
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmide cyclodextrin that recognises monophosphate anions in harmony with water molecules†
Takashi
Nakamura
 *a,
Hayato
Takayanagi
b,
Masaki
Nakahata
*c,
Takumi
Okubayashi
d,
Hitomi
Baba
e,
Yoshiki
Ishii
f,
Go
Watanabe
*efg,
Daisuke
Tanabe
d and
Tatsuya
Nabeshima
a
*a,
Hayato
Takayanagi
b,
Masaki
Nakahata
*c,
Takumi
Okubayashi
d,
Hitomi
Baba
e,
Yoshiki
Ishii
f,
Go
Watanabe
*efg,
Daisuke
Tanabe
d and
Tatsuya
Nabeshima
a
aInstitute of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan. E-mail: nakamura@chem.tsukuba.ac.jp
bDegree Programs in Pure and Applied Sciences, Graduate School of Science and Technology, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan
cGraduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan. E-mail: nakahata@chem.sci.osaka-u.ac.jp
dSchool of Science and Engineering, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan
eSchool of Science, Kitasato University, 1-15-1 Kitazato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan
fSchool of Frontier Engineering, Kitasato University, 1-15-1 Kitazato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan. E-mail: go0325@kitasato-u.ac.jp
gKanagawa Institute of Industrial Science and Technology, 705-1 Shimoimaizumi, Ebina, Kanagawa 243-0435, Japan
First published on 4th November 2024
Abstract
Anion recognition in water by synthetic host molecules is a popular and challenging topic. It has been considered difficult because the water molecules compete for the recognition units. In this study, we have successfully created a novel macrocycle that achieves precise recognition through multipoint hydrogen bonding in harmony with water molecules. Specifically, an N-methylpyridinium amide β-cyclodextrin (β-CD) derivative 1(OTf)7 was synthesized, whose amide groups are directly attached to each pyranose ring. The pyridinium amide CD encapsulated a monophosphate anion in water, but it did not show interactions with sulfonates or carboxylates, thus a remarkable selectivity was demonstrated. Two monophosphates with different substituents, phenyl phosphate (PhOPO32−) and adamantyl phosphate (AdOPO32−), exhibited interesting contrasting pictures in the inclusion process, which were revealed by a combination of NOESY experiments, ITC measurements, and MD simulations. PhOPO32− was positioned slightly “upper” (closer to the pyridinium amide side) in 17+ with the oxygen atom of the phosphate ester R–O–P involved in the hydrogen bonds with the amide N–H, and configurational entropy plays a key role in the inclusion. Meanwhile, AdOPO32− was positioned “lower” (closer to the methoxy rim of CD) with the terminal –PO32− forming hydrogen bonds with the amides, and the hydrophobic effect is a major contributing driving force of the inclusion. The molecular design presented herein to achieve the precise recognition in water and clarification of the detailed mechanisms including the hydration phenomenon greatly contribute to the development of functional molecules that work in aqueous environments.
Introduction
Hydrogen bonds are widely employed in the recognition units of synthetic receptors because of their directionality and reversibility.1–3 In particular, amide N–H groups are useful in the design of anion receptors, not only for their hydrogen bonding ability with anions but also for their synthetic ease.4,5 Various artificial receptors have been developed for anion recognition, but many of them have been investigated in organic solvents. On the other hand, one major target has been to achieve the precise recognition of anions6–11 and other molecules12–22 in water. There is an intrinsic difficulty to molecular recognition by hydrogen bonding in water, which stems from the hydration and competition with water molecules of the guest and recognition units, and from the fact that electrostatic interactions are weaker in water, a solvent with a high dielectric constant. In this context, new molecular designs to overcome these challenges need to be developed. Moreover, understanding the mechanism of the recognition process, such as the hydrogen bonding pattern, the number of involved water molecules, and the binding energy components, would contribute to establishing an integrated scientific view and design guideline for functional materials that work in aqueous environments.The combination of experimental and theoretical methods is effective in investigating the binding modes and hydration of artificial host molecules. 1H NMR measurements are used to evaluate the state of hydrogen bonds between the recognition units and a target guest from their chemical shifts. Furthermore, rich structural information offered by NOESY NMR accurately determines the position of the bound molecule together with the orientation of the functional groups in solution, and thereby delineates the detailed hydrogen bonding patterns of the host and guest molecules.23–25 Isothermal titration calorimetry (ITC) is an important method that allows the discussion about not only the enthalpy aspects, such as the strength of bonding between host and guest, but also the entropy aspects, such as the change in the structural degrees of freedom associated with the bonding and hydration/dehydration processes.21,26 The molecular perspectives regarding the thermodynamics of host–guest chemistry have been further investigated in biophysics and supramolecular science using molecular dynamics (MD) simulations,27–29 as well as experimental techniques. The potential of mean force (PMF) is a representative quantity to describe the binding mechanism of a host–guest pair with a specific reaction coordinate by following the theory of statistical mechanics.30,31 The energetic property can be equivalent to a free energy in the elementary process of host–guest chemistry. Quantitative agreement of energetics between experiments and simulations is sometimes challenging because of the simplification of host–guest binding thermodynamics as the elementary process in the MD simulation30 as well as the theoretical validity of intermolecular interaction in condensed phases.32–35 Nevertheless, computational chemistry plays a key role in the bridging of the structural and energetic properties of the host, guest, and their complex molecules to macroscopic quantities obtained through various experimental measurements.35–40
We now report the synthesis, structure, and anion recognition properties of N-methylpyridinium amide cyclodextrin 17+ (Fig. 1a) in harmony with water molecules. 17+ was designed to be obtained by the methylation of 3-pyridylamide cyclodextrin 2. We have previously reported cyclodextrin derivatives whose multiple amide groups are directly attached to cyclic oligopyranose frameworks.23–25 They work as receptors to capture anionic species in organic solvents. For example, the p-tolylamide derivative 3 selectively bound hydrogen phosphonate (RPO3H−) anions in solvents such as dichloromethane, chloroform, acetonitrile, and DMSO (Fig. 1b). In contrast to the strong inclusion of monophosphonate and monophosphate anions, halide ions did not show any interaction with the amide cyclodextrin, and carboxylates and sulphonates were only weakly bound. Thus, its remarkable recognition ability was demonstrated.23 Compared to other examples of the native and modified cyclodextrins to bind anions,26,41–43 our amide cyclodextrin derivatives have a carboxamide group directly attached at the 5 position for each of the 7 pyranose rings. With this design, the amide groups can direct inside the macrocycle's cavity and exert multipoint hydrogen bonds to the included species. In an aqueous mixed solvent (10% water–90% acetonitrile (vol%)), however, the host–guest complex of the p-tolylamide derivative 3 dissociated.23 Thus, like other examples of hydrogen-bonding receptors, the recognition of anions in water is a challenge with this amide cyclodextrin receptor.
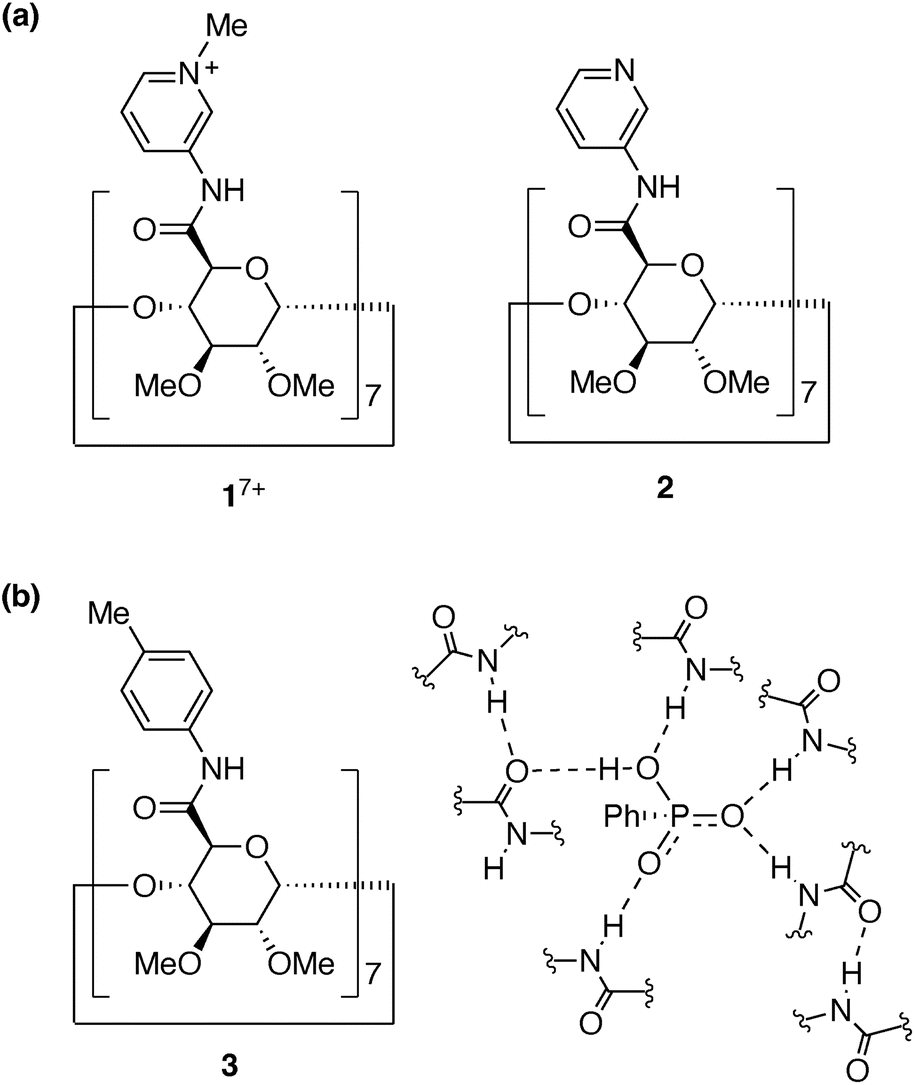 | ||
| Fig. 1 (a) Structures of pyridinium and pyridyl amide cyclodextrins, 17+ and 2. (b) Structure of p-tolylamide cyclodextrin 3 and recognition of PhPO3H− by multipoint hydrogen bonds in the previous study.23 | ||
In order to generate the anion binding properties in water, we postulated that the introduction of cationic and electron-deficient groups on the amide substituents would strengthen the multipoint interactions with the target anions. Then, the pyridinium amide cyclodextrin 1(OTf)7 was designed. N-Methyl-3-pyridylamide44,45 was chosen in anticipation of its strong interactions with anions, as well as its stability compared to the methyl-pyridiniums at the other substituted positions. In this study, it has been found that 1(OTf)7 selectively recognises monophosphate anions in water. To delineate the detailed picture of the anion recognition mechanism, we have focused on two monophosphate anions with different substituents, PhOPO32− (G12−) and AdOPO32− (G22−), in the latter half of this study. Intriguingly, the combination of a structural analysis by 1H–1H NOESY experiments, a thermodynamic analysis by ITC measurements, and a computational analysis by MD simulations, has revealed that the recognition mode, the hydration behaviours, and the driving force of inclusion significantly varied between the two phosphate anions in spite of their slight difference in the hydrophobic group.
