
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystallization-integrated mandelate racemase-catalyzed dynamic kinetic resolution of racemic mandelic acid†
Feodor
Belov
a,
Alexandra
Lieb
b and
Jan
von Langermann
 *a
*a
aInstitute of Chemistry, Biocatalytic Synthesis Group, Otto-von-Guericke University Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: jan.langermann@ovgu.de
bInstitute of Chemistry, Industrial Chemistry, Otto-von-Guericke University Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany
First published on 18th February 2025
Abstract
Classical approaches for the preparation of enantiopure mandelic acid conventionally employ chiral resolution methods like diastereomeric crystallization or kinetic resolution. Those are, however, limited by their theoretical yield of 50% of the utilized racemate. Dynamic kinetic resolution solves this challenge by the addition of a racemization step for the unprocessed enantiomer, maximizing yields. For mandelic acid, a special enzyme class of mandelate racemases can perform this racemization step. In this study, we combine enzymatic racemization of mandelic acid with diastereomeric salt crystallization of (R)-mandelic acid to achieve a chemoenzymatic dynamic kinetic resolution of mandelic acid at mild conditions in water.
Introduction
The synthesis of chiral chemical substances is one of the most important areas of research and the basis for their application in e.g. pharmaceutical and agrochemical products.1–9 Normally, the preparation of enantiomerically pure compounds is divided into two general routes. Firstly, asymmetric synthesis, which typically converts an achiral compound into one of the desired enantiomers using an enantioselective catalyst. Due to the high demands to the enantiomeric purity of many chemical compounds, the use of a highly enantioselective catalyst is necessary. Secondly, the separation of usually inexpensive racemic mixtures, whereby separation methods such as chromatography and crystallization can be used in addition to catalytic methods. This concept is usually referred to as chiral resolution (CR), while the utilization of catalysts in the separation process is called kinetic resolution (KR). In KR approaches, different reaction kinetics of the two enantiomers with a selective (bio)catalyst enable the separation of the racemic compound, ideally with a strong difference in reaction rates (conversion for one enantiomer much faster than for the other enantiomer).1–3,7–10Enzymes usually possess a high substrate specificity and enantioselectivity, making them an already tailored catalyst for the desired reaction and broadly applied in KRs. Additionally, enzymes can perform reactions at very mild conditions, thus allowing for aqueous reaction media and environmentally friendly processes as a bonus.10–15 Nevertheless, the main limitation of “standard-issue” chemical and kinetic resolutions is its highest achievable theoretical yield: only 50% of the utilized racemate are theoretically able to be converted to the desired enantiomeric product, while leaving 50% of the “undesired” enantiomer unprocessed. Here, a dynamic kinetic resolution (DKR) approach can improve yield beyond 50% in favour of the desired enantiomer in which the kinetic resolution step is combined with the continuous racemization of the remaining, typically undesired enantiomer, allowing for theoretical yields of up to 100%.1,3,7,9,16,17 However, the additional racemization step needs to be accommodated by the process requirements within the DKR to function in parallel to the enantioselective synthesis reaction. To be more specific, ideally the racemization needs to be faster than the conversion of the undesired enantiomer into the product, effectively preventing the accumulation of the undesired enantiomer.1,3,9 Racemization itself can be achieved spontaneously, e.g. by labile stereocenters incl. keto-enol tautomerism, but typically needs to be induced by specific chemicals or (bio)catalytic reaction systems.1,3,18–20 Even though less utilized, enzymatic (biocatalytic) racemization through racemases (E.C. 5.1.X.X) is of particular interest. Although, opposed to most other enzymes, their stereoselectivity is essentially non-existent, their sole purpose is to be able to bind both enantiomers of its substrate in order to convert it into the other.21 Thus, racemases also found their place in DKR-based processes as a the racemizing agent.21,22
An example, where resolution methods are often utilized, is the obtainment of enantiopure mandelic acid. Classical chemical methods usually produce a racemate of mandelic acid, which then has to be separated.23,24 For this purpose some KR- and DKR-based chemoenzymatic approaches are found in literature.24–36
Other alternatives for enantiopure mandelic acid production include aforementioned asymmetric synthetic approaches (both enzymatic and chemical)37–39 and diastereomeric salt crystallization as means of chiral resolution. The latter approach utilizes different chemical properties of the corresponding diastereomeric salts, thus (co-)crystallizing the different mandelate enantiomers with a chiral resolving agent.40–50
This study focuses on the enzymatic racemization of mandelic acid and its integration into a diastereoselective crystallization step. This is achieved by mandelate racemases that are mostly utilized as a racemizing agent in DKR-based approaches towards enantiopure mandelic acid (derivatives) and require a secondary (bio)catalytic reaction system to derivatize mandelic acid in a biocatalytic cascade51–53 or a separation method, as shown for a chromatography approach in the works of Wrzosek et al.54–56 The presented combined biocatalysis-crystallization DKR is designed to efficiently convert racemic mandelic acid to (R)-mandelic acid without the need to form the above mentioned mandelic acid derivative that eventually needs to be converted back to mandelic acid with extra process steps. It enables high yields and eases the downstream processing by synchronous in situ product crystallization (ISPC) of the desired mandelate enantiomer as its diastereomeric salt (see Fig. 1). The concept includes a simple fed-batch approach with high substrate concentrations and the removal of enantioenriched (R)-mandelic acid salt after the reaction process.
