
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence570 nm/770 nm light-excited deep-red fluorescence switch based on dithienylethene derived from BF2-curcuminoid†
Ziyong
Li‡
 *a,
Xiaoxie
Ma‡
b,
Jinzhao
Song‡
a,
Qilian
Wang
a,
Yongliang
Feng
a,
Haining
Liu
a,
Pei
Zhang
*a,
Hui
Guo
*a and
Jun
Yin
*b
*a,
Xiaoxie
Ma‡
b,
Jinzhao
Song‡
a,
Qilian
Wang
a,
Yongliang
Feng
a,
Haining
Liu
a,
Pei
Zhang
*a,
Hui
Guo
*a and
Jun
Yin
*b
aLuoyang Key Laboratory of Organic Functional Molecules, College of Food and Drug, College of Chemistry and Chemical Engineering, Department of Life Science, Luoyang Normal University, Luoyang, 471934, P. R. China. E-mail: liziyong@mails.ccnu.edu.cn; zhangpei8877@126.com; guohui@lynu.edu.cn
bNational Key Laboratory of Green Pesticide, International Joint Research Center for Intelligent Biosensor Technology and Health, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: yinj@ccnu.edu.cn
First published on 18th December 2024
Abstract
Developing dithienylethene (DTE)-based fluorescence switches triggered by biocompatible visible light has always been a long-term goal in view of their potential in numerous biological scenarios. However, their practical availability is severely limited by the short visible light (generally less than 500 nm) required for photocyclization, their inability to achieve red or near-infrared emission, and their short fluorescence lifetimes. Herein, we present a novel DTE derivative featuring a dimethylamine-functionalized BF2-curcuminoid moiety (NBDC) by using an “acceptor synergistic conjugation system” strategy. The dimethylamine group not only enables a red shift in the absorption and emission wavelengths of the open isomer but also endows NBDC with unique acid/base-gated photochromism. As expected, as-prepared NBDC presents 570 nm/770 nm light-driven photochromic properties, red-emissive fluorescence, and thermally activated delayed fluorescence (TADF) switching in toluene. To our knowledge, this represents the first instance of a yellow-green- and NIR light-controlled red fluorescence DTE switch with the longer fluorescence lifetime. Specifically, NBDC, which shows weak photochromic activity in CHCl3, demonstrates enhanced photochromic performance when gated by TFA/TEA. Ultimately, this non-toxic deep-red fluorescence switch has been successfully applied for photoswitchable imaging in vivo of living cells and zebrafish, further proving its versatility in life sciences.
Introduction
Fluorescent molecular photoswitches, which integrate switch units and fluorophores through covalent bonds, have recently garnered significant interest due to their potential in optical information storage, anti-counterfeiting, super-resolution imaging, and more.1–7 Among these, DTE switches featuring ring-open/-closed isomerization are particularly appealing due to their unique bistability, rapid response to light, and robust fatigue resistance.8–17 Typically, DTE-based fluorescence switches require operation with UV light with high energy, which is known to be harmful to cells and unfavorable for penetration into biological tissues. Therefore, developing DTE switches triggered by biocompatible visible light is of great significance and an attractive goal.Substantial efforts have been devoted to developing various strategies for visible-light-driven DTE derivatives.18–22 These strategies include direct photoexcitation requiring elaborate molecular design/synthesis (e.g., extending π-conjugation systems,23–26 donor/acceptor–acceptor structures,27–31 and intramolecular proton transfer32) and indirect photoexcitation via energy or electron transfer (e.g., triplet sensitization,33–38 upconverting nanoparticles,39,40 multiphoton absorption,41 and intermolecular charge transfer42). For instance, Zhang and Tian et al. developed a facile triplet sensitization strategy to fabricate visible-light DTEs by mixing a triplet sensitizer with a narrow singlet–triplet energy gap (ΔEST)36 or employing a sensitizer with a narrow ΔEST as a building block in the DTE skeleton.37 However, the distance-dependence and environment-sensitivity of triplet sensitization limit their practical applications, especially in biological scenarios. Recently, the same group demonstrated a nanoconfinement strategy to achieve enhanced visible light-triggered photochromic performance in fully aqueous media by self-assembling the DTE photoswitch and sensitizer into nanoconfined micelles,38 protecting the triplet sensitizer from external quenchers like O2 and water. Compared to these various strategies, extending the π-conjugation is a straightforward approach commonly employed to develop visible light-driven DTEs by reducing the HOMO–LUMO energy gap of their ring-open form.23–26 For example, Chi and Li's groups have recently developed all-visible-light triggered dithienylethene derivatives with molecular conformation changes excess 5 Å (ref. 25) and the ring-closure reaction yield exceeding 96.3% (ref. 26) by using this strategy. However, it is noteworthy that these conjugated DTEs often exhibit lower cycloreversion quantum yield, shorter excitation light wavelength, and may even lose photochromic activity due to the decreased contribution of the singlet excited state to the hexatriene fragment on the DTE core as the conjugated system extends. Addressing this “Achilles' heel,” our group recently introduced a promising strategy, referred to as the “acceptor synergistic conjugation system” strategy, to explore a series of efficient DTE derivatives triggered by visible light.43–49 For example, one conjugated DTE (CDB1) containing the electron-withdrawing difluoroboron β-diketonate (BF2bdk) moiety exhibited maximum absorption and emission at 470 nm and 542 nm in toluene before irradiation, respectively.43 When irradiated with blue light (λirr. = 470 nm) and NIR light (λirr. = 770 nm), CDB1 demonstrated reversible photoswitching behaviors in toluene. Additionally, BFBDTE with a further extended π-conjugation displayed efficient 530 nm/770 nm-induced photochromism (λmax = 727 nm for closed isomer) and fluorescence switching behavior (λem = 556 nm for open isomer) in toluene.47 It is noteworthy that the extension of the conjugation significantly redshifts the excitation and emission wavelengths of these fluorescence switches without affecting their photoswitching performance. Here, we speculate that it is possible to develop longer wavelength visible light-driven DTEs using this strategy. It is also worth noting that light toxicity and interference from sunlight decrease, and light penetration increases as the visible light wavelength extends.50 To date, no red-emissive fluorescence switch based on DTE triggered beyond 530 nm in visible light through the strategy of extending π-conjugation has been reported. Moreover, most DTEs suffer from the disadvantage of shorter fluorescence lifetimes. Therefore, developing a longer visible light-driven DTE system with red emission and longer fluorescence lifetime is highly desirable for functional materials and photopharmacology, but remains a severe challenge.
In this contribution, we have successfully prepared a novel DTE derivative featuring a BF2 complex of a dimethylamine-functionalized curcumin analogue (BF2-curcuminoid), referred to as NBDC (as shown in Fig. 1b). The strong electron donor dimethylamine enhances the intramolecular charge transfer (ICT) process compared to BFBDTE, resulting in a red shift in the absorption and emission wavelengths for the open isomer of NBDC. Additionally, it endows NBDC with a typical TADF feature, characterized by a smaller energy difference (ΔEST) between the lowest singlet excited state (S1) and the lowest triplet state (T1).51 Furthermore, a unique acid/base-gated photochromism for NBDC can be obtained considering that the dimethylamine moiety can respond to a variety of acids.52,53 As anticipated, as-prepared NBDC exhibits 570 nm/770 nm light-driven photochromic properties, red-emissive fluorescence, and TADF switching in toluene. To our knowledge, this represents the first instance of a yellow-green-light and NIR light-controlled red fluorescence switch with longer fluorescence lifetime based on DTE. Specifically, NBDC with weak photochromic activity in CHCl3 can perform an acid/base-gated enhanced photochromic performance.
