
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,4-Dihydropyrrolo[3,2-b]pyrrole modified with dibenzoxazepine: a highly efficient core for charge-transfer-based OLED emitters†
Krzysztof Górski a,
Steve Sheltonb,
Jaijanarthanan Lingagouderc,
Przemyslaw Data*c,
Denis Jacquemin*de and
Daniel T. Gryko*a
a,
Steve Sheltonb,
Jaijanarthanan Lingagouderc,
Przemyslaw Data*c,
Denis Jacquemin*de and
Daniel T. Gryko*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
bThe Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cDepartment of Molecular Physics, Faculty of Chemistry, Lodz University of Technology, 90-543 Lodz, Poland
dNantes Université, CNRS, CEISAM UMR 6230, F-44000 Nantes, France. E-mail: Denis.Jacquemin@univ-nantes.fr
eInstitut Universitaire de France (IUF), F-75005 Paris, France
First published on 12th February 2025
Abstract
The present work is focused on designing novel heteroaromatic systems, formally a hybrid of dibenzo[b,f]oxazepines and 1,4-dihydropyrrolo[3,2-b]pyrroles (DHPPs). Straightforward synthesis affords a family of rigid, centrosymmetric, π-expanded aromatic heterocycles amenable to facile post-functionalization. The rigidified molecular architecture is responsible for several key photophysical features including (1) the excellent blue colour purity (full width at half maximum parameter = 0.435 eV) and (2) stronger emission compared to analogous DHPPs capable of free rotation. Comparison of dyes possessing nitro groups at various positions reveals that if NO2 groups are located at distant positions the quadrupolar dye shows strong yellow fluorescence in non-polar solvents, whereas the same group at position 3 versus the DHPP core leads to a poorly emitting dye. Computational studies suggest that the key difference lies in relative energies of dark and bright excited states. It was shown that the DHPP core offers unique advantages as a high-emission energy system, serving as a foundation for charge-transfer (CT)-based efficient emitters. These features, combined with the ability to modulate electronic properties via peripheral functionalization, highlight the potential of the DHPP core in advanced optoelectronic devices, including new-generation OLEDs.
Introduction
Since the first synthesis of [7]circulene in 1983 by Yamamoto,1 numerous developments have taken place in the physicochemistry of nonalternant π-extended systems. The distortion induced by the presence of a nonhexagonal ring leads to unusual molecular architectures2–6 often possessing unique physicochemical properties such as specific supramolecular organization,7 open-shell character,8 or chiroptical activity.9–11 An equally important way of modifying physicochemical properties is heteroatom displacement, which also allows modulation of the electronic structure.12–15 However, due to the limited possibilities of the heteroatom incorporation into π-expanded nanographenes, this approach is not widely used in the chemistry of nonalternant π-extended dyes.16–18The goal of this study was to expand the chemical space of large nonalternant π-conjugated architectures employing the unique possibilities offered by 1,4-dihydropyrrolo[3,2-b]pyrroles (DHPPs). DHPPs are a family of electron-rich heterocycles19 exhibiting a variety of interesting properties such as solvatofluorochromism,20 two-photon absorption,21 and aggregation-induced emission.22–24 For this reason, they have found many applications, including H-binding probes,25 photochromic sensors,26 mitochondria imaging fluorescent dyes,27 solar cells,28,29 and OLEDs.30,31 Their popularity results from straightforward synthesis from readily available aldehydes and amines, which in situ form the corresponding imine, taking part in a further multicomponent process.32 This reaction is one of the very few processes that (a) involves five molecules and (b) leads to a centrosymmetric aromatic heterocycle. This unique feature enables a quick build-up of molecular complexity.33–35
To this end, we resolved to use dibenzo[b,f]oxazepines, consisting of two peripheral benzene rings and a central 7-membered ring, in which oxygen and nitrogen atoms are incorporated. In our concept, dibenzo[b,f]oxazepine serves as a source of nonalternancy. Currently, many synthetic pathways leading to this heterocyclic system are known; however, the simplest approach is based on cross-condensation between commercially available 2-fluoro- or 2-chlorobenzaldehyde and the corresponding 2-hydroxyanilines.36–41 Another important route toward constructing a heterocyclic 7-membered ring is an intramolecular, Vilsmeier-type reaction.42 Nevertheless, modern chemical development did not omit the methodology of dibenzo[b,f]oxazepine synthesis, which can be obtained in the presence of a Pd-based catalyst.43,44 While many dibenzo[b,f]oxazepine derivatives exhibit potential bioactivity and can act as pharmaceuticals,45–50 data regarding their use in functional materials chemistry are, unfortunately, lacking.
Herein, we present a straightforward synthetic methodology leading to complex, nonalternant multiheteroatom nanographenes from readily available cyclic imines, namely dibenzo[b,f]oxazepines. π-Extended 1,4-dihydropyrrolo[3,2-b]pyrrole and dibenzo[b,f]oxazepine hybrids exhibit reversible oxidation and large fluorescence quantum yields exceeding 80%. Moreover, the peripheral modification of the parent electron-rich core with electron-accepting subunits results in dyes exhibiting near-infrared (NIR) emission. In this study, compound 2a was chosen as a model emitter due to its high fluorescence quantum yield and excellent colour purity. This work highlights the synthesis, photophysical characteristics, and device performance of DHPP derivatives, emphasizing their potential as high-efficiency cores for CT emitters.
Results and discussion
Synthesis
Our strategy towards DHPPs bearing two dibenzo[b,f]oxazepine scaffolds relied on utilizing parent dibenzo[b,f]oxazepine (1a), as a cyclic imine, in our multicomponent reaction and exploiting typical imine reactivity, via the Mannich reaction.51 The choice of this approach relies on the proven fact that Schiff base formation is the first step in the formation of DHPPs. At the same time, we underline that cyclic imines were never used in this reaction and our earlier attempts to employ phenanthridine failed. The model reaction with 1a carried out under standard conditions32 led to the formation of desired DHPP 2a, with, however, a small yield of 3.6% (Table 1, entry 1). Notably, the formation of 2a seems to be much faster than typical 1,4-dihydropyrrolo[3,2-b]pyrrole prepared from acyclic imines, which may be related to the Z configuration of the double bond in 1a. Nevertheless, changing the solvent system (Table 1, entry 2) or the acidity of the reaction environment (Table 1, entry 4) was ineffective for increasing the yield of the desired product. Moreover, even utilizing conditions under which substrate 1a undergoes the Mannich reaction51 doesn't affect the reaction course toward the formation of DHPP 2a (Table 1, entry 5).
|
||||
|---|---|---|---|---|
| Entry | Conditions | T [°C] | t [h] | Yield [%] |
| 1 | 1 ml AcOH, 1 ml toluene, butanedione (1 mmol), Fe(ClO4)3 (3% mol) | 50 | 24 | 3.6 |
| 2 | 1 ml AcOH, butanedione (1 mmol), Fe(ClO4)3 (3% mol) | 50 | 24 | 2.2 |
| 3 | 1 ml AcOH, butanedione (1 mmol), after 24 h additional butanedione (1 mmol) | 50 | 48 | 4.2 |
| 4 | 1 ml AcOH, 1 ml toluene, 1 ml 1,2-dichloroethane, butanedione (1 mmol), CH3SO3H (0.1 mmol), Fe(ClO4)3 (3% mol) | 50 | 24 | 0.0 |
| 5 | 1 ml AcOH, 6 ml DMSO, butanedione (1 mmol), L-proline (0.6 mmol), Fe(ClO4)3 (3% mol) | 50 | 24 | 0.0 |
| 6 | 2 ml AcOH, 2 ml toluene, butanedione (4 mmol) | 80 | 24 | 6.0 |
| 7 | 2 ml AcOH, 2 ml toluene, butanedione (1 mmol) | 40 | 72 | 3.1 |
| 8 | 2 ml AcOH, 2 ml toluene, butanedione (4 mmol) | 100 | 24 | 3.0 |
An aspect that needs to be taken into account is the intrinsic electron-rich character of the central 1,4-dihydropyrrolo[3,2-b]pyrrole core, which is enhanced in dye 2a, due to the partial planarization of peripheral substituents leading to increased conjugation over the entire molecule (see calculations below). Thus, the presence of an oxidant, such as Fe(ClO4)3, which is necessary under standard 1,4-dihydropyrrolo[3,2-b]pyrrole synthesis conditions,32 in this case, may enhance the decomposition of dye 2a. Thus, we decided to exclude Fe(ClO4)3, returning to the initially published conditions.52 The reaction time however was prolonged up to 48 h and the whole process required the addition of another portion of butane-2,3-dione after 24 h, which resulted in a slight increase in the yield of product 2a (Table 1, entry 3). Gratifyingly, increasing four times the amount of butane-2,3-dione simultaneously with increasing the reaction temperature up to 80 °C led to a sharp increase in the reaction yield to ≈6% (Table 1, entry 6). Further changes in the reaction conditions did not lead to any improvements (Table 1, entry 7 and 8). Having suitable reaction conditions in hand we resolved to probe the scope of this reaction to embrace electron-withdrawing substituents such as bromine atoms potentially opening the pathway towards larger architectures via post-functionalization. Thus we investigated the scope of dibenzo[b,f]oxazepines able to form 1,4-dihydro-pyrrolo[3,2-b]pyrroles (Scheme 1). The introduction of an alkyl chain, such as t-butyl, doesn't affect the reaction yield (2b – 6%), whilst, using brominated dibenzo[b,f]oxazepines 1c, d distinctly increases the reaction yield (2c – 13% and 2d – 18%). This rather unusual result is plausibly a direct consequence of the decreased solubility of 2c and 2d, caused by the presence of bromine atoms (a smaller amount of the desired product is prone to oxygen-promoted decomposition). A perfect illustration of this effect is the transformation of 1e into 2e, which occurs with 45% yield. Interestingly, for dibenzo[b,f]oxazepines containing a nitro group (1f, g) or a tri-i-propylsilyl protected acetylene (1h), the formation of the corresponding 1,4-dihydro-pyrrolo[3,2-b]pyrroles 2f–h ranges from 10% to 19%.