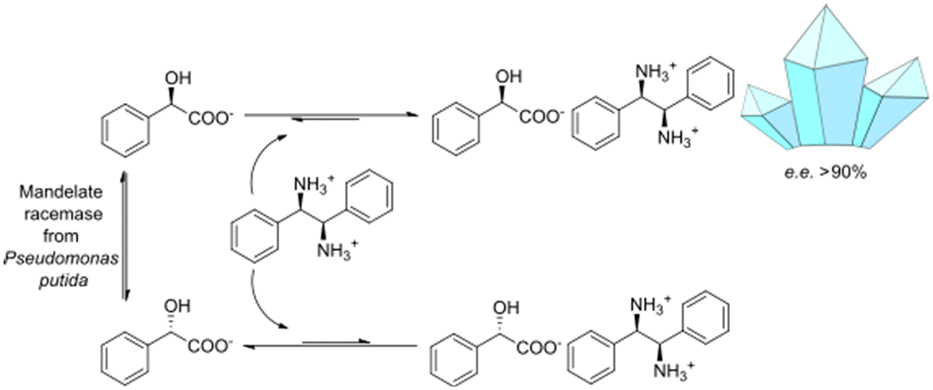 | ||
| Fig. 1 Concept scheme for the proposed crystallization-based DKR of mandelic acid. | ||
Results and discussion
Due to the need for high water activity for the selected mandelate racemase from Pseudomonas putida (E.C. 5.1.2.2), the racemization needs to be performed in aqueous media,53 since its utilization in organic solvents has shown a complete loss of catalytic activity, although the enzyme itself retains activity when extracted back into aqueous media.57 Unfortunately, most of the available literature about diastereomeric crystallization of mandelic acid (MA) makes use of organic solvents, while mandelic acid itself already possesses a very high solubility in water (approx. 1 M). Thus, the search for an enantiospecific crystallization partner for diastereomeric salt resolution would need to have a “bulky”, preferentially hydrophobic structural base (thus lowering the solubility of the diastereomeric product salt) and ideally be commercially available for a broader applicability. Based on those ground rules, (1R,2S)-2-amino-1,2-diphenylethanol ((1R,2S)-ADPE) from prior works by Hirose et al.45–47 was chosen as a bulky chiral amine to crystallize with mandelic acid. To further possible options for crystallization partners, several structurally similar and commercially available bulky amines were selected: (S)-1-phenylethylamine ((S)-1PEA), (1R,2R)-1,2-diphenylethanediamine ((1R,2R)-DPEN) and (R)-1,2,2-triphenylethylamine ((R)-122TPEA).Solubility screening
The main criterium towards the choice of the amine-based resolving agent was decided to be the solubility since it directly relates to the maximal yield. The solubilities of the above selected amines as diastereomeric salts with (R)- and (S)-mandelic acid were investigated in the aqueous reaction medium and then compared according to the overall solubility difference between the corresponding diastereomeric salts and to the other salt pairs (Table 1).| Amine counterion | Structure of amine counterion | Solubility of (R)-MA salt, mM | Solubility of (S)-MA salt, mM |
|---|---|---|---|
| The solubility screening was performed at 30 °C in pure ddH2O at pH 7, the pH was adjusted with weak NaOH and HCl solutions, when needed. (1R,2R)-DPEN salts were measured as monoamine salts. | |||
| (1R,2S)-ADPE |
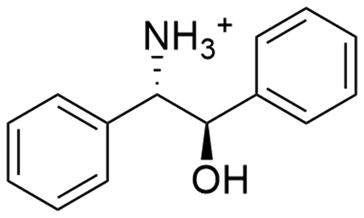
|
67.1 ± 6.8 | 56.3 ± 2.7 |
| (1R,2R)-DPEN |
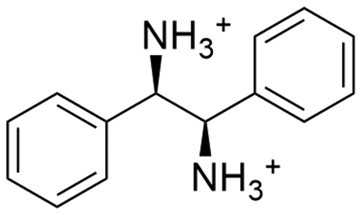
|
33.6 ± 4.8 | 88.5 ± 3.1 |
| (S)-1PEA |
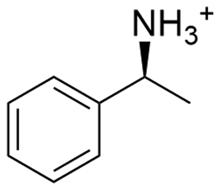
|
6138.9 ± 370.9 | 204.1 ± 12.1 |
| (R)-122TPEA |
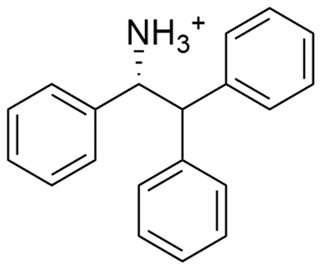
|
4.9 ± 1.1 | 9.3 ± 0.9 |
Two potential crystallization partners were eliminated right away. (R)-122TPEA had very low overall solubilities of both diastereomeric mandelate salts, while (1R,2S)-ADPE did not have a sufficient solubility gap between its diastereomeric mandelate salts. The remaining two of the selected diastereomeric salt pairs exhibited a high discrepancy between the different enantiomer salts of mandelic acid with (1R,2R)-DPEN at low concentrations and (S)-1PEA at relatively high concentrations. Interestingly, those two salt pairs showed a preference towards crystallizing different enantiomers of mandelic acid. The most promising option appears to be (1R,2R)-DPEN with diastereomeric salt solubilities of 33.6 and 88.5 mM, respectively, which facilitates a sufficiently low concentration towards the targeted (R)-enantiomer of mandelic acid. With (S)-1PEA, the less soluble salt of (S)-mandelic acid possessed a relatively high solubility of ca. 200 mM, however due to its counterpart being very soluble at >6 M and the selected mandelate racemase being able to work at high substrate concentrations, it still presented a viable option.
Optimizing diastereomeric salt crystallization conditions
Having identified two potential resolving amine counterions, it was decided to attempt to form a first impression for the efficacy of the resolution of mandelic acid with those counterions. Therefore, simple crystallization experiments on a small scale of 1 ml were executed with varying racemic mandelate and amine counterion concentrations. It has to be noted, that both mandelate and the amine counterions were utilized as sodium (mandelate) or hydrochloride/di-hydrochloride salts ((S)-1PEA and (1R,2R)-DPEN respectively). Since the ability to selectively crystallize only one enantiomer of mandelic acid was deemed the most important parameter of the experiments, the yield was not measured, focusing solely on the enantiopurity of the crystallization phase. The resulting enantiomeric excesses of the formed product salts are shown in Table 2.| Racemic mandelate, mM | Amine counterion | Counterion, mM | ee of product salt |
|---|---|---|---|
| Enantiomeric excess was determined by HPLC analysis of dried precipitated salts, which were harvested after 3 h at RT and 750 rpm. Where no product salt precipitate was obtained, the experiments are marked with “n.d.”. | |||
| 1000 | (S)-1PEA | 500 | 66.7 ± 7.0% (S) |
| 500 | (S)-1PEA | 500 | 67.6 ± 1.6% (S) |
| 250 | (S)-1PEA | 500 | n.d. |
| 100 | (S)-1PEA | 500 | n.d. |
| 1000 | (S)-1PEA | 400 | 63.9 ± 1.8% (S) |
| 1000 | (S)-1PEA | 300 | 74.9 ± 9.4% (S) |
| 1000 | (S)-1PEA | 250 | 77.5 ± 11.1% (S) |
| 1000 | (S)-1PEA | 200 | 77.2 ± 0.7% (S) |
| 1000 | (S)-1PEA | 100 | n.d. |
| 200 | (1R,2R)-DPEN | 150 | 79.9 ± 2.6% (R) |
| 200 | (1R,2R)-DPEN | 100 | 84.3 ± 2.8% (R) |
| 200 | (1R,2R)-DPEN | 75 | 86.8 ± 4.3% (R) |
| 200 | (1R,2R)-DPEN | 50 | n.d. |
Although the (S)-1PEA mandelate salts showed an exorbitant solubility difference between its respective (S)- and (R)-MA salts, it failed to reach enantiomeric excesses over 80% for its precipitated product, nearing this limit only for relatively low counterion concentrations, which would mean low yields of the enantiomerically enriched product phase. On the contrary, (1R,2R)-DPEN demonstrated fairly good enantiomeric excesses of up to 87% although its diastereomeric salts solubility limits were not that much apart. An increase in counterion concentrations led to a slight decrease of enantiomeric excess of the crystallized mandelate salts from 87% at 75 mM of (1R,2R)-DPEN to 80% at 150 mM of (1R,2R)-DPEN. But since those were only preliminary experiments with a short time for crystallization of 3 hours, those values were acceptable and open to adjustment via longer crystallization times. Hence, it was chosen to continue with (1R,2R)-DPEN as the crystallization partner for mandelic acid resolution.