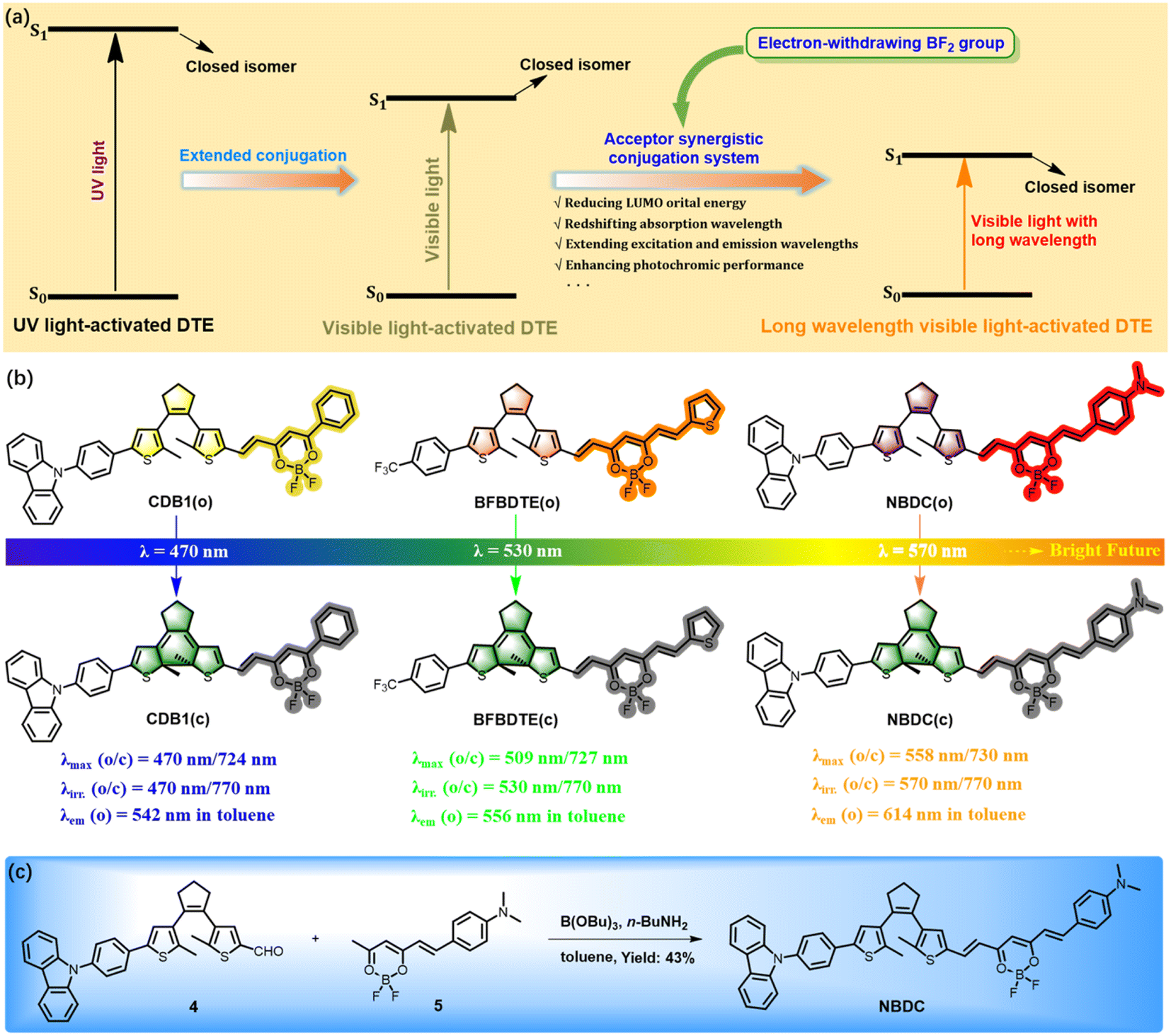 | ||
| Fig. 1 The energy diagram for photochromism triggered by UV and visible light irradiation in the traditional DTEs and DTEs with extended conjugation, and the strategy of acceptor synergistic conjugation system to construct DTEs triggered by visible light with long wavelength (S0: ground state, S1: first singlet excited state) (a). The chemical structures and photochromic reaction of conjugated DTEs modified with BF2 groups (CDB1, BFBDTE and NBDC) upon alternating irradiation with 470 nm, 530 nm and 570 nm, respectively (b). The corresponding synthetic route of NBDC (c). | ||
Results and discussion
As depicted in Fig. 1c and Scheme S1,†NBDC was synthesized via the Knoevenagel condensation reaction between intermediates 4 and 5 with n-butylamine as the catalyst in the yield of 43% in anhydrous toluene. The chemical structure of NBDC was well characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) (Fig. S1–S3†). The 1H NMR spectrum of NBDC revealed coupling constants of approximately 15.1 Hz for the double bonds, indicating a stable trans-configuration for both ethylene bonds in NBDC.With this BF2-curcuminoid-based DTE derivative in hand, the photochromism of NBDC was first explored in toluene. As illustrated in Fig. 2a, an intense absorption band centered at 558 nm (ε = 5.97 × 104 M−1 cm−1) in the yellow-green light region was observed in toluene, which arose from intramolecular charge transfer (ICT) transition from the dimethylaniline group to the BF2bdk moiety of the ring-open isomer NBDC(o).54 When irradiated with yellow-green light at 570 nm (8.9 mW cm−2) the pink solution gradually turned brown, accompanied by the appearance of a new NIR absorption band centered at 730 nm (ε = 1.13 × 104 M−1 cm−1) (Fig. 2a, inset), indicating the formation of the ring-closed isomer NBDC(c) (Scheme S2†). Although it took 600 seconds to reach the photostationary state (PSS), it represented the DTE system triggered by the longest wavelength visible light to date. Notably, a distinct isosbestic point at 596 nm was detected, indicating a single photoconversion mechanism for NBDC.55 The cycloreversion reaction occurred after continuous irradiation with NIR light at 770 nm (8.9 mW cm−2) for 120 s (Fig. 2b), resulting in the regeneration of the open isomer. Admittedly, the response rate of the closed isomer to NIR light was significantly faster than that of the open isomer to yellow-green light, as depicted in Fig. 2c. Particularly, no significant degradation was observed over ten cycles of alternating irradiation with yellow-green light and NIR light (Fig. 2d and S4†), implying decent fatigue resistance for NBDC, probably due to the inhibition of photobyproduct generation pathways under mild irradiation with 570 nm and 770 nm light. Moreover, the cyclization and cycloreversion quantum yields of NBDC in toluene were recorded as φo–c = 0.12 and φc–o = 0.007, respectively (Table 1).