Results and discussion
Synthesis and structures of pyridinium amide cyclodextrin
3-Pyridylamide cyclodextrin 2 was synthesized by the condensation reaction of per(5-carboxy-5-dehydroxymethyl)-β-cyclodextrin46 and 3-aminopyridine (Scheme S1, Fig. S1, S2 and S10, ESI†). 2 was isolated in good yield (79%), given the seven-point reaction and the weak nucleophilicity of 3-aminopyridine due to the electron-deficient nature of the 3-pyridyl group. Next, the seven 3-pyridyl rings of 2 were methylated. Several sets of conditions were investigated, and MeOTf as a methylating reagent and DMF as a solvent resulted in complete conversion. As the combination of MeOTf/DMF gave the O-methylated amidium salt of DMF47 as a byproduct, 1(OTf)7 was purified by size exclusion chromatography followed by reprecipitation. 1(OTf)7 was characterized by 1H, 13C, 19F NMR, ESI-MS, IR, and elemental analysis (Scheme S2, Fig. S3–S9 and S11, ESI†). The ESI-TOF mass measurement of 1(OTf)7 confirmed that all the seven pyridyl groups were methylated, and that the counter anion is TfO− (Fig. S9, ESI†).The NMR spectra of 1(OTf)7 were relatively sharp in CD3OD, and all the 1H and 13C signals were assigned using the 1H–1H COSY, NOESY, 1H–13C HSQC, and HMBC measurements (Fig. S5–S8, ESI†). However, the 1H NMR measurement of 1(OTf)7 in D2O at 298 K gave broad signals. To investigate the reason for the broad signals, the 1H NMR spectra of the D2O solution of 1(OTf)7 were measured at different temperatures (Fig. S12, ESI†). The signals coalesced at 363 K, and they correspond to the structure of 17+ with seven-fold symmetry. Meanwhile, at 278 K, the signals became sharper than those at 298 K, but many signals with low intensities were also observed. This suggests the existence of the C1-symmetric self-inclusion conformer in which one of the aromatic substituents is tilted inside.23,48,49 This self-inclusion conformer was also supported from the observation of the amide N–H protons. Fig. 2a and b show the comparisons of the NMR spectra of 1(OTf)7 at 278 K in D2O and H2O/D2O = 9/1 (WATERGATE). The signals at 10.5–11.6 ppm were observed only in H2O/D2O = 9/1, and are thus assigned to the exchangeable amide N–H protons of 1(OTf)7. There are 8 N–H signals (h), one relatively large signal (filled triangle in Fig. 2b) and seven small signals with equal intensities (open squares in Fig. 2b). The one large signal was assigned to the C7-symmetric conformer in which all the substituents are out, and the seven small signals were assigned to the C1-symmetric self-included conformer. The diffusion coefficients D [m2 s−1] obtained from a 1H DOSY measurement at 278 K (D2O) are similar for these two species (Fig. S14, ESI†). The observed values are log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −10.0 for the C7 conformer and logD = −10.1 for the C1 conformer. The results discussed above as well as the fact that 1(OTf)7 exhibits different 1H NMR spectra in different solvents (Fig. S13, ESI†) all support the fact that there are two conformers, not a monomer and assembled products. The ratio of these two conformers was evaluated to be C7:C1 ∼ 4:6 (278 K, D2O) based on the 1H integral ratio.
D = −10.0 for the C7 conformer and logD = −10.1 for the C1 conformer. The results discussed above as well as the fact that 1(OTf)7 exhibits different 1H NMR spectra in different solvents (Fig. S13, ESI†) all support the fact that there are two conformers, not a monomer and assembled products. The ratio of these two conformers was evaluated to be C7:C1 ∼ 4:6 (278 K, D2O) based on the 1H integral ratio.
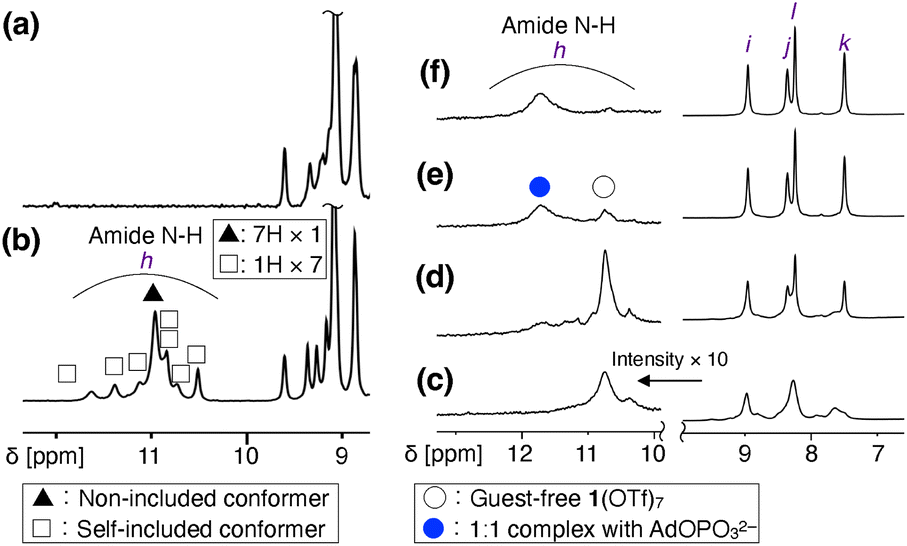 | ||
| Fig. 2 Investigation of structures of 1(OTf)7 and change in amide N–H signals upon binding a monophosphate. (a and b) Comparison of 1H NMR spectra of 1(OTf)7 in D2O (a) and H2O/D2O = 9/1 (b, WATERGATE) ([1(OTf)7] = 2 mM, 600 MHz, 278 K). (c–f) 1H NMR titration experiment for AdOPO3Na2 (G2Na2) against 1(OTf)7 in H2O/D2O = 9/1 ([1(OTf)7] = 2 mM, 600 MHz, 298 K, WATERGATE). Amide N–H and aromatic regions are shown. (c) 1(OTf)7. (d–f) 1(OTf)7 + AdOPO3Na2. (d) AdOPO3Na2 0.5 equiv.; (e) 1.0 equiv.; (f) 1.5 equiv. [/1(OTf)7]. | ||
Inclusion of phosphate anions
The anion recognition properties of 1(OTf)7 in water were then investigated by NMR measurements. When PhOPO3Na2 (G1Na2) was titrated into the D2O solution of 1(OTf)7, a new set of 1H NMR signals appeared in the 0–1 equiv. range (Fig. 3a–e and S15, ESI†). This result suggested the formation of a 1:1 complex between 17+ and PhOPO32−. The formation/dissociation is slow on the 1H NMR timescale, which is in contrast to the usual cyclodextrin-guest complexation driven by the hydrophobic effect. The slow exchange allows evaluation of the 1H integral ratio between the pyranose moiety of 17+ and aromatic proton of PhOPO32−, which supports the 1:1 composition of the host–guest complex.