Small-scale DKR experiments and enantiomeric excess optimization
As the highest enantiomeric excess was obtained using 200 mM of racemic mandelic acid and 75 mM of (1R,2R)-DPEN as the chiral resolving agent, those concentrations were chosen for the initial DKR-based experiments. A corresponding control experiment was also performed and compared in triplicates of DKR- and chiral resolution based (controls without enzyme) reactions. The resulting yields and enantiomeric excesses are shown in Fig. 2 (left). This data shows, that both the CR and DKR-based approaches show similar results. The DKR-based yield might have been slightly better with 32.4% versus 28.5% for CR-based control, while the CR-based ee of 90.3% slightly surpassed the DKR-based ee of 89.9%. | ||
| Fig. 2 Left: Yield and enantiomeric excess of DKR-based and CR-based fed-batch experiments after 96 h; 200 mM racemic mandelate, 75 mM (1R,2R)-DPEN, 10 U ml−1 mandelate racemase extract (for DKR only), 50 mM HEPES-buffer with 3.3 mM MgCl2, pH 7.5, RT (22–23 °C), 96 h, 5 ml. Right: Yield and enantiomeric excess of DKR-based and CR-based fed-batch experiments after 96 h with doubled counterion concentration; 200 mM racemic mandelate, 150 mM (1R,2R)-DPEN, 10 U ml−1 mandelate racemase extract (for DKR only), 50 mM HEPES-buffer with 3.3 mM MgCl2, pH 7.5, RT (22–23 °C), 96 h, 5 ml. Experiments were performed in triplicates. Substrates and crystallization counteragents were refilled after 24 and 48 h. | ||
Based on the presented observations, it was decided to retry the DKR- to CR-comparison with a higher concentration of 150 mM (1R,2R)-DPEN according to the procedure utilized with 75 mM (1R,2R)-DPEN on a fed-batch basis, the results shown in Fig. 2 (right). As can be seen, while the enantiomeric excess stays on the same fairly high level of 90%, the yield is improved significantly to reach slightly over 60% with the DKR-based approaches. For the CR-based controls, the yield reaches nearly 40%, while the ee starts to experience a slight decrease towards 87%. XRPD analysis of the harvested product material showed the presence of the monoamine salt as the dominant crystallized solid phase. XRPD results, including mono- and diamine reference salts of both mandelate enantiomers, are provided in the ESI† file (see Fig. S2 and S3). The yields were calculated based on HPLC (showing the mandelic acid remaining in solution) and XRPD investigations.
Compared to the initial crystallization experiments, the ees of the product salts were higher with both 75 mM and especially 150 mM (1R,2R)-DPEN concentrations, with the only difference between the experiments being the time spent stirring (2–3 h vs. 24 h between refills). Thus, it was decided to monitor the change in enantiomeric excess of the formed product salt over time.
For the purpose of simpler monitoring, 72 h long DKR-batches (without refilling) bearing 150 mM of (1R,2R)-DPEN were done as a triplicate and monitored only for ee over the course of 24 h. The progression of the ee is shown in Fig. 3. The shown progression explains the discrepancy between the initial crystallization screening and the outcome of DKR- and CR-based batches in terms of enantiomeric excess. A simple waiting period of 24 h elevates ee about 10% from 80 to 90% ee. However, additional waiting does not significantly contribute to the enantiopurity of the obtained product salt any further.
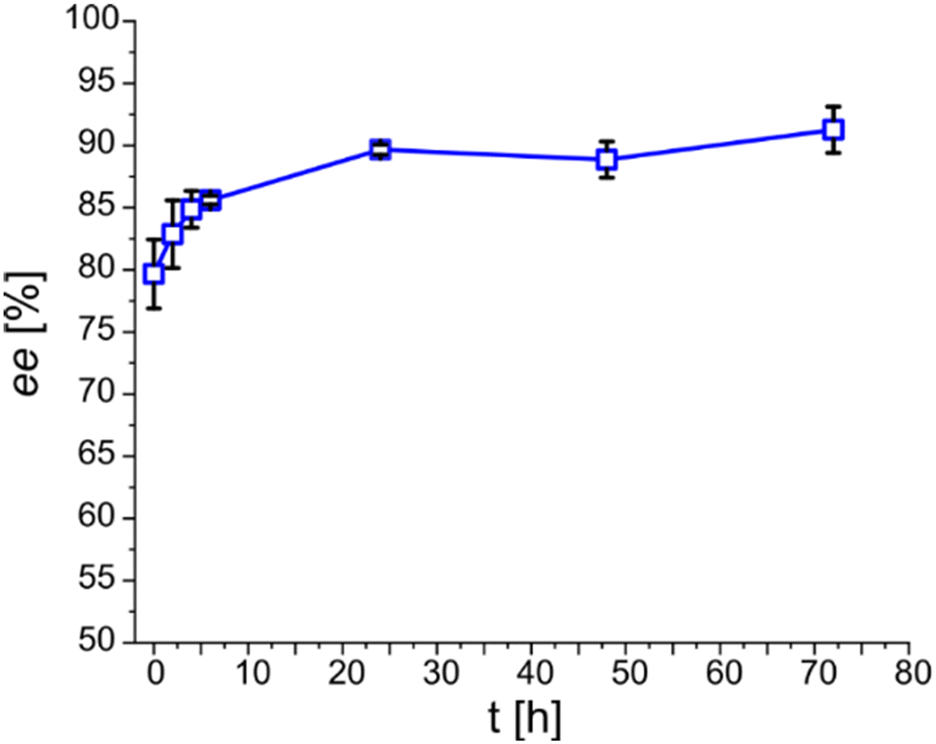 | ||
| Fig. 3 Enantiomeric excess of the product salt over time in DKR-based 72 h batch with 150 mM (1R,2R)-DPEN. 200 mM racemic mandelate, 150 mM (1R,2R)-DPEN, 10 U ml−1 mandelate racemase extract, 50 mM HEPES-buffer with 3.3 mM MgCl2, pH 7.5, RT (22–23 °C), 72 h, 5 ml. | ||
Thus, a DKR-based system for resolution of mandelic acid can be established through the proposed approach on a continuous basis, presenting high yields and a fairly high enantiomeric excess of one product enantiomer. Finally, a proof-of-concept needed to be obtained to evaluate the scalability of the process. Therefore, the system was tested on a preparative scale.