 | ||
| Fig. 2 The absorption spectra changes of NBDC in toluene (2.0 × 10−5 mol L−1) upon alternating irradiation with yellow-green light at 570 nm (8.9 mW cm−2) (a) and NIR light at 770 nm (8.9 mW cm−2) (b). The insets show the corresponding color changes upon photoirradiation. The optical response rate of NBDC upon irradiation with 570 nm and 770 nm in toluene by monitoring changes in the maximum absorption wavelength (λmax) of NBDC(c) (c). The fatigue resistance of NBDC in toluene for ten cycles (d). The photostability of the open isomer NBDC(o) in toluene under continued ambient sunlight irradiation (e). The thermal stability of the closed isomer NBDC(c) in toluene at 40 °C by monitoring changes in λmax of NBDC(c) at PSS (f). | ||
| Compounds | Solvents | λ max (nm) (ε × 10−4, M−1 cm−1) | λ max (nm) (ε × 10−4, M−1 cm−1) | φ o–c | φ c–o | λ em (nm) | τ PF (ns) | τ DF (μs) | Φ f (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption maxima of ring-open isomers. b Absorption maxima of ring-closed isomers. c Cyclization quantum yields. d Cycloreversion quantum yields. e Fluorescence emission maxima. f The prompt fluorescence lifetime. g The TADF lifetime. h Fluorescence quantum yield determined by a standard method with rhodamine 6G in water (Φf = 0.75, λex = 488 nm) as reference before irradiation with 570 nm light. | |||||||||
| NBDC | Toluene | 558 (5.97) | 730 (1.13) | 0.12 | 0.007 | 614 | 2.22 | 435.75 | 48 |
| CHCl3 | 572 (6.88) | 756 (0.25) | 0.03 | 0.005 | 643 | 1.49 | NA | 32 | |
| DMSO | 592 (7.99) | NA | NA | NA | 679 | 0.83 | NA | 2.4 | |
| NBDC-H | Toluene | 506 (5.18) | 760 (1.63) | 0.34 | 0.008 | 610 | 0.90 | NA | 8.2 |
| CHCl3 | 510 (7.88) | 810 (3.40) | 0.14 | 0.006 | 630 | 0.77 | NA | 1.6 | |
| DMSO | NA | NA | NA | NA | 679 | 0.54 | NA | 1.1 | |
Subsequently, the photochromism of NBDC was further evaluated in solvents with larger polarity (CHCl3 and DMSO). When exposed to yellow-green light and NIR light, NBDC exhibited weak photochromic properties in CHCl3, as shown in Fig. S5–S8† and Table 1. In sharp contrast with toluene, both open and closed isomers showed a distinct bathochromic shift for the absorption maximum in CHCl3 (Δλ = 14 nm for the open isomer and Δλ = 26 nm for the closed isomer) (Table 1). Additionally, it took longer for the open isomer to reach PSS in CHCl3 (ca. 27 min) compared to 10 min in toluene, implying a slower optical response rate in CHCl3. As expected, an extremely small cyclization quantum yield (φo–c = 0.03) was obtained (Table 1). Unfortunately, NBDC exhibited no photochromic activity in other solvents with different polarity (CH2Cl2, THF, Acetone, CH3CN, DMF and DMSO) under the same irradiation conditions (Fig. S9†). In brief, NBDC exhibited unique photochromic performance with solvent-dependent features, consistent with some previous cases of BF2-incorporated DTE derivatives.43,44,47,49 Additionally, 1H NMR spectral variations of NBDC in CDCl3 were used to evaluate its photoisomerization (Fig. S10†), revealing a photocyclization conversion ratio at PSS of only 15.3% due to the weak photochromism in CHCl3. Photostability and thermal stability are crucial criteria for evaluating the performance of photoswitchable fluorescent DTE switches for potential applications in switchable sensing, super-resolution imaging, and photopharmacology.10,17,56 The photostability of the open isomer NBDC(o) was first explored in toluene under continuous ambient light irradiation. Fig. 2e demonstrated negligible variation in the absorption spectra centered at 558 nm when NBDC(o) was continuously exposed to ambient light for 600 min, revealing excellent photostability, ensuring unaffected switch operation by ambient sunlight. The thermal stability of the closed isomer NBDC(c) was then investigated under dark conditions. As illustrated in Fig. 2f, the NIR absorption intensity at 730 nm for NBDC(c) remained essentially unchanged after 600 min in the dark at 40 °C, indicating good thermal stability and a larger ground state activation energy difference detrimental for thermal cycloreversion reaction. Accordingly, this efficient 570 nm/770 nm-driven DTE derivative, which combines decent fatigue resistance, excellent photostability against ambient sunlight, and thermal stability, is expected to be applied in biological systems.
Considering the intense emission performance of BF2-curcuminoid complexes, we subsequently explored the photoluminescence (PL) performance of NBDC in both prompt and delayed modes across three different solvents. As illustrated in Fig. 3a, upon excitation at 560 nm, NBDC exhibited bright red fluorescence centered at 614 nm in toluene, with a relative fluorescence quantum yield estimated at Φf = 48%. Time-resolved decay measurements at 614 nm revealed a prompt fluorescence lifetime of 2.22 ns at room temperature (Fig. S11a and Table 1†). In the delayed mode, a new emission peak at 630 nm and a long delayed fluorescence lifetime (τ = 435.75 μs) were detected in toluene at room temperature (λex = 560 nm) (Fig. 3a and b), whereas very weak delayed fluorescence was observed in CHCl3 and DMSO (Fig. S12†). To further investigate whether NBDC featured thermally activated delayed fluorescence (TADF), its phosphorescence spectra were measured in toluene at 77 K, showing a weakened intensity compared to room temperature. Additionally, the difference in wavelengths between the prompt fluorescence (at room temperature) and phosphorescence spectra (at 77 K) (Fig. 3c and S13†) indicated a small ΔEST between the excited S1 (614 nm, 2.02 eV) and T1 (630 nm, 1.97 eV) states, calculated to be 0.05 eV. This small energy gap (ΔEST < 0.2 eV) suggests classical TADF performance for NBDC. Upon irradiation with yellow-green light at 570 nm, the emission intensity at 614 nm gradually attenuated, and the red fluorescence faded (Fig. 3d), due to the ration reduction of NBDC(o) and efficient Förster-type resonance energy transfer (FRET) between the excited BF2-curcuminoid and the closed DTE core (Fig. 3j).57 After reaching the photostationary state (PSS), the emission intensity of NBDC(o) was quenched by approximately 51% due to partial overlap between the emission spectrum of NBDC(o) and the absorption spectrum of NBDC(c) (Fig. 3e). Irradiation with 770 nm NIR light restored the original emission intensity, resulting from the reformation of NBDC(o) (Fig. 3d). Notably, this fluorescent switching process can be operated reversibly for ten cycles with some noticeable attenuation (Fig. 3f).