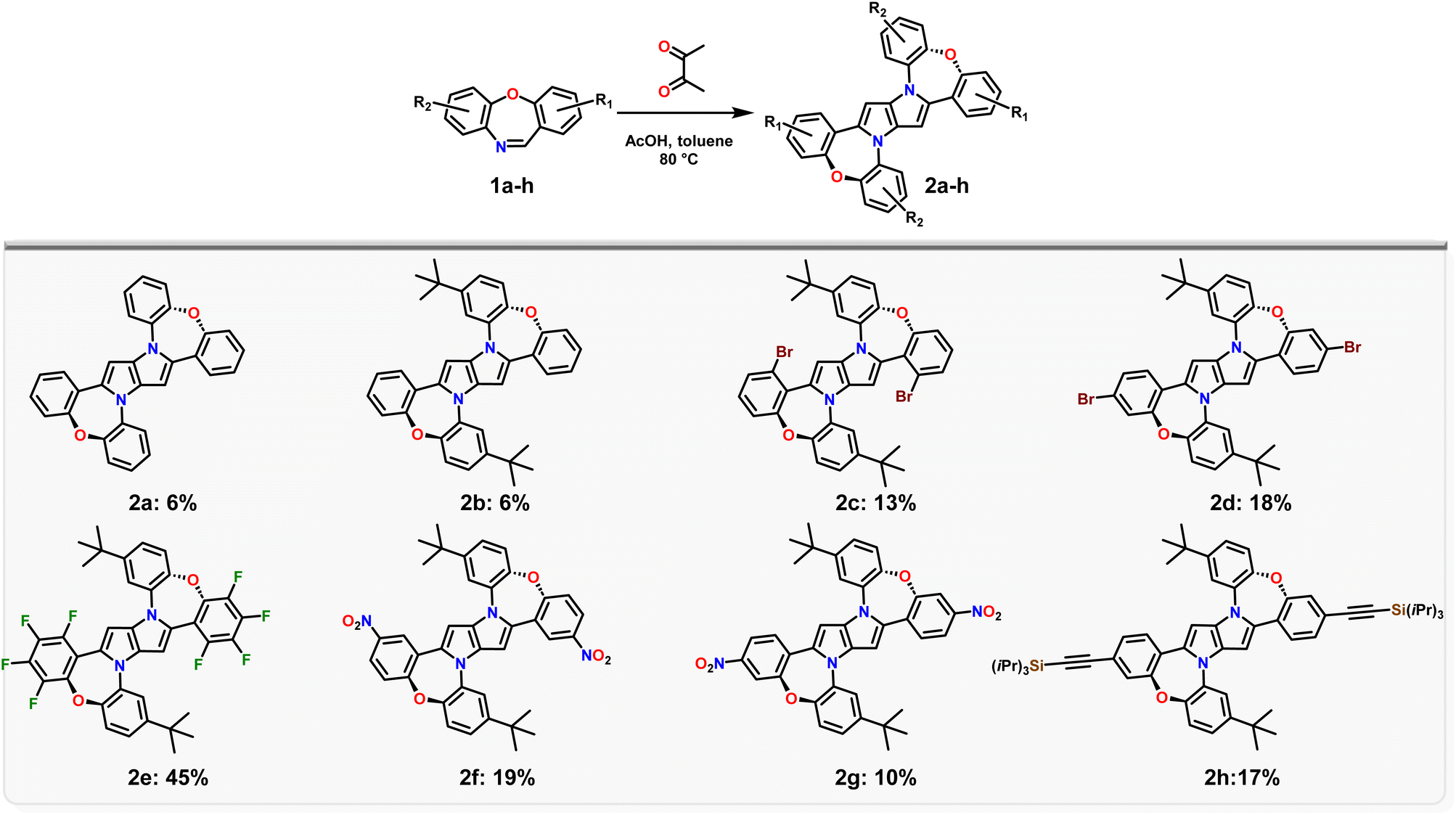 | ||
| Scheme 1 The scope of dibenzo[b,f]oxazepines able to form 1,4-dihydropyrrolo[3,2-b]pyrroles (2a–g). | ||
Having in our hands a panel of DHPPs bearing two dibenzo[b,f]oxazepine units, we embarked on their post-functionalisation. Since the most interesting photophysical effects in DHPPs were found in centrosymmetric quadrupolar A–D–A architectures,19,20 the general post-functionalization strategy was directed towards attaching electron-deficient units at peripheral positions of the new core. Dye 2d when heated in N-methylpyrrolidone in the presence of CuCN undergoes conversion into mono- and dicyanoderivatives 3a, b (Scheme 2). DHPPs possessing cyano groups but in different arrangements were obtained via a Suzuki reaction with 4-cyanophenylboronic acid, resulting in the formation of 4a, b with moderate yields. On the other hand, dye 2h under sila-Sonogashira reaction conditions53 can be directly transformed into elongated donor–acceptor architectures 5a–d, in which the electron-rich 1,4-dihydro-pyrrolo[3,2-b]pyrrole core is peripherally decorated with acceptor subunits such as nitrile (DHPP 5a), 5-thiatruxene sulfone (DHPP 5b),54 naphthalimide (DHPP 5c) or perylenediimide (DHPP 5d) (Scheme 3).