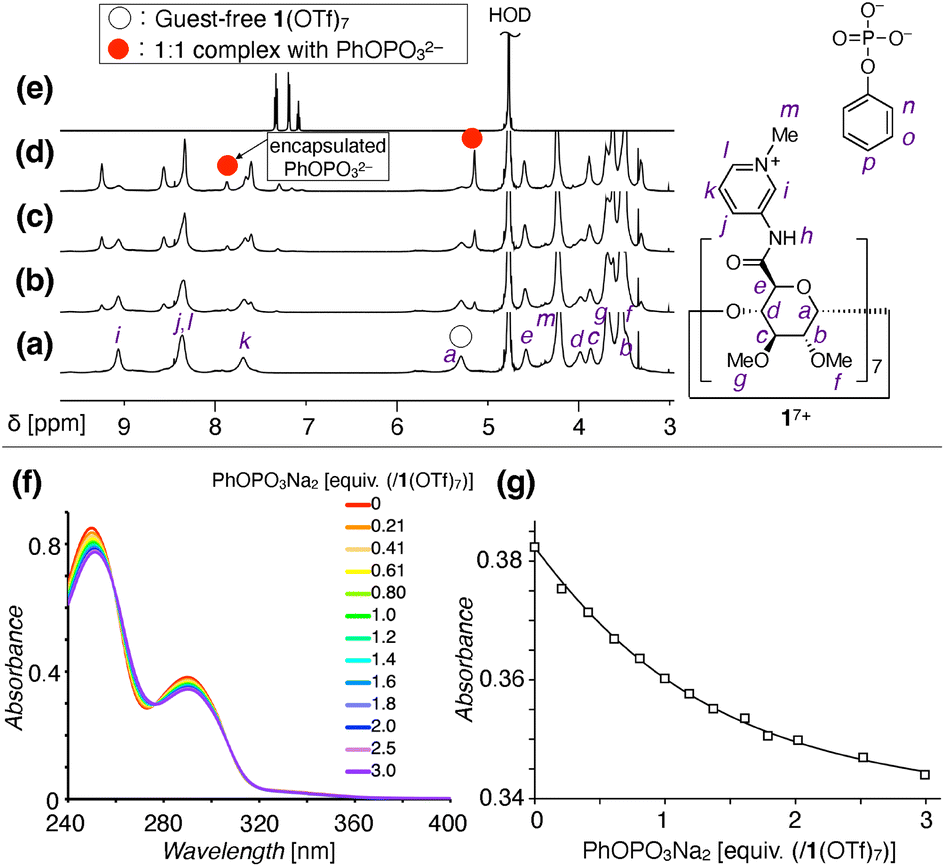 | ||
| Fig. 3 Binding of PhOPO3Na2 (G1Na2) and 1(OTf)7. (a–e) 1H NMR titration experiment of PhOPO3Na2 against 1(OTf)7 in D2O ([1(OTf)7] = 10 mM, 600 MHz, D2O, 298 K). Aromatic and sugar regions are shown. (a) 1(OTf)7. (b–d) 1(OTf)7 + PhOPO3Na2. (b) PhOPO3Na2 0.25 equiv.; (c) 0.50 equiv.; (d) 1.0 equiv. [/1(OTf)7]. (e) PhOPO3Na2. (f and g) UV-vis titration experiment of PhOPO3Na2 against 1(OTf)7 ([1(OTf)7] = 12 μM, H2O, 298 K). (f) UV-vis absorbance spectra. (g) A least squares fitting to determine the binding constant K (data at λabs = 290 nm). | ||
31P NMR observation during the course of the titration experiments revealed a signal assigned to the encapsulated PhOPO32− at δ = −2.6 ppm, which is shifted upfield compared to the free PhOPO32− (δ = 0 ppm) (Fig. S16, ESI†). Meanwhile, the 19F NMR signal of TfO− as a counter anion of 17+ did not change before and after the inclusion of PhOPO32−, thus TfO− is not suggested to be encapsulated inside the cavity of 17+ (Fig. S17, ESI†). The UV-vis and circular dichroism spectra of 1(OTf)7 also changed in the presence of PhOPO3Na2, indicating some conformational changes upon the inclusion of PhOPO32− (Fig. 3f and S18, ESI†). The binding constant K [M−1] of 1(OTf)7 with PhOPO3Na2 in H2O was determined to be logK = 5.10 ± 0.05 (Fig. 3g).
Involvement of the amide N–H in the anion binding was investigated by 1H NMR measurement in H2O/D2O = 9/1. PhOPO3Na2 was initially investigated as a guest, but the amide protons disappeared upon complexation, possibly due to the exchange process having a similar timescale to the NMR (Fig. S19, ESI†). The use of another phosphate, AdOPO3Na2 (G2Na2, see Schemes S3–S5 and Fig. S20–S25 in the ESI† for its synthesis and characterization), led to their successful observation (Fig. 2c–f, S26 and S27, ESI†). The amide N–H signals of the guest-free 1(OTf)7 were observed as a broad signal at 10.74 ppm, but the N–H signals assigned to the inclusion complex of AdOPO32− were observed at 11.72 ppm. This downfield shift supports the existence of hydrogen bonds of the N–H group with the –PO32− group of AdOPO32−.
Structural analysis by 1H–1H NOESY
1H–1H NOESY NMR provided important information and intriguing insights about the structures of the inclusion complexes (Fig. 4). 1H–1H NOESY measurement of the sample of 1(OTf)7 and PhOPO3Na2 revealed correlations between the protons of Ph (n–p in Fig. 4a) and those of the 3- and 5-positions of the pyranose ring as well as that of the methoxy group at the 3-position (c, e, and g in Fig. 4a). This supports the fact that the Ph group of PhOPO32− is included inside the cavity, since the protons c and e are positioned at the internal face of the cyclodextrin torus. As only the ortho proton of Ph (n) showed NOE correlations with the proton at the 5-position of pyranose (e), the direction of PhOPO32− was determined as depicted in Fig. 4a with its –OPO32− group positioned near the amide N–H groups. The NOE correlation pattern between the Ph group and the pyranose ring as well as the absence of NOE between the Ph group and the methyl group of the pyridinium (m) also supported the conclusion that the anionic –OPO32− groups are not near the cationic N-methylpyridinium groups.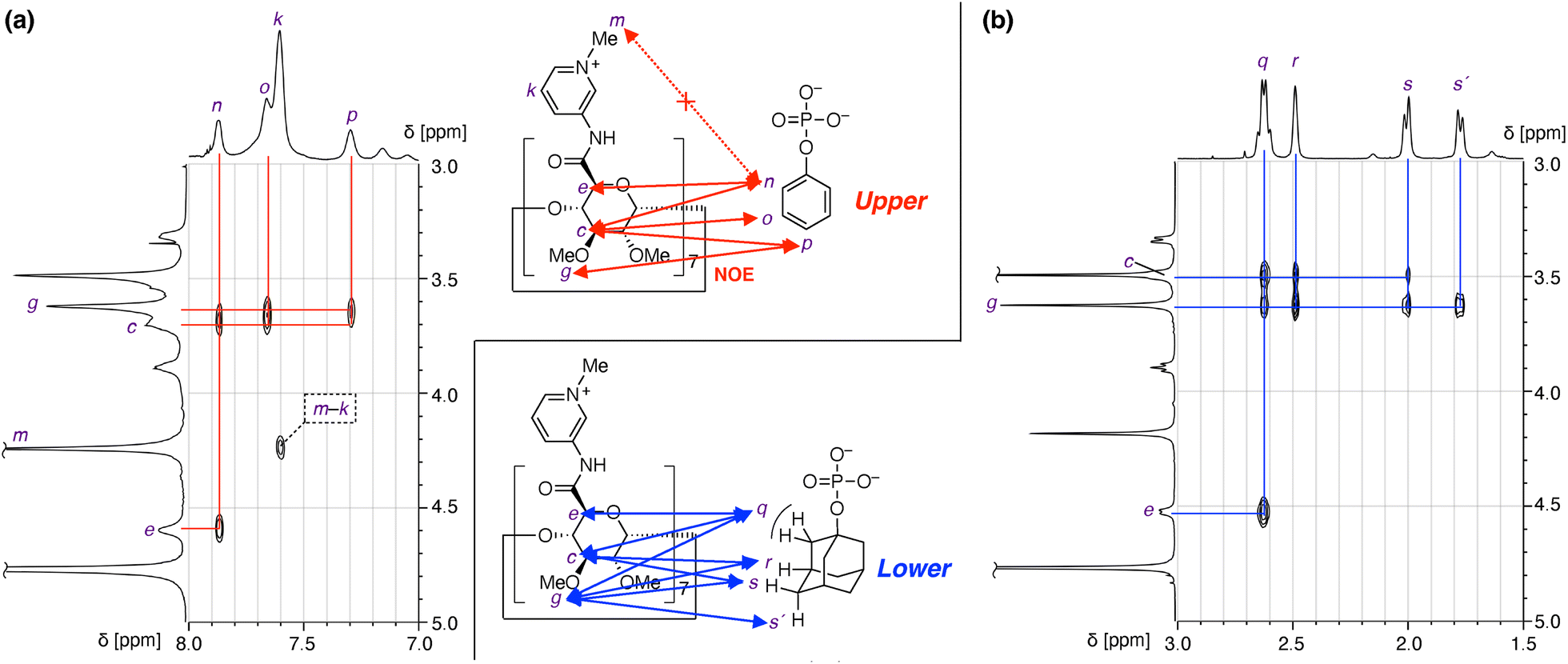 | ||
| Fig. 4 Structural investigation of inclusion complexes of 1(OTf)7 and phosphate anions by 1H–1H NOESY analysis (600 MHz, D2O, 298 K). (a) PhOPO3Na2 (G1Na2). (b) AdOPO3Na2 (G2Na2). | ||
A 1H–1H NOESY spectrum of the sample of 1(OTf)7 and AdOPO3Na2 is shown in Fig. 4b. As in the case of PhOPO32−, the NOE correlations confirmed the inclusion of AdOPO32− in the cyclodextrin cavity of 17+. A notable point is that the methoxy proton (g) showed NOE correlations to all the protons of the adamantyl group, including the methylene proton (q) adjacent to the phosphate group. This result suggested that the position of the encapsulated AdOPO32− is closer to the methoxy rim of the cyclodextrin cavity (“lower” when the orientation of 17+ is as in Fig. 4) than that of PhOPO32−.
Selectivity in molecular recognition
Next, the recognition properties of 1(OTf)7 were investigated with various guests using UV-vis titration (Fig. S29–S34, ESI†) and 1H NMR experiments (Fig. S35 and S36, ESI†). In order to quantitatively compare the binding constants K [M−1] (Fig. 5), the MES (2-morpholinoethanesulfonic acid) buffer at pH 7.0 was used as a medium for the titration experiments. It was checked by 1H NMR that the interaction between MES and 1(OTf)7 was weak (Fig. S28, ESI†). In MES buffer (pH 7.0), the binding constant of PhOPO3Na2 (G1Na2) to 1(OTf)7 (12 μM) was logK = 4.71 ± 0.07, thus slightly lower than H2O (logK = 5.10 ± 0.05), but the inhibition effect was low.