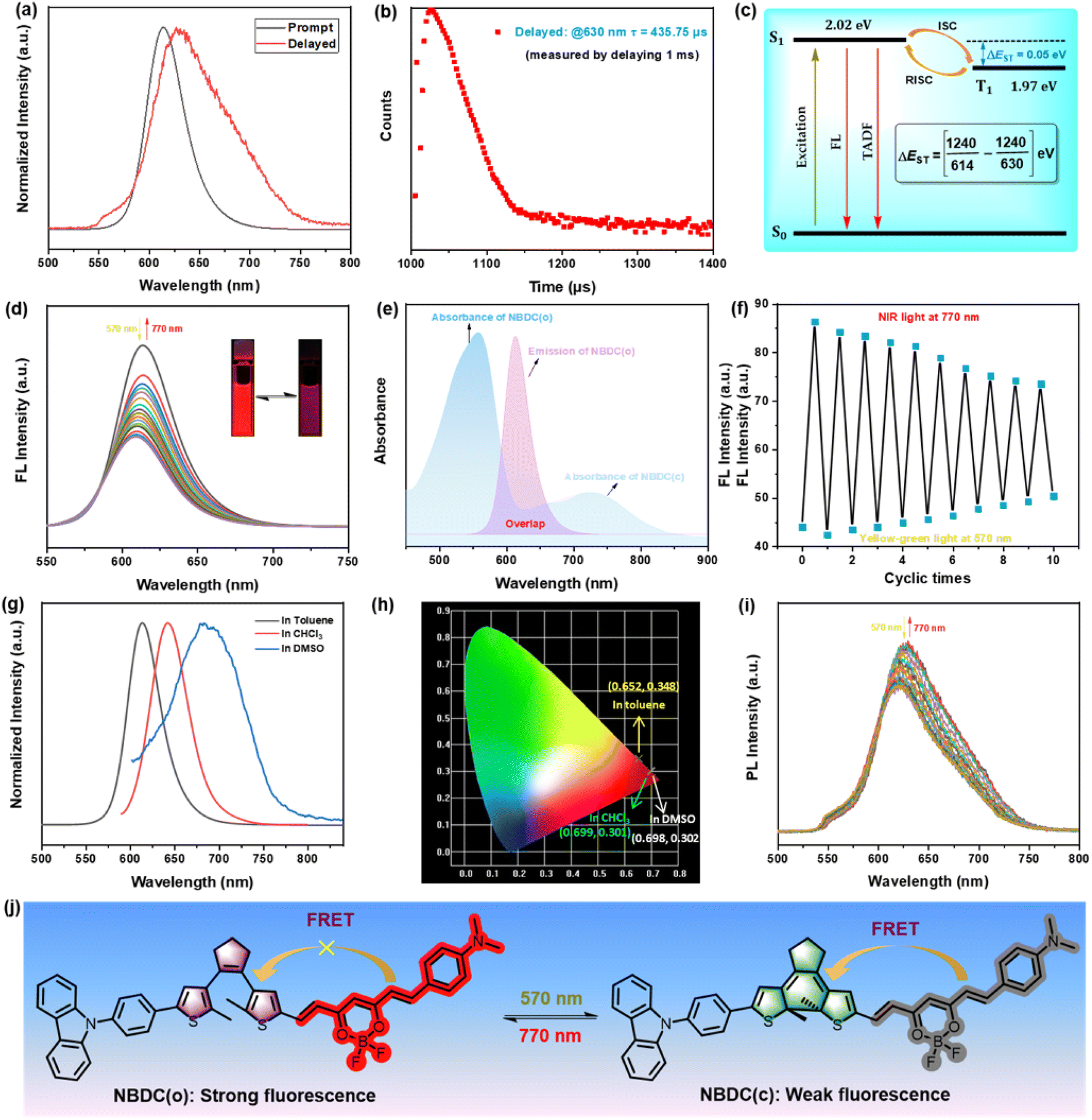 | ||
| Fig. 3 PL spectra of NBDC measured under prompt and delayed mode in toluene (2.0 × 10−5 mol L−1) at room temperature (λex = 560 nm) (a). The delayed fluorescence decay curves (measured by delaying 1 ms) of NBDC in toluene (b). Simplified Jablonski diagram of TADF performance for NBDC and the corresponding ΔEST based on the prompt fluorescence at room temperature and phosphorescence spectra at 77 K (c). The prompt fluorescence spectra changes of NBDC in toluene (2.0 × 10−5 mol L−1) upon alternating irradiation with 570 nm light (8.9 mW cm−2) and NIR light at 770 nm (8.9 mW cm−2) (d). (Inset) Corresponding fluorescent color changes upon photoirradiation. Overlap between the emission spectrum of NBDC(o) and absorption spectrum of NBDC(c) (e). The fluorescence fatigue resistance of NBDC in toluene for ten cycles (f). The prompt fluorescence spectra of NBDC in different solvents (2.0 × 10−5 mol L−1) before irradiation (g). The corresponding CIE chromaticity diagram in different solvents (h). Delayed fluorescence spectra changes of NBDC in toluene (2.0 × 10−5 mol L−1) upon alternating irradiation with 570 nm light and NIR light at 770 nm (i). Photochromic reaction of NBDC along with changes in fluorescence due to FRET process (j). FRET: Förster-type resonance energy transfer. | ||
Similar fluorescent switching behavior was detected in CHCl3 under the same light irradiation (Fig. S14 and S15†). Compared to toluene, NBDC exhibited a significant bathochromic shift in the emission peak in CHCl3 (λem = 643 nm, τPF = 1.49 ns) and DMSO (λem = 679 nm, τPF = 1.83 ns) (Fig. 3g and Table 1), consistent with the changes in absorption spectra. NBDC emitted standard red or deep-red fluorescence according to the CIE chromaticity coordinates in toluene (0.652, 0.348), CHCl3 (0.699, 0.301), and DMSO (0.698, 0.302) (Fig. 3h), demonstrating solvent-dependent fluorescent switching behavior. Surprisingly, NBDC displayed distinctive deep-red TADF switching performance in toluene. As depicted in Fig. 3i, irradiation with yellow-green light gradually decreased the delayed fluorescence intensity at 630 nm, which returned to its original intensity under NIR light irradiation. In summary, the as-prepared NBDC showed an unprecedented dual-mode deep-red fluorescence and TADF switching performance triggered by yellow-green and NIR light, which holds great potential for photo-controlled biological imaging and super-resolution imaging application.
Inspired by the unique structural features of the dimethylamine moiety, which can respond to various acids, and leveraging the intense fluorescence observed in both toluene and CHCl3, we delved into the photophysical response of NBDC to trifluoroacetic acid (TFA) and its potential to regulate NBDC's photoswitching performance in solutions. As shown in Fig. 4a, upon the addition of TFA (0–5.0 eq.), the maximal absorption intensity at 572 nm (ε = 8.94 × 104 M−1 cm−1) for NBDC gradually decreased, accompanied by the emergence of a new absorption peak at 510 nm (ε = 7.88 × 104 M−1 cm−1) in CHCl3, resulting in noticeable color transitions from purple to orange. Additionally, an isobestic point at 524 nm indicated the formation of a new species (NBDC-H) induced by TFA. Moreover, the corresponding deep-red fluorescence emission at 643 nm in CHCl3 progressively diminished along with a fluorescence lifetime decreased from 1.49 ns to 0.77 ns with increasing TFA concentration (Fig. 4b and Table 1), attributable to the inhibition of the intramolecular charge transfer (ICT) process upon proton recognition (Fig. 4g). Consequently, NBDC shows promise as a novel colorimetric and fluorescent sensing material for monitoring pH changes across various biological processes.
