
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodynamic therapy photosensitizers and photoactivated chemotherapeutics exhibit distinct bioenergetic profiles to impact ATP metabolism†
Richard J.
Mitchell
a,
Dmytro
Havrylyuk
b,
Austin C.
Hachey
c,
David K.
Heidary
*b and
Edith C.
Glazer
 *b
*b
aNational Cancer Institute Frederick, USA
bNorth Carolina State University at Raleigh, USA. E-mail: eglazer@ncsu.edu
cUniversity of Kentucky, USA
First published on 21st November 2024
Abstract
Energy is essential for all life, and mammalian cells generate and store energy in the form of ATP by mitochondrial (oxidative phosphorylation) and non-mitochondrial (glycolysis) metabolism. These processes can now be evaluated by extracellular flux analysis (EFA), which has proven to be an indispensable tool in cell biology, providing previously inaccessible information regarding the bioenergetic landscape of cell lines, complex tissues, and in vivo models. Recently, EFA demonstrated its utility as a screening tool in drug development, both by providing insights into small molecule–organelle interactions, and by revealing the peripheral and potentially undesired off-target effects small molecules have within cells. Surprisingly, technologies to quantify cellular bioenergetics have not been systematically applied in phototherapy development, leaving open several questions about how the mechanism of action of a compound can impact essential cellular functions. Here, we utilized the Seahorse analyzer to address this question for photosensitizers (PSs) for photodynamic therapy (PDT) and contrast these systems to molecules that photo-release a ligand and thus act as photocages or photoactivated chemotherapeutics (PACT), intending to understand the influence these two classes of compounds have on cellular bioenergetics. EFA results show that acute treatment of A549 lung adenocarcinoma cells with PDT agents induces a quiescent bioenergetic response as a result of mitochondrial respiration shutdown. The loss of oxidative phosphorylation is followed by disruption of glycolysis, which occurs after an initial increase in glycolytic respiration is unable to compensate for the interruption of the electron transport chain (ETC). In contrast, the PACT agents tested had little impact on cellular respiration, and the minor inhibition of these metabolic processes was not related to the mechanism of action, as reflected by a lack of correlation with photoejection efficiency. Notably, a system capable of both generating 1O2 and photo-releasing a ligand exhibited the dominant profile of a PDT agent and induced the quiescent bioenergetic state, indicating potential implications on cellular bioenergetics for so-called dual-action agents. These findings are presented with the aim to provide the necessary groundwork for expanding the application and utility of EFA to phototherapeutics and to highlight the role of metabolic alterations in PDT.
Introduction
Chemotherapy is part of the treatment regimen for approximately 60% of patients with cancer.1 However, traditional chemotherapies generally have dose-limiting adverse effects which have been associated with poor outcomes. Photodynamic therapy (PDT) and photoactivated chemotherapy (PACT) have been developed to limit the tissues exposed to cytotoxic agents. In both types of phototherapy, light is used to activate a prodrug molecule. For Ru(II) polypyridyl complexes, irradiation of the inactive complex induces excitation and population of the singlet metal to ligand charge transfer state (1MLCT), which then undergoes intersystem crossing, yielding the 3MLCT state. In PDT, the chemical agents are photosensitizers (PSs) that undergo energy transfer from their triplet excited state to catalytically convert ground-state 3O2 to reactive oxygen species (ROS), including 1O2, via type I or type II electron and energy transfer processes.2 PDT agents have shown significant translational utility, with porphyrin-based PSs in routine use in the clinic, and a new Ru(II)-based photosensitizer, TLD-1433, currently under evaluation in Phase II clinical trials3 with the FDA designating it with Fast Track status. In contrast to generating ROS, PACT relies on the concept of photocaging, where light is used to release an active molecule from a protecting group. We and others have developed Ru(II)-based PACT agents based on intrinsically strained systems, where the distortion of the complex brings the 3MLCT state close in energy to a triplet metal-centered state 3MC, facilitating population of this formally antibonding orbital and resulting in photoejection of a ligand.4–7 This allows for the selective delivery of targeted agents that are intended to precisely regulate specific biological processes, often without inducing systemic cytotoxicity.8–12Despite considerable interest in the development of compounds for PDT and PACT, the relationship between the mechanisms of action of these agents and cellular bioenergetics remains largely unexplored. Moreover, bioenergetic interactions of metal-containing agents are generally not well characterized, with some notable exceptions. There have been studies of a small number of metal complexes containing iridium,13 cobalt,14 copper,14,15 gold,15–18 platinum,19 ruthenium,20–22 rhenium,23 and iron24 that include assessments of the influence of the compounds on bioenergetics. However, to our knowledge, none of these complexes leverage light-mediated mechanisms of activation. We believe this constitutes a large knowledge gap, as light-activated compounds are being considered for various pre-clinical and clinical applications, including direct cell killing, immune priming, and delivery of targeted, highly specific biological effectors, but without knowledge of the impacts they may have on the essential energy producing pathways of oxidative phosphorylation and glycolysis. Herein, we investigate PACT and PDT agents and their respective effects on cellular bioenergetics using the Seahorse extracellular flux analyzer, and identify noteworthy differences between these two treatment approaches.
Quantifying cellular respiration was once a cumbersome process.25 The introduction of the Seahorse extracellular flux analyzer, as well as other multi-sample flux analyzers, has greatly expanded the throughput of these technologies to assess impacts on oxidative phosphorylation and glycolysis.26 This has facilitated the characterization of these key processes, and made bioenergetic analysis an accessible part of the drug discovery pipeline.27–29 By running high-throughput bioenergetics assays with drug candidates, their effects on oxidative phosphorylation and glycolysis on adenosine triphosphate (ATP) production can be evaluated, providing key information as a part of the desired mechanism of action or revealing a potentially dangerous off-target effect. Biological samples that can used for analysis include cells lines from 2D tissue culture, 3D organoid models,30,31ex vivo samples,32 and even in vivo systems such as zebrafish embryos33,34 and Caenorhabditis elegans.35 Strong correlations have been shown for metabolic disruptions detected in the Seahorse analyzer for compounds that are toxins in vivo,36–38 drugs that can induce metabolic reprograming,39,40 and even antibiotics.41
Deregulation of cellular metabolism is a hallmark of cancer, making it an attractive target for anticancer therapy.42,43 Most cancer cells are considered to be addicted to glycolysis for energy production, a phenomenon first reported by Otto Warburg in the 1920s,44 and exhibit varying degrees of mitochondrial dysfunction. Furthermore, recent work has shown that mitochondria play an important role in cancer development, particularly in metastasis.45–47 Thus, it is important to understand how potential anti-cancer agents can impact central metabolic processes and mitochondrial function, particularly as inhibition of mitochondrial function is considered a partially selective cancer treatment strategy. The mitochondrial membrane potential of cancer cells is 80 mV more negative than healthy cells.48 This negative mitochondrial membrane potential attracts lipophilic cations, making cancer cells more susceptible to treatment with molecules with these physiochemical properties, as these compounds perturb mitochondrial functions.48 Since a substantial portion of Ru(II)-based molecules are both lipophilic and cationic, they possess the appropriate characteristics to disrupt mitochondrial function and impair oxidative phosphorylation. Investigating the impact of Ru(II) molecules on these energy-producing processes could provide key information regarding structural and functional considerations (i.e. photoinert vs. photolabile complexes) when attempting to target – or preserve – cellular bioenergetic functions. As a result, we performed a systematic analysis of the effects of photostable Ru(II) complexes that can act as photosensitizers, and are potential PDT agents, and compared the effects of Ru(II) complexes that can eject a ligand, and thus are potential PACT agents. Notably, the photoejecting metal centers are capable of forming adducts to biomolecules. We further expanded the study to include several structurally related compounds that contain thermally labile chloride ligands and are capable of similar covalent interactions with biomolecules, but without the light activation step. These were termed “traditional” agents, in contrast to the photoactive systems.
We found that both lipophilic Ru(II) complexes and an organic PDT agent rapidly terminate cellular bioenergetics in lung adenocarcinoma (A549) cells. Treatment with these photosensitizers causes the cells to become first glycolytic, and then quiescent, after they fail to compensate for the lost energy that was previously provided by oxidative phosphorylation. Interestingly, the PACT compounds exhibited a radically different profile. While the Ru(II) systems are structurally similar, the PACT systems generally do not influence cellular bioenergetics, which could be attributed to their alternate mechanisms of action.4,49,50 We also demonstrate, in concurrence with previous work on traditional agents that are not prodrugs,51 that this platform works exceedingly well as a screening tool, expanding the utility of flux analyses to phototherapeutics. This foundational study establishes a workflow for studying phototherapeutic and bioenergetic interactions, facilitating future studies for characterizing the necessary structural features and mechanisms of Ru(II)-based agents that target cancer metabolism.
Results and discussion
Correlation between cytotoxicity and hydrophobicity
A focused library of previously synthesized and reported Ru(II) molecules4,49,50,52–55 was used in this analysis. While other metal complexes are of interest to select research groups, we focused on Ru(II) polypyridyl complexes, due to their long-standing prominence in photochemical and photobiological studies, and their growing clinical significance for phototherapy applications, as evidenced by the success of TLD-1433. Moreover, in contrast to most other metal coordination compounds, Ru(II) complexes can act as either PDT or PACT agents with only slight structural changes. As a result, this small collection of molecules allowed for comparison of disparate photochemical mechanisms of action within closely related chemical structures. Characterization data is provided in these published works4,49,50,52–55 and in the ESI,† and all compounds investigated exhibited ≥95% purity. Additional studies of the biological effects of the individual molecules can be found in these prior publications.Notably, all compound structures are closely related (Chart 1A and B), with 1–12 being tris- and bis-homoleptic octahedral agents containing polypyridyl ligands with no reactive functional groups or moieties likely to engage in hydrogen-bonding or polar interactions. Compounds 13–15 (Chart 1B) are also octahedral, and contain one tridentate polypyridyl ligand, a bidentate ligand, and an exchangeable chloride ligand. These compounds are not potential phototherapeutics so the effect of light on cytotoxicity was not determined. All compounds studied carry a +2 charge (1–12), a +1 charge (13 and 15) or 0 charge (14). The compounds that were chosen as PACT agents contained strain-inducing ligands, which incorporate groups that induce steric clash when assembled around the octahedral metal center. These ligands are 6,6′-dimethyl-2,2′-bipyridine (dmbpy), 2,9-dimethyl-1,10′-phenanthroline (dmphen), and 2,2′-biquinoline (biq), along with the pyridyl-pyrazole ligand (in compounds 2, 4, 5, 7, 11, and 12). In contrast, complexes that are not strained are expected to act as PDT agents, and incorporate 2,2′-bipyridine (bpy), 1,10′-phenanthroline (phen), and 4,7-diphenyl-1,10-phenanthroline (DIP) ligands (compounds 1, 3, 6, 8, 9, and 10).
