the reaction survey was the importance of pre-stirring the reaction mixture. As reported in the original protocol,14 the agitation of the reaction mixture for 10 minutes prior to electrolysis improved the formation of the product. However, we found an extended time or 30 minute pre-stirring more beneficial and reproducible with these coupling partners (entries 2 and 3). Upon deeper study of this observation, it was noticed that this induction phase was extremely beneficial in the release of one ligand unit from the precatalyst, indicating that this activation is essential for the success of the reaction (see the ESI† for further details).
 | ||
| Fig. 2 Batch optimization reaction, 0.2 mmol scale. aDetermined using calibrated HPLC analysis against biphenyl as the standard. Isolated yields in brackets. Py = pyridine, bpy = 2,2′-bipyridine, phen = phenanthroline. | ||
Next, we evaluated the electrode material (entries 4–6). In this case, the use of reticulate vitreous carbon (RVC) both as a cathode and an anode in the electrochemical cell provided better results compared to all the other combinations tested (entry 6, 62%). Other modifications such as solvent, base, and ligand screenings showed lower performances (entries 7–9). To further assess the characteristics of the reaction, moisture sensitivity tests were carried out (entries 10 and 11). The addition of water resulted in a much lower yield, as suggested by the need of molecular sieves in the original protocol,14 while the reaction could be carried out in open air with only a slight decrease of performance. Finally, the necessity of electricity and an appropriate catalytic system was confirmed via control experiments. Surprisingly, when the same substrates were subjected to the state-of-the-art photochemical conditions for the synthesis of C–O bonds employing a metallaphotoredox approach, employing 4CzIPN as a photocatalyst with NiBr2·dme under 465 nm light irradiation, only traces of products could be observed, showing the complementarity between photochemical and electrochemical systems (see the ESI† for further details).15 The optimized electrochemical reaction conditions involved, along with the aryl halide and 3 equivalents of the alcohol, are 10 mol% of Ni(dttbbpy)3Cl2, 2 equivalents of DBU, 0.2 M of tetrabutylammonium bromide as supporting electrolyte and N,N-dimethyl acetamide (DMA) as solvent. This stock solution, after 30 minutes of pre-stirring, was subjected to 10 mA of constant current until 6.0 F mol−1 of charge had been passed through the solution in a cell equipped with RVC electrodes as both cathodes and anodes. At the end of the reaction, full conversion of the aryl halide was observed, with side products such as phenol, diaryl ether, and biaryl detected via GC-MS analysis.
During the optimization process, severe reproducibility issues were observed, which affected the development of our experimental protocol. Even when brand-new RVC electrodes were used, dramatically inconsistent results were recorded, ranging from ∼60% yield to traces of 4a (Fig. 3). We decided to investigate this uncommon behavior, and after a few experiments it was evident that this outcome could be attributed to the electrodes (Fig. 3, see the ESI† for further details). Analysis of the electrodes via Scanning Electron Microscopy (SEM) revealed that some new electrodes had particles deposited on the carbon surface, and we hypothesized that these particles could be attributed to polymeric residues. Under this assumption, a thorough pretreatment to employ clean electrodes was developed, which involved washing the electrodes in acetone or pentane under sonication and subsequent drying at 100 °C for 12 hours (Fig. 3A). By applying this method, the irreproducibility problem was overcome, and consistent data could be obtained across different batches of RVC electrodes. This set of experiments moved our curiosity on the evaluation of the electrodes at the end of the electrolysis.