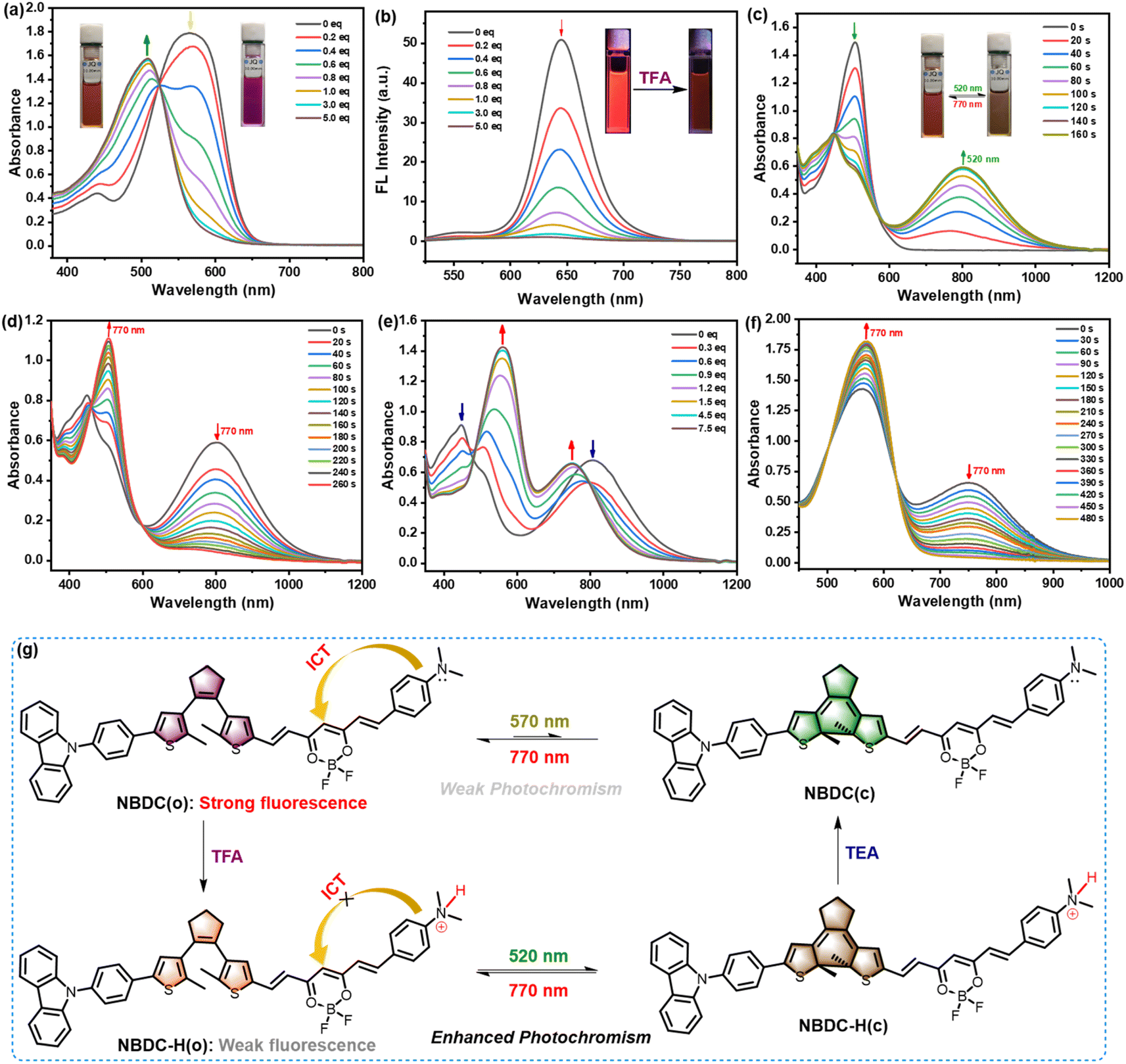 | ||
| Fig. 4 The absorption (a) and fluorescence (b) spectra changes of NBDC(o) in the presence of TFA (0–5.0 eq.) in CHCl3 (2.0 × 10−5 mol L−1). The insets show the corresponding color and fluorescence changes before and after addition of 5.0 eq. TFA. The absorption spectra changes of as-protonated DTE NBDC-H in CHCl3 (2.0 × 10−5 mol L−1) upon irradiation with green light at 520 nm (8.9 mW cm−2) (c) and NIR light at 770 nm (8.9 mW cm−2) (d). The insets show the corresponding color changes upon photoirradiation. The absorption spectra changes of NBDC-H(c) in the presence of TEA (0–7.5 eq.) in CHCl3 (2.0 × 10−5 mol L−1) (e). The absorption spectra changes of NBDC-H(c) after addition of TEA (7.5 eq.) in CHCl3 upon irradiation with NIR light at 770 nm (f). Schematic representation of protonation of NBDC(o) and deprotonation of NBDC-H(o) upon addition of TFA (5.0 eq.) and TEA (7.5 eq.), respectively, and photochromic reaction of NBDC and NBDC-H upon alternating irradiation with 570 nm (or 520 nm) light and NIR light at 770 nm (g). | ||
Recent advancements have focused on developing gated-photochromic DTEs, identified as an optimal strategy to prevent destructive readouts.58–60 In this context, we explored the gated photochromism of NBDC, which exhibits notably weak photochromic activity in CHCl3. Intriguingly, upon adding 5.0 eq. TFA, the photochromic activity of NBDC was significantly enhanced in CHCl3. As depicted in Fig. 4c, the protonated DTE derivative, NBDC-H, displayed an intense absorption peak at 510 nm (ε = 7.88 × 104 M−1 cm−1) in CHCl3 before photoirradiation, marking a significant blue shift from its unprotonated counterpart due to ICT perturbation upon protonation. Upon 520 nm green light irradiation (8.9 mW cm−2), a lower-energy NIR absorption band at 810 nm (ε = 3.40 × 104 M−1 cm−1) emerged, changing the solution color from orange to brown, indicative of the generation of the closed form NBDC-H(c) (Fig. 4g). The λmax of NBDC-H(c) experienced a 54 nm redshift compared to NBDC(c) (λmax = 756 nm), due to conjugation system elongation after protonation. Moreover, the photoresponse rate of NBDC-H(o) to reach the photostationary state (PSS) notably increased compared to NBDC(o) (Fig. S16†). Subsequent 770 nm NIR light irradiation triggered the cycloreversion reaction, regenerating the initial open form (Fig. 4d). The φo–c and φc–o of NBDC-H in CHCl3 were determined to be 0.14 and 0.006, respectively (Table 1), suggesting that protonation of the dimethylamine group enhances photoswitching performance, providing an innovative approach for designing novel efficient long-wavelength visible light-regulated DTE derivatives. As depicted in Fig. 4e, the gradual addition of 0–7.5 eq. triethylamine (TEA) into the brown solution of NBDC-H(c) caused the maximum absorption peak at 810 nm to fade, and dual absorption peaks at 564 nm and 756 nm emerged, indicative of the closed isomer NBDC(c) formation, which is otherwise challenging to obtain in large quantities in CHCl3 (Fig. 4g). When irradiated with 770 nm NIR light, the 756 nm absorption band diminished, accompanied by a redshift of the 564 nm peak to 572 nm, consistent with the absorption peak of the open isomer NBDC(o) in untreated CHCl3 (Table 1). In toluene, NBDC exhibited similar sensing performance towards TFA and TFA/TEA-regulated photochromism, as evidenced in Fig. S17–S21, Table 1 and Scheme S3.† However, negligible changes were observed in the absorption spectra in DMSO (Fig. S22† and Table 1), which may be because large polar DMSO solvent is not conducive for stabilizing protonated species in the acid–base equilibrium reactions of dimethylamine moiety and TFA. Thus, NBDC presents a distinctive acid/base-gated photochromic performance triggered by green and NIR light in CHCl3 and toluene, positioning it as a potential dual-controlled molecular switch.