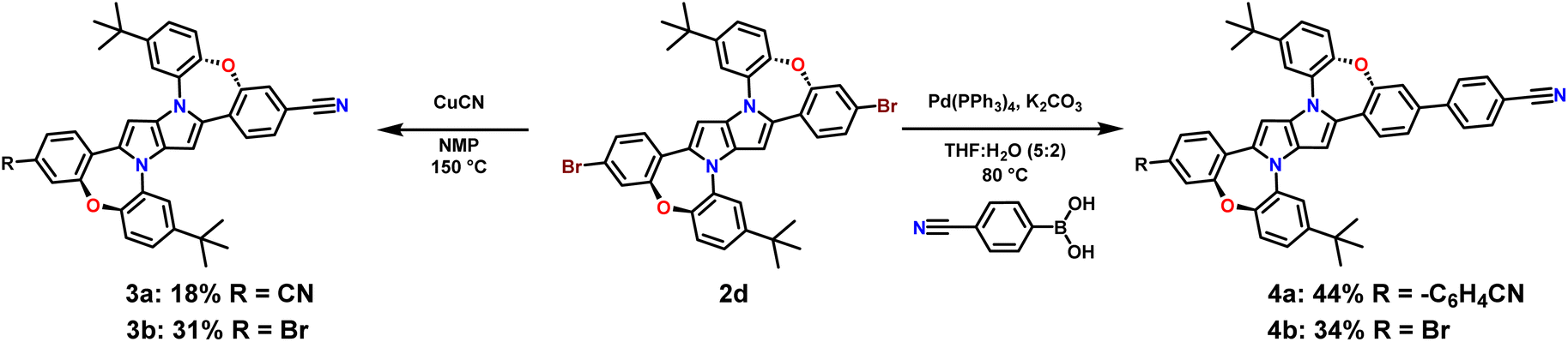 | ||
| Scheme 2 Post-functionalization of DHPP 2d. | ||
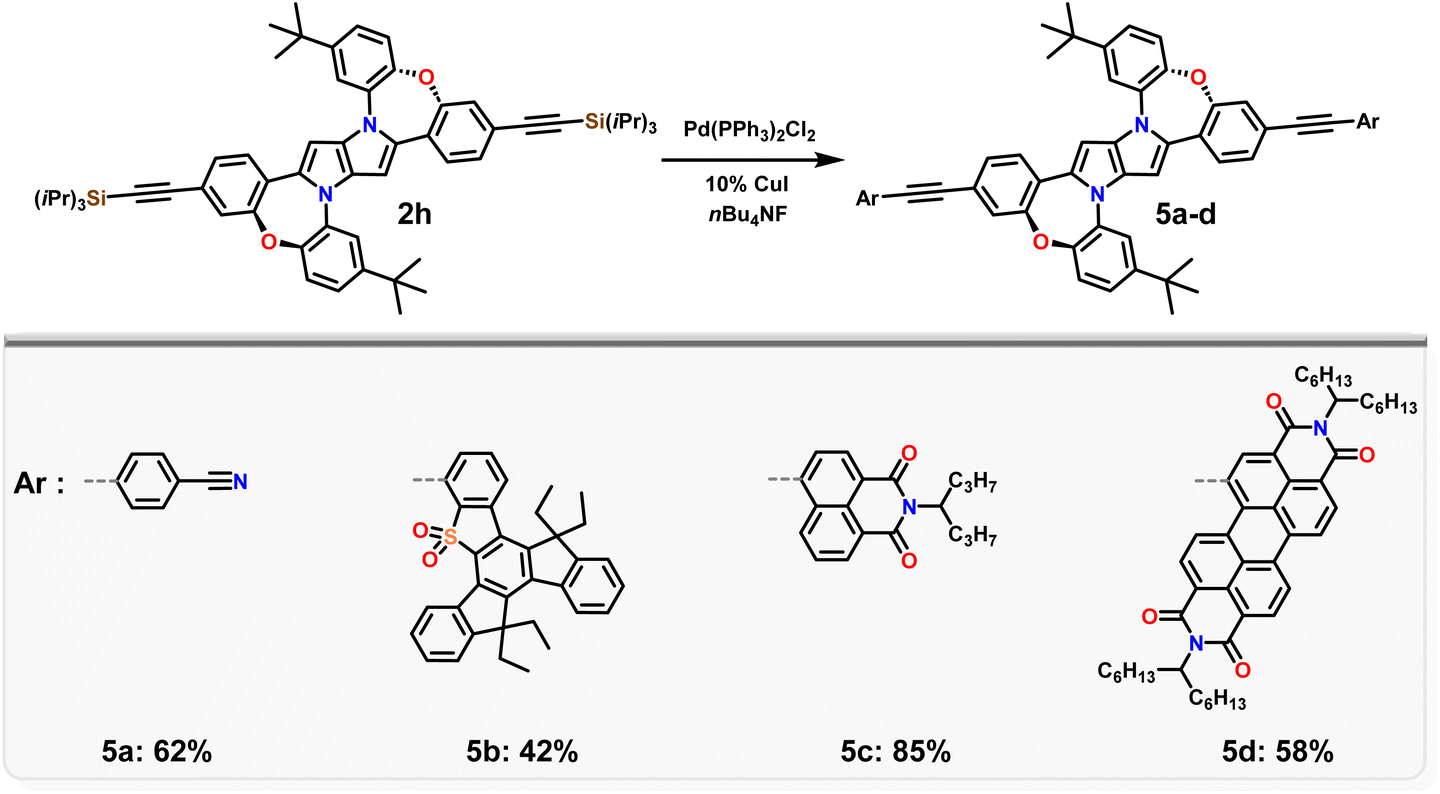 | ||
| Scheme 3 Post-functionalization of DHPP 2h via the sila-Sonogashira protocol. | ||
Crystal structure
While most of the obtained dyes tend to crystalize, only 2e provided X-ray quality crystals. As shown in Fig. 1a and c, the molecule consists of a planar DHPP-based centre, decorated peripherally with a dibenzo[b,f]oxazepine subunit, characteristically bending the molecule around an oxygen atom with a distortion angle of about 126°. While many π-expanded dyes suffer from low solubility, this deformation acts oppositely; thus, 2e exhibits sufficient solubility in most non-polar and moderately polar solvents. DFT calculations suggest that the obtained dyes can exist in two major forms: Ci-centrosymmetric and C2-symmetric, with the molecule containing an inversion center being ca. 0.42 kcal mol−1 more stable. This is consistent with the 2e structures, which crystallize in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, though we cannot exclude a mixture of both forms in solution.
space group, though we cannot exclude a mixture of both forms in solution.
 | ||
| Fig. 1 X-ray structure of 2e, (a) single molecule – top view, (b) interaction between two molecules – top view, (c) single molecule – side view, and (d) interaction between two molecules – side view. Depth hue was used to present the spatial orientation of atoms in the molecule and crystal lattice. | ||
Distortion within the 7-membered ring (Fig. 1c) hinders strong π-stacking; however, fluorinated rings tend to interact (d = 3.410 Å), leading to aggregates within the entire crystal lattice (Fig. 1b and d). Consequently, new hybrids of 1,4-dihydropyrrolo[3,2-b]pyrrole with dibenzo[b,f]oxazepine can be considered potential semiconductors. Furthermore, strong association modifies the electronic structure; thus, the described intermolecular interactions are directly responsible for the distinct changes of spectroscopic properties observed during passing from solution to solid, in the synthesized dyes.
Experimental and theoretical spectroscopic studies
The unsubstituted parent heterocycle 2a and its alkyl derivative 2b and halogenated derivatives 2c–e exhibit strong absorption with a λmaxabs around 375–382 nm in toluene (Table 2, Fig. 2 and S52–S56†), which corresponds to a S0 → S1 transition characterized by high oscillator strengths exceeding 1 according to TD-DFT calculations (Table S1†). Nevertheless, in the case of dye 2e, using dichloromethane induces a nearly twofold drop in the observed absorption molecular coefficient (ε). Similar behaviour was also noticed in some 4,5,6,7-tetrafluorobenzo[b]furane derivatives.55 On the other hand, a small expansion of the π-electron system caused by the introduction of two (tri-i-propylsilyl)acetylene moieties (dye 2h) distinctly changes the absorption spectrum. First of all, it is bathochromically shifted by about 2800 cm−1 (Table 2), in both solvents in comparison to DHPP 2a. Secondly, an enormous oscillator strength (f ≈ 2, Table S1†) is computed for the S0 → S1 electron transition, consistent with the observed near doubling of the ε which reaches about 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.