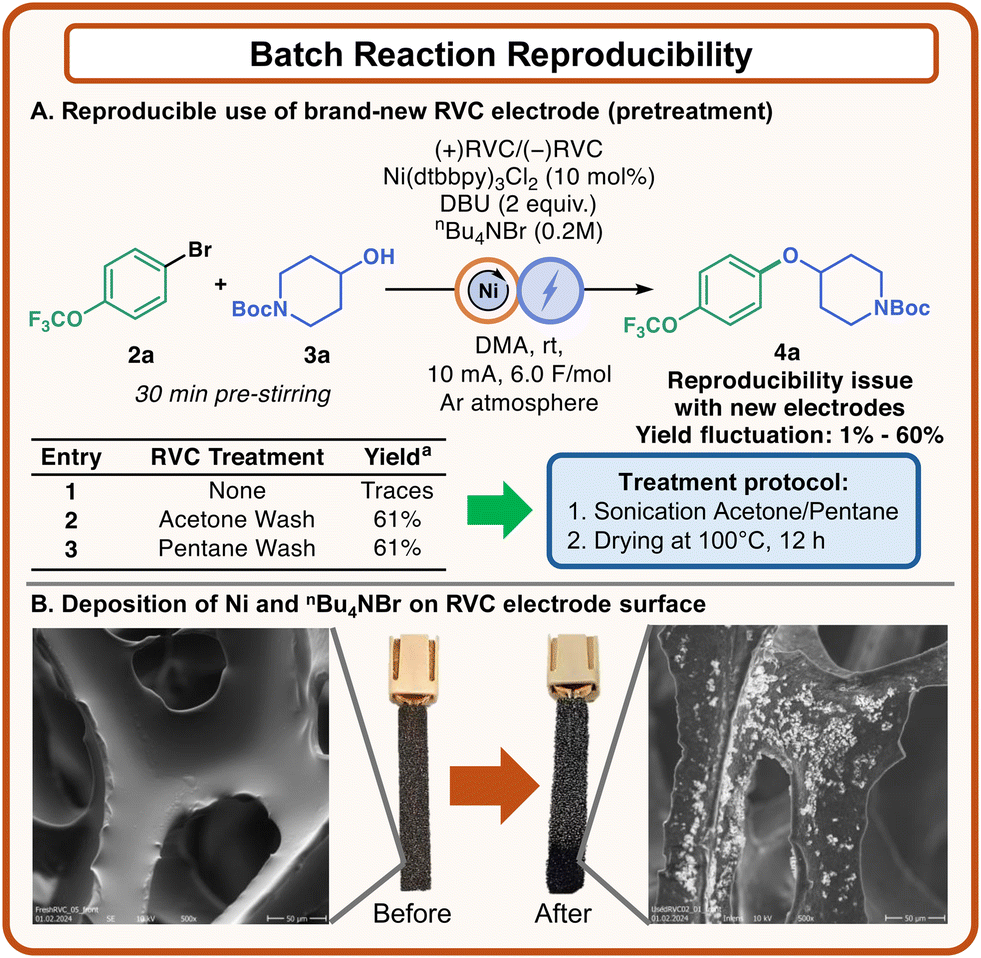 | ||
| Fig. 3 Reproducibility of batch reactions. (A) Development of an electrode pretreatment to ensure reproducible data. (B) Electrode material before and after the reaction. | ||
Interestingly, SEM analysis demonstrated the presence of solid deposits on the surface of the electrodes (Fig. 3B). The analysis of a used electrode with Energy Dispersive X-Ray (EDX) showed that these solid particles contained Ni and Br, which were present in solution as the catalyst and the supporting electrolyte, respectively (see the ESI† for further details).
With all the valuable information in hand, we proceeded to evaluate the translation of the reaction to a flow setting. Instead of RVC, graphite felt electrodes were installed in the flow electrolysis cell, as they are easy-to-handle and cost-effective materials, more suitable for the utilization of large quantities. However, felt materials are very porous and absorb a consistent amount of the reaction mixture, compromising the solution stream at low flow rates and affecting reaction reproducibility. For these reasons, we employed a reactor design that could allow the use of felt electrodes both in a single-pass and recirculation manner to ensure prompt scalability of the nickel electrocatalyzed reaction (Fig. 4).16–18
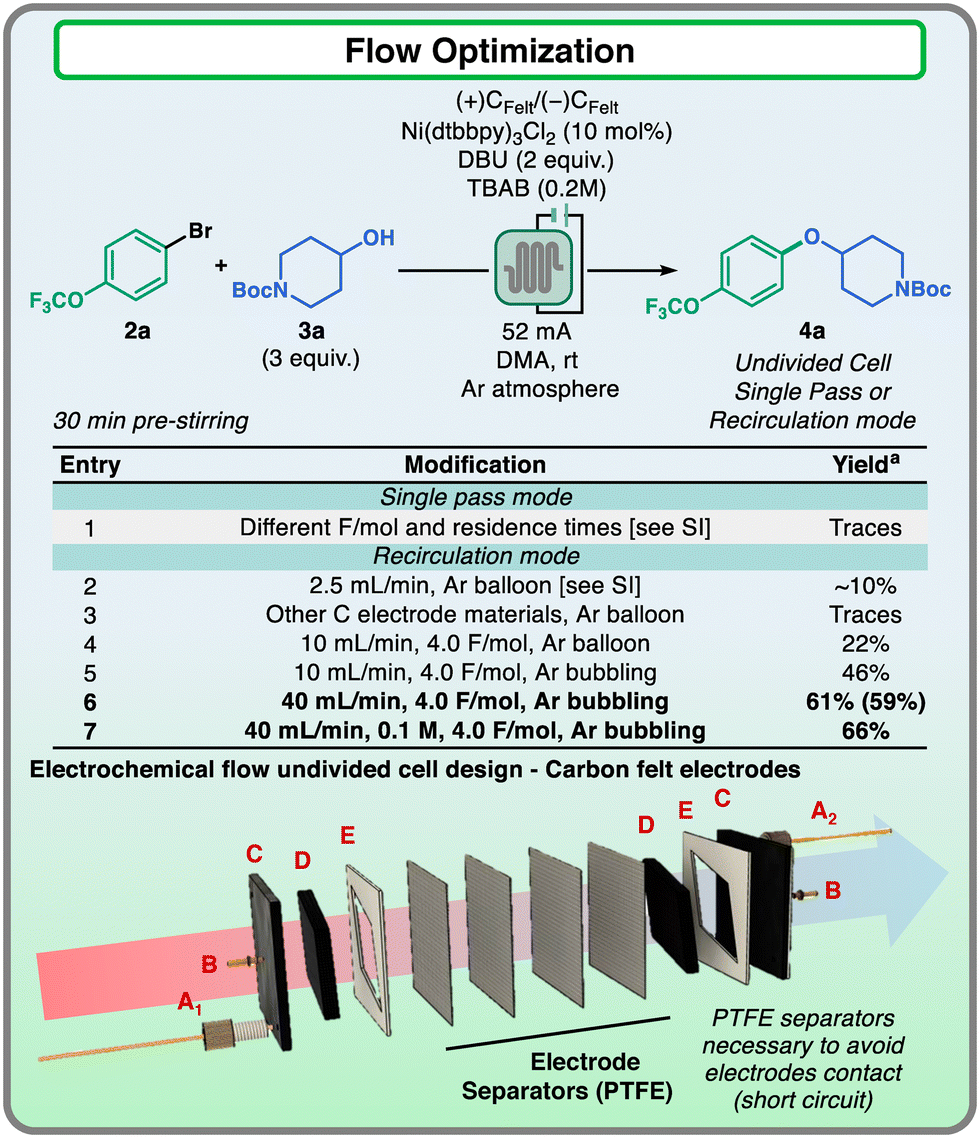 | ||
| Fig. 4 Flow optimization reaction and exploded view of the flow-reactor design. Reaction scale: 1.33 mmol scale, 2.0 mmol scale with 0.1 M. aDetermined using calibrated HPLC analysis against biphenyl as the standard. Isolated yields in brackets. Reactor design: (A1) inlet, (A2) outlet, (B) electrical connections, (C) graphite plate used as current collectors, (D) carbon felt electrodes, (E) PTFE gaskets, and electrode separators. | ||
The flow reactor consisted in a parallel “plate and frame” type design, in which two rhombus-shape graphite felt electrodes (3 mm thickness, Fig. 4D) were separated by a stack of 4 PTFE mesh sheets (thickness: 150–180 μm each) to avoid short circuit, without affecting the flow of the mixture across the apparatus. The electrical connection to the