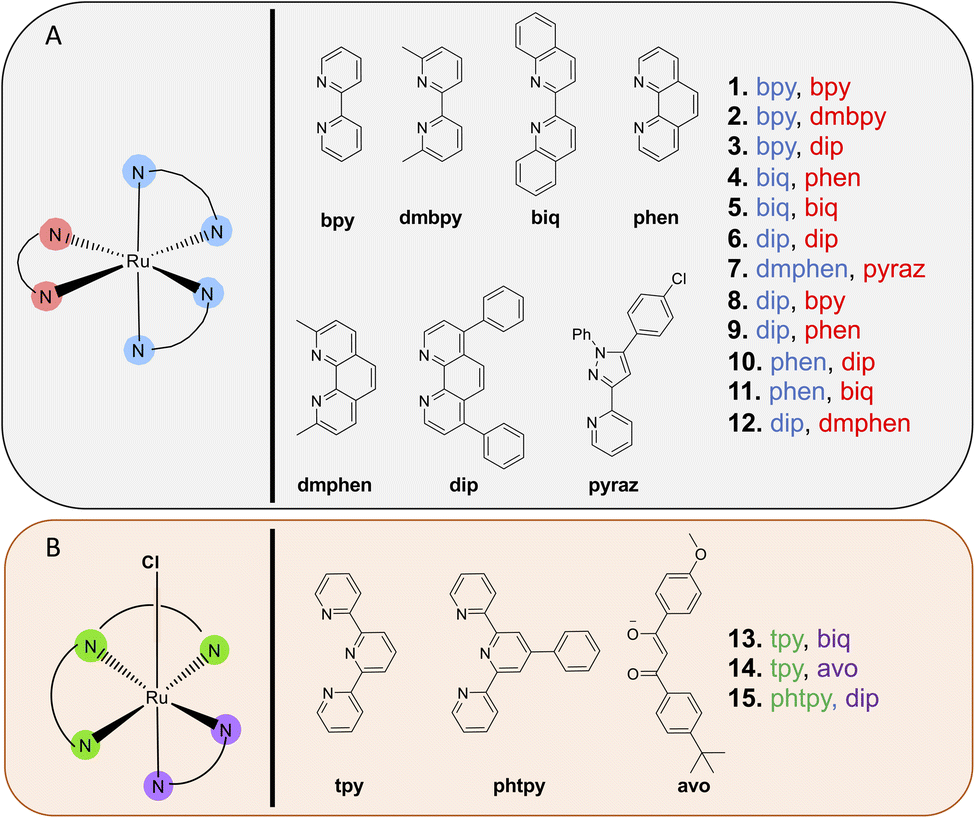 | ||
| Chart 1 Ruthenium(II)-based compounds investigated in this study. (A) Ru(II) PACT (2, 4, 5, 7, and 11) and PDT compounds (1, 3, 6, 8, 9, and 10) investigated. (B) Thermally activated Ru(II) scaffolds investigated. | ||
The cytotoxicity of the various compounds was assessed prior to performing the extracellular flux analysis to determine an appropriate range of concentrations to use when probing cellular bioenergetics. The cytotoxicity was determined for all compounds in the dark and for 1–12 after irradiation with 450 nm light (29.1 J cm−2) in the A549 lung cancer cell line. In general, PDT compounds were significantly more photo-cytotoxic than photocaged compounds in this study (Fig. 1A and Table 1). The difference in efficacy between PACT and PDT agents is apparent when comparing their phototoxicity indices (PI; the ratio of the cytotoxicity in the dark to the light), as PDT agents were up to 50-fold more potent whereas PACT compounds were only up to 7.2-fold more potent upon irradiation. This is attributed to distinct mechanisms of action, and the fact that photosensitizers are catalysts that generate ROS, inducing DNA damage,56–60 lipid peroxidation,61,62 and notably for this study, mitochondrial dysfunction.20,63,64 In contrast, while photoactivated chemotherapeutics are capable of acting via multiple mechanisms, either via metal-based damage to biomolecules or the release of organic ligands that are cytotoxic, they are still intrinsically limited by the stoichiometric quantities of cytotoxins produced.
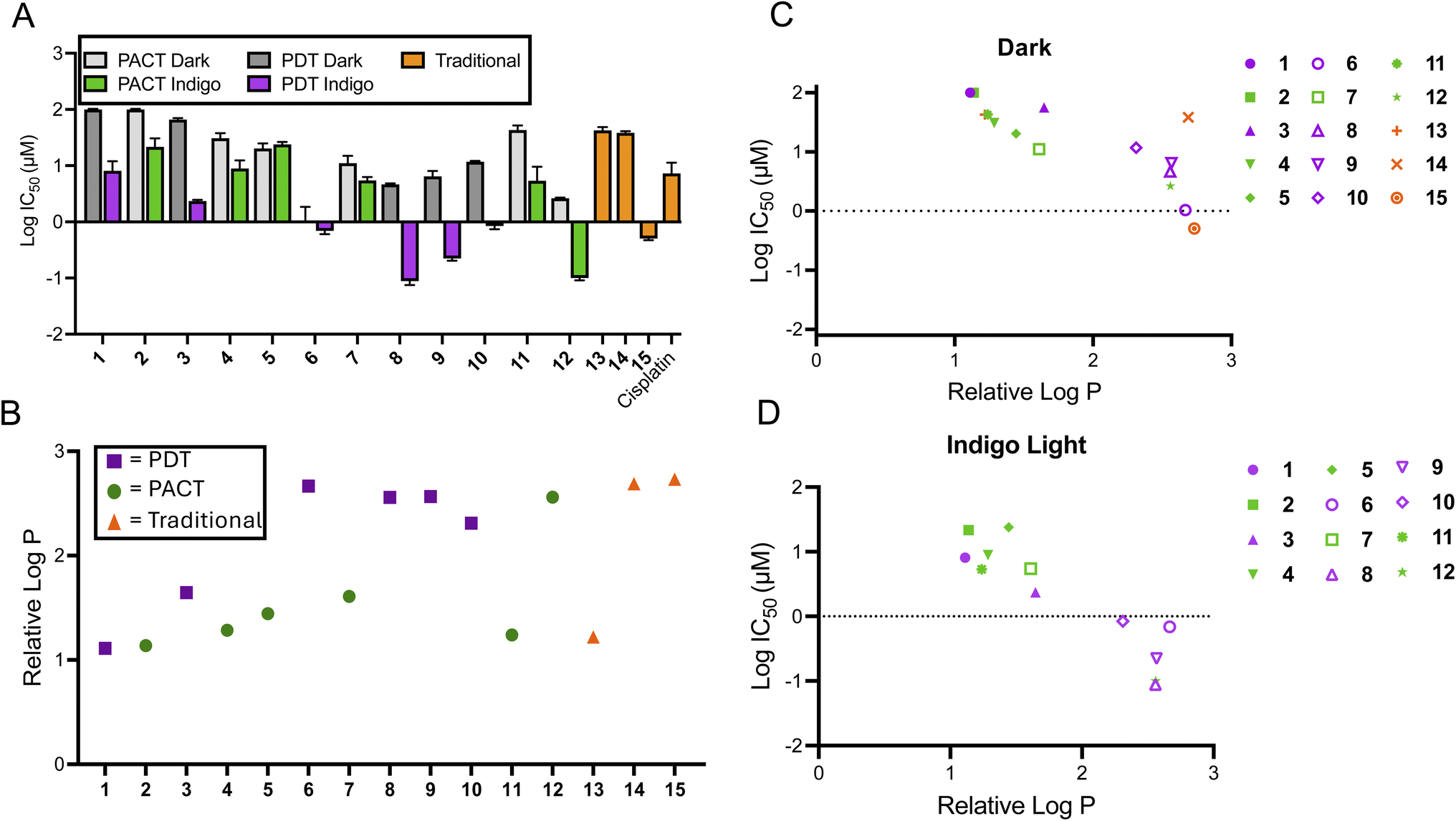 | ||
Fig. 1 Correlation of cytotoxicity and lipophilicity of compounds 1–15. (A) The average log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50 of compounds 1–15. Cytotoxicity values were determined 72 h after treatment; irradiated samples were treated with 450 nm light (29.1 J cm−2) and dark samples were protected from light (n = 3). (B) The relative logP values of 1–15 as determined by the chromatography method described in the ESI.† (C) The comparison between relative logP and cytotoxicity of 1–15 in the dark. (D) The comparison between relative logP and cytotoxicity of 1–12 following irradiation. IC50 of compounds 1–15. Cytotoxicity values were determined 72 h after treatment; irradiated samples were treated with 450 nm light (29.1 J cm−2) and dark samples were protected from light (n = 3). (B) The relative logP values of 1–15 as determined by the chromatography method described in the ESI.† (C) The comparison between relative logP and cytotoxicity of 1–15 in the dark. (D) The comparison between relative logP and cytotoxicity of 1–12 following irradiation. | ||
| Compound | IC50 (±St. Dev.) [μM] | PI | Lowest concentration for inhibition of OCR [μM] | ||
|---|---|---|---|---|---|
| Dark | Light | Dark | Light | ||
| a Indigo = 450 nm light (29.1 J cm−2; 485 mW cm−2). b Red light = 660 nm (57.8 J cm−2; 16 mW cm−2). c All data is representative of triplicate or quadruplicate measurements. | |||||
| 1 | >100 | 8.6 (3.6) | >11.6 | >25 | >25 |
| 2 | >100 | 22.6 (8.4) | >4.4 | >25 | >25 |
| 3 | 66.4 (3.9) | 2.34 (0.11) | 28.4 | >1 | 0.3 |
| 4 | 31.3 (6.8) | 9.2 (2.8) | 3.4 | >10 | 10 |
| 5 | 20.5 (4.0) | 24.0 (2.3) | 1.0 | >10 | >10 |
| 6 | 1.1 (0.6) | 0.7 (0.1) | 1.6 | 1 | 0.1 |
| 7 | 11.4 (3.2) | 5.5 (0.7) | 2.1 | >10 | 10 |
| 8 | 4.6 (0.2) | 0.09 (0.01) | 51.1 | 1 | 0.1 |
| 9 | 6.6 (1.4) | 0.22 (0.02) | 30.0 | >1 | 0.1 |
| 10 | 11.8 (0.4) | 0.8 (0.1) | 14.8 | >1 | 1 |
| 11 | 43.4 (7.8) | 6.0 (3.6) | 7.2 | >25 | 10 |
| 12 | 2.6 (0.1) | 0.10 (0.01) | 26.0 | 10 | 1 |
| 13 | 42.5 (5.6) | N.D. | N.D. | 10 | N.D. |
| 14 | 38.5 (2.6) | N.D. | N.D. | 10 | N.D. |
| 15 | 0.48 (0.03) | N.D. | N.D. | >3.3 | N.D. |
| HPPHb | >90 | 3.7 (1.5) | ∼27 | 3 | 0.4 |
| Cisplatin | 8.1 (2.9) | N.D. | N.D. | >20 | N.D. |
In order to assess a physiochemical property that could impact subcellular localization and alter metabolic processes, the logarithm of the partition coefficient (logP) for each compound was determined, which is correlated to the lipophilicity. A chromatographic method was used that established the correlation between retention time and logP using small molecules with known logP values. Based on this data set, the relative logP values for the Ru(II) compounds was determined.65,66 The chromatogram for each compound is shown in Fig. S2,† and plots of their relative logP values are provided in Fig. 1B, S3 and Table S1.† There was a positive correlation between logP and cytotoxicity for the series of compounds (Fig. 1C and D), consistent with other studies that show that hydrophobic metal complexes can have cytotoxic effects, sometimes due to association with mitochondria. An exception was compound 14, which was inactive, despite having the second highest lipophilicity. Thus, the thermally activated compounds 13–15 did not fit the general trend for the relationship between cytotoxicity and lipophilicity. There were no other obvious structural features to consider in structure–activity relationships, as the chemical structures are coordination complexes of non-polar polyaromatic ligands.