Preparative-scale DKR experiment
After the successful initial experiments, the combined racemization-crystallization reaction concept was validated at preparative scale to showcase its synthetic potential. A reaction in the 50 ml format was prepared, operated in a fed-batch mode for 96 h and the reaction progress monitored periodically via HPLC (see Fig. 4). The product salt was harvested and its purity was analyzed via NMR. The ee of this DKR-based approach was determined at 94.9% ((R)-mandelic acid), with an overall yield of 60.3% (4.61 g of product salt) determined in accordance with isolated product mass and HPLC analysis. Afterwards, mandelic acid was extracted from the product salt (4.547 g after analytic procedures) to yield 1.78 g (56% overall yield based on racemic mandelic acid) of pure mandelic acid, with a purity of 94% determined via NMR, although it has to be noted, that 4.4% of the impurities can be attributed to the spontaneously formed isopropyl ester of the mandelic acid due to residual acid prior to extraction with isopropanol. The counterion was also extracted and yielded 2.24 g with a purity of 98.7%.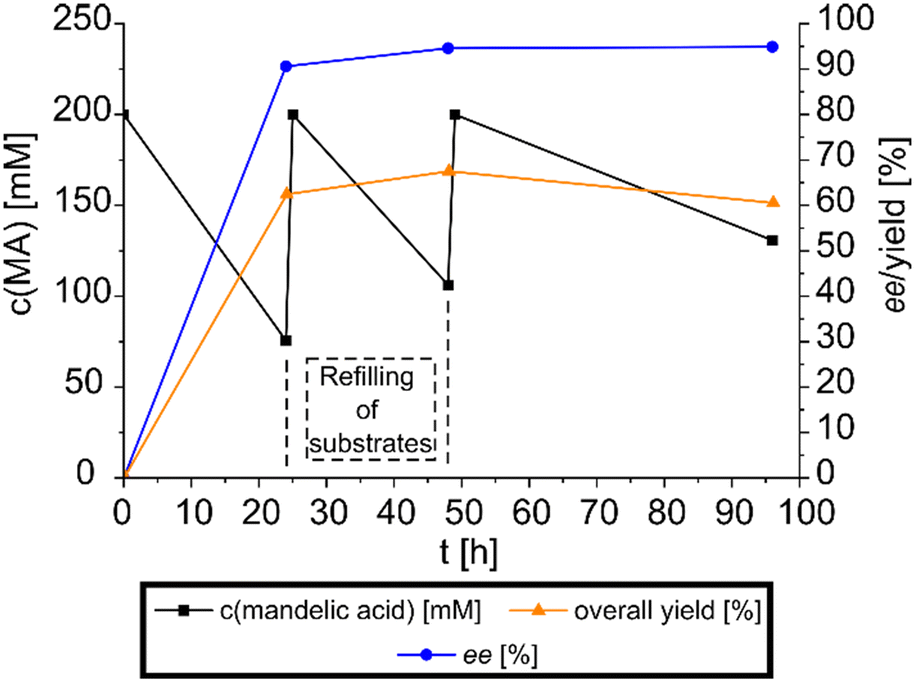 | ||
| Fig. 4 Monitoring of the preparative DKR fed-batch. The black curve shows the concentration of mandelic acid at the measurement points, while the blue curve shows the enantiomeric excess of the product salt. The overall yield progression is shown as the orange curve. 200 mM racemic mandelate, 150 mM (1R,2R)-DPEN, 30 U ml−1 mandelate racemase extract, 50 mM HEPES-buffer with 3.3 mM MgCl2, pH 7.5, RT (22–23 °C), 96 h, 50 ml, fed-batch. | ||
Experimental
Transformation
Chemocompetent E. coli BL21(DE3) cells were thawed on ice in 50 μl aliquots. To the thawed cell suspension on ice, 1 μl of the plasmid solution of pET-52b(+), carrying the gene for the mandelate racemase from Pseudomonas putida (N-terminal StrepII-tag and a C-terminal His10-tag) and an ampicillin resistance gene,58 was added. The cell aliquot was incubated for 30 min on ice. After that, a heat shock was applied by placing the cell aliquot into a prewarmed heating block at 42 °C for 30 s. Immediately after, the cells were returned onto the ice and incubated there for another 5 min. 950 μl of sterile LB medium were added to the cells. The cell suspension was incubated at 37 °C and 750 rpm for 1 h and variable volumes were plated onto selective LB-agar plates supplemented with 0.1 mg ml−1 ampicillin. The plates were incubated at 37 °C overnight.Overnight cultures and Cryostocks
Overnight cultures were prepared in 5 ml of sterile LB medium and supplemented with ampicillin to a final concentration of 0.1 mg ml−1. The cultures were inoculated from single colonies from successful transformations or cryostocks and grown overnight at 37 °C and 180 rpm. Cryostocks were prepared by adding 800 μl of an overnight culture to 200 μl of sterile glycerol (resulting in 20% v/v glycerol stocks) and frozen at −80 °C.Protein expression
Expression cultures were grown in 500 ml of LB medium in 2 l cultivation flasks without induction. 1 ml of 50 mg ml−1 ampicillin stock solution (0.1 mg ml−1 final concentration) was added. Furthermore, 500 μl of a sterilized 1:10 dilution of antifoam B (by Sigma Aldrich) were added to prevent foaming. The flasks were inoculated to a starting OD600 of approximately 0.05 from overnight cultures. The resulting cultures were incubated at 37 °C for 4–5 hours under constant shaking at 200 rpm until reaching an OD600 of approximately 1. At this point, the cultures were harvested via centrifugation at 4000 × g for 10 min at 4 °C. The pellets were resuspended in 50 ml of 50 mM HEPES buffer (pH 7.5) containing 3.3 mM MgCl2 and centrifuged again at 4000 × g for 10 min at 4 °C as a washing step, the liquid was discarded. After resuspension in 5 ml of 50 mM HEPES buffer (pH 7.5) containing 3.3 mM MgCl2, the pellet suspensions were unified and frozen at −20 °C.Lysis
The frozen harvested cells were thawed on ice. To the thawed suspension, 0.5 ml of a 10 mg ml−1 lysozyme stock solution (0.25 mg ml−1 final concentration) and 200 μl of a 1 mg ml−1 DNAse I stock solution (0.1 mg ml−1 final concentration) were added per 20 ml of suspension. Afterwards, the cells were left on ice for 30 min. As the next step, lysis was performed via ultrasonication on a Sonics & Materials Inc. VibraCell VC750 ultrasonic processor equipped with a 3 mm in diameter tapered ultrasonic tip. For the time program, the cell suspension (on ice for cooling) was pulsed for 5 s at an amplitude of 30% and left idle for 10 s. This cycle was repeated, until an overall pulse time of 3 min was reached. After that, the lysed cells were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 4 °C for 45 min. The cleared supernatant was unified and shock-frosted with liquid nitrogen. The resulting cell extract was lyophilized overnight. The dried cell extract was then subjected to an activity assay to determine its specific activity.