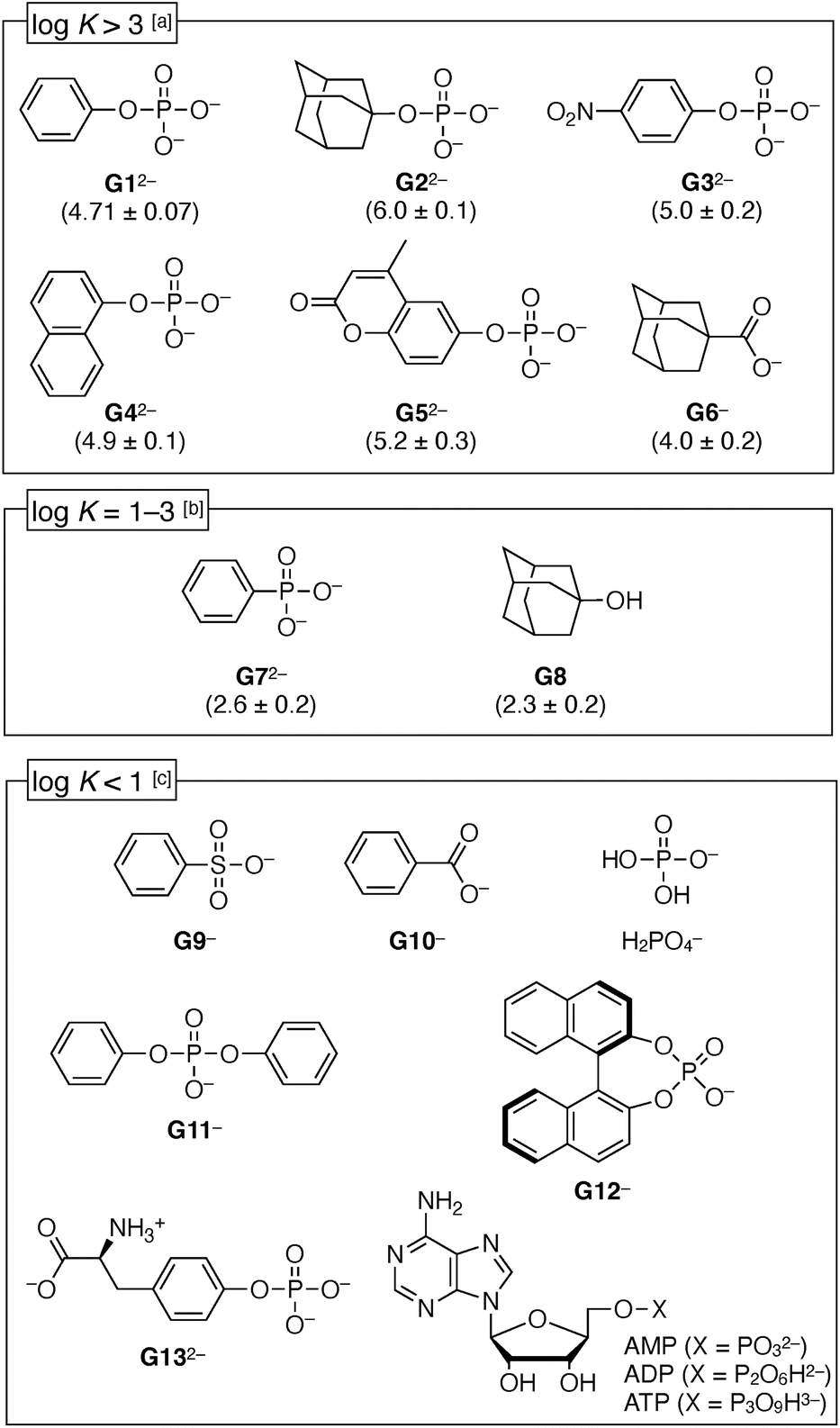 | ||
| Fig. 5 Structures of anions and categorization in terms of binding constant K [M−1] with 1(OTf)7. The logK value for each anion is shown in parentheses (error = SD (n = 3)). Conditions: [a] UV-vis titration (H2O, [1(OTf)7] = 12 μM, [MES buffer] = 0.38 mM, pH = 7.0). [b] 1H NMR titration (D2O, [1(OTf)7] = 1 mM, [MES buffer] = 32 mM, pH = 7.0 (pD = 7.5)). [c] 1H NMR titration (D2O or MES buffer in D2O (pH = 7.0 (pD = 7.5))). | ||
First, the aromatic phosphates (G32−–G52−) were investigated, and they exhibited similar high logK values (4.9–5.2) as G12−, regardless of the electron-deficient group or steric hindrance around the –OPO32− group. It was suggested that the –OPO32− group is an important factor for the anion binding. Meanwhile, benzenesulfonate (G9−) or benzoate (G10−) did not bind with 1(OTf)7, thus a remarkable selectivity depending on the functional groups has been demonstrated. Phenylphosphonate (G72−) was in the medium strength range (logK = 2.6 ± 0.2). Meanwhile, the inorganic phosphate (H2PO4−) and hydrophilic phosphates (G13−, AMP, ADP, ATP) were not bound, so it is certain that the hydrophobic effect plays a partial role. Diphenyl phosphate (G11−) and binaphthyl phosphate G12− were also not bound, probably because their bulkiness hindered access of the phosphate group to the amides inside the cavity of the cyclodextrin 17+. The highest value among the investigated guests was observed for AdOPO32− (G22−) with logK = 6.0 ± 0.1. In the case of the adamantane derivatives, the contribution of the hydrophobic effect is apparently higher, as can be seen from the fact that the adamantyl carboxylate G6− also showed a substantial value (logK = 4.0 ± 0.2). However, the –OPO32− group in G22− surely plays a substantial role, which can be concluded from its comparison with a neutral guest, adamantanol G8, whose binding was weaker than the phosphate G22− by more than 3 orders of magnitude (logK = 2.3 ± 0.2).
The pKa2 value of PhOPO3H2 (H2G1) is 5.85,50 thus it mostly exists as a dianion at pH = 7.0 (G12−:HG1− = 93:7). In order to determine the protonation state of the phenyl phosphate bound in the pyridinium amide cyclodextrin 17+, the UV-vis titration was also performed at pH = 5.5 (where G12−:HG1− = 31:69) instead of pH = 7.0. As a result, the overall binding constant was the same, logK = 4.6 ± 0.1 (Fig. S37, ESI†). This result that the binding constant is not dependent on the pH suggests that (1) both the dianion G12− and monoanion HG1− can bind to 17+, and (2) dianion G12− is the dominant state to bind to 17+ at pH = 7.0, where 93% of the unbound phenyl phosphate exists as a dianion.
Calorimetric analysis
Next, the thermodynamics of the binding of phosphate anions with pyridinium amide CD 1(OTf)7 were investigated by ITC (isothermal titration calorimetry) measurements (Fig. 6, S42 and S43, ESI†). PhOPO3Na2 (G1Na2) and AdOPO3Na2 (G2Na2) were investigated, and these two phosphates showed interesting contrasting results. | ||
| Fig. 6 Isothermal titration calorimetry measurements of the binding of phosphates (PhOPO3Na2 (G1Na2) and AdOPO3Na2 (G2Na2)) with 1(OTf)7 in H2O. Conditions: cell: 1(OTf)7, 0.2 mM, 200 μL; syringe: PhOPO3Na2 or AdOPO3Na2, 4.0 mM; injection: 2.0 μL each. Error bars, SD (n = 3). (a and b) Enthalpograms and ΔH change per injection [kJ mol−1 inj.] observed in measurements at different temperatures: (a) PhOPO3Na2; (b) AdOPO3Na2. (c and d) ΔG, ΔH, and −TΔS for the binding of phosphates: (c) PhOPO3Na2; (d) AdOPO3Na2. (e and f) ΔH–T plot to evaluate ΔCp = ∂H/∂T: (e) PhOPO3Na2; (f) AdOPO3Na2. | ||
First, the binding of PhOPO3Na2 was found to be endothermic in the measured temperature range (5–45 °C) (Fig. 6a). Fitting with a 1:1 binding model yielded the thermodynamic parameters (ΔG, ΔH, ΔS) as shown in Fig. 6c, which demonstrated that the reaction is entropy-driven (ΔH > 0, ΔS > 0). The temperature dependence of the enthalpy change indicated that the molar heat capacity change ΔCp (= ∂H/∂T) had a positive value, +160 ± 50 J mol−1 K−1 (Fig. 6e). In general, it is known that the binding of hydrophobic molecules to unmodified cyclodextrins shows negative values for both ΔH and ΔCp.26 Thus, the result obtained here for the pyridinium amide CD 1(OTf)7 and PhOPO3Na2 is in contrast to this native cyclodextrin-guest binding, and it suggested that the normal hydrophobic effect is not the main driving force.
The standard entropy ΔS° can be expressed as follows:51–53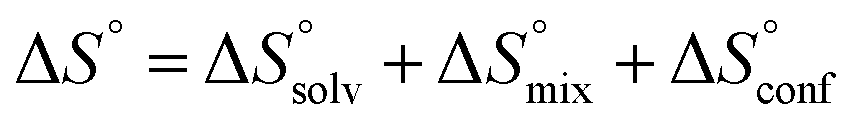 , where
, where 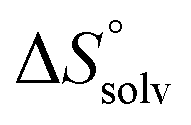 is the change in the solvation entropy,
is the change in the solvation entropy, 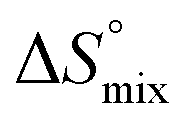 is the change in the mixing entropy, and
is the change in the mixing entropy, and 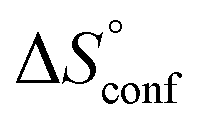 is the change in the configurational entropy.