Utility of the Seahorse platform for PDT agents
Mitochondrial biology and metabolism can be assessed with extracellular flux analysis tools that detect the changes in extracellular O2 and H+ levels that are associated with different perturbations within the electron transport chain (ETC).67–69 The mitochondrial stress test (MitoStress Test) utilizes established inhibitors of various components of the ETC to obtain information about ATP biosynthesis, changes in cellular respiration, and overall influences on mitochondrial health. Following incubation with a compound of interest, the cells in the MitoStress Test are treated with a series of validated inhibitors of metabolic processes to obtain well-characterized changes in respiration. The first compound used is oligomycin, which inhibits ATP synthase (Fig. 2A), resulting in a decrease in cellular respiration.70 After data is obtained under conditions where ATP synthase is inhibited, an injection of carbonyl cyanide p-(trifluoromethoxy)phenyl-hydrazone (FCCP), a mitochondrial membrane uncoupler,67 is performed. This mimics an energy-demanding stimulus, resulting in a ramp in mitochondrial respiration. The next injection is a rotenone/antimycin A cocktail. Rotenone inhibits Complex I, while antimycin A inhibits Complex III.71,72 The ETC is shut down when these two compounds are added to cells, resulting in the elimination of mitochondrial-associated respiration (Fig. 2B).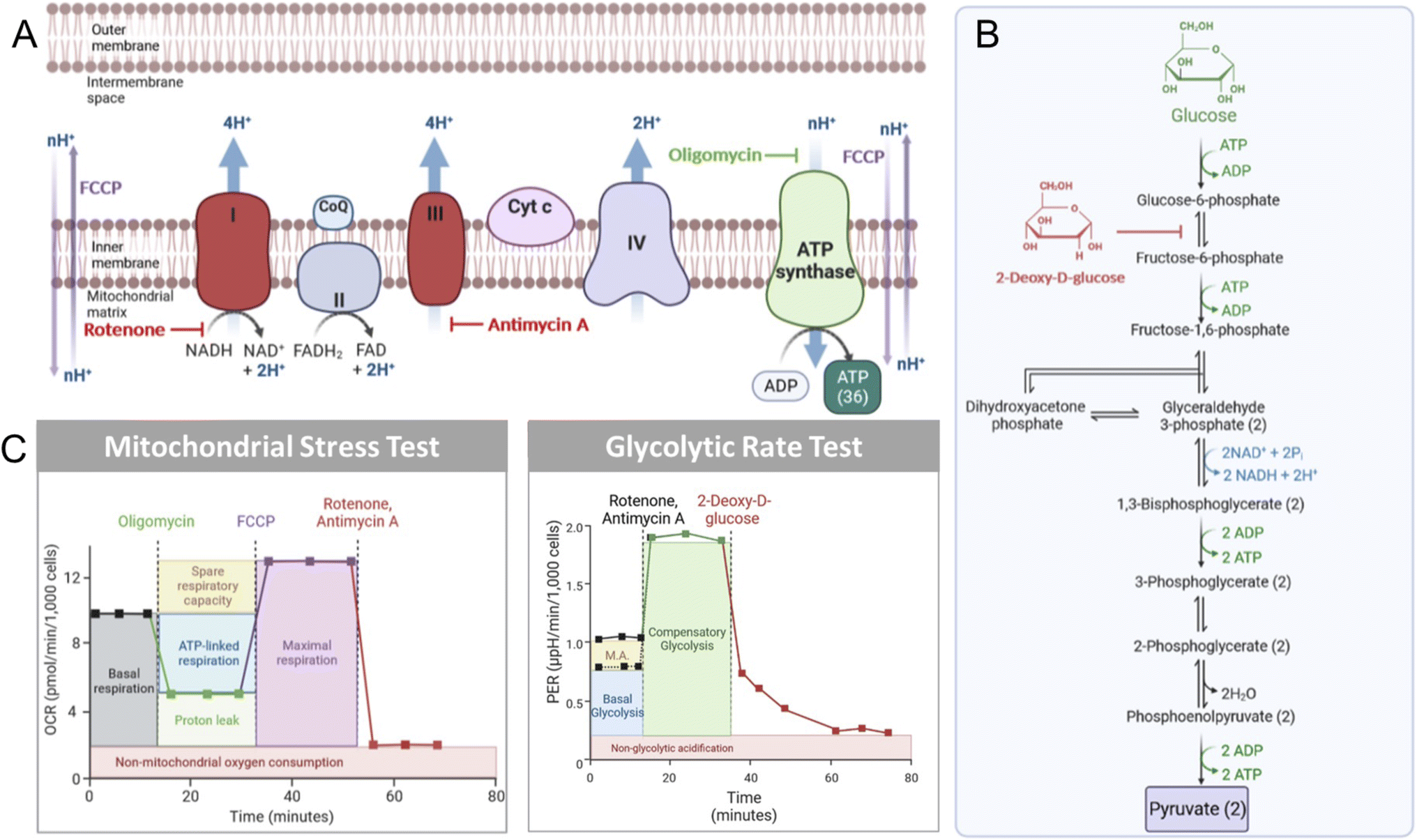 | ||
| Fig. 2 Processes involved in extracellular flux analysis. (A) The inhibitory actions of small molecules oligomycin, FCCP, rotenone, and antimycin A used in the MitoStress Test. (B) The key parameters that can be extracted from the MitoStress Test and the Glycolytic Rate Test. (C) The role of 2-deoxyglucose (2-DG) in inhibiting glycolysis. Mitochondrial acidification is represented by “M.A.” in the Glycolytic Rate test. | ||
Glycolysis can also be monitored by extracellular flux technology. For this, basal glycolysis and mitochondrial acidification are measured and then the ETC is shutdown by the injection of rotenone and antimycin A. Glycolysis is then upregulated in the cells in an attempt to compensate for the loss in oxidative phosphorylation, providing a reading of compensatory glycolysis. Next, glycolysis is blocked by the injection of 2-deoxyglucose (2-DG; Fig. 2C), which inhibits phosphoglucose isomerase preventing the conversion of 2-deoxy-D-glucose-6-phosphate to fructose-6-phosphate.73 After 2-DG injection in the glycolytic rate assay, non-glycolytic acidification is measured. This provides a reading of the acidification that occurs independently of glycolysis, such as due to the TCA cycle.
For this study, A549 cells were selected as the model cell line due in part to the fact that they have a similar dependency upon glycolysis and oxidative phosphorylation for ATP biosynthesis.74 The light and dark IC50 values for the various compounds were evaluated in order to determine the appropriate concentrations to ensure cell health and viability during the cellular bioenergetics experiments. In addition, a short incubation time (1 h) was applied to avoid pleiotropic mitochondrial toxicity. The choice of cell line, the use of sub-cytotoxic concentrations, and the short incubation time are all essential experimental choices to ensure valid data would be obtained from the extracellular flux analysis.
Prior to evaluating the full collection of compounds, two models were chosen to assess the utility of the Seahorse platform for PDT agents. Compound 3, a photosensitizer activated by indigo light, and 2-(1-hexyloxyethyl)-2-devinylpyropheophorbide-a (HPPH, Photochlor), a red-light active photosensitizer with a logP of ∼6.0,75,76 were studied. The cellular respiration remained unaffected by both compounds in the absence of light (Fig. 3A and C), but a marked concentration-dependent decrease was observed following irradiation, with indigo light for 3 (Fig. 3B) and red light (660 nm) for HPPH (Fig. 3D). Next, the key parameters of the MitoStress Test (basal respiration, maximal respiration, and non-mitochondrial respiration) were quantified for 3- and HPPH-treated cells in the dark and following irradiation (Fig. 3E–P). Basal respiration remained unaffected in the dark (Fig. 3E), but decreased in dose response for 3 with irradiation (Fig. 3F). A similar trend was observed for HPPH-treated cells (Fig. 3G and H). Both maximal and non-mitochondrial respiration were not significantly influenced by each compound in the dark, but decreased once irradiated (Fig. 3I–P). The fact that the same effects were observed with blue and red light activated systems demonstrates that the phenotype is not associated with a particular light treatment, but rather, the ability of the PSs to generate ROS.
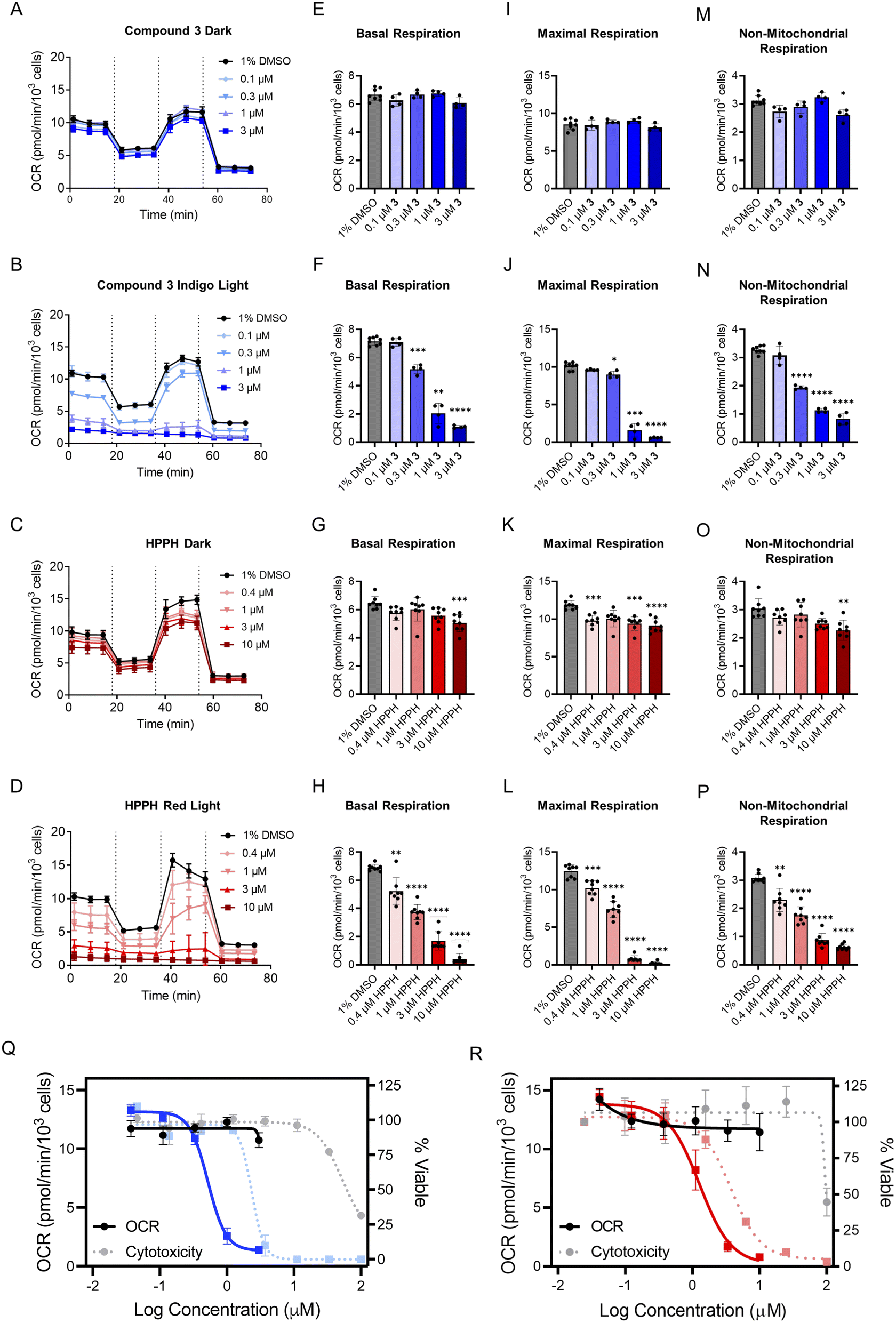 | ||
| Fig. 3 Extracellular flux and cytotoxicity of HPPH and compound 3 in A549 cells. For HPPH, the light treatment was irradiation with red light (660 nm, 57.8 J cm−2); for compound 3, the light treatment was irradiation with indigo light (450 nm, 29.1 J cm−2) (A–D) quantification of OCR following treatments: (A) compound 3 for 1 h in the absence of light; (B) compound 3 following irradiation; (C) HPPH for 1 h in the absence of light; (D) HPPH following irradiation. (E–H) Basal respiration measured in cells following treatments: (E) compound 3 in the absence of light; (F) compound 3 following irradiation; (G) HPPH in the absence of light; (H) HPPH following irradiation. (I–L) Maximal respiration for A549 cells after treatments: (I) 3 in the absence of light; (J) following irradiation; (K) HPPH in the absence of light; (L) HPPH following irradiation. (M–P) Non-mitochondrial respiration following treatments: (M) 3 in the absence of light; (N) 3 following irradiation; (O) HPPH in the absence of light; (P) HPPH following irradiation. For (A–P), n ≥ 4. (Q) Correlation for 3 between maximal respiration and cytotoxicity. (R) Correlation for HPPH between maximal respiration and cytotoxicity. In (Q and R), the darker colors and solid lines indicate OCR measurements and the dotted line and lighter color represent cytotoxicity. The right y-axis corresponds to the cytotoxicity (n = 3). The left y-axis corresponds to the OCR (solid lines and darker coloring) (n ≥ 4); *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001. | ||
The oxygen consumption rate (OCR) range for basal and non-mitochondrial respiration measurements was 7 picomoles per minute per thousand cells and 3 picomoles per minute per thousand cells, respectively. Maximal respiration exhibited an OCR range of approximately 12 picomoles per minute per thousand cells. Since maximal respiration was the parameter with the most dynamic OCR range, it was selected for determining an OCR IC50 for each dose–response. This gave values for HPPH-treated A549 cells of 1.2 μM and 0.5 μM for 3 in the treated cells (Fig. 3Q and R). Next, the OCR and cytotoxicity dose response curves were overlaid for comparison (Fig. 3Q and R). Notably, there was a 3- to 5-fold shift to higher potency for the effect of each compound on the OCR compared to the cytotoxicity in each condition. This finding illustrates the importance of conducting bioenergetic analyses at concentrations below the cytotoxicity IC50, as treating cells with too high of a concentration, even at short exposure times, results in near-baseline readings and misinterpretation of mechanistic implications.
Bioenergetic impacts of phototherapeutics with different mechanisms of action
Following the establishment of these parameters, extracellular flux analysis was conducted for each of the agents at two different concentrations in the dark and following irradiation (Fig. S5 and S6†). The PACT agents 4, 5, 7, and 11 reduced cellular respiration by less than 30% as shown in Fig. 4A. In marked contrast, the PDT agents 3, 6, 8, 9, and 10 reduced maximal respiration by at least 90% (Fig. 4B and S5†). Notably, compound 1, a hydrophilic complex that is able to photogenerate 1O2, did not fit the trend (Fig. S6 and Table S1†), possibly due to reduced accumulation within the cell because of its lower lipophilicity. Compound 12 also exhibited unanticipated inhibition of respiration, which was inconsistent with the activities of the other PACT systems. Values for the lowest concentrations where inhibition of OCR were observed are provided in Table 1; compounds 3, 6, 8, 9, and HPPH all suppressed OCR at concentrations below 1 μM.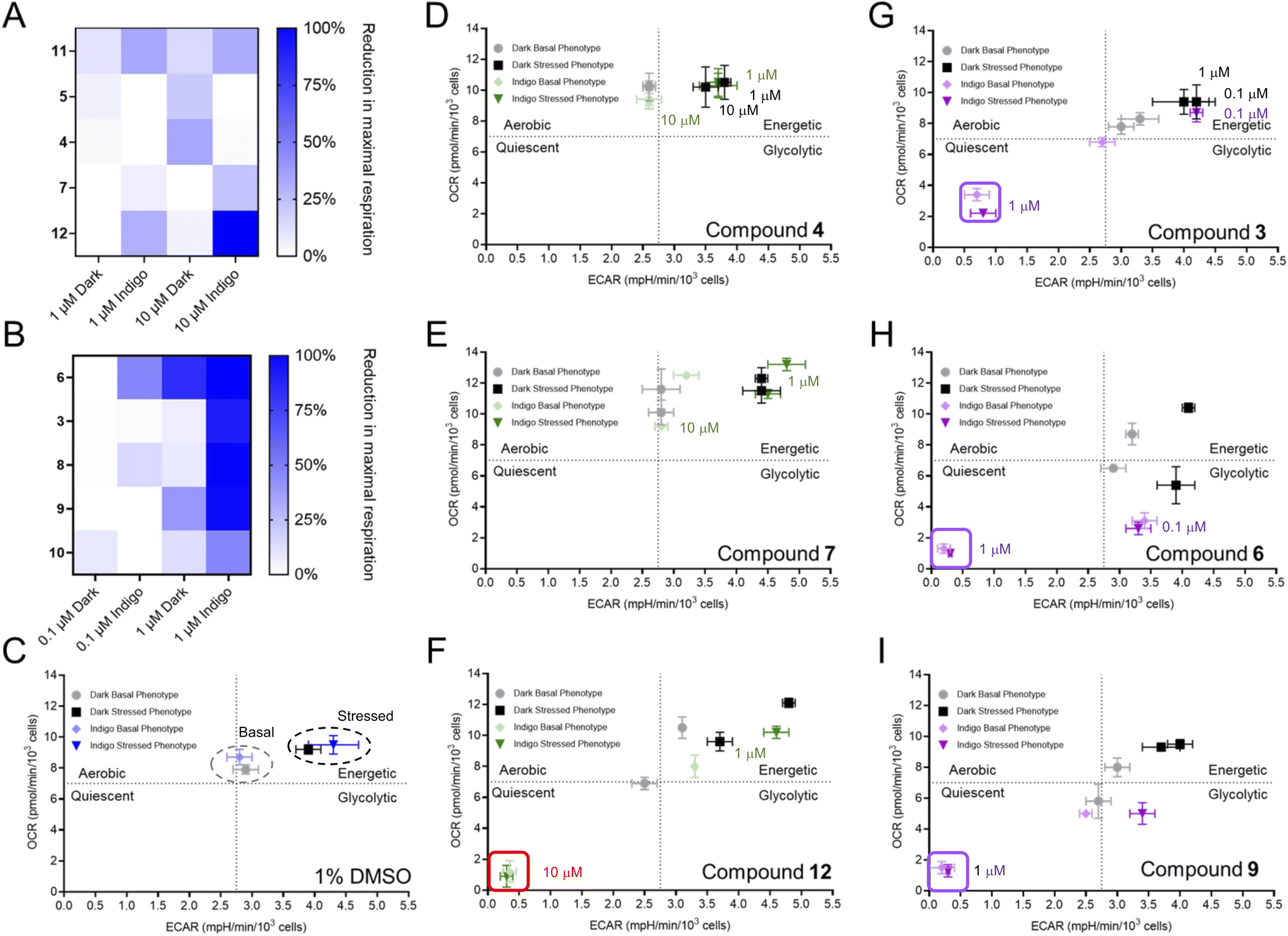 | ||
| Fig. 4 Bioenergetic impacts of phototherapeutics in A549 cells. The reduction in maximal respiration following treatment with (A) PACT agents and (B) PDT agents at the indicated concentrations. The bioenergetic phenotype of cells treated with (C) vehicle, (D) 4, (E) 7, (F) 12, (G) 3, (H) 6, (I) 9. In (C–I), the lighter colors reflect the basal phenotype (prior to the addition of oligomycin and FCCP), while darker colors represent the stressed phenotype (after oligomycin and FCCP but before addition of rotenone/antimycin A). n > 3 for Seahorse data. The trend is that compounds that photogenerate 1O2 result in a quiescent phenotype (see the boxed data for 3, 6, 9, and 12) while compounds that photoeject have a minimal effect on metabolic reprograming. | ||
To gain greater understanding, the glycolytic rate test was also performed (Fig. S8 and S9†). In the dark, PACT agent 2 and PDT agent 6 had little impact on the non-glycolytic acidification. However, 6 increased basal glycolysis and decreased compensatory glycolysis at a concentration of 1 μM, consistent with the IC50 for cytotoxicity in the dark (1.1 μM). This complex has previously been shown to decouple the mitochondrial membrane potential in the dark, suppressing oxidative phosphorylation,53 so it is consistent that the cells try to compensate with increased glycolysis. Interestingly, a similar effect was seen for 6 with light treatment at a concentration of 0.1 μM, followed by a complete abrogation of all glycolytic activity at 1 μM, as indicated by the glycolytic proton efflux rate (PER) falling to zero. A similar concentration-dependent decrease was observed for HPPH with irradiation with red light, with the PER showing no glycolytic activity at a concentration of 10 μM (Fig. S9†). This data demonstrates that PDT agents induce disruption of glycolysis, in contrast to PACT agents.
A bioenergetic map was generated to compare the basal and stressed phenotypes with each compound. Oxidative phosphorylation is reflected by the basal OCR and glycolytic function is discerned by the basal extracellular acidification rate (ECAR). Metabolic switching occurs when the cells are placed under “stressed conditions” by the addition of the stressor compounds oligomycin and FCCP, which force mitochondrial respiration into overdrive without producing additional ATP. This induces an energy demand and forces cells to produce ATP solely by glycolysis, increasing ECAR. For vehicle-treated cells in the dark and following irradiation, the values for OCR and ECAR were comparable for both basal and stressed cells, indicating that light treatment alone did not impact energetics and that cells increased the ECAR by accelerating glycolysis when stressed (Fig. 4C). Notably, a quiescent phenotype, associated with low OCR and ECAR, was observed for PDT-treated cells (Fig. 4G–I), demonstrating a loss of ATP production. This is consistent with the findings for both the mitochondrial stress test and the glycolytic rate test, demonstrating that PDT agents suppress both oxidative phosphorylation and glycolysis and thereby abrogate the ability of the cell to generate ATP. In contrast, the PACT-treated cells exhibited a similar phenotype as the vehicle control (Fig. 4C–E and S12†) and were able to respond to increased energy demand.
The bioenergetic profile seen for PDT agents was dependent on concentration, as shown in Fig. 4, S10 and S11,† with some compounds exhibiting alterations in OCR and ECAR at significantly lower concentrations than the IC50. For example, HPPH induced the quiescent phenotype at a concentration 0.37 μM, 10-fold lower than the IC50 for cytotoxicity of 3.7 μM (Fig. S10†). While HPPH had little impact in the dark, the effect of some compounds with light treatment appeared correlated to the effects observed without irradiation. Comparison of compounds 3 and 6 provides an illustrative example. There is a large difference in the cytotoxicity of these two compounds in the dark (IC50 of 66 μM for 3vs. 1 μM for 6) which is also reflected by a shift in bioenergetic profile. This is paralleled by the data for the light-induced decrease in OCR and ECAR, which occurred for 6 at a concentration of 0.1 μM, while these values were essentially unaffected at the same concentration for compound 3 (Fig. 4, S10 and S11†). Thus, it is imperative to decouple bioenergetic effects that are driven by photoactivity vs. those that are present (albeit to a lesser degree) for the compound in the absence of irradiation, and impacts on both OCR and ECAR should be assessed.
The only PACT compound that hampered cellular bioenergetics was 12 (Fig. 4F). This unexpected finding might be attributed an additional mechanism of action. It was considered that 12 could be active both as a PDT and PACT agent, which has been described for other Ru(II) compounds.49,77 Given the inclusion of the strain-inducting dmphen ligand, the compound photoejects (Fig. S13, S14 and S19†), but the perturbation in mitochondrial activity suggested it was also a PDT agent. Thus, we investigated if this molecule also acted as a photocatalyst and generated 1O2. As shown in Fig. S20,† compound 12 generates high levels of 1O2 as detected by singlet oxygen sensor green (SOSG). This demonstrates that the metabolic alterations observed for 12 can be attributed to the photosensitizing effects of the molecule acting as a PDT agent, despite its capacity to also function as a PACT agent.
The glycolytic rate for cells following treatment with 2 or 6 was measured to demonstrate how PACT and PDT agents influence glycolysis (Fig. S8†). No change was observed for 2 both in the dark and following irradiation. However, compound 6 elevated the glycolytic rate after the 1 h incubation in the dark at a concentration of 1 μM, and at 0.1 μM with irradiation. The elevation in PER at basal measurements suggests that 6 shuts down the ETC, resulting in a compensatory response in glycolysis. This, in conjunction with previous reports,53 reflects the high affinity of 6 for mitochondria, as well as demonstrating how glycolytic measurements can inform on the mitochondrial-damaging efficacy of these metal complexes. Furthermore, this assay demonstrates that bioenergetic stress associated with 6 and the other PDT agents may be multimodal, as previously reported, but might be initiated by mitochondrial disfunction.78 The reduction in glycolysis occurs after cells are unable to compensate for the loss of energy produced via oxidative phosphorylation. We also find that this effect is seen with HPPH, indicating that the phenotype is independent of compound structure, as HPPH is not a metal complex.
In contrast, minimal mitochondrial toxicity was observed with PACT agents. We considered that this could potentially be attributed to low photosubstitution yields, so the photoejection kinetics of each PACT were measured and the photoproducts were identified (Fig. S13–S19†). When ligand exchange was measured by ultraviolet-visible spectroscopy, all compounds appeared to have photoejected, with almost no parent compound remaining (Fig. S13†). Ligand dissociation was also quantified by liquid chromatography-mass spectrometry (LC-MS). Interestingly, compounds 7, 11, and 12 did not completely photodissociate (Fig. S13 and Table S2†) but exhibited PI values higher than compounds 2, 4, and 5 which fully ejected the ligand (Table 1). This indicated that cytotoxicity was not wholly dependent on ligand loss and formation of a new ruthenium species.
The biquinoline (biq)-containing compounds 4, 5, and 11 presented an interesting series, as the intramolecular strain in the molecules increased as the number of biquinoline ligands increased. Compound 4 underwent two dissociation steps, resulting in high abundances of [Ru(biq)(MeCN)4]2+ and [Ru(biq)(H2O)4]2+ (Fig. S16,† note that the ligand exchange product depends on the solvent used). These tetra-substituted photoproducts were only present at quantifiable abundances for 4, suggesting that the strain introduced with a mono- or bis–biq complex (compound 11 or 5) results in photodissociation of one ligand (Fig. S17 and S18†), while a tris–biq system (compound 4) facilitates the loss of multiple bidentate ligands. This series of compounds demonstrated that improved photodissociation and lipophilicity of a scaffold is not sufficient for the creation of a highly active complex. Compounds 2 and 12 each ejected one ligand (Fig. S15 and S19†). Given that 2, 4, 5, and 7 exhibited >90% photoactivation and poor mitochondrial activity, mitochondrial dysfunction can be eliminated as the mechanism of action for these PACT agents. Given the incomplete photodissociation of 12 and its ability to generate 1O2, compound 12 likely acts partially via a PDT mechanism, as reflected by the impact on ECAR and OCR. However, the concentration dependence of this phenotype (Fig. 4F and S10†) is notably different from other PDT agents, as the impact on metabolic processes was only apparent at concentrations well above the cytotoxicity IC50 value (0.1 μM). Thus, the cytotoxicity may be driven by the activity of the released dmphen ligand, which is active in the mid-nM range.79
As another point of comparison, a library of Food and Drug Administration (FDA)-approved small molecules agents with non-light mediated mechanisms of action were also assessed as mitochondrial stress inducers (Fig. S21†). Cycloheximide, a translation inhibitor, and actinomycin D, a ribosome biogenesis stress inducer, were the only two drugs that were found to reduce maximal respiration. Cycloheximide-treated A549 cells have exhibited reduced ATP synthesis in a previous report, which could correlate to the reduction in maximal respiration observed here (Fig. S21†).80 This could be a result from mitochondrial dysfunction induced by elevated ROS, which has been shown for leukemias treated with actinomycin D.81 These results show how two previously-studied FDA-approved drugs impact cellular respiration, likely as a secondary mechanism to inhibition of protein translation. Additionally, the negative results from multiple other FDA-approved drugs after acute treatment further support our finding that PDT agents elicit unusual and notable effects on cellular bioenergetics.
Recommended parameters for use of seahorse platform for phototherapeutics and other agents
Inappropriate compound dosing can result in altered baseline readings and incorrect mechanistic characterization. Our findings indicate that 72 h growth inhibition curves can be utilized to help determine the appropriate concentration range for bioenergetic analyses. One of the most significant findings from this study is data supporting a recommendation to include concentrations 3–5-fold below the IC50, as this is a concentration range where effects on the OCR and ECAR should be observed if the compounds do in fact directly impact mitochondrial function. This is in marked contrast to most studies we have observed in the literature, where concentrations either at or above the cytotoxicity IC50 are used. Moreover, important information can be gained by performing the analysis at several concentration points, and these dose responses add confidence to the determination of a trend in behavior. Interestingly, in some cases low concentrations of an agent have been shown to enhance OCR, which is then reversed with higher compound doses.82 This highlights the sensitivity of cellular systems to compound effects that are highly concentration dependent.Information on the timeframe for cellular damage and induction of cell death processes is also required for each compound under assessment, and short incubation times provide the benefit of ensuring that the observed impacts on mitochondrial function are not simply downstream effects of global processes, including the induction of cell death. We noted significant variation in literature reports for incubation times, but most of the bioenergetic measurements have been taken after several hours' co-incubation with the mitochondrial-targeting small molecules. There are some notable exceptions where short incubation times were utilized, including a recent study showing that tamoxifen-conjugated coordination complexes increased reactive oxygen species and reduced maximal cellular respiration within 3 hours of treatment,15 and another study that observed reduced oxygen consumption and impaired oxidative phosphorylation after a 2 hours incubation.13 However, the other studies that utilize extended incubation times, such as 24 hours, may present misleading results, where mitochondria function impairment is not a direct effect of the compound under study.
While the in vitro conditions of concentration and time are clearly important variables that can result in significant alterations in effects, there are several other factors that will likely influence the measurable outcomes. For any light-activated molecule, the irradiance is a key factor. We used relatively low values (485 mW cm−2 for the 450 nm light, 16 mW cm−2 for red light). With phototherapy approaches that utilize endogenous oxygen, higher irradiance can be associated with oxygen depletion, resulting in reduced efficacy.83 For promising potential PDT agents, it may be worthwhile to perform bioenergetic analysis as a function of irradiance.
It must be stated that the biological effects of metal complexes, like organic molecules, can be highly sensitive to batch-to-batch variation, so promising compounds should be re-synthesized and reevaluated as a matter of course before advanced studies, such as evaluations of in vivo efficacy, are performed. Moreover, the robustness of biological results can only be verified with multiple biological replicates, and these studies are most compelling when consistency is found across experiments performed in multiple cell lines. Our goal with this study was to compare a collection of structurally related compounds from different functional classes to determine if there was a general trend in their impact on mitochondrial function, based on their photochemical mechanisms of action; it is not to advance any single molecule towards in vivo testing. As a result, we did not undertake extensive studies to confirm reproducibility with multiple compound batches, and did not perform broad investigations such as proteomics to probe for additional mechanistic insights on individual molecules. Moreover, as our studies were focused on the acute effects of compounds on metabolic processes after short (1 h) incubation times, the potential downstream effects on gene expression or protein production processes are not likely to be relevant to this work. We believe the consistent behavior we observed on cellular bioenergetics across the PDT agents is highly noteworthy, and the different behavior of the PACT agents indicates that these compounds do not disrupt mitochondrial function as part of their mechanism of action. We hope this report will have the benefit of motivating appropriately focused studies for promising new agents based on their photochemical mechanism of action.
Conclusions
This study provides the foundational groundwork for bioenergetic investigations of photoactivatable chemotherapeutics and photodynamic therapeutics, especially those that have been identified as dual-active agents. We evaluated twelve photoactive compounds with different mechanisms of action and varying physiochemical properties. Three Ru(II) compounds that are not photoactive were also evaluated to decipher whether mitochondrial toxicity was an associated mechanism for phototherapy, or simply a byproduct of cellular treatment with lipophilic cationic cytotoxins. We found that photosensitizers effectively inhibit oxidative phosphorylation and glycolysis, and furthermore, demonstrate that this phenotypic response can be measured in dose response, revealing that impacts on metabolism are observed at concentrations below those required for cytotoxicity.The glycolytic rate assay demonstrated that PDT treatment is primarily associated with mitochondrial dysfunction, and reduced glycolysis occurs once the cell is unable to compensate for the reduction in energy production resulting from ETC shutdown. This phenotype was exhibited by compounds 3, 6, 9, 10, and 12. Compound 12 is a notable example of a so-called “dual action” compound that both generates 1O2 and photoejects a ligand, and it exhibits a metabolic disruption profile consistent with 1O2 generation. The behavior of the organic photosensitizer HPPH shows that this is not limited to Ru(II) systems, but rather appears to be a general feature of PDT agents. Thus, it may be that mitochondrial dysfunction and suppression of glycolysis is a significant component of PDT efficacy. In stark contrast, Ru(II) compounds that were PACT agents (compounds 2, 4, 5, 7, and 11) and compounds that are toxic without light activation (13–15), exhibited minimal mitochondrial toxicity.
Moreover, these finding are highly relevant in the context of recent studies evaluating metabolic reprograming in combination with PDT.84–86 It is recognized that mitochondrial oxidative phosphorylation leads to depletion of cellular oxygen, resulting in hypoxia, while increased glycolysis and the production of lactic acid contribute to an immunosuppressive tumor microenvironment. As PDT relies on oxygen and is less effective under hypoxic conditions, it is highly significant that sub-lethal compound- or light-doses in PDT could facilitate metabolic reprograming and increase oxygen levels. It is interesting to note several studies showing that inhibition of glycolysis, oxidative phosphorylation, and other metabolic processes can potentiate PDT,87 while other reports have demonstrated metabolic reprogramming induced by PDT itself, though this was presented as a potential means for synergy with chemotherapeutics with other mechanisms of action.88 The suppression of glycolysis and the resulting decrease in extracellular acidification also can be anticipated to facilitate the development of antitumor immunity. Thus, it is only logical to conclude that PDT can impact essential energy producing metabolic processes, which in turn will potentiate PDT, and likely enhance the efficacy of combination treatments with immunotherapy.89 This motivates repeating PDT treatments over a period of minutes to days, similar to the fractionation used in radiation therapy. The effects can create a positive feedback loop if the timing of these effects is optimized.
This hypothesis is supported in part by efficacy studies, as fractionation has been recently utilized in PDT with good results.90 While this report did not attempt sub-lethal dosimetry, it demonstrated an improvement in long-term survival in a mouse model from 20% to 80% with fractionation of a light dose using a 2 h dark period. The mechanism behind the improved survival is not known, but the authors reported that a lower concentration of reactive oxygen species was associated with the fractionated treatment (0.78 mM) vs. single fraction treatment (1.08 mM). This is strongly suggestive of enhanced sensitivity of the treated tumor, allowing for improved outcomes with lower doses.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
RJM conceived of the study, synthesized some of the compounds, performed the Seahorse analysis, evaluated and interpreted the data, and wrote most of the manuscript. DH and ACH synthesized some of the compounds and performed some of the chemical analysis. DH performed the SOSG study and edited the manuscript. DKH conceived of the study, performed cytotoxicity studies, evaluated and interpreted the data, and edited the manuscript. ECG performed literature analysis, interpreted the data, and wrote and edited the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the National Institutes of Health (Grant GM107586) for funding this research. This research was supported by the Redox Metabolism Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558).References
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed
.
- S. Y. Lee, C. Y. Kim and T. G. Nam, Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS
- M. Ankathatti Munegowda, A. Manalac, M. Weersink, S. A. McFarland and L. Lilge, Ru(II) containing photosensitizers for photodynamic therapy: A critique on reporting and an attempt to compare efficacy, Coord. Chem. Rev., 2022, 470, 214712 CrossRef CAS
- B. S. Howerton, D. K. Heidary and E. C. Glazer, Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS
- D. Havrylyuk, K. Stevens, S. Parkin and E. C. Glazer, Toward Optimal Ru(II) Photocages: Balancing Photochemistry, Stability, and Biocompatibility Through Fine Tuning of Steric, Electronic, and Physiochemical Features, Inorg. Chem., 2020, 59, 1006–1013 CrossRef CAS PubMed
- S. Bonnet, Why develop photoactivated chemotherapy?, Dalton Trans., 2018, 47, 10330–10343 RSC
- S. Bonnet, Ruthenium-Based Photoactivated Chemotherapy, J. Am. Chem. Soc., 2023, 145, 23397–23415 CrossRef CAS
- D. Havrylyuk, A. C. Hachey, A. Fenton, D. K. Heidary and E. C. Glazer, Ru(II) photocages enable precise control over enzyme activity with red light, Nat. Commun., 2022, 13, 3636 CrossRef CAS PubMed
- G. He, M. He, R. Wang, X. Li, H. Hu, D. Wang, Z. Wang, Y. Lu, N. Xu, J. Du, J. Fan, X. Peng and W. Sun, A Near-Infrared Light-Activated Photocage Based on a Ruthenium Complex for Cancer Phototherapy, Angew. Chem., Int. Ed., 2023, 62, e202218768 CrossRef CAS
- L. Bretin, Y. Husiev, V. Ramu, L. Zhang, M. Hakkennes, S. Abyar, A. C. Johns, S. E. Le Dévédec, T. Betancourt, A. Kornienko and S. Bonnet, Red-Light Activation of a Microtubule Polymerization Inhibitor via Amide Functionalization of the Ruthenium Photocage, Angew. Chem., Int. Ed., 2024, 63, e202316425 CrossRef CAS PubMed
- E. Rafic, C. Ma, B. B. Shih, H. Miller, R. Yuste, T. Palomero and R. Etchenique, RuBi-Ruxolitinib: A Photoreleasable Antitumor JAK Inhibitor, J. Am. Chem. Soc., 2024, 146, 13317–13325 CrossRef CAS
- M. K. Cahill, M. Collard, V. Tse, M. E. Reitman, R. Etchenique, C. Kirst and K. E. Poskanzer, Network-level encoding of local neurotransmitters in cortical astrocytes, Nature, 2024, 629, 146–153 CrossRef CAS
- M. L. Ruan, W. X. Ni, J. C. H. Chu, T. L. Lam, K. C. Law, Y. Zhang, G. Yang, Y. He, C. Zhang, Y. M. E. Fung, T. Liu, T. Huang, C. N. Lok, S. L. Chan and C. M. Che, Iridium(III) carbene complexes as potent girdin inhibitors against metastatic cancers, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316615121 CrossRef CAS PubMed
- D. R. Alajroush, C. B. Smith, B. F. Anderson, I. T. Oyeyemi, S. J. Beebe and A. A. Holder, A Comparison of In Vitro Studies between Cobalt(III) and Copper(II) Complexes with Thiosemicarbazone Ligands to Treat Triple Negative Breast Cancer, Inorg. Chim. Acta, 2024, 562, 121898 CrossRef CAS PubMed
- V. Scalcon, R. Bonsignore, J. Aupič, S. R. Thomas, A. Folda, A. A. Heidecker, A. Pöthig, A. Magistrato, A. Casini and M. P. Rigobello, Exploring the Anticancer Activity of Tamoxifen-Based Metal Complexes Targeting Mitochondria, J. Med. Chem., 2023, 66, 9823–9841 CrossRef CAS PubMed
- R. T. Mertens, S. Parkin and S. G. Awuah, Cancer cell-selective modulation of mitochondrial respiration and metabolism by potent organogold(iii) dithiocarbamates, Chem. Sci., 2020, 11, 10465–10482 RSC
- R. T. Mertens, W. C. Jennings, S. Ofori, J. H. Kim, S. Parkin, G. F. Kwakye and S. G. Awuah, Synthetic Control of Mitochondrial Dynamics: Developing Three-Coordinate Au(I) Probes for Perturbation of Mitochondria Structure and Function, JACS Au, 2021, 1, 439–449 CrossRef CAS
- C. Olelewe, J. H. Kim, S. Ofori, R. T. Mertens, S. Gukathasan and S. G. Awuah, Gold(III)-P-chirogenic complex induces mitochondrial dysfunction in triple-negative breast cancer, iScience, 2022, 25, 104340 CrossRef CAS PubMed
- Z. Zhu, Z. Wang, C. Zhang, Y. Wang, H. Zhang, Z. Gan, Z. Guo and X. Wang, Mitochondrion-targeted platinum complexes suppressing lung cancer through multiple pathways involving energy metabolism, Chem. Sci., 2019, 10, 3089–3095 RSC
- A. Notaro, A. Frei, R. Rubbiani, M. Jakubaszek, U. Basu, S. Koch, C. Mari, M. Dotou, O. Blacque, J. Gouyon, F. Bedioui, N. Rotthowe, R. F. Winter, B. Goud, S. Ferrari, M. Tharaud, M. Řezáčová, J. Humajová, P. Tomšík and G. Gasser, Ruthenium(II) Complex Containing a Redox-Active Semiquinonate Ligand as a Potential Chemotherapeutic Agent: From Synthesis to In Vivo Studies, J. Med. Chem., 2020, 63, 5568–5584 CrossRef CAS PubMed
- A. Gandioso, A. Vidal, P. Burckel, G. Gasser and E. Alessio, Ruthenium(II) Polypyridyl Complexes Containing Simple Dioxo Ligands: a Structure-Activity Relationship Study Shows the Importance of the Charge, ChemBioChem, 2022, 23, e202200398 CrossRef CAS
- D. Baier, B. Schoenhacker-Alte, M. Rusz, C. Pirker, T. Mohr, T. Mendrina, D. Kirchhofer, S. M. Meier-Menches, K. Hohenwallner, M. Schaier, E. Rampler, G. Koellensperger, P. Heffeter, B. Keppler and W. Berger, The Anticancer Ruthenium Compound BOLD-100 Targets Glycolysis and Generates a Metabolic Vulnerability towards Glucose Deprivation, Pharmaceutics, 2022, 14, 238 CrossRef CAS PubMed
- L. E. Enslin, K. Purkait, M. D. Pozza, B. Saubamea, P. Mesdom, H. G. Visser, G. Gasser and M. Schutte-Smith, Rhenium(I) Tricarbonyl Complexes of 1,10-Phenanthroline Derivatives with Unexpectedly High Cytotoxicity, Inorg. Chem., 2023, 62, 12237–12251 CrossRef CAS PubMed
- S. Benetti, M. Dalla Pozza, L. Biancalana, S. Zacchini, G. Gasser and F. Marchetti, The beneficial effect of cyclohexyl substituent on the in vitro anticancer activity of diiron vinyliminium complexes, Dalton Trans., 2023, 52, 5724–5741 RSC
- D. A. Ferrick, A. Neilson and C. Beeson, Advances in measuring cellular bioenergetics using extracellular flux, Drug Discovery Today, 2008, 13, 268–274 CrossRef CAS
- C. A. Schmidt, K. H. Fisher-Wellman and P. D. Neufer, From OCR and ECAR to energy: Perspectives on the design and interpretation of bioenergetics studies, J. Biol. Chem., 2021, 297, 101140 CrossRef CAS
- R. Wang, S. J. Novick, J. B. Mangum, K. Queen, D. A. Ferrick, G. W. Rogers and J. B. Stimmel, The acute extracellular flux (XF) assay to assess compound effects on mitochondrial function, J. Biomol. Screening, 2015, 20, 422–429 CrossRef CAS PubMed
- N. Biesemann, J. S. Ried, D. Ding-Pfennigdorff, A. Dietrich, C. Rudolph, S. Hahn, W. Hennerici, C. Asbrand, T. Leeuw and C. Strübing, High throughput screening of mitochondrial bioenergetics in human differentiated myotubes identifies novel enhancers of muscle performance in aged mice, Sci. Rep., 2018, 8, 9408 CrossRef
- I. Yoo, I. Ahn, J. Lee and N. Lee, Extracellular flux assay (Seahorse assay): Diverse applications in metabolic research across biological disciplines, Mol. Cells, 2024, 47, 100095 CrossRef PubMed
- S. Russell, J. Wojtkowiak, A. Neilson and R. J. Gillies, Metabolic Profiling of healthy and cancerous tissues in 2D and 3D, Sci. Rep., 2017, 7, 15285 CrossRef PubMed
- G. Campioni, V. Pasquale, S. Busti, G. Ducci, E. Sacco and M. Vanoni, An Optimized Workflow for the Analysis of Metabolic Fluxes in Cancer Spheroids Using Seahorse Technology, Cells, 2022, 11, 866 CrossRef CAS
- D. Schniertshauer, D. Gebhard and J. Bergemann, A New Efficient Method for Measuring Oxygen Consumption Rate Directly ex vivo in Human Epidermal Biopsies, Bio-Protoc., 2019, 9, e3185 Search PubMed
-
S. T. Bond, K. A. McEwen, P. Yoganantharajah and Y. Gibert, in Teratogenicity Testing: Methods and Protocols, ed. L. Félix, Springer New York, New York, NY, 2018, pp. 393–401, DOI:10.1007/978-1-4939-7883-0_21
- E. Rollwitz and M. Jastroch, Plate-Based Respirometry to Assess Thermal Sensitivity of Zebrafish Embryo Bioenergetics in situ, Front. Physiol., 2021, 12, 746367 CrossRef
- G. D. Preez, H. Fourie, M. Daneel, H. Miller, S. Höss, C. Ricci, G. Engelbrecht, M. Zouhar and V. Wepener, Oxygen consumption rate of Caenorhabditis elegans as a high-throughput endpoint of toxicity testing using the Seahorse XFe96 Extracellular Flux Analyzer, Sci. Rep., 2020, 10, 4239 CrossRef
- C. C. Beeson, G. C. Beeson and R. G. Schnellmann, A high-throughput respirometric assay for mitochondrial biogenesis and toxicity, Anal. Biochem., 2010, 404, 75–81 CrossRef CAS
- J. Eakins, C. Bauch, H. Woodhouse, B. Park, S. Bevan, C. Dilworth and P. Walker, A combined in vitro approach to improve the prediction of mitochondrial toxicants, Toxicol. in Vitro, 2016, 34, 161–170 CrossRef CAS
- A. Prado, G. A. Petroianu, D. E. Lorke and J. W. Chambers, A trivalent approach for determining in vitro toxicology: Examination of oxime K027, J. Appl. Toxicol., 2015, 35, 219–227 CrossRef CAS PubMed
- V. M. Gohil, S. A. Sheth, R. Nilsson, A. P. Wojtovich, J. H. Lee, F. Perocchi, W. Chen, C. B. Clish, C. Ayata, P. S. Brookes and V. K. Mootha, Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis, Nat. Biotechnol., 2010, 28, 249–255 CrossRef CAS PubMed
- Y. Tan, J. Li, G. Zhao, K.-C. Huang, H. Cardenas, Y. Wang, D. Matei and J.-X. Cheng, Metabolic reprogramming from glycolysis to fatty acid uptake and beta-oxidation in platinum-resistant cancer cells, Nat. Commun., 2022, 13, 4554 CrossRef CAS
- M. A. Lobritz, P. Belenky, C. B. Porter, A. Gutierrez, J. H. Yang, E. G. Schwarz, D. J. Dwyer, A. S. Khalil and J. J. Collins, Antibiotic efficacy is linked to bacterial cellular respiration, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8173–8180 CrossRef CAS
- D. Hanahan, Hallmarks of Cancer: New Dimensions, Cancer Discovery, 2022, 12, 31–46 CrossRef CAS PubMed
- G. Cheng, J. Zielonka, D. McAllister, S. Tsai, M. B. Dwinell and B. Kalyanaraman, Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation, Br. J. Cancer, 2014, 111, 85–93 CrossRef CAS PubMed
- M. V. Liberti and J. W. Locasale, The Warburg Effect: How Does it Benefit Cancer Cells?, Trends Biochem. Sci., 2016, 41, 211–218 CrossRef CAS PubMed
- S. Fulda, L. Galluzzi and G. Kroemer, Targeting mitochondria for cancer therapy, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed
- P. E. Porporato, N. Filigheddu, J. M. B. Pedro, G. Kroemer and L. Galluzzi, Mitochondrial metabolism and cancer, Cell Res., 2018, 28, 265–280 CrossRef CAS
- J. G. Baldwin and L. Gattinoni, Cancer cells hijack T-cell mitochondria, Nat. Nanotechnol., 2022, 17, 3–4 CrossRef CAS
- M. D. Forrest, Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy, bioRxiv, 2015, 025197, DOI:10.1101/025197
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Light-activated ruthenium complexes photobind DNA and are cytotoxic in the photodynamic therapy window, Chem. Commun., 2012, 48, 9649–9651 RSC
- D. Havrylyuk, D. K. Heidary, Y. Sun, S. Parkin and E. C. Glazer, Photochemical and Photobiological Properties of Pyridyl-pyrazol(in)e-Based Ruthenium(II) Complexes with Sub-micromolar Cytotoxicity for Phototherapy, ACS Omega, 2020, 5, 18894–18906 CrossRef CAS PubMed
-
G. W. Rogers, S. E. Burroughs and B. P. Dranka, Direct Measurements of Cellular Metabolism for Identification of Mitochondrial Drug Targets, Application Note 5994-0454, 2018 Search PubMed
- R. T. Ryan, D. Havrylyuk, K. C. Stevens, L. H. Moore, D. Y. Kim, J. S. Blackburn, D. K. Heidary, J. P. Selegue and E. C. Glazer, Avobenzone incorporation in a diverse range of Ru(ii) scaffolds produces potent potential antineoplastic agents, Dalton Trans., 2020, 49, 12161–12167 RSC
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Modifying Charge and Hydrophilicity of Simple Ru(II) Polypyridyl Complexes Radically Alters Biological Activities: Old Complexes, Surprising New Tricks, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed
- R. J. Mitchell, S. M. Kriger, A. D. Fenton, D. Havrylyuk, A. Pandeya, Y. Sun, T. Smith, J. E. DeRouchey, J. M. Unrine, V. Oza, J. S. Blackburn, Y. Wei, D. K. Heidary and E. C. Glazer, A monoadduct generating Ru(II) complex induces ribosome biogenesis stress and is a molecular mimic of phenanthriplatin, RSC Chem. Biol., 2023, 4, 344–353 RSC
- P. Gajda-Morszewski, I. Gurgul, E. Janczy-Cempa, O. Mazuryk, M. Łomzik and M. Brindell, Inhibition of Matrix Metalloproteinases and Cancer Cell Detachment by Ru(II) Polypyridyl Complexes Containing 4,7-Diphenyl-1,10-phenanthroline Ligands—New Candidates for Antimetastatic Agents, Pharmaceuticals, 2021, 14(10), 1014 CrossRef CAS
- R. T. Ryan, K. C. Stevens, R. Calabro, S. Parkin, J. Mahmoud, D. Y. Kim, D. K. Heidary, E. C. Glazer and J. P. Selegue, Bis-tridentate N-Heterocyclic Carbene Ru(II) Complexes are Promising New Agents for Photodynamic Therapy, Inorg. Chem., 2020, 59, 8882–8892 CrossRef CAS
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Efficient DNA photocleavage by [Ru(bpy)2(dppn)]2+ with visible light, Chem. Commun., 2010, 46, 2426–2428 RSC
- V. Pierroz, R. Rubbiani, C. Gentili, M. Patra, C. Mari, G. Gasser and S. Ferrari, Dual mode of cell death upon the photo-irradiation of a RuII polypyridyl complex in interphase or mitosis, Chem. Sci., 2016, 7, 6115–6124 RSC
- C. S. Burke, A. Byrne and T. E. Keyes, Targeting Photoinduced DNA Destruction by Ru(II) Tetraazaphenanthrene in Live Cells by Signal Peptide, J. Am. Chem. Soc., 2018, 140, 6945–6955 CrossRef CAS
- S. Monro, J. Scott, A. Chouai, R. Lincoln, R. Zong, R. P. Thummel and S. A. McFarland, Photobiological Activity of Ru(II) Dyads Based on (Pyren-1-yl)ethynyl Derivatives of 1,10-Phenanthroline, Inorg. Chem., 2010, 49, 2889–2900 CrossRef CAS PubMed
- L.-P. Zhao, S.-Y. Chen, R.-R. Zheng, X.-N. Rao, R.-J. Kong, C.-Y. Huang, Y.-B. Liu, Y. Tang, H. Cheng and S.-Y. Li, Photodynamic Therapy Initiated Ferrotherapy of Self-Delivery Nanomedicine to Amplify Lipid Peroxidation via GPX4 Inactivation, ACS Appl. Mater. Interfaces, 2022, 14, 53501–53510 CrossRef CAS
- S. Shui, Z. Zhao, H. Wang, M. Conrad and G. Liu, Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway, Redox Biol., 2021, 45, 102056 CrossRef CAS
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Ruthenium(II) polypyridyl complexes as mitochondria-targeted two-photon photodynamic anticancer agents, Biomaterials, 2015, 56, 140–153 CrossRef CAS
- E. Zafon, I. Echevarría, S. Barrabés, B. R. Manzano, F. A. Jalón, A. M. Rodríguez, A. Massaguer and G. Espino, Photodynamic therapy with mitochondria-targeted biscyclometallated Ir(iii) complexes. Multi-action mechanism and strong influence of the cyclometallating ligand, Dalton Trans., 2022, 51, 111–128 RSC
- K. M. Deo, J. Sakoff, J. Gilbert, Y. Zhang and J. R. Aldrich Wright, Synthesis, characterisation and potent cytotoxicity of unconventional platinum(iv) complexes with modified lipophilicity, Dalton Trans., 2019, 48, 17217–17227 RSC
- J. M. Sangster, Octanol-Water Partition Coefficients of Simple Organic Compounds, J. Phys. Chem. Ref. Data, 1989, 18, 1111–1229 CrossRef CAS
- B. Plitzko and S. Loesgen, Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism, Bio-Protoc., 2018, 8, e2850 CAS
- J. Zhang and Q. Zhang, Using Seahorse Machine to Measure OCR and ECAR in Cancer Cells, Methods Mol. Biol., 2019, 1928, 353–363 CrossRef CAS
- C. A. Schmidt, K. H. Fisher-Wellman and P. D. Neufer, From OCR and ECAR to energy: Perspectives on the design and interpretation of bioenergetics studies, J. Biol. Chem., 2021, 297 Search PubMed
- J. Symersky, D. Osowski, D. E. Walters and D. M. Mueller, Oligomycin frames a common drug-binding site in the ATP synthase, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 13961–13965 CrossRef CAS
- L.-s. Huang, D. Cobessi, E. Y. Tung and E. A. Berry, Binding of the Respiratory Chain Inhibitor Antimycin to the Mitochondrial bc1 Complex: A New Crystal Structure Reveals an Altered Intramolecular Hydrogen-bonding Pattern, J. Mol. Biol., 2005, 351, 573–597 CrossRef CAS
- S. Heinz, A. Freyberger, B. Lawrenz, L. Schladt, G. Schmuck and H. Ellinger-Ziegelbauer, Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation, Sci. Rep., 2017, 7, 45465–45478 CrossRef CAS
- D. Zhong, X. Liu, K. Schafer-Hales, A. I. Marcus, F. R. Khuri, S.-Y. Sun and W. Zhou, 2-Deoxyglucose induces Akt phosphorylation via a mechanism independent of LKB1/AMP-activated protein kinase signaling activation or glycolysis inhibition, Mol. Cancer Ther., 2008, 7, 809–817 CrossRef CAS
- N. Romero, P. Swain, G. W. Rogers and B. P. Dranka, Bioenergetic profiling and fuel dependencies of cancer cell lines: quantifying the impact of glycolytic and mitochondrial ATP production on cell proliferation, Cancer Res., 2018, 78, 3487 CrossRef
- S. Yano, S. Hirohara, M. Obata, Y. Hagiya, S.-i. Ogura, A. Ikeda, H. Kataoka, M. Tanaka and T. Joh, Current states and future views in photodynamic therapy, J. Photochem. Photobiol., C, 2011, 12, 46–67 CrossRef CAS
- X. Zheng, J. Morgan, S. K. Pandey, Y. Chen, E. Tracy, H. Baumann, J. R. Missert, C. Batt, J. Jackson, D. A. Bellnier, B. W. Henderson and R. K. Pandey, Conjugation of 2-(1′-Hexyloxyethyl)-2-devinylpyropheophorbide-a (HPPH) to Carbohydrates Changes its Subcellular Distribution and Enhances Photodynamic Activity in Vivo, J. Med. Chem., 2009, 52, 4306–4318 CrossRef CAS PubMed
- K. Arora, M. Herroon, M. H. Al-Afyouni, N. P. Toupin, T. N. Rohrabaugh Jr, L. M. Loftus, I. Podgorski, C. Turro and J. J. Kodanko, Catch and Release Photosensitizers: Combining Dual-Action Ruthenium Complexes with Protease Inactivation for Targeting Invasive Cancers, J. Am. Chem. Soc., 2018, 140, 14367–14380 CrossRef CAS PubMed
- N. Alatrash, F. H. Issa, N. S. Bawazir, S. J. West, K. E. Van Manen-Brush, C. P. Shelor, A. S. Dayoub, K. A. Myers, C. Janetopoulos, E. A. Lewis and F. M. MacDonnell, Disruption of microtubule function in cultured human cells by a cytotoxic ruthenium(ii) polypyridyl complex, Chem. Sci., 2020, 11, 264–275 RSC
- A. C. Hachey, D. Havrylyuk and E. C. Glazer, Biological activities of polypyridyl-type ligands: implications for bioinorganic chemistry and light-activated metal complexes, Curr. Opin. Chem. Biol., 2021, 61, 191–202 CrossRef CAS PubMed
- N. Romero, G. Rogers, A. Neilson and B. P. Dranka, Quantifying Cellular ATP Production Rate Using Agilent Seahorse XF Technology, Application Note 5991-9303EN, 2018, pp. 1–17 Search PubMed.
- H.-C. Wu, D. Rérolle, C. Berthier, R. Hleihel, T. Sakamoto, S. Quentin, S. Benhenda, C. Morganti, C. Wu, L. Conte, S. Rimsky, M. Sebert, E. Clappier, S. Souquere, S. Gachet, J. Soulier, S. Durand, J. J. Trowbridge, P. Bénit, P. Rustin, H. El Hajj, E. Raffoux, L. Ades, R. Itzykson, H. Dombret, P. Fenaux, O. Espeli, G. Kroemer, L. Brunetti, T. W. Mak, V. Lallemand-Breitenbach, A. Bazarbachi, B. Falini, K. Ito, M. P. Martelli and H. de Thé, Actinomycin D Targets NPM1c-Primed Mitochondria to Restore PML-Driven Senescence in AML Therapy, Cancer Discovery, 2021, 11, 3198–3213 CrossRef CAS PubMed
- C. E. Greif, R. T. Mertens, G. Berger, S. Parkin and S. G. Awuah, An anti-glioblastoma gold(i)-NHC complex distorts mitochondrial morphology and bioenergetics to induce tumor growth inhibition, RSC Chem. Biol., 2023, 4, 592–599 RSC
- H. D. Cole, A. Vali, J. A. Roque III, G. Shi, G. Kaur, R. O. Hodges, A. Francés-Monerris, M. E. Alberto, C. G. Cameron and S. A. McFarland, Ru(II) Phenanthroline-Based Oligothienyl Complexes as Phototherapy Agents, Inorg. Chem., 2023, 62, 21181–21200 CrossRef CAS PubMed
- Z. Alkarakooly, Q. A. Al-Anbaky, K. Kannan and N. Ali, Metabolic reprogramming by Dichloroacetic acid potentiates photodynamic therapy of human breast adenocarcinoma MCF-7 cells, PLoS One, 2018, 13, e0206182 CrossRef PubMed
- L. Zuo, W. Nie, S. Yu, W. Zhuang, G. Wu, H. Liu, L. Huang, D. Shi, X. Sui, Y. Li and H.-Y. Xie, Smart Tumor-Cell-Derived Microparticles Provide On-Demand Photosensitizer Synthesis and Hypoxia Relief for Photodynamic Therapy, Angew. Chem., Int. Ed., 2021, 60, 25365–25371 CrossRef CAS PubMed
- G. Lin, L. Tillman, T. Luo, X. Jiang, Y. Fan, G. Liu and W. Lin, Nanoscale Metal-Organic Layer Reprograms Cellular Metabolism to Enhance Photodynamic Therapy and Antitumor Immunity, Angew. Chem., Int. Ed., 2024, e202410241 CAS
- Q. Yu, X. Li, J. Wang, L. Guo, L. Huang and W. Gao, Recent Advances in Reprogramming Strategy of Tumor Microenvironment for Rejuvenating Photosensitizers-Mediated Photodynamic Therapy, Small, 2024, 20, 2305708 CrossRef CAS
- Y. Li, D. Wang, Z. Zhang, Y. Wang, Z. Zhang, Z. Liang, F. Liu and L. Chen, Photodynamic therapy enhances the cytotoxicity of temozolomide against glioblastoma via reprogramming anaerobic glycolysis, Photodiagn. Photodyn. Ther., 2023, 42, 103342 CrossRef CAS PubMed
- D. F. Boreel, P. N. Span, S. Heskamp, G. J. Adema and J. Bussink, Targeting Oxidative Phosphorylation to Increase the Efficacy of Radio- and Immune-Combination Therapy, Clin. Cancer Res., 2021, 27, 2970–2978 CrossRef CAS
- H. Sun, W. Yang, Y. Ong, T. M. Busch and T. C. Zhu, Fractionated Photofrin-Mediated Photodynamic Therapy Significantly Improves Long-Term Survival, Cancers, 2023, 15, 5682 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05393a |
| This journal is © The Royal Society of Chemistry 2025 |