To gain a deeper understanding of the relationship between the photoswitching behaviors and electronic properties of NBDC and NBDC-H, density functional theory (DFT) calculations were conducted to examine electronic densities and the optimized geometries of the open and closed isomers at the B3LYP/6-31G* level using the Gaussian 09 program.61 As depicted in Fig. 5a, the highest occupied molecular orbital (HOMO) of NBDC(o) was predominantly localized around the dimethylphenylamine (DMPA) group, while the lowest unoccupied molecular orbital (LUMO) was extensively distributed around the BF2-curcuminoid moiety, emphasizing the electron-deficient nature of the BF2 group. This separation of HOMO and LUMO orbitals facilitates an intramolecular charge transfer (ICT) transition between the BF2 acceptor and DMPA donor in the excited state, promoting photocyclization under yellow-green light. Conversely, for NBDC(c), the HOMO was concentrated on the central DTE core, and the LUMO encompassed the entire molecular skeleton, leading to a narrower energy gap (Eg = 1.77 eV) compared to NBDC(o) (Eg = 2.47 eV) due to the extended π-conjugation system. In sharp contrast, for NBDC-H(o), the HOMO was spread across the carbazole and adjacent thiophene moieties, attributed to the electron-withdrawing effect of the formed ammonium salt upon protonation, disrupting the original ICT process within the BF2-curcuminoid fragment. This resulted in significant fluorescence quenching and a notable hypsochromic shift in the absorption wavelength of the open isomer (e.g., λmax = 558 nm in toluene and 572 nm in CHCl3 for NBDC; 506 nm in toluene and 510 nm in CHCl3 for NBDC-H). Additionally, NBDC-H(c) exhibited a smaller energy gap (Eg = 1.64 eV) compared to NBDC(c) (Eg = 1.77 eV), contributing to the absorption redshift for NBDC-H(c) (Δλmax = 30–54 nm). As shown in Fig. 5b, the energy-minimized structure of NBDC(o) demonstrated a classical antiparallel conformation conducive to photocyclization. From a side view, the DMPA and thiophene-substituted BF2-curcuminoid framework appeared nearly coplanar, indicating a well-suited conjugate structure for facilitating the ICT transition. NBDC(c) maintained a coplanar structure except for the carbazole group, which was angled slightly. The optimized structures of NBDC-H(o) and NBDC-H(c) remained largely unaffected by protonation. Furthermore, as depicted in Fig. 5c, an enhanced dipole moment for NBDC(c) was calculated at 20.77 D, compared to 18.58 D for NBDC(o), highlighting a significant polarity change during photoisomerization. Remarkably, the protonated forms of NBDC exhibited much larger dipole moment changes (i.e., μ = 69.98 D for NBDC-H(o); μ = 65.58 D for NBDC-H(c)), with the positive π-charge predominantly located in the dimethyl ammonium group (blue region). These DFT calculations not only corroborated the experimental results observed in photochromic and TFA-gated studies but also provided a promising foundation for designing red light-excited DTE derivatives.
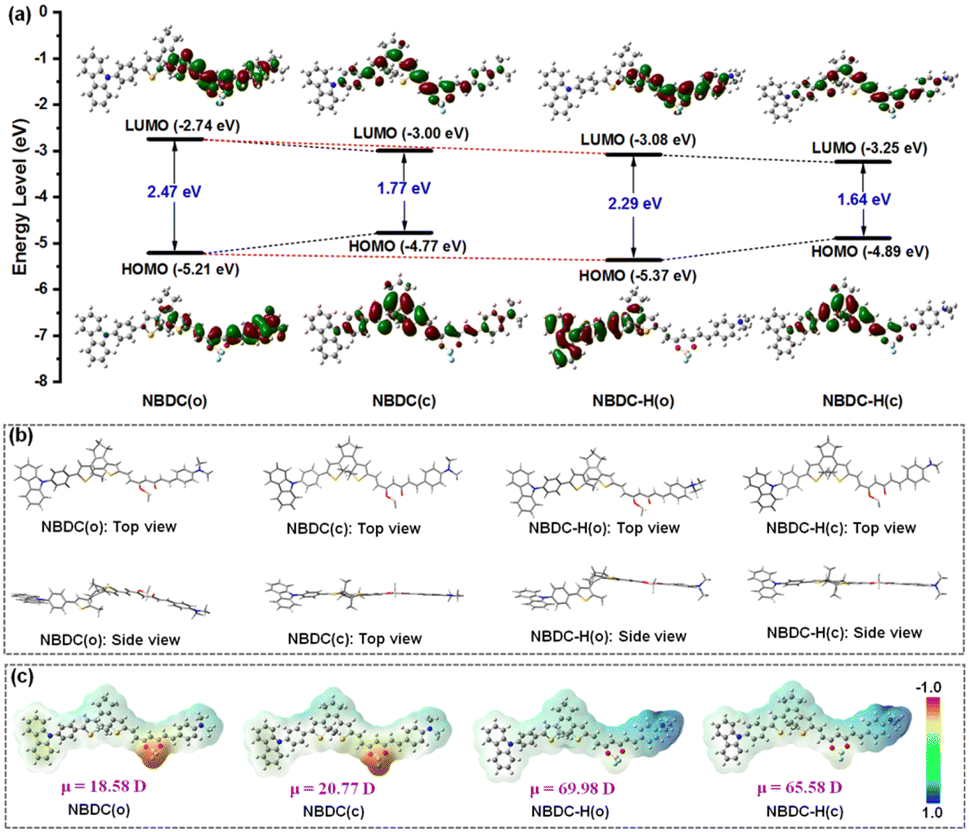 | ||
| Fig. 5 Frontier molecular orbital profiles of NBDC(o), NBDC(c), NBDC-H(o), NBDC-H(c) (a), the corresponding energy-minimized structures (b), and their calculated electrostatic potential surfaces (blue region represents the positive work and the red region represents the negative work) (c) based on DFT calculations at the B3LYP/6-31G* level using the Gaussian 09 program. | ||
Encouraged by the dual-mode deep-red fluorescence and TADF switching performance, as well as the long fluorescence lifetime of NBDC and its biocompatible yellow-green/NIR-excited photochromism, we preliminarily evaluated the applicability of NBDC for photoswitchable imaging in living B16 (mouse melanoma) cells. To increase the water solubility of the hydrophobic NBDC, nanoparticles loaded with NBDC (referred to as NBDC NPs) were fabricated using the amphiphilic Pluronic F127 as the encapsulating matrix. The resulting nanoparticles displayed an average hydrodynamic diameter of approximately 118 nm (Fig. S23†), which favors efficient cellular uptake and accumulation due to the enhanced permeability and retention effect.62
When incubated with NBDC NPs at concentrations of 20–100 μg mL−1 for 24 hours, the MTT assay revealed minimal cytotoxicity (Fig. S24†), suggesting good biocompatibility for bio-imaging applications. Unexpectedly, NBDC NPs exhibited relatively weak photochromic and fluorescence switching properties in water, and the emission wavelength of NBDC NPs redshifted to the NIR region (λem = 848 nm), as illustrated in Fig. S25.† In contrast to the absence of photochromic activity in large polar organic solvents, the recovery of photoswitching performance in nano-aqueous solutions may be because the encapsulation of NBDC inside the hydrophobic and small-sized micelle enables a distinct nanoconfinement effect that shields the system from solvent polarity38 and can stabilize antiparallel conformation in favor of photocyclization. Subsequently, B16 cells were incubated with NBDC NPs (60 μg mL−1) for 3 hours. As shown in Fig. 6a, the cells exhibited moderate red fluorescence under fluorescence microscopy, indicating successful uptake of NBDC NPs. Upon irradiation with 570 nm yellow-green light for 10 minutes, the intracellular red fluorescence was largely quenched. Subsequent irradiation with 770 nm NIR light restored the fluorescence signals to their initial state. Remarkably, a higher fluorescence on–off contrast (2.2) was observed in living cells compared to bulk solutions in a cuvette, likely due to differences in the intrinsic physicochemical properties (e.g., pH, salts, temperature, etc.) of the cellular microenvironment compared to in vitro solutions.63 Importantly, no significant fluorescence attenuation or cellular damage was noted after several cycles of the photoswitching process. Few studies have evaluated the reversible fluorescence switching performance in biological organisms.63,64 We selected zebrafish as an in vivo model to explore the photoswitching of NBDC NPs. Three-day-old zebrafishes were placed in water containing NBDC NPs (60 μg mL−1), which they presumably absorbed through cutaneous permeation. Intense red fluorescence was detected throughout the fish body upon treatment with NBDC NPs. Asexpected, a 570 nm/770 nm-triggered fluorescence switching performance with high fluorescence on-off contrast (2.5) was achieved in living zebrafish without any noticeable morphological defects (Fig. 6b). These results suggest that yellow-green/NIR light-controlled deep-red fluorescence switching holds potential for applications in photo-controlled precision fluorescence bio-imaging.