is the change in the configurational entropy.  at 25 °C has a relationship with the molar heat capacity change ΔCp as in the following equation:51–53
at 25 °C has a relationship with the molar heat capacity change ΔCp as in the following equation:51–53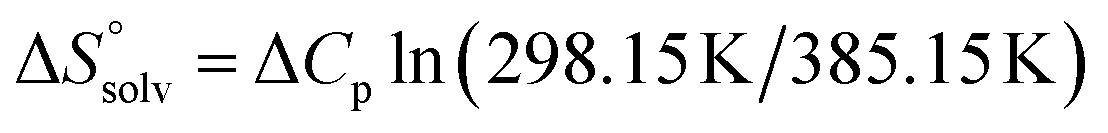 . This leads to
. This leads to 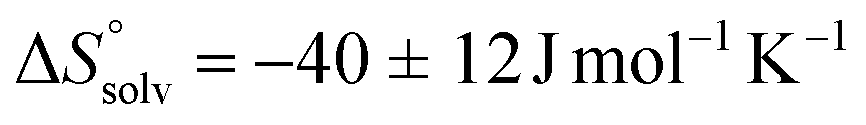 , which means that water molecules become structured when PhOPO3Na2 and 1(OTf)7 form the host–guest complex. For a bimolecular reaction (A + B => P),
, which means that water molecules become structured when PhOPO3Na2 and 1(OTf)7 form the host–guest complex. For a bimolecular reaction (A + B => P), 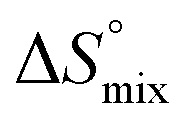 is −33 J mol−1 K−1.51–53 Thus,
is −33 J mol−1 K−1.51–53 Thus, 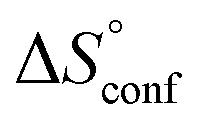 can be estimated to be +245 ± 12 J mol−1 K−1. This high positive value is in contrast to the negative
can be estimated to be +245 ± 12 J mol−1 K−1. This high positive value is in contrast to the negative  seen for the host–guest systems of unmodified cyclodextrins.54
seen for the host–guest systems of unmodified cyclodextrins.54
Meanwhile, the binding of AdOPO3Na2 was endothermic at low temperature (5 and 15 °C), but exothermic at high temperature (35 and 45 °C) (Fig. 6b). ΔH decreases as the temperature increases (Fig. 6d), and ΔCp = −280 ± 20 J mol−1 K−1 was obtained from the ΔH vs. T plot (Fig. 6f). The trend in the thermodynamic parameters 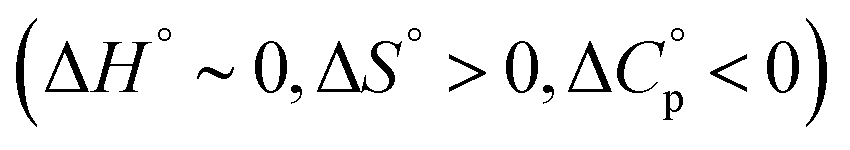 is similar to that of the normal hydrophobic effects, thus the hydrophobicity of the adamantyl group contributes to the binding in the case of AdOPO3Na2. The treatment of ΔS° in the same manner as already described yielded
is similar to that of the normal hydrophobic effects, thus the hydrophobicity of the adamantyl group contributes to the binding in the case of AdOPO3Na2. The treatment of ΔS° in the same manner as already described yielded 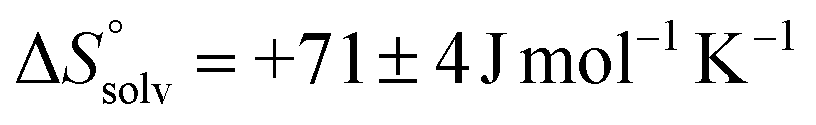 and
and 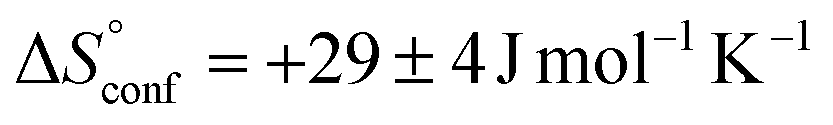 , which are substantially different from the values for PhOPO32−
, which are substantially different from the values for PhOPO32−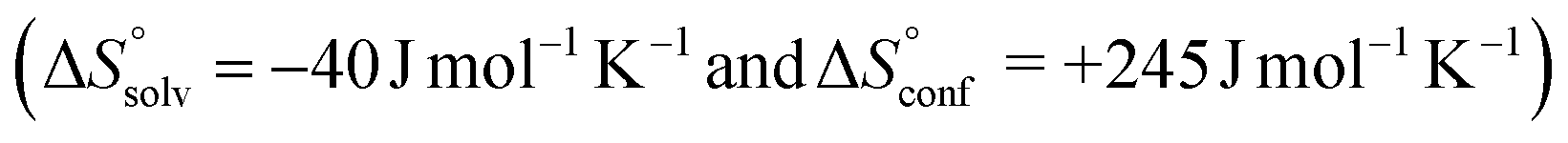 .
.
Thus, by comparison with the results of AdOPO32−, it was indicated that an interesting phenomenon more than the normal hydrophobic effects seen in the unmodified CDs is involved in the recognition of PhOPO32−. One possible explanation might be that partial conversion of the self-included conformer of 1(OTf)7 to the non-included conformer upon inclusion of PhOPO32− leads to exposure of the self-included pyridinium amide group to the outside, which leads to hydrophobic ordering of H2O around the pyridinium group  and increased flexibility of the host framework
and increased flexibility of the host framework 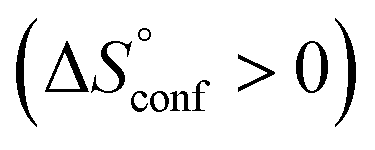 .
.
Comparison of binding constants
The binding constants K between pyridinium amide CD 1(OTf)7 and phenyl phosphonate G1Na2 determined by the UV-vis, NMR, and ITC measurements are summarized in Table 1. The binding constant K has a tendency to increase under dilute conditions (see also Fig. S40 and S41, and Table S1† for a dilution experiment of an NMR sample of the host–guest complex). The weak but competitive effect of counter anions (TfO−) is a possible factor (as discussed, the negligible change in 19F NMR signal of TfO− before and after the inclusion of G12− suggested that TfO− was not encapsulated inside the cavity of 17+ (Fig. S17, ESI†)). The binding constant K determined by ITC was slightly smaller than the value determined by UV-vis (entries 2 and 3). This might be because the exchange of the outer counter anions, which does not appear in the UV-vis spectral change of 17+, is involved in the latter half of the ITC titration. The H/D isotope effect was not seen in the values of the binding constants (entries 6 and 7).| Entry | Method | Solvent | [1(OTf)7] [μM] | logK [M−1] |
|---|---|---|---|---|
| 1 | 1H NMR | D2O | 2.0 × 103 | 3.3 ± 0.2 |
| 2 | ITC | H2O | 2.0 × 102 | 4.29 ± 0.03 |
| 3 | UV-vis | H2O | 2.0 × 102 | 5.0 ± 0.1 |
| 4 | UV-vis | 0.38 mM MES buffer (pH = 7.0) | 3.6 × 10 | 4.6 ± 0.1 |
| 5 | UV-vis | 0.38 mM MES buffer (pH = 7.0) | 1.2 × 10 | 4.71 ± 0.07 |
| 6 | UV-vis | H2O | 1.2 × 10 | 5.10 ± 0.05 |
| 7 | UV-vis | D2O | 1.2 × 10 | 5.2 ± 0.1 |
Molecular dynamics simulation
To further clarify the anion recognition mechanism of the pyridinium amide CD 17+, an all-atom MD simulation and its free-energy analysis in an aqueous environment were utilized in this study. Three guests are chosen here: PhOPO32−, which was included in an endothermic process; AdOPO32−, which was supposedly recognised by the normal hydrophobic effects; and PhCO2−, which did not show any interaction in the experiment. The distance between the centres of mass of the host and guest was initially set to 0 nm, and was increased to generate the initial structures of the MD simulations (Fig. 7a). The obtained complex of the 17+-guest pair was equilibrated with seven TfO− counter anions in the aqueous solution of 18000 water molecules. The MD system consisted of approx. 72000 particles, which corresponds to a concentration [1(OTf)7] = 3.0 mM. The atomic charges in the host 1(OTf)7 and guest molecules were determined at the DFT level of HF/6-31G+(d,p), and the force–field parameters were assigned within the modern framework of a second-generation general amber force field (GAFF2).55,56 An umbrella-sampling analysis was performed using the model of TIP4P/Ew with the position restraint of the harmonic potential (see the ESI† for the detailed settings).