000 M−1 cm−1.
| Dye | λmaxabs [nm] (ε/103 [M−1 cm−1]) | λmaxem [nm] | Δν [cm−1] | Φfl [%] |
|---|---|---|---|---|
| a λmaxabs – absorption maximum wavelength, ε – molar absorption coefficient, λmaxem – emission maximum wavelength, Δν – Stokes shift, Φfl – fluorescence quantum yield. 9,10-Diphenylanthracene in toluene was used as the quantum yield standard (Φfl = 70%). | ||||
| 2a | 363 (33.2), 375 (31.6) | 407, 424 | 2100 | 80 |
| 361 (37.0), 372 (35.0) | 407, 424 | 2300 | 69 | |
| 2b | 362 (41.2), 373 (39.4) | 410, 425 | 2400 | 69 |
| 361 (39.1), 371 (37.0) | 410, 425 | 2600 | 69 | |
| 2c | 369 (38.4), 382 (36.6) | 425 | 2600 | 0.1 |
| 366 (34.8), 376 (33.1) | 421 | 2800 | 0.07 | |
| 2d | 372 (45.5), 383 (44.0) | 421, 437 | 2400 | 15 |
| 372 (47.1), 383 (45.2) | 421, 437 | 2400 | 5 | |
| 2e | 358 (44.9), 374 (43.7) | 404, 419 | 2000 | 64 |
| 357 (21.6), 371 (20.6) | 404, 419 | 2200 | 28 | |
| 2f | 371 (33.5) | 629 | 11100 |
2 |
| 370 (40.8) | — | — | — | |
| 2g | 465 (39.0) | 572 | 4000 | 93 |
| 477 (47.0) | 716 | 7000 | 6 | |
| 2h | 402 (69.9), 420 (71.2) | 450, 474 | 1600 | 82 |
| 401 (69.9), 413 (69.3) | 453, 474 | 2100 | 85 | |
| 3a | 404 (27.5), 421 (29.1) | 451, 474 | 1600 | 87 |
| 409 (33.3), 421 (34.4) | 460, 480 | 2000 | 86 | |
| 4a | 418 (29.7) | 484, 506 | 3300 | 90 |
| 417 (33.6) | 531 | 5100 | 95 | |
| 5a | 434 (57.3) | 494, 517 | 2800 | 100 |
| 435 (72.1) | 544 | 4600 | 96 | |
| 5b | 376 (26.7), 456 (68.4) | 504, 531 | 2100 | 79 |
| 380 (33.1), 449 (79.8) | 555 | 4200 | 81 | |
| 5c | 377 (40.4), 475 (54.7) | 581 | 3800 | 77 |
| 377 (47.3), 474 (63.4) | 793 | 8500 | 1 | |
| 5d | 532 (94.3), 611 (26.4) | 836 | 4400 | <0.001 |
| 530 (88.5), 591 (26.1) | — | — | — | |
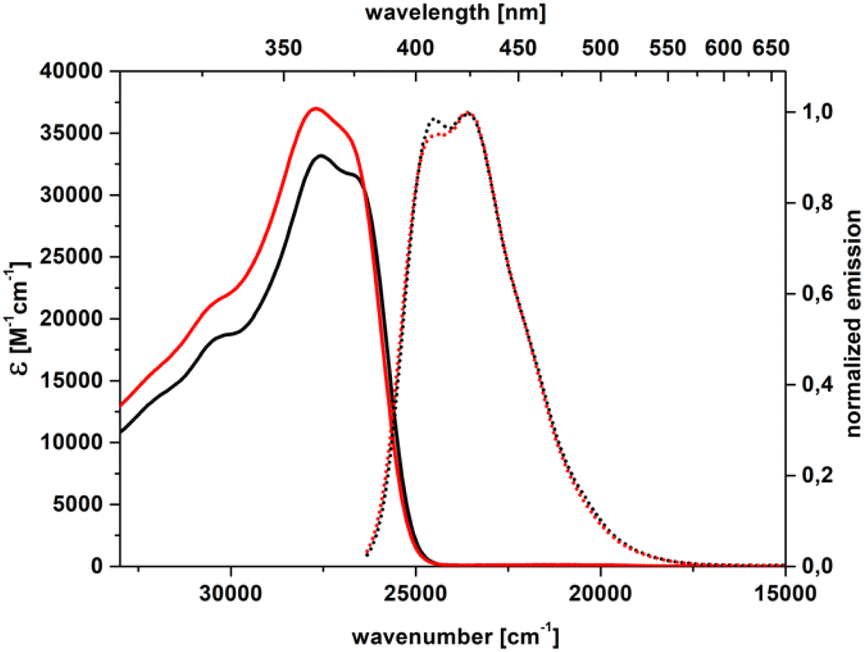 | ||
| Fig. 2 Absorption (solid) and emission (dot) spectra of 2a in toluene (black) and dichloromethane (red). | ||
However, the lack of essential changes in the absorption spectra upon solvent polarity increasing, for compounds 2a–e and 2h, indicates a centrosymmetric molecular structure, which is in line with the X-ray results as well as DFT results.
For dyes 2a–e, the fluorescence appears in the blue region, with λmaxem ranging around 407 nm to 425 nm (Table 2). The fluorescence of dye 2a characterized by the particular purity of the blue emission (CIE 1931: 0.156; 0.032) and large fluorescence quantum yield (Φfl) reaching 80% in toluene deserves special attention. On comparing the electron density difference (EDD) plots of 2a to that of its unbridged counterpart, i.e. 1,2,4,5-tetraphenyl-1,4-dihydropyrrolo[3,2-b]pyrrole, one notices quite similar topologies (Fig. 3). Nevertheless, the Φfl in toluene for the former is more than twice that of the latter.52 Undoubtedly, the observed differences have to be related to more effective nonradiative dissipation of energy in the excited state in the case of 1,2,4,5-tetraphenyl-1,4-dihydropyrrolo[3,2-b]pyrrole compared with DHPP 2a. Analyses of the S0 and S1 geometries for both molecules reveal that the relaxation of 1,2,4,5-tetraphenyl-1,4-dihydropyrrolo[3,2-b]pyrrole in the S1 state involves rotation of the benzene rings attached to positions 2 and 5 by ca. 20°, while in 2a planarization of the entire molecule is observed, yet with smaller changes in the dihedral angle, consistent with an increased emission yield (Fig. 3). This improved rigidity may also explain the excellent blue colour purity which is directly correlated with a rather narrow full width at half maximum parameter (FWHM = 0.435 eV). Consistently, a comparison of the photophysics of dye 2a and 2,5-bis(2-methoxyphenyl)-1,4-bis(4-methylphenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole56 indicates that the λmaxabs of 2a is bathochromically shifted by ca. 30 nm in CH2Cl2 whereas the λmaxem values are essentially the same.
 | ||
| Fig. 3 Optimal ground (a) and excited state (b) structures of 1,2,4,5-tetraphenyl-1,4-dihydropyrrolo[3,2-b]pyrrole with key dihedral angles represented, together with the EDD plot (c). Optimal ground (d) and excited state (e) structures of 2a with key dihedral angles represented, together with the EDD plot (f). In the EDD plots, the blue and red lobes correspond to a decrease and increase in electron density, respectively. The contour threshold is 0.001. | ||
As stated above, the optical spectra of dyes 2a–e are also rather equivalent. This is consistent with the fact that the EDD involved in S0 → S1 and in S1 → S0 transitions are located on the same part of the molecules 2a–e (Fig. S82†). Dye 2h differs somewhat, in which the excited-state reorganization also partially involves the (tri-i-propylsilyl)acetylene subunits that act as weak electron acceptors (Fig. S82†), consistent with the observed redshifts (Table 2). However, no distinct solvatofluorochromism is found.
Nevertheless, while the introduction of a t-butyl group (2b) does not significantly impact either λmaxem or Φfl (Table 2), the presence of bromine atoms in 2c, d leads to a significant drop of Φfl in toluene, to 0.1% and 15%, and in dichloromethane to 0.07% and 5%, respectively. While the lowering of the fluorescence quantum yield can be easily explained by an increased population of triplet states (intersystem crossing), caused by the heavy atom effect, distinct differences between Φfl for 2c and 2d suggest the presence of another nonradiative relaxation channel of the S1 state for 2c. Indeed, 1H NMR spectra of dye 2c, recorded at different temperatures, indicate that the presence of bromine atoms raises the energy barrier between the Ci and C2 conformations (Fig. S23†), which might explain the different photophysical behaviour of the two compounds. Unlike 2a–d, the fluorescence of 2h is distinctly bathochromically shifted to ≈450 nm, for both solvents (Table 2); nevertheless, the redshift of the emission doesn't affect Φfl, which ranges around 82%.
The enhanced electron-donating character of the new heterocyclic core in DHPP 2a opened the pathway for exploring the photophysics for derivatives possessing two peripheral electron-accepting subunits. The first class of such systems is nitro-aromatics 2f, g. The nitro group in 2f slightly influences the absorption, practically not changing the λmaxabs, compared to 2a (Table 2). On the other hand, the effect of the NO2 moiety in the case of 2g is much more pronounced, shifting the λmaxabs by about 100 nm toward lower energy. Such changes are qualitatively reproduced by theory (Table S1†). Despite the acceptor–donor–acceptor structure of dyes 2f, g, no changes in absorption spectra are observed upon solvent polarity increasing (Fig. S57 and S58†), indicating the centrosymmetric quadrupolar character of these nitro-aromatics.57 The NO2 group is strongly involved in the S0 → S1 excitation process (Fig. S82†), causing the relocation of electron density from the center of the molecule toward the electron-deficient moiety. Dye 2g exhibits both strong fluorescence in toluene in the yellow region and strong solvatofluorochromism indicating excited-state symmetry breaking58–60 (Table 2). Indeed, upon increasing solvent polarity, the emission spectrum becomes dramatically redshifted (λmaxem = 716 nm in DCM versus λmaxem = 572 nm in toluene) with a simultaneous, nearly 16-times decrease in fluorescence quantum yield. In contrast, the emission spectrum of 2f is extremely redshifted, with Δν = reaching 11000 cm−1 in toluene, while for 2g it is only 4000 cm−1. This behaviour is directly related to charge delocalization at the S1 state. In both compounds the first electron excitation possesses a distinct charge transfer character; however, the mutual orientation of the 1,4-dihydropyrrolo[3,2-b]pyrrole core and the NO2 group in 2g leads to significant delocalization, while in the case of 2f, the charge becomes more localized. In toluene, the Φfl values are markedly different for these two dyes (2% for 2f and 93% for 2g). This effect could be rationalized using theory, since in 2f there are two nearly degenerated states (one bright and one dark) and the fluorescence is likely quenched by the presence of this dark state, whereas in 2g, the dark state is significantly shifted up in energy and plays no role in the photophysics.61 In the context of fluorescent nitroaromatics, it is interesting to compare 2f and 2g to their analogs lacking rigidification imparted by dibenzoxazepine.62 Whereas λmaxabs and λmaxem of 2,5-bis(4-nitrophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole and 2,5-bis(3-nitrophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole are located at almost the same wavelengths as those of dyes 2f and 2g, the emission intensity is enhanced after rigidification of the structure. In particular in the case of dye 2f the Φfl is 10 times higher than that of 2,5-bis(3-nitrophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole in toluene.
A more typical picture for the behaviour of quadrupolar, centrosymmetric dyes presents a series of nitrile derivatives 3a, 4a and 5a, which structurally differ from each other in the distance between the electron-withdrawing CN group and electron-rich core. In comparison to dye 2a, the absorption spectra of all nitrile derivatives are significantly bathochromically shifted, while the absorption maximum increases along with donor–acceptor separator length, reaching 434 nm for 5a (Table 2). As in other discussed systems, first electron excitation mostly involves the 1,4-dihydropyrrolo[3,2-b]pyrrole core; however, in the series of 3a → 4a → 5a, a distinct rearrangement of electron density toward the accepting moiety is observed (Fig. S82 and S83†). While ε values for 3a and 4a at the λmaxabs range from 29 × 103 M−1 cm−1 to 34 × 103 M−1 cm−1, elongation of the donor–acceptor separator by the introduction of the triple bond increases ε more than twice, in the case of 5a (Table 2). Moreover, similar changes, as observed in 2h, are also found in dyes 5b–d; thus ε increasing can be directly related to π-electron expansion, caused by the presence of carbon–carbon triple bonds. Despite some differences in ε values, solvent polarity increasing slightly affects absorption spectra of 3a, 4a and 5a–d. On the other hand, distinct changes are observed in the emissive properties of nitrile derivatives. While 3a, 4a and 5a exhibit pronounced solvatofluorochromism, the strongest response to the increasing solvent dielectric constant is observed for DHPP 5a (Table 2 and Fig. S60–S62†). A larger distance between the DHPP core and electron-accepting moieties leads to more pronounced charge separation upon excitation, making the S1 → S0 transition energy noticeably smaller. Nevertheless, for all nitrile derivatives in both solvents, a large Φfl is maintained, ranging between 90 and 100% (Table 2). Similar to previously described nitroderivatives, rigidification slightly affects λmaxabs and λmaxem when comparing 5a to corresponding unbridged 1,4-dihydropyrrolo[3,2-b]pyrrole;63 however, Φfl of 5a is nearly 5 times higher.
Replacing the 4-cyanophenyl substituent with stronger electron-accepting subunits, such as 5-thiatruxene sulfone (5b),64 naphthalimide (5c), or perylenediimide (5d), leads to an even more pronounced bathochromic shift of λmaxabs reaching 611 nm for 5d (Table 2, Fig. 4 and S63–S65†). Similarly to nitrile derivatives, the EDD plots corresponding to an S0 → S1 excitation in 5b show that the electronic reorganization takes place in the molecule's center, with distinct displacement toward the sulfone groups (Fig. S83†). Moreover, by increasing the electron-accepting character of the electron-withdrawing moiety (5c and d), the charge-transfer character becomes more pronounced. The visible solvatofluorochromic response of 5b is accompanied by maintaining a large Φfl of about 80%, for both solvents (Table 2). The strong fluorescence redshifts observed for 5c and 5d, with λmaxem = 793 nm and 836 nm, respectively, with a fluorescence tail reaching >1100 nm, clearly show NIR emission (Fig. S63 and S64†). However, such a lowering of the S1 → S0 energy band gap increases the efficiency of the nonradiative deactivation of the excited S1 state (according to the so-called energy gap law), and the values of Φfl become rather small (Table 2).
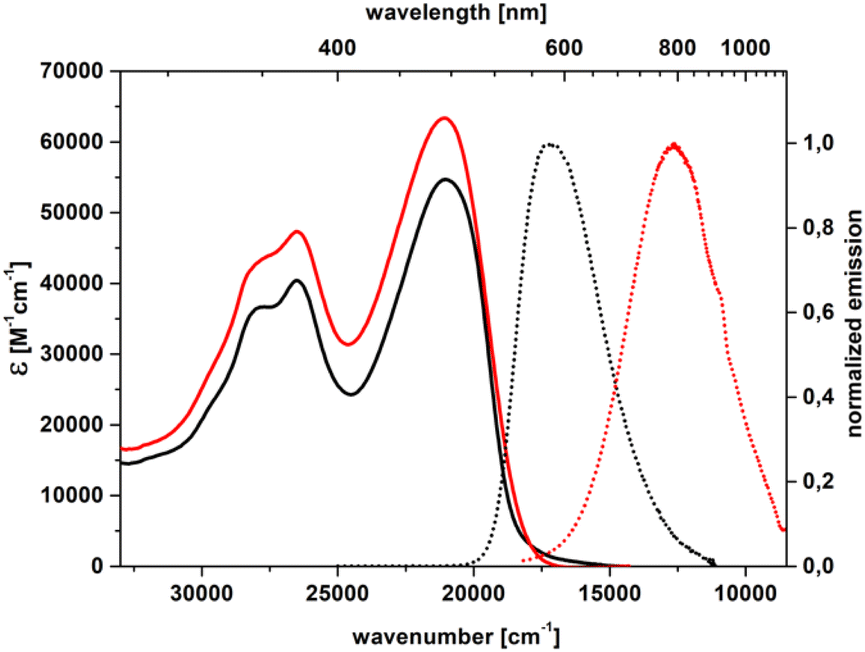 | ||
| Fig. 4 Absorption (solid) and emission (dot) spectra of 5c in toluene (black) and dichloromethane (red). | ||
Photostability and thermal stability
Due to 2a representing a new class of nonalternant multiheteroatom doped nanographenes and single photon spectroscopy revealing interesting properties of this system and its derivatives, we decided to estimate the photostability of 2a by quantitatively determining the photodegradation quantum yield (Φdeg). To this end, we used an already well-established protocol,65 that considers both the range of light inducing the decay and the absorption spectrum of the compound, which is not often the case in the literature. Irradiation was performed in an aerated toluene solution at around 20 °C. Absorption spectra were recorded at intervals of 60 seconds, during which time the solution was exposed to 363 nm light, which was sufficient to observe clear changes (Fig. S66a†). Moreover, the linear course of the A0/A(t) relationship as a function of F(t) indicates the lack of absorption bands of the photodecomposition product and allows the determination of the slope coefficient necessary to calculate the photodegradation quantum yield (Fig. S66b†). Φdeg was estimated to be ca. 0.014%, corresponding to the photodecomposition of one molecule upon absorption of around 7200 photons, which stands in line with the electron-rich nature of 2a.To evaluate the thermal properties of 2a thermogravimetry analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed under a nitrogen atmosphere. TGA reveals significant changes in sample mass, around 10%, in the range of 360 to 400 °C (Fig. S85a†). On the other hand, DSC shows the endothermic process with a peak of 395 °C corresponding to the melting point (Fig. S85b†). Thus mass drop is correlated to slow evaporation of the sample rather than decomposition, indicating high thermal stability of 2a. Interestingly, two different exothermic processes can be observed at 262 °C and 202 °C upon cooling (Fig. S86†). The significant difference between the melting point and overcooled recrystallization process (133 °C) indicates difficulties related to the appropriate orientation of molecules within the forming crystal lattice, likely related to distortions caused by the 7-membered ring hindering strong intermolecular interactions. On the other hand, the process appearing at 202 °C is probably correlated to reorganization within the crystal lattice formed at 262 °C, leading to other crystalline structure. Confirmation of this assumption can be found during the analysis of the next heating cycle which reveals changes in the melting point (382 °C). The broad peak at 382 °C suggests inhomogeneity of the crystalline structure formed at 202 °C, while the difference in the melting point between the first heating cycle and the second one (13 °C) points to significant changes in the crystalline structure.
Electrochemistry
Anticipating their application in optoelectronic devices, we decided to study the electrochemical properties for all novel heterocyclic dyes using cyclic voltammetry. Indeed, the energy of the highest occupied molecular orbital (HOMO) and the reversibility of the redox process are particularly relevant parameters. All compounds exhibit a reversible first oxidation process, while a second oxidation can be observed in both 2h and 5b (Fig. S74 and S78†). Except for 2g and 5b, d, the first oxidation takes place in the 0.7 V to 1.0 V vs. the Fc/Fc+ range. The determined EHOMO = −5.51 eV of 2a insignificantly changes upon attachment of an alkyl chain (2b), a bromine atom (2d) or (tri-i-propylsilyl)acetylene moieties (2h) (Table 3). However, the stabilization of the HOMO level for 2c by ca. 0.1 eV indicates that some steric, as well as electronic effects, can modulate its energy. Nevertheless, the strongest stabilization is caused by the presence of 8 fluorine atoms (2e), logically inducing a lowering of the HOMO level by ca. 0.3 V as compared to 2a, down to −5.80 eV.| Dye | Eox1/2 [V] | EHOMO [eV] | ΔEopt [eV] | ELUMO [eV] |
|---|---|---|---|---|
| a Eox1/2 – oxidation half-wave potential measured in dichloromethane vs. Fc/Fc+, EHOMO – the energy of the HOMO calculated from the first oxidation potential as −e(4.8 + Eox1/2), ΔEopt – optical band gap in toluene, estimated from absorption and emission spectra cross-section (* – determined from absorption spectra onset), and ELUMO – the energy of the LUMO estimated using ΔEopt + EHOMO. | ||||
| 2a | 0.71 | −5.51 | 3.17 | −2.34 |
| 2b | 0.72 | −5.52 | 3.17 | −2.35 |
| 2c | 0.82 | −5.62 | 3.07 | −2.55 |
| 2d | 0.73 | −5.53 | 3.08 | −2.45 |
| 2e | 1.00 | −5.80 | 3.19 | −2.61 |
| 2f | 0.98 | −5.78 | 2.92* | −2.86 |
| 2g | 0.34 | −5.14 | 2.39 | −2.75 |
| 2h | 0.72 | −5.52 | 2.85 | −2.67 |
| 0.83 | −5.63 | |||
| 3a | 0.91 | −5.71 | 2.84 | −2.87 |
| 4a | 0.80 | −5.60 | 2.75 | −2.85 |
| 5a | 0.74 | −5.54 | 2.67 | −2.87 |
| 5b | 0.27 | −5.07 | 2.58 | −2.49 |
| 0.72 | −5.52 | |||
| 5c | 0.75 | −5.55 | 2.35 | −3.20 |
| 5d | 0.25 | −5.05 | 1.71 | −3.34 |
On the other hand, a fascinating relation can be observed when quadrupolar dyes, 3a, 4a and 5a, are compared. Increasing the distance between the electron-rich core and the accepting nitrile group in one-photon spectroscopy leads to a gradual redshift of the emission spectra (see above). Yet the direction of the impact of the CN moiety completely differs for the ground electronic state. Upon increasing the length between the donor and acceptor subunits, the HOMO undergoes significant destabilization, up to −5.54 eV for 5a, which is only 0.03 eV lower than HOMO energy of the benchmark 2a (Table 3). This indicates that, despite the conjugation between the electron-rich centre and the electron-withdrawing CN group, the influence of a nitrile moiety in the ground state is significantly weakened.
OLED device characteristics
To evaluate the applicability of the new compounds in optoelectronic devices,66–72 OLEDS were fabricated and characterized. Compound 2a was selected based on its favourable molecular design, characterized by a π-conjugated core integrating the dibenzo[b,f]oxazepine and 1,4-dihydropyrrolo[3,2-b]pyrrole frameworks. This hybrid structure confers high photophysical performance, including a large fluorescence quantum yield (Φfl ≈ 80%) and excellent colour purity. The energy levels of compound 2a, determined from electrochemical and spectroscopic studies, indicated compatibility with commonly used host materials, leading to the choice of CBP (4,4′-bis(N-carbazolyl)-1,1′-biphenyl) as the host. The device structure was designed carefully, considering energy alignment and charge transport properties. The optimized architecture comprised ITO (Indium Tin Oxide) as the anode for hole injection, NPB (N,N′-bis(naphthalene-1-yl)-N,N′-bis(phenyl)-benzidine) (30 nm) as the hole transport layer, TSBPA (4,4′-(diphenylsilanediyl)bis(N,N-diphenylaniline)) (10 nm) as a hole-blocking layer to confine excitons, 10% of compound 2a in CBP (20 nm) as the emissive layer, TPBi (2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)) (50 nm) as the electron transport layer, and LiF (1 nm)/Al (100 nm) as the cathode. The device architecture, depicted in Fig. 5a, was inspired by previously reported designs but tailored to maximize the performance of the DHPP-based emitter. This multilayer structure ensured effective charge injection, recombination, and exciton confinement, key to achieving high efficiency. The fabricated OLEDs demonstrated excellent performance. The electroluminescence spectrum, shown in Fig. 5b, revealed a peak emission at 440 nm, corresponding to pure blue light. Nevertheless, the Commission Internationale de l’Éclairage (CIE) chromaticity coordinates (Fig. 5c) of the device (x = 0.167, y = 0.127) confirmed the high colour purity of the emission, a critical feature for display applications. The current density–voltage and luminance characteristics, presented in Fig. 5d and f, highlight the device's efficient charge injection and light output. The OLED exhibited a low turn-on voltage of approximately 3.5 V and reached a luminance peak of 1285 cd m−2. These results underscore the suitability of compound 2a for energy-efficient light-emitting applications. However, significant roll-off in luminance and efficiency was observed at higher current densities, likely caused by triplet–triplet annihilation or charge imbalance. The device's external quantum efficiency (EQE), as shown in Fig. 5e and Table 4, reached a maximum of 3.59%, validating the high emissive efficiency of compound 2a. This performance is attributed to the molecular rigidity of 2a, which reduces non-radiative decay and its optimal energy alignment with the CBP host. Furthermore, the planar and π-conjugated structure of 2a facilitates effective charge transport and exciton generation, contributing to its high EQE. The design and performance of compound 2a in the OLEDs are deeply rooted in its molecular structure. The dibenzo[b,f]oxazepine units introduce rigidity and prevent vibrational relaxation, while the central DHPP core contributes to its electron-donating properties. Combined with a well-matched host–guest energy alignment, these structural features result in efficient exciton formation and radiative recombination. Despite these successes, efficiency roll-off at voltages exceeding 9 V remains challenging, likely due to bimolecular quenching processes. Future work could address these issues by optimizing the layer thicknesses, introducing advanced host materials, or employing tandem architectures to mitigate exciton–polaron interactions. | ||
| Fig. 5 OLED device characteristic: (a) schematic of an OLED device structure with a 2a active layer, (b) electroluminescence spectrum of the studied device, (c) CIE 1931 chromaticity diagram indicating blue emission, (d) current density – potential, (e) external quantum efficiency-current density and (f) luminance-current density characteristics of the studied device. | ||
| Device | Turn-on voltage [V] | λem [nm] | Lmax [cd m−2] | ηmax [cd A−1] | CIE coordinates (x, y) | EQE (%) | ||
|---|---|---|---|---|---|---|---|---|
| Max | 100 cd m−2 | 1000 cd m−2 | ||||||
| a λem – emission wavelength, Lmax – maximal luminance, ηmax – current efficiency, and EQE – external quantum yield. | ||||||||
| 2a | 3.5 | 440 | 1285 | 13.01 | 0.168, 0.127 | 3.59 | 3.21 | 1.02 |
Conclusions
We have proven for the first time that cyclic imines can be transformed into 1,4-dihydropyrrolo[3,2-b]pyrroles as long as imine functionality is not embedded into an aromatic core. The resulting electron-rich dyes possessing two dibenzo[b,f]oxazepine scaffolds fused with a DHPP core are rigid and more planar compared to typical tetraarylpyrrolo[3,2-b]pyrroles. Their unusual, curved yet rather rigid architecture is responsible for appreciable photophysical properties which include large fluorescence quantum yields exceeding 80% in almost all cases regardless of the substitution pattern. Moreover rigidification also results in more narrow emission spectra and higher colour purity for the non-substituted, parent blue-emitting dye. Moreover, peripheral functionalization with strong electron-accepting moieties bathochromically shifts emission toward the near-infrared region (NIR). The optoelectronic device performance results demonstrate the potential of compound 2a as a highly efficient blue emitter for OLED applications. The combination of excellent photophysical properties, molecular stability, and device performance of this DHPP-based compound exemplifies its efficiency in blue emission and device compatibility, paving the way for novel OLED technologies. Future work will explore structural diversification to broaden the application scope of DHPP-based materials in optoelectronics.Data availability
The data supporting the article entitled ‘1,4-Dihydropyrrolo[3,2-b]pyrrole modified with dibenzoxazepine: A Highly Efficient Core for Charge-Transfer-Based OLED Emitters’ have been included as part of the ESI.†Author contributions
Conceptualization: K. G.; investigation: K. G., J. L., S. S., and D. J.; supervision: D. T. G., D. J., and P. D.; visualization: K. G., S. S., J. L., and D. J.; writing – original draft: K. G., S. S., D. J., and D. T. G.; writing – review & editing: K. G., S. S., P. D.; D. J., and D. T. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Polish National Science Center, Poland (grants OPUS 2020/37/B/ST4/00017, 2022/45/B/ST5/03712 and HARMONIA 2018/30/M/ST5/00460). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowka-Curie grant agreement no. 101007804. This research used resources from the GLiCID Computing Facility (Ligerien Group for Intensive Distributed Computing, https://doi.org/10.60487/glicid, Pays de la Loire, France). J. L. was the Doctoral Candidate in the Interdisciplinary Doctoral School at the Łódź University of Technology, Poland. We thank Joseph Milton for amending the manuscript.Notes and references
- K. Yamamoto, T. Harada, M. Nakazaki, T. Naka, Y. Kai, S. Harada and N. Kasai, J. Am. Chem. Soc., 1983, 105, 7171–7172 CrossRef CAS.
- I. R. Márquez, N. Fuentes, C. M. Cruz, V. Puente-Muñoz, L. Sotorrios, M. L. Marcos, D. Choquesillo-Lazarte, B. Biel, L. Crovetto, E. Gómez-Bengoa, M. T. González, R. Martin, J. M. Cuerva and A. G. Campaña, Chem. Sci., 2017, 8, 1068–1074 RSC.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- S. K. Pedersen, K. Eriksen and M. Pittelkow, Angew. Chem., Int. Ed., 2019, 58, 18419–18423 CrossRef CAS PubMed.
- A. Caruso, M. A. Siegler and J. D. Tovar, Angew. Chem., Int. Ed., 2010, 49, 4213–4217 CrossRef CAS PubMed.
- R. E. Messersmith, S. Yadav, M. A. Siegler, H. Ottosson and J. D. Tovar, J. Org. Chem., 2017, 82, 13440–13448 CrossRef CAS PubMed.
- K. Kato, K. Takaba, S. Maki-Yonekura, N. Mitoma, Y. Nakanishi, T. Nishihara, T. Hatakeyama, T. Kawada, Y. Hijikata, J. Pirillo, L. T. Scott, K. Yonekura, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2021, 143, 5465–5469 CrossRef CAS PubMed.
- J. Liu, S. Mishra, C. A. Pignedoli, D. Passerone, J. I. Urgel, A. Fabrizio, T. G. Lohr, J. Ma, H. Komber, M. Baumgarten, C. Corminboeuf, R. Berger, P. Ruffieux, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 12011–12020 CrossRef CAS PubMed.
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC.
- S. Qiu, A. C. Valdivia, W. Zhuang, F.-F. Hung, C.-M. Che, J. Casado and J. Liu, J. Am. Chem. Soc., 2024, 146, 16161–16172 CrossRef CAS PubMed.
- C. M. Cruz, I. R. Márquez, I. F. A. Mariz, V. Blanco, C. Sánchez-Sánchez, J. M. Sobrado, J. A. Martín-Gago, J. M. Cuerva, E. Maçôas and A. G. Campaña, Chem. Sci., 2018, 9, 3917–3924 RSC.
- R. Li, B. Ma, S. Li, C. Lu and P. An, Chem. Sci., 2023, 14, 8905–8913 RSC.
- K. Górski, J. Mech-Piskorz, B. Leśniewska, O. Pietraszkiewicz and M. Pietraszkiewicz, J. Org. Chem., 2020, 85, 4672–4681 CrossRef PubMed.
- K. Górski, J. Mech-Piskorz, K. Noworyta, B. Leśniewska and M. Pietraszkiewicz, New J. Chem., 2018, 42, 5844–5852 RSC.
- M. Más-Montoya, L. Usea, A. E. Ferao, M. F. Montenegro, C. Ramírez de Arellano, A. Tárraga, J. N. Rodríguez-López and D. Curiel, J. Org. Chem., 2016, 81, 3296–3302 CrossRef PubMed.
- T. Fujikawa, Y. Segawa and K. Itami, J. Org. Chem., 2017, 82, 7745–7749 CrossRef CAS PubMed.
- D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D. Yang, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS PubMed.
- M. Żyła, E. Gońka, P. J. Chmielewski, J. Cybińska and M. Stępień, Chem. Sci., 2016, 7, 286–294 RSC.
- S. Stecko and D. T. Gryko, JACS Au, 2022, 2, 1290–1305 CrossRef CAS PubMed.
- D. H. Friese, A. Mikhaylov, M. Krzeszewski, Y. M. Poronik, A. Rebane, K. Ruud and D. T. Gryko, Chem.–Eur. J., 2015, 21, 18364–18374 CrossRef CAS PubMed.
- M. Tasior, G. Clermont, M. Blanchard-Desce, D. Jacquemin and D. T. Gryko, Chem.–Eur. J., 2019, 25, 598–608 CrossRef CAS PubMed.
- Y. Ji, Z. Peng, B. Tong, J. Shi, J. Zhi and Y. Dong, Dyes Pigm., 2017, 139, 664–671 CrossRef CAS.
- K. Li, Y. Liu, Y. Li, Q. Feng, H. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258–7267 RSC.
- Y. Ma, Y. Zhang, L. Kong and J. Yang, Molecules, 2018, 23, 3255 CrossRef PubMed.
- B. Dereka and E. Vauthey, Chem. Sci., 2017, 8, 5057–5066 RSC.
- J.-Y. Wu, C.-H. Yu, J.-J. Wen, C.-L. Chang and M. Leung, Anal. Chem., 2016, 88, 1195–1201 CrossRef CAS PubMed.
- M. Tasior, O. Vakuliuk, A. Wrzosek, V. I. Vullev, A. Szewczyk, D. Jacquemin and D. T. Gryko, ACS Org. Inorg. Au, 2024, 4, 248–257 CrossRef CAS PubMed.
- J. Wang, Z. Chai, S. Liu, M. Fang, K. Chang, M. Han, L. Hong, H. Han, Q. Li and Z. Li, Chem.–Eur. J., 2018, 24, 18032–18042 CrossRef CAS PubMed.
- R. Domínguez, N. F. Montcada, P. de la Cruz, E. Palomares and F. Langa, Chempluschem, 2017, 82, 1096–1104 CrossRef PubMed.
- Y. Zhou, M. Zhang, J. Ye, H. Liu, K. Wang, Y. Yuan, Y.-Q. Du, C. Zhang, C.-J. Zheng and X.-H. Zhang, Org. Electron., 2019, 65, 110–115 CrossRef CAS.
- P. P. Abatti, N. O. Decarli, S. Gogoc, P. Data, I. H. Bechtold, E. Westphal and H. Gallardo, ChemPlusChem, 2023, 88, e202300539 CrossRef CAS PubMed.
- M. Tasior, O. Vakuliuk, D. Koga, B. Koszarna, K. Górski, M. Grzybowski, Ł. Kielesiński, M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2020, 85, 13529–13543 CrossRef CAS PubMed.
- M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Chem. Sci., 2023, 14, 2353–2360 RSC.
- M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Angew. Chem., Int. Ed., 2021, 133, 15125–15132 CrossRef.
- M. Tasior, P. Kowalczyk, M. Przybył, M. Czichy, P. Janasik, M. H. E. Bousquet, M. Łapkowski, M. Rammo, A. Rebane, D. Jacquemin and D. T. Gryko, Chem. Sci., 2021, 12, 15935–15946 RSC.
- N. I. Petrenko, M. M. Kozlova, T. N. Gerasimova and J. Fluor, Chem, 1987, 36, 93–98 CAS.
- P. K. Gutch and J. Acharya, Heterocycl. Commun., 2007, 13, 393–396 CAS.
- Y. R. Jorapur, G. Rajagopal, P. J. Saikia and R. R. Pal, Tetrahedron Lett., 2008, 49, 1495–1497 CrossRef CAS.
- Y. Lin, N. Li and Y. Cherng, J. Heterocycl. Chem., 2014, 51, 808–814 CrossRef CAS.
- M. Ghafarzadeh, E. S. Moghadam and F. Faraji, J. Heterocycl. Chem., 2013, 50, 754–757 CrossRef CAS.
- F. M. García-Valle, V. Tabernero, T. Cuenca, M. E. G. Mosquera and J. Cano, Organometallics, 2019, 38, 894–904 CrossRef.
- A. W. H. Wardrop, G. L. Sainsbury, J. M. Harrison and T. D. Inch, J. Chem. Soc., Perkin Trans. 1, 1976, 563, 1279–1285 RSC.
- W. Hu, F. Teng, H. Hu, S. Luo and Q. Zhu, J. Org. Chem., 2019, 84, 6524–6535 CrossRef CAS PubMed.
- D. Tsvelikhovsky and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 14228–14231 CrossRef CAS PubMed.
- P. P. M. A. Dols, B. J. B. Folmer, H. Hamersma, C. W. Kuil, H. Lucas, L. Ollero, J. B. M. Rewinkel and P. H. H. Hermkens, Bioorg. Med. Chem. Lett., 2008, 18, 1461–1467 CrossRef CAS PubMed.
- E. A. Hallinan, T. J. Hagen, R. K. Husa, S. Tsymbalov, S. N. Rao, J. P. VanHoeck, M. F. Rafferty, A. Stapelfeld, M. A. Savage and M. Reichman, J. Med. Chem., 1993, 36, 3293–3299 CrossRef CAS PubMed.
- J. M. Klunder, K. D. Hargrave, M. West, E. Cullen, K. Pal, M. L. Behnke, S. R. Kapadia, D. W. McNeil, J. C. Wu, G. C. Chow and J. Adams, J. Med. Chem., 1992, 35, 1887–1897 CrossRef CAS PubMed.
- M. Binaschi, A. Boldetti, M. Gianni, C. A. Maggi, M. Gensini, M. Bigioni, M. Parlani, A. Giolitti, M. Fratelli, C. Valli, M. Terao and E. Garattini, ACS Med. Chem. Lett., 2010, 1, 411–415 CrossRef CAS PubMed.
- H. Umemiya, H. Fukasawa, M. Ebisawa, L. Eyrolles, E. Kawachi, G. Eisenmann, H. Gronemeyer, Y. Hashimoto, K. Shudo and H. Kagechika, J. Med. Chem., 1997, 40, 4222–4234 CrossRef CAS PubMed.
- T. A. Ban, M. Fujimori, W. M. Petrie, M. Ragheb and W. H. Wilson, Int. Pharmacopsychiatr., 1982, 17, 18–27 CrossRef CAS PubMed.
- Y. Ren, Y. Wang, S. Liu and K. Pan, ChemCatChem, 2014, 6, 2985–2992 CrossRef CAS.
- A. Janiga, E. Glodkowska-Mrowka, T. Stoklosa and D. T. Gryko, Asian J. Org. Chem., 2013, 2, 411–415 CrossRef CAS.
- R. Stężycki, M. Grzybowski, G. Clermont, M. Blanchard-Desce and D. T. Gryko, Chem.–Eur. J., 2016, 22, 5198–5203 CrossRef PubMed.
- K. Górski, J. Mech-Piskorz, B. Leśniewska, O. Pietraszkiewicz and M. Pietraszkiewicz, J. Org. Chem., 2019, 84, 11553–11561 CrossRef PubMed.
- K. Górski, T. Ostojić, M. Banasiewicz, E. T. Ouellette, L. Grisanti and D. T. Gryko, Chem.–Eur. J., 2023, 29, e2022034 CrossRef PubMed.
- M. Krzeszewski, B. Thorsted, J. Brewer and D. T. Gryko, J. Org. Chem., 2014, 79, 3119–3128 CrossRef CAS PubMed.
- K. Górski, D. Kusy, S. Ozaki, M. Banasiewicz, R. Valiev, S. R. Sahoo, K. Kamada, G. Baryshnikov and D. T. Gryko, J. Mater. Chem. C, 2024, 12, 1980–1987 RSC.
- B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- T. Kim, J. Kim, H. Mori, S. Park, M. Lim, A. Osuka and D. Kim, Phys. Chem. Chem. Phys., 2017, 19, 13970–13977 RSC.
- K. Górski, I. Deperasińska, G. V. Baryshnikov, S. Ozaki, K. Kamada, H. Ågren and D. T. Gryko, Org. Lett., 2021, 23, 6770–6774 CrossRef PubMed.
- Y. M. Poronik, G. V. Baryshnikov, I. Deperasińska, E. M. Espinoza, J. A. Clark, H. Ågren, D. T. Gryko and V. I. Vullev, Commun. Chem., 2020, 3, 190 CrossRef CAS PubMed.
- A. Janiga, D. Bednarska, B. Thorsted, J. Brewer and D. T. Gryko, Org. Biomol. Chem., 2014, 12, 2874–2881 RSC.
- K. Górski, K. Noworyta and J. Mech-Piskorz, RSC Adv., 2020, 10, 42363–42377 RSC.
- B. Golec, A. Gorski, R. P. Thummel, M. Sierakowski and J. Waluk, Photochem. Photobiol. Sci., 2023, 22, 333–344 CrossRef CAS PubMed.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- G. Meng, D. Zhang, J. Wei, Y. Zhang, T. Huang, Z. Liu, C. Yin, X. Hong, X. Wang, X. Zeng, D. Yang, D. Ma, G. Li and L. Duan, Chem. Sci., 2022, 13, 5622–5630 RSC.
- X. Li, J. Zhang, Z. Zhao, L. Wang, H. Yang, Q. Chang, N. Jiang, Z. Liu, Z. Bian, W. Liu, Z. Lu and C. Huang, Adv. Mater., 2018, 30, 1705005 CrossRef PubMed.
- J. Lee, H. F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92–98 CrossRef CAS PubMed.
- S. Nam, J. W. Kim, H. J. Bae, Y. M. Maruyama, D. Jeong, J. Kim, J. S. Kim, W. Son, H. Jeong, J. Lee, S. Ihn and H. Choi, Adv. Sci., 2021, 8, 2100586 CrossRef CAS PubMed.
- X. Cai and S. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- S. Izawa, M. Morimoto, K. Fujimoto, K. Banno, Y. Majima, M. Takahashi, S. Naka and M. Hiramoto, Nat. Commun., 2023, 14, 5494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, UV-Vis spectra, cyclic voltammograms, OLED fabrication details and characteristics, computational details, Cartesian coordinates of ground and excited states, and X-ray structure details. CCDC 2364052 (2e). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04272g |
| This journal is © The Royal Society of Chemistry 2025 |