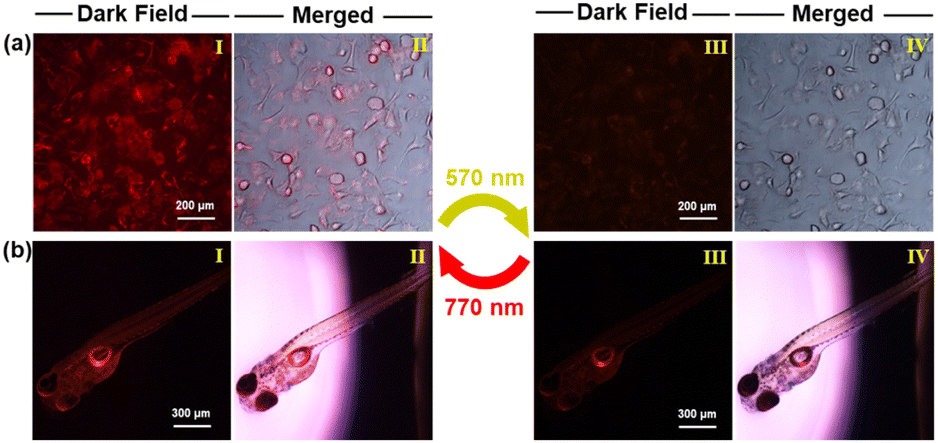 | ||
| Fig. 6 Fluorescence microscopy images of living B16 cells (a) and zebrafish (b) treated with NBDC NPs (60 μg mL−1) upon alternating irradiation with yellow-green light at 570 nm (8.9 mW cm−2) and NIR light at 770 nm (8.9 mW cm−2). Fluorescence images (I and III); merged images of fluorescence and bright field (II and IV). | ||
Conclusions
In summary, we have successfully developed a novel DTE-based deep-red fluorescence switch, NBDC, featuring a dimethylamine-functionalized BF2-curcuminoid moiety, utilizing an “acceptor synergistic conjugation system” strategy. The integration of photoswitching experiments and theoretical calculations has highlighted several key findings: First, incorporating the BF2-curcuminoid with a dimethylaniline moiety significantly redshifts the absorption of the open isomer into the longer wavelengths of the visible spectrum, thereby enabling DTE systems to be triggered by such light. Second, the enhanced intramolecular charge transfer (ICT) between the dimethylaniline donor and the boron difluoride acceptor shifts the emission of the open isomer towards the red or even deep red light region. Third, the active nonbonding p-electrons on the BF2-curcuminoid facilitate spin–orbital coupling between the singlet and triplet excited states, enhancing intersystem crossing and endowing the system with a typical TADF feature characterized by a smaller ΔEST. Finally, the unique acid/base-gated photochromism is facilitated by the responsive nature of the dimethylamine moiety to various acids. Photoswitching experiments demonstrated that NBDC presented an unprecedented 570 nm/770 nm light-driven photochromism, red-emissive fluorescence and TADF switching in toluene. This marks the first instance of a dual-mode red fluorescence and TADF switch based on DTE, triggered by yellow-green and NIR light, featuring a prolonged delayed fluorescence lifetime (τ = 435.75 μs). This is highly significant for applications in photo-controlled bioimaging and super-resolution imaging. Notably, in CHCl3, where NBDC shows minimal photochromic activity, it can achieve enhanced photochromic performance through TFA/TEA gating. Ultimately, the proof-of-concept applications of such nontoxic red fluorescence switch in photoswitchable imaging in vivo of living cells and zebrafish were successfully realized, thereby affirming its versatility in life sciences. Therefore, we anticipate that this deep-red fluorescence switch will have potential applications in fluorescence imaging requiring accurate detection, especially in living environments rich in autofluorescence. Besides, this successful example paves the way for further exploration of red-/NIR light-controlled DTEs for a broad range of biological and photopharmacological applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. Li, X. Ma and J. Song synthesized the target compound, performed photoswitching property measurements, and DFT calculations. Q. Wang, Y. Feng assisted in the synthesis of some intermediates and some photochromic property measurements. H. Liu and P. Zhang incubated B16 cells, cultured zebrafish and conducted the imaging test. Z. Li, P. Zhang, H. Guo and J. Yin designed and supervised the research and wrote the paper. All of the authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from National Natural Science Foundation of China (No. 32101150), the Science and Technology Project of Henan Province (No. 242102230119), and the Key Scientific Research Project of Higher Education of Henan Province (No. 22A430007), and Innovation and Entrepreneurship Training Program for College students in China (No. 202410482001 and 202310482001).Notes and references
- H. B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef PubMed.
- J. Volarić, W. Szymanski, N. A. Simeth and B. L. Feringa, Chem. Soc. Rev., 2021, 50, 12377–12449 RSC.
- N. M.-W. Wu, M. Ng and V. W.-W. Yam, Nat. Commun., 2022, 13, 33 CrossRef PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- R. Usui, K. Yamamoto, H. Okajima, K. Mutoh, A. Sakamoto, J. Abe and Y. Kobayashi, J. Am. Chem. Soc., 2020, 142, 10132–10142 CrossRef PubMed.
- T. Fukaminato, S. Ishida and R. Métivier, NPG Asia Mater., 2018, 10, 859–881 CrossRef CAS.
- H. Y. Jung, B. Kim, M. H. Jeon and Y. Kim, Small, 2022, 18, 2103523 CrossRef CAS PubMed.
- H. Tian and J. Zhang, Photochromic Materials: Preparation, Properties and Applications, Wiley-VCH, 2016, pp. 393–416 Search PubMed.
- H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS.
- M. Li and W. H. Zhu, Acc. Chem. Res., 2022, 55, 3136–3149 CrossRef CAS PubMed.
- Z. Li, S. Chen, Y. Huang, H. Zhou, S. Yang, H. Zhang, M. Wang, H. Guo and J. Yin, Chem. Eng. J., 2022, 450, 138087 CrossRef.
- Z. G. Zheng, R. S. Zola, H. K. Bisoyi, L. Wang, Y. Li, T. J. Bunning and Q. Li, Adv. Mater., 2017, 29, 1701903 CrossRef PubMed.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catal., 2018, 1, 516–522 CrossRef.
- Z. Li, X. Zeng, C. Gao, J. Song, F. He, T. He, H. Guo and J. Yin, Coord. Chem. Rev., 2023, 497, 215451 CrossRef.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- C. C. Ko and V. W. W. Yam, Acc. Chem. Res., 2018, 51, 149–159 CrossRef PubMed.