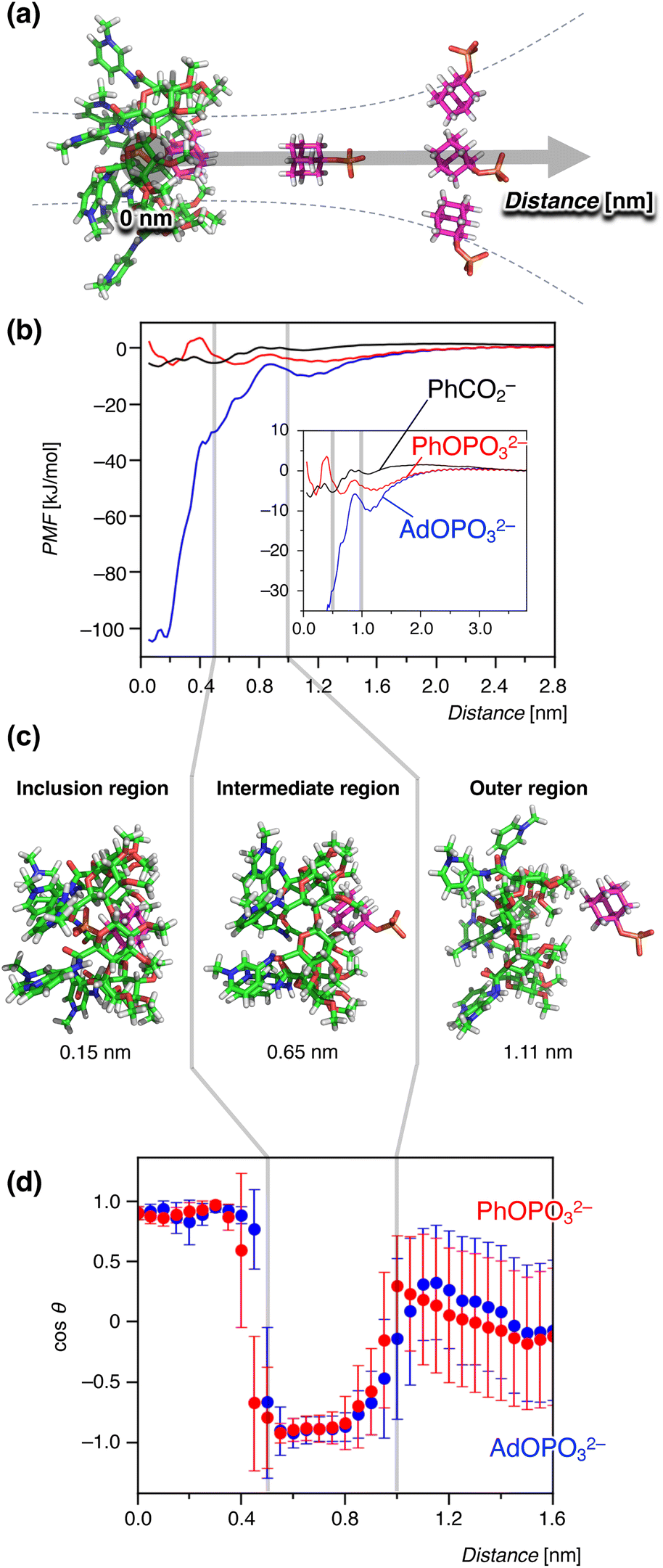 | ||
| Fig. 7 Binding free energetics during the host–guest interaction between 1(OTf)7 and anions observed in the MD simulation. The gray lines in (b–d) represent the boundaries of the inclusion-intermediate and intermediate-outer regions. The error bars are standard deviations. (a) A schematic diagram of umbrella-sampling analysis as a reaction coordinate with the distances of the centers of mass of host and guest. (b) Potentials of mean force (PMF) for the anion guests PhCO2−, PhOPO32−, and AdOPO32−. Inset: an enlarged view in the range of 0–3.7 nm. (c) Representative structures observed at distances of 0.15, 0.65, and 1.11 nm. (d) Orientation of the phosphate guests during the binding process. The angle θ is defined by a vector perpendicular to the oligopyranose torus of β-cyclodextrin and another one in the direction of the phosphate group (see Fig. S45 in the ESI† for details). | ||
Fig. 7b shows the potential of mean force (PMF) in the inclusion process of 17+. Representative structures of the host–guest binding regions are shown together in Fig. 7c. The PMF reflects the free energy ΔG′ derived from the following equation:31,35,57–59
| ΔG = −RTlnC°K = −RTlnC°V + ΔG′. | (1) |
lnC°V behaves as the modification for the difference in concentration, in which C° is the standard concentration and V is the effective volume of the binding site in the inclusion complex. Note that the PMF is not identical to the experimentally determined binding constant K because of the term −RTlnC°V. The PMF profiles in Fig. 7b indicated that ΔG′ was strongly negative for AdOPO32− in the region of 0–0.50 nm. The profile of PhOPO32− showed a small but clear minimum at around 0.2 nm. That of PhCO2− also showed a minimum at around 0.1 nm, but PhCO2− showed a long-range repulsive interaction when the host–guest distance became smaller. These results suggest that, in line with the experiments, the bindings of AdOPO32− and PhOPO32− were favourable for the inclusion distance, and that of PhCO2− was relatively unfavourable in the formation of the host–guest complex. It should be noted that the binding behaviour of the phosphate guests with the host 17+ was qualitatively captured in the present MD simulation, while the quantitative agreement of the binding free energy with the experimental measurements was not achieved even when considering the correction term of −RTlnC°V. Although free energetics for neutral CD derivatives has been estimated in the preceding study,35 the difference in this study may be due to the force field issue for the ionic groups of 17+.33,34 ΔG′ was much smaller for AdOPO32− than the experimental value of ΔG. Considering that −RTlnC°V was estimated to be around 10 kJ mol−1 when the effective volume of the binding site in 17+ was that of a sphere with a radius of 0.2 nm, this difference can be interpreted to be the result of a very large −RTlnC°V, indicating the formation of a tightly-bound AdOPO32− complex with a small V, like a pinning state.
We further focused on the structural mechanism of inclusion in the range of 0–1.6 nm. Fig. 7d shows the orientation dependence for the phosphate anions with respect to the host–guest distance (see the ESI† for the energetic profiles in response to the reaction coordinate). As seen in Fig. 7d, the inclusion profile can be divided into three regions with energy gaps between them; i.e., the outer region (>1.0 nm), intermediate region (0.5–1.0 nm), and inclusion region (0–0.5 nm). Interestingly, these three stages were characterized by the orientation of the phosphate anions (Fig. 7d). The phosphate anion moved freely without any orientational restriction from the host 17+ in the outer region. When the distance became less than 1.0 nm (intermediate region), the phosphate group was orientated in the opposite direction from the host (Fig. 7c and d). This corresponded to the partial inclusion of the hydrophobic group and the exposure of the –OPO32− group to the exterior water. As the guest became included deeper, hydration of the phosphate groups decreased while the interaction with the host CD 17+ increased (Fig. S46†). When the anion got closer to the inner cavity (0–0.5 nm, inclusion region), the orientation of the phosphate group reversed to the side of the pyridinium groups of 17+ (Fig. 7c and d). This orientation flip was permitted by the favorable intermolecular interaction competitively overcoming the solvent-reorganization effect in the aqueous environment (Fig. S46†), which leads to an overall stabilization of ΔG′ inside the inner CD cavity (Fig. 7b). The change in the orientation then allowed the formation of hydrogen bonds with the amide N–H groups as described in the next section (Fig. 8). Thus, this boundary of the intermediate and inclusion regions can be interpreted as the point where the stabilization by host 17+ (hydrophobic effects, hydrogen bonds, and electrostatic interactions) surpasses the hydration of the phosphate group.
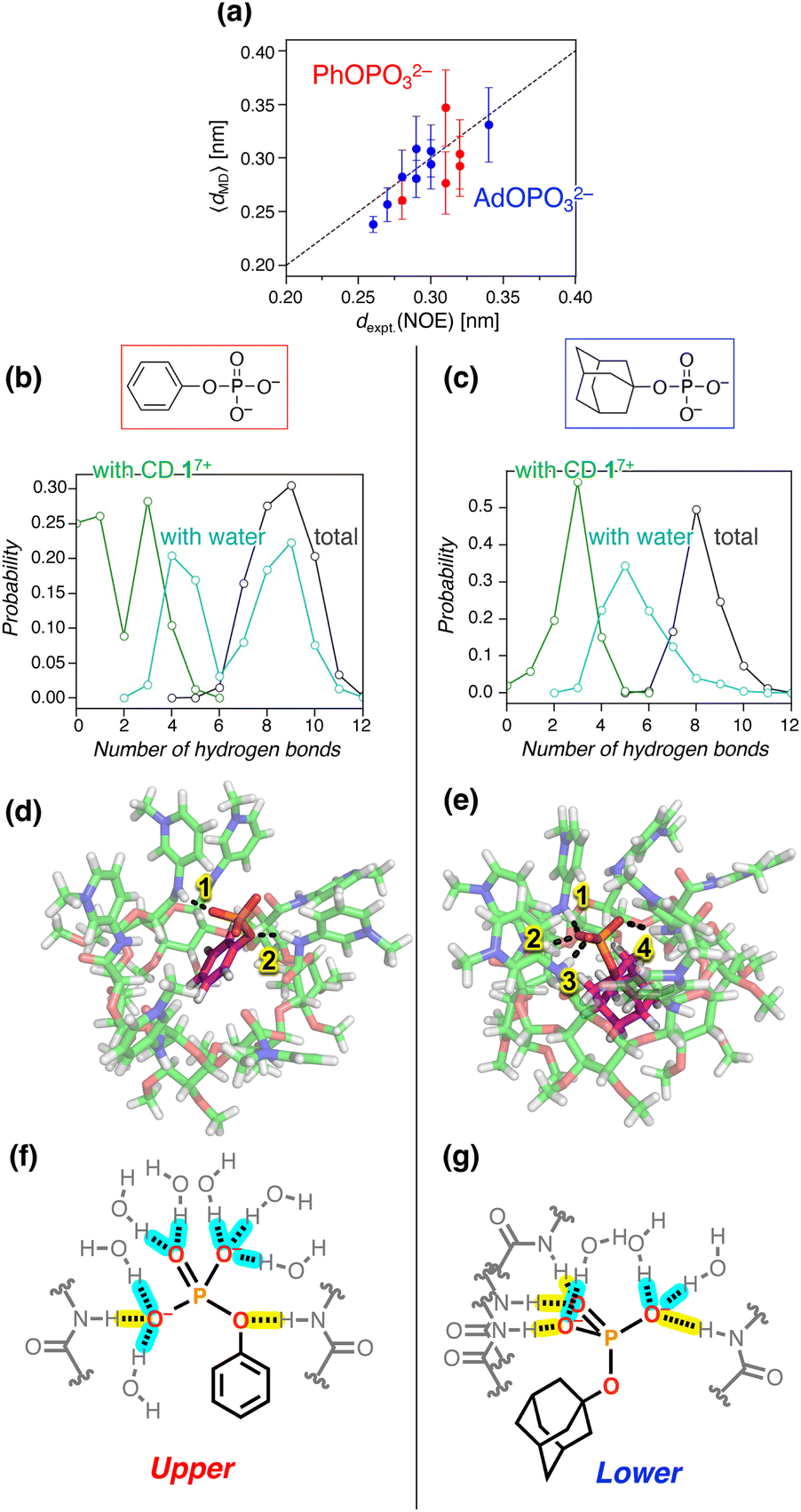 | ||
| Fig. 8 Structures and hydrogen bonding patterns for the inclusion complex of 17+. The error bars in (a) are standard deviations. (a) Comparison of dexpt.(NOE) and 〈dMD〉 for the H–H distances in the inclusion complexes of PhOPO32− and AdOPO32−. The values for each pair of hydrogen atoms are summarized in Tables S2 and S3, ESI.† (b and c) Probability distribution of the number of hydrogen bonds of the phosphate anions with amide CD 17+, water, and the sum of both: (b) PhOPO32−; (c) AdOPO32−. The hydrogen-bonding states for PhOPO32− and AdOPO32− were only sampled around the first minimum of PMF as 0.22 ± 0.01 and 0.10 ± 0.01 nm, respectively, observed from all the umbrella windows. (d and e) 3D snapshots and (f and g) corresponding schematic views of the inclusion complexes: (d and f) PhOPO32−; (e and g) AdOPO32−. The dotted lines in (d and e) depict the hydrogen bonds between the phosphate oxygens and the amide N–H. Hydrogen bonds in (f and g) of the phosphate group with amide N–H and water are highlighted in yellow and blue, respectively. | ||
Next, the difference in the inclusion process of the two phosphates, AdOPO32− and PhOPO32−, was analysed in terms of the energy barriers for ΔG′ at the boundaries as well as the interaction energy with water. For the recognition process of AdOPO32−, only a small energy barrier could be identified across the boundary of the outer–intermediate regions and the PMF monotonically decreased as the deep inclusion complex formed (Fig. 7b). In the case of PhOPO32−, the energy barrier at around 1.0 nm was also low and thus a partial inclusion complex could be easily formed. However, PhOPO32− had a high energy barrier across the boundary of the intermediate–inclusion regions (Fig. 7b). As already discussed, this free energy barrier before forming the deep inclusion complex is interpreted as the dehydration step of the host and the guest. For the partial inclusion state in the intermediate region, the small hydrophobic group of PhOPO32− offered enough space to co-include a water molecule in the CD cavity, which implies an increase of the effective volume of the binding site included in eqn (1). The water molecule could bind to the phosphate ester oxygen R–O–P of PhOPO32−via a hydrogen bond in the intermediate region, which disturbed the orientation flip of the phosphate group directing outward from the CD cavity (Fig. 7d). This extra hydration of PhOPO32− then resulted in the high energy barrier of ΔG′ during the formation of the deep inclusion complex upon dehydration (Fig. 7b). In other words, the dehydration step to form a deep inclusion complex with the orientational flip of the phosphate would behave as a rate-determining step in the inclusion process.
The significant PMF decrease for ΔG′ in the inclusion region of AdOPO32− (Fig. 7b) can be considered to correspond to the exothermic process seen in the ITC measurement. Meanwhile, PhOPO32− exhibited a relatively low PMF minimum of ΔG′, the high free energy barrier across the boundary of the inclusion–intermediate regions (Fig. 7b). These features can be reasonably interpreted as being derived from the same root as the endothermic and entropy-driven inclusion of PhOPO32−. Thus, the contribution of hydrophobicity in the MD simulation matched well with the experimental results obtained in the ITC investigations.
Hydrogen bonding structures
A more detailed structural analysis using MD simulations and their comparison with the NOESY experiments gave a clear picture of the hydrogen bonds (Fig. 8). Quantitative analysis of NOESY (Fig. 4) using the relationship between the NOE intensity and distance (the NOE intensity being inversely proportional to the sixth power of the 1H–1H distance) provided the experimental distances (dexpt.(NOE)) between the protons of 17+ and PhOPO32−/AdOPO32− (Tables S2 and S3, ESI†). In our MD simulation, the corresponding H–H distances in the deep inclusion complex of 17+ were quantified as 〈dMD〉 averaged from the sampled trajectories under the restriction of the umbrella potential using the following equation:60 | (2) |
The structural analysis then focused on the number of hydrogen bonds formed by the amide N–H of 17+ or water molecules with the phosphate anions. As the anion was transferred from the intermediate region to the inclusion one, the number of hydrogen bonds of PhOPO32− with the amide N–Hs converged to 1.7 on average, while that of AdOPO32− was 3.0 (Fig. S47, ESI†). In the inclusion states extracted from all the umbrella windows (Fig. 7b and S44, ESI†), the most probable number of observed hydrogen bonds with the amide N–H was three for both PhOPO32− and AdOPO32− (Fig. 8b and c). However, in the case of PhOPO32−, the inclusion state with the amide N–H remained weak, and the hydrogen-bonding donors of the phosphate guest were observed to be switched to water molecules. As a result, the numbers of hydrogen bonds with water molecules were distributed bimodally in the inclusion complex with PhOPO32− (Fig. 8b). This difference depending on the phosphates arises from the distinct patterns in which the –OPO32− group was involved in the hydrogen-bonding states. In PhOPO32−, the oxygen atom of the phosphate ester R–O–P as well as those of the terminal –PO32− acted as hydrogen bond acceptors to the amide N–H. The average O⋯H distance 〈dMD〉 with the amide N–H of 17+ calculated using eqn (2) was 0.260 nm for the phosphate ester R–O–P, which was shorter than 0.294 nm for the terminal –PO32− (Fig. 8d and f). The phenyl group loosely interacted with the inner CD cavity (Fig. 7b), which enabled PhOPO32− to shift more towards the pyridinium amide side. Thus, water molecules had easy access to the phosphate groups (Fig. S47 and S48, ESI†), and the hydrogen-bonding cage by the amide N–H of 17+ was sometimes broken down even in the inclusion region (Fig. 8b). These structural behaviours can be interpreted to enlarge the effective volume of the binding site inside the complex and reduce the contribution of −RTlnC°V in eqn (1). Note that the bimodal fraction of the hydrogen-bonding states observed in the deep-inclusion region did not reflect the existence of a bimodal complexation60 for the host–guest configuration (Fig. 7d), but can be just interpreted as the result of thermal fluctuations inside the loosely enclosed environment by the amide N–H of 17+. In contrast, AdOPO32− dominantly formed hydrogen bonds with 17+ at the terminal –PO32− (Fig. 8e and g) and the hydration number of AdOPO32− remained lower than that of PhOPO32− (Fig. 8c, g and S47, ESI†). The terminal oxygens of AdOPO32− were closely bound to the amide N–H of 17+ (O⋯H, 〈dMD〉 = 0.180 nm), and the corresponding distance for the phosphate ester R–O–P was as large as 0.330 nm (Fig. 8e and g). The radial distribution function for the oxygen atoms of the two phosphates also implies that the amide N–H of 17+ was preferably bound to the terminal oxygens in the case of AdOPO32− (Fig. S48, ESI†). Hence, the anionic phosphate group of AdOPO32− inside the CD cavity was in weaker contact with the water molecules. This behaviour can be explained as the result of tight trapping of the adamantyl group by the hydrophobic effect in the CD cavity, which is in agreement with the interpretation of both the large contributions of not only ΔG′ but also −RTlnC°V in eqn (1).
The differences in the position of the guests (upper or lower) according to the involvement of the ester oxygen during hydrogen bonding are in good agreement with the positions indicated by the NOESY experiments (Fig. 4). Furthermore, the relatively low stabilization of PhOPO32− in PMF (Fig. 7b) and the high energy and structural fluctuations of PhOPO32− compared to AdOPO32− (Fig. 7d, 8b and c) can be interpreted as originating from the entropy-driven molecular recognition of PhOPO32− revealed by the ITC experiments. In summary, the differences between the hydrophobic groups, Ph and Ad, provided interesting characteristics of the recognition modes that had not previously been considered.
Conclusions
A new amide β-cyclodextrin derivative 17+ with the N-methyl-pyridinium group has been synthesized as a trifluoromethanesulfonate salt. 1(OTf)7 selectively bound organic monophosphate anions in water. 1H NMR titration experiments indicated the involvement of hydrogen bonds of the amide N–H with the phosphates. Detailed NMR studies confirmed that the hydrophobic group of the monophosphates is encapsulated in the cavity of the cyclodextrin torus with the –PO32− group located near the amide N–H. Selectivity in the anion binding experiments can be explained by the contribution of both the hydrophobic effects and hydrogen bonding to the phosphate group. A comparison of two phosphates, PhOPO32− (G12−) and AdOPO32− (G22−), gave contrasting results and a deeper understanding. It was suggested from a NOESY analysis that the position of the encapsulated PhOPO32− is “upper” (closer to the pyridinium amide side) compared to that of AdOPO32−. ITC measurements revealed that the inclusion of PhOPO32− is endothermic and entropy-driven (ΔH > 0, ΔS > 0, ΔCp > 0), which is unusual and different from examples of the normal hydrophobic effect seen in unmodified CDs. Temperature-dependent ITC measurements revealed that the increase in the configurational entropy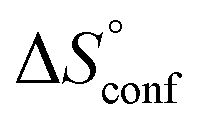 is a major factor for the inclusion of PhOPO32−. In contrast, the results for AdOPO32− suggested that the hydrophobic effect is the main contributing driving force. An all-atom MD simulation has provided clear pictures of the inclusion process. PhOPO32− was trapped in the “upper” position with a larger structural fluctuation, and the oxygen of the phosphate ester R–O–P was involved in the hydrogen bonds with the amide N–H and water molecules. Meanwhile, the adamantyl group of AdOPO32− was firmly trapped in the CD cavity and excluded more water molecules around the phosphate group, and the amide N–H formed hydrogen bonds to the terminal –PO32−. Thus, the combination of the anion selectivity study, NOESY analysis, ITC measurements, and MD simulation has not only revealed the detailed inclusion structures of a new amide cyclodextrin derivative, but also provided a successful demonstration in which an artificial receptor works in harmony with water molecules and achieves precise molecular recognition in aqueous environments. The host effectively utilizes the hydrophobic effects, the networks of multipoint hydrogen bonds, and the configurational entropy gains, which is reminiscent of biomacromolecules. This study significantly contributes to the design guidelines of anion receptors and the science of functional molecules in aqueous environments. Further developments and applications using amide cyclodextrin frameworks are currently in progress.
is a major factor for the inclusion of PhOPO32−. In contrast, the results for AdOPO32− suggested that the hydrophobic effect is the main contributing driving force. An all-atom MD simulation has provided clear pictures of the inclusion process. PhOPO32− was trapped in the “upper” position with a larger structural fluctuation, and the oxygen of the phosphate ester R–O–P was involved in the hydrogen bonds with the amide N–H and water molecules. Meanwhile, the adamantyl group of AdOPO32− was firmly trapped in the CD cavity and excluded more water molecules around the phosphate group, and the amide N–H formed hydrogen bonds to the terminal –PO32−. Thus, the combination of the anion selectivity study, NOESY analysis, ITC measurements, and MD simulation has not only revealed the detailed inclusion structures of a new amide cyclodextrin derivative, but also provided a successful demonstration in which an artificial receptor works in harmony with water molecules and achieves precise molecular recognition in aqueous environments. The host effectively utilizes the hydrophobic effects, the networks of multipoint hydrogen bonds, and the configurational entropy gains, which is reminiscent of biomacromolecules. This study significantly contributes to the design guidelines of anion receptors and the science of functional molecules in aqueous environments. Further developments and applications using amide cyclodextrin frameworks are currently in progress.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Nakamura conceived the project. H. T., T. O., and D. T. performed the organic synthesis and spectroscopic studies. M. N. performed the ITC measurements. H. B., Y. I., and G. W. performed the MD simulations. T. Nakamura, M. N., Y. I., and G. W. wrote the manuscript with discussion and contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI (Grant Numbers JP19H05714, JP19H05718, JP19H05719, JP20H05202, JP22H04519 (Aquatic Functional Materials), JP21H01946, and JP22K05124), JST ACT-X (Grant Number JPMJAX23D3) and CREST (Grant Number JPMJCR23O1), the Kurita Water and Environment Foundation (Grant Numbers 23H033 and 24H021), and the Asahi Glass Foundation. Y. I. was also supported by the Kitasato University Research Grant for Young Researchers. The MD simulations were implemented by using the supercomputers of the Research Center for Computational Science in Okazaki (22-IMS-C043 and 23-IMS-C038), Grand Chariot at Hokkaido University, and OCTOPUS at Osaka University partially through the HPCI System Research Project (hp230083 and hp230132).References
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- N. Y. Meredith, S. Borsley, I. V. Smolyar, G. S. Nichol, C. M. Baker, K. B. Ling and S. L. Cockroft, Angew. Chem., Int. Ed., 2022, 61, e202206604 CrossRef CAS PubMed.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- S. K. Dey, A. Basu, R. Chutia and G. Das, RSC Adv., 2016, 6, 26568–26589 RSC.
- S. Kubik, C. Reyheller, S. Stüwe and J. Inclusion Phenom, Macrocyclic Chem., 2005, 52, 137–187 CrossRef CAS.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- S. Kubik, Acc. Chem. Res., 2017, 50, 2870–2878 CrossRef CAS.
- L. K. Macreadie, A. M. Gilchrist, D. A. McNaughton, W. G. Ryder, M. Fares and P. A. Gale, Chem, 2022, 8, 46–118 CAS.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2011, 133, 7344–7347 CrossRef CAS.
- R. U. Lemieux, Acc. Chem. Res., 1996, 29, 373–380 CrossRef CAS.
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS.
- F. Biedermann, W. M. Nau and H. J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS.
- A. M. Agafontsev, A. Ravi, T. A. Shumilova, A. S. Oshchepkov and E. A. Kataev, Chem.–Eur. J., 2019, 25, 2684–2694 CrossRef CAS PubMed.
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS.
- S. J. C. Lee, J. W. Lee, H. H. Lee, J. Seo, D. H. Noh, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2013, 117, 8855–8864 CrossRef CAS PubMed.
- F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879–14888 CrossRef CAS PubMed.
- K. Caprice, M. Pupier, A. Kruve, C. A. Schalley and F. B. L. Cougnon, Chem. Sci., 2018, 9, 1317–1322 RSC.
- J. W. Barnett, M. R. Sullivan, J. A. Long, D. Tang, T. Nguyen, D. Ben-Amotz, B. C. Gibb and H. S. Ashbaugh, Nat. Chem., 2020, 12, 589–594 CrossRef CAS.
- D. Van Eker, S. K. Samanta and A. P. Davis, Chem. Commun., 2020, 56, 9268–9271 RSC.
- T. Nakamura, S. Yonemura and T. Nabeshima, Chem. Commun., 2019, 55, 3872–3875 RSC.
- S. Yonemura, T. Nakamura and T. Nabeshima, Chem. Lett., 2020, 49, 493–496 CrossRef CAS.
- T. Nakamura, S. Yonemura, S. Akatsuka and T. Nabeshima, Angew. Chem., Int. Ed., 2021, 60, 3080–3086 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed.
- E. Flood, C. Boiteux, B. Lev, I. Vorobyov and T. W. Allen, Chem. Rev., 2019, 119, 7737–7832 CrossRef CAS PubMed.
- B. Ding, Y. Yu, S. Geng, B. Liu, Y. Hao and G. Liang, J. Agric. Food Chem., 2022, 70, 2466–2482 CrossRef CAS PubMed.
- X. Kou, D. Su, F. Pan, X. Xu, Q. Meng and Q. Ke, Carbohydr. Polym., 2024, 324, 121524 CrossRef CAS PubMed.
- W. You, Z. Tang and C. E. A. Chang, J. Chem. Theory Comput., 2019, 15, 2433–2443 CrossRef CAS.
- K. Kasahara, R. Masayama, K. Okita and N. Matubayasi, J. Chem. Phys., 2021, 155, 204503 CrossRef CAS PubMed.
- N. M. Henriksen and M. K. Gilson, J. Chem. Theory Comput., 2017, 13, 4253–4269 CrossRef CAS PubMed.
- K. G. Sprenger, V. W. Jaeger and J. Pfaendtner, J. Phys. Chem. B, 2015, 119, 5882–5895 CrossRef CAS PubMed.
- Y. Ishii and N. Matubayasi, J. Chem. Theory Comput., 2020, 16, 651–665 CrossRef.
- L. Wickstrom, P. He, E. Gallicchio and R. M. Levy, J. Chem. Theory Comput., 2013, 9, 3136–3150 CrossRef CAS PubMed.
- S. Mieda, A. Ikeda, Y. Shigeri and W. Shinoda, J. Phys. Chem. C, 2014, 118, 12555–12561 CrossRef CAS.
- C. Ueda, J. Park, K. Hirose, S. Konishi, Y. Ikemoto, M. Osaki, H. Yamaguchi, A. Harada, M. Tanaka, G. Watanabe and Y. Takashima, Supramol. Mater., 2022, 1, 100001 Search PubMed.
- Z. Wu, S. Wang, Z. Zhang, Y. Zhang, Y. Yin, H. Shi and S. Jiao, RSC Adv., 2022, 12, 30495–30500 RSC.
- Y. Kawashima, T. Hamachi, A. Yamauchi, K. Nishimura, Y. Nakashima, S. Fujiwara, N. Kimizuka, T. Ryu, T. Tamura, M. Saigo, K. Onda, S. Sato, Y. Kobori, K. Tateishi, T. Uesaka, G. Watanabe, K. Miyata and N. Yanai, Nat. Commun., 2023, 14, 1056 CrossRef CAS.
- J. Park, Y. Sasaki, Y. Ishii, S. Murayama, K. Ohshiro, K. Nishiura, R. Ikura, H. Yamaguchi, A. Harada, G. Matsuba, H. Washizu, T. Minami and Y. Takashima, ACS Appl. Mater. Interfaces, 2023, 15, 39777–39785 CrossRef CAS.
- W. C. Cromwell, K. Byström and M. R. Eftink, J. Phys. Chem., 1985, 89, 326–332 CrossRef CAS.
- Y. Liu, Y.-M. Zhang, S.-X. Sun, Y.-M. Li and R.-T. Chen, J. Chem. Soc., Perkin Trans. 2, 1997, 1609–1613 RSC.
- M. Ghosh, R. Zhang, R. G. Lawler and C. T. Seto, J. Org. Chem., 2000, 65, 735–741 CrossRef CAS.
- H. V. Huynh and J. T. Vossen, Inorg. Chem., 2020, 59, 12486–12493 CrossRef CAS PubMed.
- A. Dorazco-González, H. Höpfl, F. Medrano and A. K. Yatsimirsky, J. Org. Chem., 2010, 75, 2259–2273 CrossRef PubMed.
- T. Kraus, M. Buděšínský and J. Závada, Eur. J. Org Chem., 2000, 3133–3137 CrossRef CAS.
- Z. J. Chen, H. W. Xi, K. H. Lim and J. M. Lee, ACS Sustain. Chem. Eng., 2015, 3, 325–333 CrossRef CAS.
- L. Catoire, V. Michon, L. Monville, A. Hocquet, L. Jullien, J. Canceill, J.-M. Lehn, M. Piotto and C. H. Du Penhoat, Carbohydr. Res., 1997, 303, 379–393 CrossRef CAS.
- J. Terao, K. Ikai, N. Kambe, S. Seki, A. Saeki, K. Ohkoshi, T. Fujihara and Y. Tsuji, Chem. Commun., 2011, 47, 6816–6818 RSC.
- D. Shamir, I. Zilbermann, E. Maimon, A. I. Shames, H. Cohen and D. Meyerstein, Inorg. Chim. Acta, 2010, 363, 2819–2823 CrossRef CAS.
- K. P. Murphy, D. Xie, K. S. Thompson, L. M. Amzel and E. Freire, Proteins: Struct., Funct., Genet., 1994, 18, 63–67 CrossRef CAS.
- B. M. Baker and K. P. Murphy, J. Mol. Biol., 1997, 268, 557–569 CrossRef CAS PubMed.
- P. Hariharan, D. Balasubramaniam, A. Peterkofsky, H. R. Kaback and L. Guan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2407–2412 CrossRef CAS PubMed.
- W. Chen, C.-E. Chang and M. K. Gilson, Biophys. J., 2004, 87, 3035–3049 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CAS.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, G. Giambasu, T. Giese, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. Le Grand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, L. Wilson, R. M. Wolf, X. Wu, L. Xiao, Y. Xiong, D. M. York and P. A. Kollman, AMBER 2019/Amber-Tools19, University of California, San Francisco, CA, USA, 2019 Search PubMed.
- M. K. Gilson, J. A. Given, B. L. Bush and J. A. McCammon, Biophys. J., 1997, 72, 1047–1069 CrossRef CAS PubMed.
- Y. Deng and B. Roux, J. Phys. Chem. B, 2009, 113, 2234–2246 CrossRef CAS.
- E. Gallicchio, M. Lapelosa and R. M. Levy, J. Chem. Theory Comput., 2010, 6, 2961–2977 CrossRef CAS PubMed.
- M. Zubiaur and C. Jaime, J. Org. Chem., 2000, 65, 8139–8145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, characterization data, and data for anion recognition and molecular dynamics calculations. See DOI: https://doi.org/10.1039/d4sc04529g |
| This journal is © The Royal Society of Chemistry 2025 |