felts was accomplished using graphite plates as current collectors (Fig. 4C), each of them connected to the power supply by a pogo pin (Fig. 4B). With this reactor design, the optimization of the flow transformation was carried out (Fig. 4). Preliminary attempts of running reactions in single pass mode did not lead to the formation of considerable amounts of the desired product. These results were attributed to the low flow rates that did not allow sufficient mass transfer,19 especially in a setup where the reaction mixture was flowing through a porous carbon felt material (entry 1, see the ESI† for further details). These results indicated that higher flow rates would be necessary to guarantee an efficient electrochemical reaction. To validate this hypothesis, the reactor was used in recirculation mode employing a peristaltic pump, and promising formation of the desired product 4a was observed, in contrast to the results obtained with the single pass approach (entry 2). Surveying different electrode materials did not show any relevant formation of the desired product (entry 3), while increasing the flow rate to 10 mL min−1 led to further improvement of the process (22%, entry 4). Nevertheless, these results were still far from the yield obtained for the optimized reaction conditions in batch cells. We reasoned that the fast recirculation of the reaction mixture could compromise the inert condition of the solution, therefore it was decided to keep the reaction mixture inert by bubbling argon through the reaction mixture during the electrolysis time. This adjustment was sufficient to enhance the product formation (46%, entry 5). Finally, a further increase in the flow rate to 40 mL min−1 led to the formation of 4a in similar yields to those observed in batch, even by using a higher reaction mixture concentration (entries 6 and 7). During the optimization, the reactor showed consistent results for 2 consecutive runs without the need of opening the reactor, and after that the electrodes could be regenerated using a treatment protocol similar to that developed for the batch cell electrodes (see the ESI† for further details).
This new flow protocol opened new opportunities for the scale-up of this electrocatalytic method, where the experimental procedure for the preparation of the reaction mixture does not differ from the optimized batch one except for the inert atmosphere ensured by inert gas bubbling instead of positive pressure on the vessel headspace.
After optimization, the developed flow protocol was then tested on different substrates to validate its robustness (Fig. 5). Regarding the alcohol coupling partner, the reaction displayed good yields with primary and secondary alcohols (4a–4f). In particular, the reaction could be efficiently employed in the presence of functional groups such as ethers (4b), extended alkyl chains (4c), protected amines (4a and 4d), alkenes (4e), and esters (4f). Other aryl bromides have also been tested, showing moderate compatibility of the method towards different arenes and heteroarenes (4g–4k). Electron poor (4g) and electron rich (4h) aryl bromides could be employed as competent partners in the coupling. Furthermore, heteroarenes like thiophenes (4i) and substituted pyridines (4j and 4k) provided the desired products with acceptable yields (up to 43%).
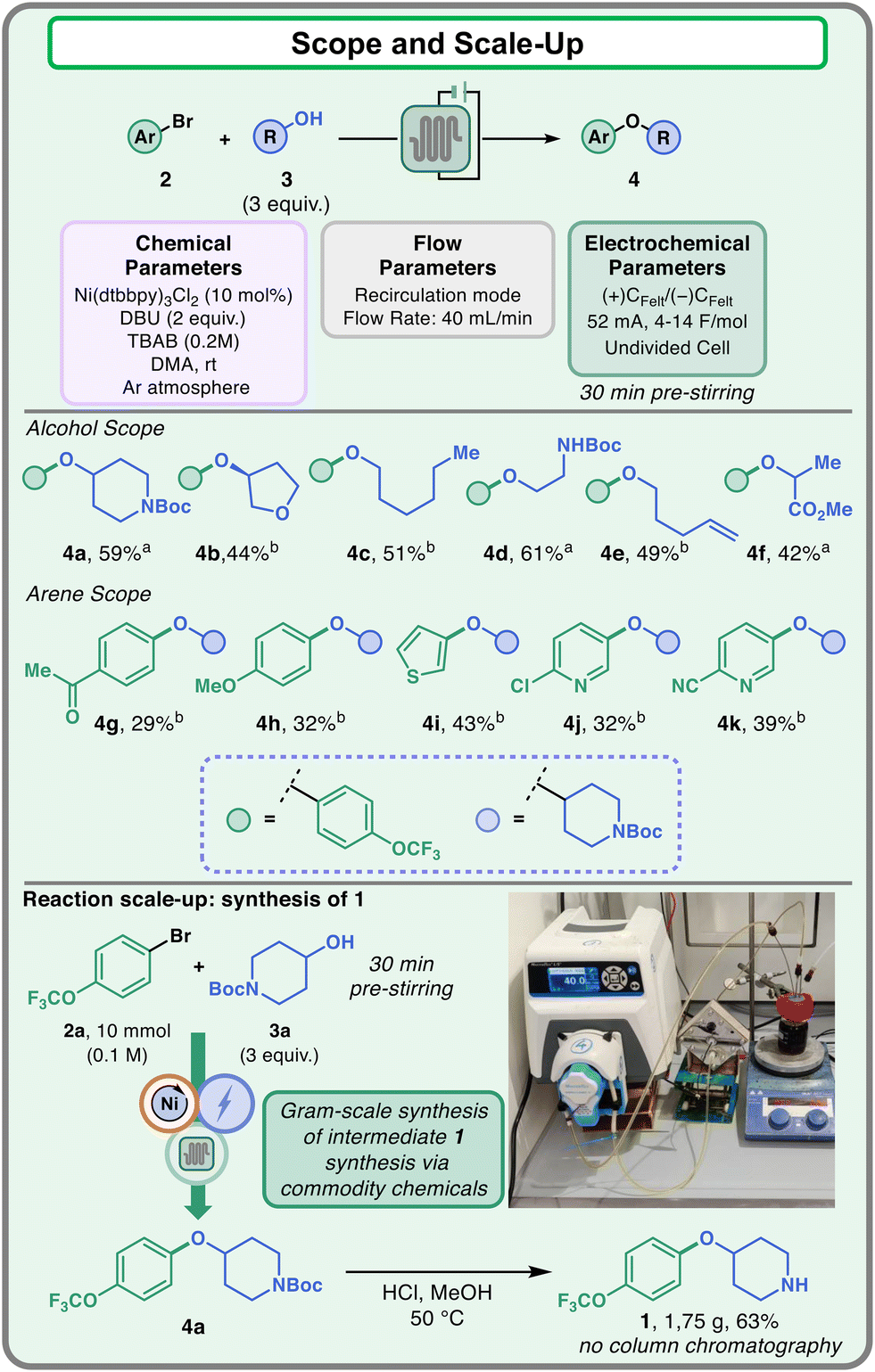 | ||
| Fig. 5 Scope of the electrochemical flow protocol. Scale-up of delamanid intermediate 1. a2.0 mmol scale (0.1 M of 2). b1.33 mmol scale (0.066 M of 2). | ||
Finally, the flow protocol was adopted for the scale-up synthesis of delamanid intermediate 1. For this reaction, the two commodity chemicals 2a and 3a have been employed as coupling partners, and the reaction has been conducted at the 10 mmol scale. The Boc-protected intermediate 4a was extracted and then subjected to mild deprotection to obtain compound 1, which could be isolated without column chromatography by simple work-up in 63% yield.
In conclusion, we evaluated the versatile, robust, and scalable formation of C–O bonds via nickel electrocatalysis. The reaction was initially investigated in batch reactors, addressing critical reproducibility issues of untreated RVC electrodes. The transformation was successfully transferred and optimized using a flow electrochemical reactor. The flow protocol was validated both in terms of robustness and scalability. Overall, we anticipate that this work provides valuable insights on the flexibility of electrochemical organic synthesis and its potential applications in medicinal and process chemistry settings.
Data availability
The data supporting this article have been shown in the figures, and they are included as part of the article and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based on research funded in part by the Bill & Melinda Gates Foundation [INV-056601]. The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation. Under the grant conditions of the foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission. The Research Center Pharmaceutical Engineering (RCPE) is funded within the framework of COMET – Competence Centers for Excellent Technologies by BMK, BMAW, Land Steiermark and SFG. The COMET program is managed by the FFG. We thank Dr. Gerald Auer (University of Graz, Institute of Earth Sciences) for recording SEM and EDX analysis. We would like to express our gratitude to Dr. Trevor Laird, Dr. John L. Dillon, and Dr. Ryan C. Nelson for the insightful discussions. We also thank Dr. Bhanwar K. Malviya for the help with the reactor design.References
- T. Y. Zhang, Chem. Rev., 2006, 106, 2583–2595 CrossRef CAS PubMed.
- M. Butters, D. Catterick, A. Craig, A. Curzons, D. Dale, A. Gillmore, S. P. Green, I. Marziano, J.-P. Sherlock and W. White, Chem. Rev., 2006, 106, 3002–3027 CrossRef CAS.
- M. Colombo and I. Peretto, Drug Discovery Today, 2008, 13, 677–684 CrossRef CAS PubMed.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef PubMed.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- M. Berton, J. M. de Souza, I. Abdiaj, D. T. McQuade and D. R. Snead, J. Flow Chem., 2020, 10, 73–92 CrossRef.
- Y. Liu, M. Matsumoto, H. Ishida, K. Ohguro, M. Yoshitake, R. Gupta, L. Geiter and J. Hafkin, Tuberculosis, 2018, 111, 20–30 CrossRef CAS PubMed.
- World Health Organization's List of Essential Medicines, https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2023.02, (accessed 09 Aug 2024).
- M. Miyake, A. Asahina and T. Okada, US10252995B2, 2019.
- H.-J. Zhang, L. Chen, M. S. Oderinde, J. T. Edwards, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2021, 60, 20700–20705 CrossRef CAS PubMed.
- I. Ghosh, N. Shlapakov, T. A. Karl, J. Düker, M. Nikitin, J. V. Burykina, V. P. Ananikov and B. König, Nature, 2023, 619, 87–93 CAS.
- J. D. García-Espinoza, I. Robles, A. Durán-Moreno and L. A. Godínez, Environ. Sci. Pollut. Res., 2022, 29, 42305–42318 CrossRef PubMed.
- L. F. Castañeda, F. C. Walsh, J. L. Nava and C. Ponce de León, Electrochim. Acta, 2017, 258, 1115–1139 CrossRef.
- T. X. Huong Le, M. Bechelany and M. Cretin, Carbon, 2017, 122, 564–591 CrossRef.
- S. Maljuric, W. Jud, C. O. Kappe and D. Cantillo, J. Flow Chem., 2020, 10, 181–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00438h |
| This journal is © The Royal Society of Chemistry 2025 |