000 rpm and 4 °C for 45 min. The cleared supernatant was unified and shock-frosted with liquid nitrogen. The resulting cell extract was lyophilized overnight. The dried cell extract was then subjected to an activity assay to determine its specific activity.
Activity assay for mandelate racemase
For the activity test, the conversion of pure (R)-mandelic acid was analyzed over time. For this purpose, a 20 mg ml−1 stock solution of (R)-mandelic acid was prepared in 50 mM HEPES buffer with 3.3 mM MgCl2 and the pH was adjusted to 7.5. The dried cell extract was also dissolved in the same buffer at pH 7.5 for a stock solution of 2 mg ml−1. 500 μl of (R)-mandelic acid stock were put in a 2 ml microcentrifuge tube and preheated to 25 °C. To initiate the reaction, 500 μl of the enzyme stock solution were added, resulting into final concentrations of 10 mg ml−1 (65.7 mM) of (R)-mandelic acid and 1 mg ml−1 enzyme (cell extract) in a final volume of 1 ml. The reactions were incubated at 25 °C and 900 rpm for 15, 30, 45 or 60 s, at which point 100 μl samples were drawn and immediately mixed with 500 μl of acetonitrile to precipitate the protein and hence stop the reaction. The resulting samples were vortexed and then centrifuged at 13000 rpm for 10 min. From the cleared supernatant 200 μl were drawn and mixed with 1300 μl of a 2 mM CuSO4 solution in ddH2O in an HPLC-vial. This sample was then measured in a chiral HPLC set-up for the enantiomeric ratio between (R)- and (S)-mandelic acid. From the measured percentage and the fixed initial (R)-mandelic acid concentration of 65.7 mM the amount of formed (S)-mandelic acid in μmol was calculated and plotted against time in min. The plot was linearized to yield an activity of the enzyme in U, which was then divided by the enzyme concentration of 1 mg ml−1 (1 ml reactions) in the reaction, resulting in a final specific activity of the analyzed dried cell extracts. For each time point, a triplicate of reactions was performed. If within 15–60 s no clear linear plot could be obtained due to lower activity of the obtained dried cell extract, the measurement points were extended to 1, 2, 4 and 8 min.Preparation of sodium mandelate, (1R,2R)-DPEN/(S)-1PEA hydrochloride salts and amine mandelate salts
To prepare sodium mandelate, 2 g (13.1 mmol) of racemic mandelic acid were dissolved in 50 ml cyclopentyl methyl ether (CPME). To the dissolved mandelate, 500 μl of a saturated NaOH solution was added. The resulting suspension was stirred for 1 h at room temperature, after which the precipitated sodium mandelate was filtered out and dried at room temperature. The purity of the obtained sodium mandelate was analyzed via NMR.(1R,2R)-DPEN and (S)-1PEA hydrochloride salts were prepared in a similar manner. An appropriate amount of the amines, 1–2 g of (1R,2R)-DPEN (4.7–9.4 mmol) or 1 ml (7.8 mmol) of (S)-1PEA, were dissolved in 50 ml of CPME. To this amine solution, 5 ml of a 3 M HCl in CPME solution (Sigma-Aldrich) was added. The resulting suspension was stirred for 1 h at room temperature, after which the precipitated amine hydrochloride (dihydrochloride for (1R,2R)-DPEN) was filtered out and dried at room temperature.
The amine mandelate salts for the solubility testing were prepared as follows. Separate solutions of (R)- or (S)-mandelic acid and their amine counterion in CPME were prepared, bearing equimolar concentrations. Thus, equimolar amounts of the stock solutions of mandelate and the amine counterion were unified. For the diamine salts of (1R,2R)-DPEN, the amount of mandelate was doubled. The resulting suspensions were stirred for 1 hour at room temperature. Monoamine salts of (1R,2R)-DPEN were prepared from water. Here, equimolar (75 mM) solutions of (R)- or (S)-mandelic acid and the dihydrochloride of (1R,2R)-DPEN in water were mixed in equal proportions, the pH was adjusted to 7. To facilitate crystallization, approximately half ((R)-salt) to two thirds ((S)-salt) of the water volume was evaporated at 40 °C under an argon stream, until first crystals were visible in the remaining aqueous phase. Afterwards, the crystallization was left for 1 h at room temperature (if necessary, the suspension was left for 15 min at 4 °C). The precipitated mandelate salts were filtered out and dried at room temperature.
Solubility screening
A small amount (between 10–20 mg) of the chosen mandelate salt was mixed into 1 ml of ddH2O. Additional salt was added until saturation was reached, if necessary. pH was kept at 7, adjusted with weak HCl and NaOH solutions, adjusted again after 24 h and afterwards every 48 h. Additional mandelate salt was added, if necessary. The saturated solutions were shaken at 30 °C and 900 rpm for 5 days or until no further pH changes occurred. After no pH change was observed, the solutions were centrifuged for 10 min at 13000 rpm and the cleared aqueous supernatant was filtered through 0.25 μm syringe filters to remove traces of crystalline salt. The filtrate was collected into previously weighed vials. Those vials were weighed again filled with the filtrate for the determination of the exact mass of the water. Then, the liquid in the vials was evaporated at 40 °C under a constant argon stream in a Thermo Scientific Pierce ReactiTherm I & ReactiVap I heating and evaporation unit. The evaporated vials were weighed and the solubility of the mandelate salts was calculated. The experiments for each salt were performed in triplicates.
Crystallization screening for enantiomeric excess
For resolution testing on small scale, solutions of sodium mandelate and its amine counterion hydrochlorides ((1R,2R)-DPEN or (S)-1PEA) in 50 mM HEPES buffer with 3.3 mM MgCl2 (pH 7.5) were prepared in double the concentrations, that were meant to be analyzed. The pH was adjusted back to 7.5 with conc. HCl and saturated NaOH solutions.Afterwards, the solutions were mixed in a 1:1 ratio to obtain 1 ml of final volume (500 μl:500 μl), effectively halving their respective stock solution concentrations. The resulting suspensions were shaken at 750 rpm and room temperature for 3 hours. Then the tubes with the samples were centrifuged for 10 min at 13000 rpm, the cleared supernatant was discarded. The obtained salt pellet was pressed onto filter paper to remove further liquid and dried for 2 hours at room temperature. The dried pellets were analyzed for enantiomeric excess via chiral HPLC according to standard procedure described below. For each analyte ratio, a triplicate was prepared.
Batches und fed-batches on a 5 ml scale
Small-scale experiments in the 5 ml format were prepared as follows. A 2.5 ml 400 mM sodium mandelate solution in 50 mM HEPES buffer with 3.3 mM MgCl2 (pH 7.5) was prepared, the pH was adjusted with conc. HCl and saturated NaOH solutions. An analogous solution of either 150 mM or 300 mM of (1R,2R)-DPEN dihydrochloride was prepared and pH-adjusted as well, although past the 150 mM mark the hydrochloride yielded rather a suspension than a solution. Both solutions were then mixed to yield a 5 ml reaction with 200 mM of mandelate and either 75 or 150 mM of (1R,2R)-DPEN. The vial of the (1R,2R)-DPEN was flushed with the mandelate solution to avoid loss of (1R,2R)-DPEN. The pH of the formed reaction solution was again adjusted to 7.5. Afterwards, 10 U ml−1 of the mandelate racemase cell extract was added to the mixture, if the reaction was to be performed in a DKR format, the pH was checked and adjusted to 7.5, if necessary. The reaction was stirred at room temperature and 750 rpm.For simple batch approaches (ee curve), the reactions were stirred for 72 h. 200 μl samples were drawn on certain time points and prepared according to standard procedure (see HPLC method) to measure ee. For fed-batch reactions, 200 μl samples were drawn after 24 h, measuring the mandelate concentration in solution and the respective ee (see HPLC method). The mandelate concentration was refilled to its initial value of 200 mM, while (1R,2R)-DPEN was refilled in a 1:2 ratio (half the molar amount) to the refilled mandelate (all on a scale of 4.8 ml), the pH was adjusted to 7.5. After another 24 h of stirring at room temperature and 750 rpm (48 h mark), the same refilling procedure (4.6 ml scale) was repeated. After refilling, another 10 U ml−1 of mandelate racemase cell extract was added into DKR-based reactions. The refilled reactions were left stirring for another 48 h (96 h reactions in total) at room temperature and 750 rpm. At the 96 h mark, further HPLC samples were drawn to calculate the yields of the reactions. The product salt was harvested by filtration, the reaction flask was flushed twice with the cleared filtrate to avoid product loss. The harvested salt was pressed into filter paper to remove residual liquid, dried at room temperature and weighed.
X-ray powder diffraction (XRPD) analysis
The analysis of a solid phase sample from the reactions and the reference salts was performed using a first-generation Empyrean diffractometer (PANalytical, Almelo, The Netherlands). Data was collected in reflection geometry (Bragg–Brentano) mode using a PIXcel3D 1 × 1 detector. The salt samples were prepared on zero background holders (silicon disks) and the measurements were performed in a 2θ range from 4–50° using Cu Kα(1+2) radiation. The step size was defined as 0.0131° and the time per step was set to 73.7 s. The setup was controlled using the PANalytical Data Controller software (vers. 5.3). The data was plotted using the Origin software.Preparative scale experiment
On preparative scale, the same approach was chosen, as with the 5 ml reactions. 25 ml of 400 mM sodium mandelate and 25 ml of 300 mM (1R,2R)-DPEN dihydrochloride solutions in 50 mM HEPES buffer with 3.3 mM MgCl2 (pH 7.5) were prepared, their pH was adjusted to 7.5 with conc. HCl and saturated NaOH solutions. The solutions were unified in a 100 ml Erlenmeyer flask, briefly stirred and their pH was adjusted. The vial of the (1R,2R)-DPEN was flushed with the mandelate solution to avoid loss of (1R,2R)-DPEN. 30 U ml−1 of the mandelate racemase cell extract were added to the mixture, the pH was checked and adjusted, if necessary. The reaction was stirred at 900 rpm and room temperature for 24 h. 200 μl samples were drawn after 24 h, the mandelate concentration in solution and the ee were measured (see HPLC method). The reaction was refilled as described for 5 ml fed-batches and stirred for another 24 h. Then, the refilling process was repeated at the 48 h mark, another 30 U ml−1 of mandelate racemase cell extract were added as well, the pH was adjusted. The reaction was stirred for another 48 h at room temperature and 900 rpm. Afterwards, 96 h HPLC samples were drawn to calculate the final yield from the residual mandelate concentration in solution. The product salt was harvested by filtration, the reaction flask being flushed twice with the cleared filtrate to avoid product loss. The harvested salt was pressed into filter paper to remove residual liquid and dried at room temperature, weighed and analyzed for enantiomeric excess via chiral HPLC and checked for impurities via NMR.The harvested product salt was suspended in 100 ml of ddH2O. 25 ml of saturated NaOH were added to the suspension, dissolving the product salt completely and obtaining a yellow precipitate of the (1R,2R)-DPEN. This solution was then extracted three times with 50 ml of previously dried CPME (24 h, 400 rpm, dried with anhydrous MgSO4). The organic phases were unified and evaporated to recover (1R,2R)-DPEN. To the aqueous phase, 50 ml of a 37% HCl solution were added, the pH was monitored to turn sour. The aqueous phase was then evaporated. The remaining solid was extracted five times with 50 ml of previously dried isopropanol (24 h, 400 rpm, dried with anhydrous MgSO4). The extractions were centrifuged to leave the undesired NaCl solid out of the product phase. The extraction phases were evaporated to yield the extracted mandelic acid. The purities of the extracted mandelic acid and (1R,2R)-DPEN were analyzed via NMR.
Chiral HPLC
Chiral HPLC analysis was performed on a Shimadzu Nexera series HPLC consisting of the following modules: SCL-40, DGU-405, LC-40D, SIL-40C, CTO-40S, SPD-M40. For separation, a Phenomenex Chirex 3126 column (150 × 4.6 mm; 5 μm, 110 Å) was used. The diluent was a mixture of 85% of 2 mM CuSO4 solution in ddH2O (pH ∼4) and 15% of HPLC-grade acetonitrile.For concentration measurements, samples were prepared as follows. From the reaction mixture, 200 μl samples were drawn and centrifuged for 10 min at 13000 rpm. From the cleared liquid phase, 100 μl were drawn into another vial. The product salt pellet was pressed onto filter paper and left to dry for 1 hour at room temperature for ee monitoring (24 and 48 h samples). To the 100 μl of liquid phase, 500 μl of acetonitrile was added to precipitate all proteins prior to measurement, the sample was vortexed and then centrifuged for 10 min at 13000 rpm. 200 μl of the cleared supernatant were added to 1300 μl of 2 mM CuSO4 solution, the sample was vortexed. 1 ml of this mixture was drawn into a HPLC-vial. 200 μl of a 15 mM solution of D-alanine in 2 mM CuSO4 was added as an internal standard for normalization. The readied samples were measured, the peak areas of (R)- and (S)-mandelic acids were normalized by the internal standard and their concentrations were calculated in accordance to an appropriate calibration curve.
For enantiomeric excess measurements of the product solid phase, samples were prepared as follows. For 24 and 48 h samples, the centrifuged pellet of the 200 μl samples was used, for the final ee measurement, the sample was taken from the dried harvested product salt. Approximately 3–10 mg of the solid phase (product salt) were dissolved in 500 μl of 2 mM CuSO4 with the addition of 10 μl of saturated NaOH solution. 500 μl of acetonitrile were added, the sample was vortexed. Afterwards, the samples were centrifuged for 10 min at 13000 rpm. 200 μl of the centrifuged sample was added to 1 ml of 2 mM CuSO4 to yield the final HPLC sample. The samples pH was adjusted to pH 4 with concentrated HCl (37% w/w). The sample was then briefly centrifuged again, if necessary, to remove possible copper-DPEN complexes precipitates and transferred into a HPLC-vial for measurement. All samples were measured in isocratic mode at a flow rate of 1 ml min−1 for 60 min. Column temperature was kept at 30 °C, the detection wavelength was 254 nm.
Summary and conclusions
This study aims to showcase the synthetic potential of dynamic kinetic resolution towards the preparation of enantiopure mandelic acid, consisting of a diastereomeric crystallization combined with enzymatic racemization using mandelic acid racemase. The presented approach for dynamic kinetic has shown great efficacy and presented a very good enantiomeric excess of the raw product phase. Furthermore, the diastereomeric crystallization was achieved at mild conditions and in a completely aqueous reaction phase, retaining high yields and enantiomeric excesses of the crystalline product salt. The system shows great potential for a continuous approach, including possible gravimetric separation of the product salt and a very good potential for the recyclability of the reaction broth, including the uncrystallized mandelic acid, since it is a racemate due to the racemase in solution, thus it would just need to be refilled to its initial concentration for process continuation. The shown dynamic kinetic resolution system using mandelate racemase may eventually outperform any form of chiral or kinetic resolution as yields of >50% are obtainable. Investigations of phase diagrams of the product salts of both enantiomers in water would help to determine ideal crystallization conditions and thus maximize possible obtainable yields from the preparative process.Data availability
The data supporting this article has been included as part of the ESI.†Author contributions
Conceptualization: F. B., J. v. L.; data curation: F. B., A. L.; formal analysis: F. B., A. L.; funding acquisition: J. v. L.; investigation: F. B., A. L.; methodology: F. B., A. L., J. v. L.; project administration: J. v. L.; resources: J. v. L.; supervision: J. v. L.; validation: F. B., A. L.; visualization: F. B.; writing – original draft: F. B.; writing – review & editing: F. B., A. L., J. v. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for F. B. and J. v. L. by Central SME Innovation Programme (ZIM, projects 16KN073233 and ZF4402103CR9) and the Heisenberg Programme of the Deutsche Forschungsgemeinschaft (project number 450014604) is gratefully acknowledged. The authors thank Dr. Katja Bettenbrock (Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg) for providing the plasmid carrying the mandelate racemase gene. The authors also thank Dr. Liane Hilfert and Sabine Hentschel (NMR division, Institute of Chemistry, Otto-von-Guericke University Magdeburg) for their help and fruitful discussions, as well as Ines Sauer (Industrial Chemistry, Institute of Chemistry, Otto-von-Guericke University Magdeburg) for her experimental support in XRPD measurements.References
- H. Pellissier, Eur. J. Org. Chem., 2022, 2022(7), e202101561 CrossRef CAS.
- G. L. Thejashree, E. Doris, E. Gravel and I. N. N. Namboothiri, Eur. J. Org. Chem., 2022, 2022(44), e202201035 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2011, 67, 3769–3802 CrossRef CAS.
- A. Garg, D. Rendina, H. Bendale, T. Akiyama and I. Ojima, Front. Chem., 2024, 12, 1398397 CrossRef CAS PubMed.
- S. Li, X. Xu, L. Xu, H. Lin, H. Kuang and C. Xu, Nat. Commun., 2024, 15, 3506 CrossRef CAS PubMed.
- G. Han, W. Ren, S. Zhang, Z. Zuo and W. He, RSC Adv., 2024, 14, 16520–16545 RSC.
- Y. Teng, C. Gu, Z. Chen, H. Jiang, Y. Xiong, D. Liu and D. Xiao, Chirality, 2022, 34, 1094–1119 CrossRef CAS PubMed.
- S. Mane, Anal. Methods, 2016, 8, 7567–7586 RSC.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed.
- M. Hall, RSC Chem. Biol., 2021, 2, 958–989 RSC.
- C. Terazzi, A. Spannenberg, J. von Langermann and T. Werner, ChemCatChem, 2023, 15(19), e202300917 CrossRef CAS.
- E. Forró and F. Fülöp, Curr. Med. Chem., 2022, 29, 6218–6227 CrossRef PubMed.
- S. S. Berry and S. Jones, Org. Biomol. Chem., 2021, 19, 10493–10515 RSC.
- M. M. Musa, F. Hollmann and F. G. Mutti, Catal. Sci. Technol., 2019, 9, 5487–5503 RSC.
- C. José, M. V. Toledo and L. E. Briand, Crit. Rev. Biotechnol., 2016, 36, 891–903 CrossRef PubMed.
- X. Wu, Y. Liu and Z. Jin, in Dynamic Kinetic Resolution (DKR) and Dynamic Kinetic Asymmetric Transformations (DYKAT), ed. J.-E. Bäckvall, Georg Thieme Verlag KG, Stuttgart, 2023 Search PubMed.
- R. Gurubrahamam, Y.-S. Cheng, W.-Y. Huang and K. Chen, ChemCatChem, 2016, 8, 86–96 CrossRef CAS.
- A. H. Kamaruddin, M. H. Uzir, H. Y. Aboul-Enein and H. N. A. Halim, Chirality, 2009, 21, 449–467 CrossRef CAS PubMed.
- L.-C. Yang, H. Deng and H. Renata, Org. Process Res. Dev., 2022, 26, 1925–1943 CrossRef CAS.
- G. A. Applegate and D. B. Berkowitz, Adv. Synth. Catal., 2015, 357, 1619–1632 CrossRef CAS PubMed.
- C. Femmer, M. Bechtold, T. M. Roberts and S. Panke, Appl. Microbiol. Biotechnol., 2016, 100, 7423–7436 CrossRef CAS PubMed.
- M. M. Musa, Chirality, 2020, 32, 147–157 CrossRef CAS PubMed.
- R. V. Singh and K. Sambyal, Crit. Rev. Biotechnol., 2023, 43, 1226–1235 CrossRef CAS PubMed.
- L. Martínková and V. Křen, Appl. Microbiol. Biotechnol., 2018, 102, 3893–3900 CrossRef PubMed.
- X. Chen, Q. Wu and D. Zhu, Process Biochem., 2015, 50, 759–770 CrossRef CAS.
- S. Wang, T. Shi, Z. Fang, C. Liu, W. He, N. Zhu, Y. Hu, X. Li and K. Guo, J. Flow Chem., 2022, 12, 227–235 CrossRef CAS.
- S. Chen, F. Liu, K. Zhang, H. Huang, H. Wang, J. Zhou, J. Zhang, Y. Gong, D. Zhang, Y. Chen, C. Lin and B. Wang, Tetrahedron Lett., 2016, 57, 5312–5314 CrossRef CAS.
- Y. Cao, S. Wu, J. Li, B. Wu and B. He, J. Mol. Catal. B: Enzym., 2014, 99, 108–113 CrossRef CAS.
- D. Yıldırım and S. S. Tükel, Biocatal. Biotransform., 2014, 32, 251–258 CrossRef.
- C. Yao, Y. Cao, S. Wu, S. Li and B. He, J. Mol. Catal. B: Enzym., 2013, 85–86, 105–110 CrossRef CAS.
- U. T. Strauss and K. Faber, Tetrahedron: Asymmetry, 1999, 10, 4079–4081 CrossRef CAS.
- M. E. Scott, X. Wang, L. D. Humphreys, M. J. Geier, B. Kannan, J. Chan, G. Brown, D. F. A. R. Dourado, D. Gray, S. Mix and A. Pukin, Org. Process Res. Dev., 2022, 26, 849–858 CrossRef CAS.
- X. Zhang, F. Chang, F. Zhu, T. Xu and Y. Zhang, J. Chem. Res., 2022, 46(4), 17475198221109155 CrossRef CAS.
- X.-H. Zhang, C.-Y. Wang, X. Cai, Y.-P. Xue, Z.-Q. Liu and Y.-G. Zheng, Bioprocess Biosyst. Eng., 2020, 43, 1299–1307 CrossRef CAS PubMed.
- L. Martínková, L. Rucká, J. Nešvera and M. Pátek, World J. Microbiol. Biotechnol., 2017, 33, 8 CrossRef PubMed.
- P. Kaul, A. Banerjee, S. Mayilraj and U. C. Banerjee, Tetrahedron: Asymmetry, 2004, 15, 207–211 CrossRef CAS.
- T. Yutthalekha, C. Wattanakit, V. Lapeyre, S. Nokbin, C. Warakulwit, J. Limtrakul and A. Kuhn, Nat. Commun., 2016, 7, 12678 CrossRef CAS PubMed.
- X.-P. Jiang, T.-T. Lu, C.-H. Liu, X.-M. Ling, M.-Y. Zhuang, J.-X. Zhang and Y.-W. Zhang, Int. J. Biol. Macromol., 2016, 88, 9–17 CrossRef CAS PubMed.
- P.-C. Yan, J.-H. Xie, X.-D. Zhang, K. Chen, Y.-Q. Li, Q.-L. Zhou and D.-Q. Che, Chem. Commun., 2014, 50, 15987–15990 RSC.
- T. Lerdwiriyanupap, R. Cedeno, P. Nalaoh, S. Bureekaew, V. Promarak and A. E. Flood, Cryst. Growth Des., 2023, 23, 2001–2010 CrossRef CAS.
- M. H. T. Kwan, J. Breen, M. Bowden, L. Conway, B. Crossley, M. F. Jones, R. Munday, N. P. B. Pokar, T. Screen and A. J. Blacker, J. Org. Chem., 2021, 86, 2458–2473 CrossRef CAS PubMed.
- S. K. Tulashie, J. von Langermann, H. Lorenz and A. Seidel-Morgenstern, Cryst. Growth Des., 2011, 11, 240–246 CrossRef CAS.
- Z. Szeleczky, P. Bagi, E. Pálovics, F. Faigl and E. Fogassy, J. Chem. Res., 2016, 40, 21–25 CrossRef CAS.
- X.-H. Pham, J.-M. Kim, S.-M. Chang, I. Kim and W.-S. Kim, J. Mol. Catal. B: Enzym., 2009, 60, 87–92 CrossRef CAS.
- K. Kodama, K. Kawasaki, M. Yi, K. Tsukamoto, H. Shitara and T. Hirose, Cryst. Growth Des., 2019, 19, 7153–7159 CrossRef CAS.
- K. Kodama, H. Shitara and T. Hirose, Cryst. Growth Des., 2014, 14, 3549–3556 CrossRef CAS.
- H. Shitara, T. Shintani, K. Kodama and T. Hirose, J. Org. Chem., 2013, 78, 9309–9316 CrossRef CAS PubMed.
- J. Wang and Y. Peng, Molecules, 2021, 26(18), 5536 CrossRef CAS PubMed.
- X. Buol, C. Caro Garrido, K. Robeyns, N. Tumanov, L. Collard, J. Wouters and T. Leyssens, Cryst. Growth Des., 2020, 20, 7979–7988 CrossRef CAS.
- Z. Szeleczky, P. Bagi, E. Pálovics and E. Fogassy, Tetrahedron: Asymmetry, 2014, 25, 1095–1099 CrossRef CAS.
- D. Li, Z. Zeng, J. Yang, P. Wang, L. Jiang, J. Feng and C. Yang, Biosci., Biotechnol., Biochem., 2013, 77, 1236–1239 CrossRef CAS PubMed.
- W. J. Choi, K. Y. Lee, S. H. Kang and S. B. Lee, Sep. Purif. Technol., 2007, 53, 178–182 CrossRef CAS.
- N. Kaftzik, J. Mol. Catal. A: Chem., 2004, 214, 107–112 CrossRef CAS.
- K. Wrzosek, M. A. G. Rivera, K. Bettenbrock and A. Seidel-Morgenstern, Biotechnol. J., 2016, 11, 453–463 CrossRef CAS PubMed.
- K. Wrzosek, I. Harriehausen and A. Seidel-Morgenstern, Org. Process Res. Dev., 2018, 22, 1761–1771 CrossRef CAS.
- I. Harriehausen, K. Wrzosek, H. Lorenz and A. Seidel-Morgenstern, Adsorption, 2020, 26, 1199–1213 CrossRef CAS.
- M. Pogorevc, H. Stecher and K. Faber, Biotechnol. Lett., 2002, 24, 857–860 CrossRef CAS.
- A. Narmandakh and S. L. Bearne, Protein Expression Purif., 2010, 69, 39–46 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00576g |
| This journal is © The Royal Society of Chemistry 2025 |