- Z. Li, C. He, Z. Lu, P. Li and Y.-P. Zhu, Dyes Pigm., 2020, 182, 108623 CrossRef.
- H. Wang, H. K. Bisoyi, X. Zhang, F. Hassan and Q. Li, Chem, 2022, 28, e202103906 CrossRef PubMed.
- Z. Zhang, W. Wang, M. O'Hagan, J. Dai, J. Zhang and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202205758 CrossRef.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef.
- N. M. Wu, M. Ng, W. H. Lam, H. L. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2017, 139, 15142–15150 CrossRef PubMed.
- M. Luo, J. Zhao, Y. Liu, Z. Mao, S. Wang and Z. Chi, Adv. Funct. Mater., 2023, 33, 2211009 CrossRef.
- P. Hong, J. Liu, K. X. Qin, R. Tian, L. Y. Peng, Y. S. Su, Z. Gan, X. X. Yu, L. Ye, M. Q. Zhu and C. Li, Angew. Chem., Int. Ed., 2024, 63, e202316706 CrossRef PubMed.
- S. Takeuchi, T. Nakagawa and Y. Yokoyama, Chem. Commun., 2020, 56, 6492–6494 RSC.
- F. Hu, M. Cao, X. Ma, S. H. Liu and J. Yin, J. Org. Chem., 2015, 80, 7830–7835 CrossRef PubMed.
- Z. Li, Y. Song, Y. Dai, Y. Pei, Z. Lu and H. Guo, Opt. Mater., 2019, 95, 109235 CrossRef.
- S. Qiu, A. T. Frawley, K. G. Leslie and H. L. Anderson, Chem. Sci., 2023, 14, 9123–9135 RSC.
- G. Long, Y. Deng, W. Zhao, G. Zhou, D. J. Broer, B. L. Feringa and J. Chen, J. Am. Chem. Soc., 2024, 146, 13894–13902 CrossRef PubMed.
- H. Xi, Z. Zhang, W. Zhang, M. Li, C. Lian, Q. Luo, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2019, 141, 18467–18474 CrossRef.
- V. W.-W. Yam, C.-C. Ko and N. Zhu, J. Am. Chem. Soc., 2004, 126, 12734–12735 CrossRef PubMed.
- S. Fredrich, R. Göstl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef PubMed.
- S. Fredrich, T. Morack, M. Sliwa and S. Hecht, Chem, 2020, 26, 7672–7677 CrossRef.
- Z. Zhang, J. Zhang, B. Wu, X. Li, Y. Chen, J. Huang, L. Zhu and H. Tian, Adv. Opt. Mater., 2018, 6, 1700847 CrossRef.
- Z. Zhang, W. Wang, P. Jin, J. Xue, L. Sun, J. Huang, J. Zhang and H. Tian, Nat. Commun., 2019, 10, 4232 CrossRef PubMed.
- W. Wang, W. Yang, Z. Zhang, J. Dai, Y. Xu and J. Zhang, Chem. Sci., 2024, 15, 5539–5547 RSC.
- J. C. Boyer, C. J. Carling, B. D. Gates and N. R. Branda, J. Am. Chem. Soc., 2010, 132, 15766–15772 CrossRef PubMed.
- K. Zheng, S. Han, X. Zeng, Y. Wu, S. Song, H. Zhang and X. Liu, Adv. Mater., 2018, 30, e1801726 CrossRef PubMed.
- K. Mori, Y. Ishibashi, H. Matsuda, S. Ito, Y. Nagasawa, H. Nakagawa, K. Uchida, S. Yokojima, S. Nakamura, M. Irie and H. Miyasaka, J. Am. Chem. Soc., 2011, 133, 2621–2625 CrossRef.
- G. Liu, Y. M. Zhang, C. Wang and Y. Liu, Chem, 2017, 23, 14425–14429 CrossRef.
- Z. Li, Y. Pei, Y. Wang, Z. Lu, Y. Dai, Y. Duan, Y. Ma and H. Guo, J. Org. Chem., 2019, 84, 13364–13373 CrossRef.
- Z. Li, Y. Dai, Z. Lu, Y. Pei, H. Chen, L. Zhang, Y. Duan and H. Guo, Chem. Commun., 2019, 55, 13430–13433 RSC.
- Z. Li, Y. Xie, M. Zhu, Y. Song, M. Qin and X. Hu, Opt. Mater., 2019, 94, 257–265 CrossRef.
- Z. Li, J. Song, X. Gao, X. Ma, K. Liu, Z. Ma, Q. Wang, X. Zeng, H. Zhang, P. Zhang, H. Guo and J. Yin, Chin. Chem. Lett., 2024 DOI:10.1016/j.cclet.2024.110073.
- Z. Li, J. R. Zhang, X. K. Tian, S. Yang, S. Chen, H. Zhou and X. G. Yang, Chem. Sci., 2022, 13, 9381–9386 RSC.
- D. Hong, J. Han, X. Cao, S. Wang, Q. Song, Y.-P. Ou, H. Guo and Z. Li, J. Lumin., 2023, 262, 119967 CrossRef.
- Z. Li, X. Gao, H. Zhang, X. Ma, Y. Liu, H. Guo and J. Yin, Chin. Chem. Lett., 2023, 34, 107645 CrossRef.
- J. G. Peak and M. Peak, J. Photochem. Photobiol., 1995, 61, 484–487 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- J. Yin, Y. Hu and J. Yoon, Chem. Soc. Rev., 2015, 44, 4619–4644 RSC.
- J. T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X. Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC.
- X. Ma, W. Chi, X. Han, C. Wang, S. Liu, X. Liu and J. Yin, Chin. Chem. Lett., 2021, 32, 1790–1794 CrossRef.
- Y. Wu, Y. Xie, Q. Zhang, H. Tian, W. Zhu and A. D. Li, Angew. Chem., Int. Ed., 2014, 53, 2090–2094 CrossRef PubMed.
- X. Hu, J. Zhao, Z. Wang, H. Li, P. Cai, J. Si, X. Yao, Q. Ai and Z. Liu, Dyes Pigm., 2022, 205, 110571 CrossRef.
- T. Kawai, T. Sasaki and M. Irie, Chem. Commun., 2001, 8, 711–712 RSC.
- C. Shi, R. Zhu, K. Guo, L. Zhang, Z. Liu, X. Liu, Q. Ai and X. Hu, Adv. Opt. Mater., 2023, 11, 2301844 CrossRef.
- X. Chai, Y. X. Fu, T. D. James, J. Zhang, X. P. He and H. Tian, Chem. Commun., 2017, 53, 9494–9497 RSC.
- S. Chen, W. Li, X. Li and W.-H. Zhu, J. Mater. Chem. C, 2017, 5, 2717–2722 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, Gaussian 09, Revision B.01. Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- J. Zhang, F. Fang, B. Liu, J. H. Tan, W. C. Chen, Z. Zhu, Y. Yuan, Y. Wan, X. Cui, S. Li, Q. X. Tong, J. Zhao, X. M. Meng and C. S. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 41051–41061 CrossRef PubMed.
- Y. Kim, H. Jung, Y. H. Choe, C. Lee, S. K. Ko, S. Koun, Y. Choi, B. H. Chung, B. C. Park and T. L. Huh, Angew. Chem., Int. Ed., 2012, 124, 2932–2936 CrossRef.
- U. Al-Atar, R. Fernandes, B. Johnsen, D. Baillie and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 15966–15967 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05473c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |