
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magic methylation with methyl-containing peroxides
Daliah
Farajat
 a,
Yuhua
Zhang
b and
Chao-Jun
Li
*ac
a,
Yuhua
Zhang
b and
Chao-Jun
Li
*ac
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 2K6, Canada. E-mail: cj.li@mcgill.ca
bAccustandard Inc., 125 Market Street, New Haven, Connecticut 06513, USA
cFRQNT Centre for Green Chemistry and Catalysis, Canada
First published on 4th December 2024
Abstract
Methyl groups rank among the most abundant carbon fragments found in natural products and small-molecule pharmaceuticals. The late-stage and environmentally friendly installation of these groups onto biologically active molecules has attracted widespread attention in both industry and academia. In 2008, we published the first use of a methyl radical derived from a peroxide toward a directed transition-metal catalysed C–H methylation. In the past sixteen years, methyl-containing peroxides have proven themselves as robust reagents for introducing methyl groups onto organic molecules. In this review, our goal is to provide a thorough summary of the research advancements achieved in this field thus far.
 Daliah Farajat | Daliah Farajat is a PhD candidate in chemistry in Prof. Chao-Jun Li's group at McGill University. She earned her first B.A. & Sc. in Cognitive Science from McGill, followed by a BSc in Biochemistry from Concordia University. Her research focuses on developing new methodologies in nickel catalysis and hydrazone umpolung chemistry. Outside of her research, Daliah is actively involved in several initiatives, including the Green Chemistry McGill (GCM) group, the Empowering Women in Organic Chemistry (EWOC) Eastern Canada chapter, and serving as a graduate student representative for the Centre in Green Chemistry and Catalysis (CGCC). |
 Yuhua Zhang | Yuhua Zhang got his PhD in 2001 from Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. He was a postdoctoral research associate in Tulane University (2003–2007), where he discovered the use of peroxides for C–H methylations while working with Prof. C.-J. Li, followed by a second postdoctural research position at Washington University in St Louis (2007–2009). Currently, Yuhua Zhang is a research scientist at AccuStandard Inc. in New Haven, CT. |
 Chao-Jun Li | Prof. Chao-Jun Li received his PhD at McGill University in 1992 and was an NSERC Postdoctoral Fellow at Stanford University (US). He was an Assistant/Associate/Full Professor at Tulane University (US). Since 2003, he has been a Canada Research Chair (Tier I) in Green Chemistry, E. B. Eddy Chair Professor and Distinguished James McGill Chair at McGill University. He has also served as the co-director for Quebec's FQRNT Center for Green Chemistry and Catalysis since 2009. Dr Li was elected as a Fellow of the Royal Society of Canada, the Royal Society of Chemistry (UK), the American Association for the Advancement of Science (AAAS), the Chemical Institute of Canada, the American Chemical Society (ACS), The World Academy of Sciences (TWAS) and the European Academy of Sciences. |
1 Introduction
Methyl groups constitute one of the most prevalent functionalities in both natural and synthetic products. Recent estimates indicate that up to 67% of the top-selling small-molecule drugs feature at least one methyl group.1,2 The deliberate installation of methyl groups is a common practice aimed at enhancing a molecule's bioavailability and drug–target interactions without drastically changing its molecular weight; a modification that often leads to a substantial increase in potency. This widely observed phenomenon is commonly dubbed “the magic methyl effect”.3,4 Consequently, methodologies involving the incorporation of methyl groups onto drug candidates attract considerable attention in the pharmaceutical industry (Scheme 1).5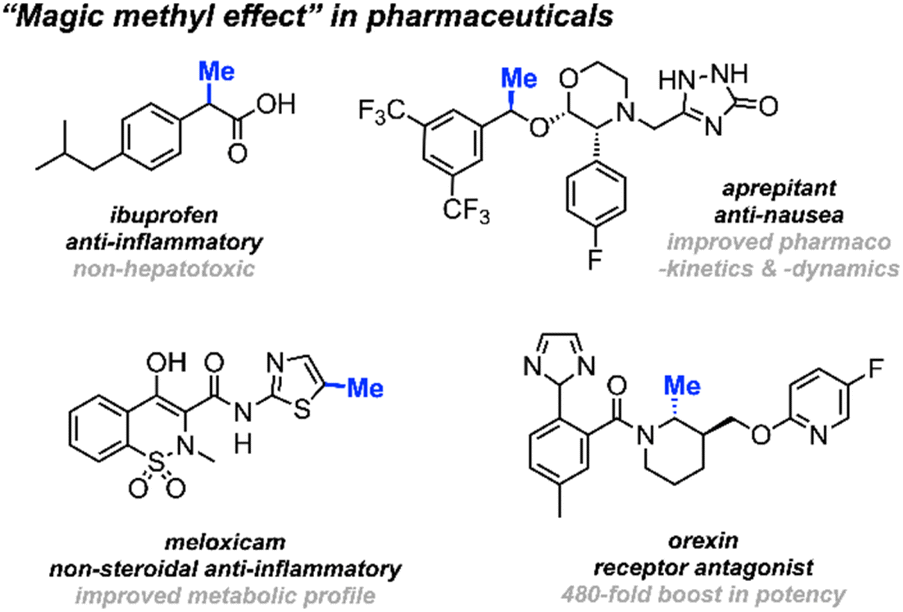 | ||
| Scheme 1 Selected examples of the “magic methyl effect” in pharmaceuticals and drug development. | ||
A wide variety of methylation reactions exist, typically employing methyl-containing reagents as precursors to methyl-anions, -cations, or -radicals, depending on the specific nature of the reaction. Frequently utilised methylating reagents include methyl iodide, dimethyl sulfate, dimethyl carbonate, methyl triflate, and various methyl organometals.6 However, each of these reagents carries its own set of caveats, hazards, and operational challenges. Common issues include the requirement for harsh reaction conditions, as well as strong bases and stoichiometric quantities of metal additives.7 To date, the ongoing endeavour of discovering innovative and environmentally friendly methylating reagents persists.
For the past two decades, our research group centred its efforts on investigating transition-metal-catalysed C–H bond activation followed by C–C bond formation, namely, Cross-Dehydrogenative Couplings (CDC).8–13 In 2008, during an exploration of phenyl C(sp2)–H bond activation of 2-phenylpyridine, we remarkably discovered the formation of o-tolypyridine when the reaction was conducted with di-tert-butyl peroxide (DTBP) as an oxidant.14 We hypothesised that the methyl group originated from a rearrangement of DTBP radicals. Through a screening process involving various methyl-containing peroxides, dicumyl peroxide (DCP) emerged as the most effective when catalysed by Pd(OAc)2 (10 mol%) at 130–150 °C, yielding methylated products in the range of 30–76% (Scheme 2).
 | ||
| Scheme 2 Palladium-catalysed ortho-methylation of 2-phenylpyridines using DCP as a methyl radical source and proposed mechanism. | ||
The proposed reaction mechanism involves a β-methyl elimination, generating a methyl-palladium intermediate while simultaneously releasing a ketone.15 Nearly stoichiometric quantities of acetophenone D and α-cumyl alcohol G were isolated by column chromatography, confirming that DCP served as not only the methylating reagent but also the hydrogen acceptor. Our publication marked the first demonstration of a transition-metal catalysed and directed methylation of a carbon–hydrogen bond using a peroxide-derived methyl radical.
Since our discovery, research groups worldwide have further studied the utilisation of various methyl-containing peroxides as powerful reagents for methylation reactions (Scheme 3). A plethora of outstanding research has been published on this chemistry, showcasing significant advancements.16 This has also broadened our mechanistic insights, allowing us to understand that using peroxides as methyl-containing precursors involves balancing several possible reaction pathways, including how the methyl radical is generated, single electron transfer (SET), and hydrogen atom transfer (HAT). Herein, we aim to present a comprehensive overview of the progress and developments made in this field to-date.
 | ||
| Scheme 3 Library of peroxides used as methyl precursors (corresponding bond dissociation energy in kcal mol−1):17,18 dicumyl peroxide (DCP), di-tert-butylperoxide (DTBP), tert-butylperacetate (TBPA), tert-butyl hydroperoxide (TBHP), tert-butyl peroxybenzoate (TBPB), tert-butyl 2-ethylhexaneperoxoate (TBEHP), and tert-butyl cyclopentyl carbonoperoxoate (TBCyCP), *based on structurally similar peroxide (top); heat, light, or metal driven generation of methyl radicals (middle); alternative applications for alkoxy radicals, including single electron transfer (SET) and hydrogen atom transfer (HAT) (bottom). | ||
2 Direct methylation
2.1 Methylation of C–H bonds
Most investigations of C–H bond methylation via peroxides were focused on C(sp2)–H and C(sp3)–H bonds; we have yet to see any reports on C(sp)–H bonds.2.1.1.1 Palladium catalysis. The Pd(OAc)2/DCP reaction system not only contributes to the methylation of phenylpyridine derivatives, it also promotes the C(sp2)–H methylation of benzenesulfonamide and benzamide compounds at the ortho position. In our 2008 study, we found that although acetanilide was reactive under the Pd(OAc)2/DCP conditions, only 33–42% yields were obtained (Scheme 4). Thus, an improved method towards methylation of acetanilide compounds by peroxide remained a desirable objective.
 | ||
| Scheme 4 Palladium-catalysed ortho-methylation of anilides using DCP as a methyl source. | ||
In 2017, Cai and co-workers reported that in HOAc as the solvent, Pd(OAc)2 (10 mol%) and DCP (3 equiv.) allowed the ortho-methylation of benzenesulfonamide and benzamide compounds in 43–65% yields (Scheme 5).19 The authors also discovered that there was no significant difference in yields whether the reaction was carried out under argon or an open air atmosphere. Although the authors only reported 4 sample results, they demonstrated progress towards ortho-methylation of arene compounds.
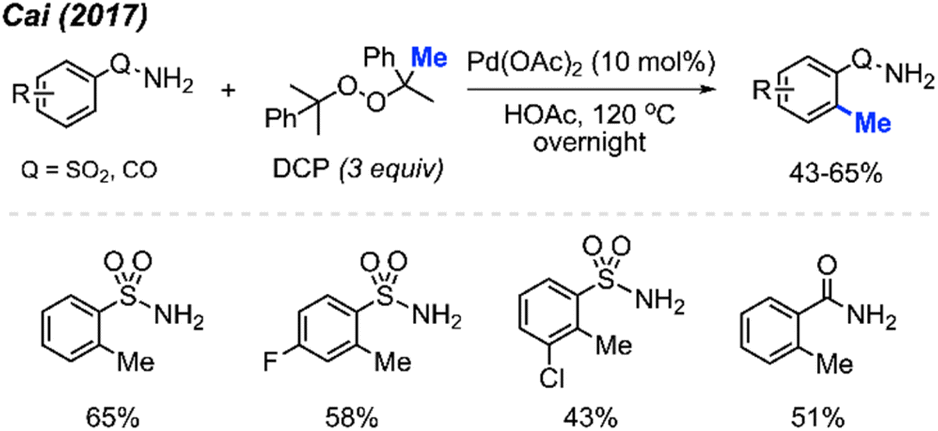 | ||
| Scheme 5 Palladium-catalysed ortho-methylation of benzenesulfonamide and benzamide compounds using DCP as a methyl source. | ||
Aside from the aforementioned ortho-aryl methylation reactions using Pd(OAc)2/DCP reagents, the Pd(OAc)2/DTBP reaction system proved to be beneficial for the methylation of benzoic acids. In 2019, Cheng and co-workers reported that combining two equivalents of DTBP and potassium acetate with 10 mol% of Pd(OAc)2 catalysed the ortho-methylation of benzoic acid derivatives in 28–79% yields using 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as a solvent heated to 80 °C (Scheme 6A).20 Compared to Li14 and Cai's19 reaction conditions, this reaction was milder (open to air and lower temperature), and the authors demonstrated the protocol on 18 functionally diverse samples. The use of HFIP proved to be vital, as the reaction does not proceed without it. Various functional groups (methyl, trifluoromethyl, halogen, and methoxy) at meta-, ortho- and para-positions on benzoic acid, as well as disubstituted benzoic acids are well tolerated in this reaction. Notably, the meta-positioned substituent directs the methylation selectively towards the less hindered activated C–H bond. It is also worth pointing out that 54% and 34% isolated yields were obtained for gram scale reactions (Scheme 6B), which demonstrated the potential of this methylation reaction towards practical applications.
 | ||
| Scheme 6 Palladium-catalysed ortho-methylation of benzoic acids using DTBP as methyl source. (A) Optimised reaction conditions, (B) gram scale reaction. | ||
When 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) was added, the reaction was completely inhibited, which indicated the involvement of a radical process. As shown in Scheme 5, Pd(OAc)2 reacts with benzoic acid A in the presence of KOAc to form Pd(II) species B. The tert-butoxy radical generated from DTBP under heat then reacts with intermediate B to yield intermediate C. Next, β-methyl elimination of species C forms intermediate D. Subsequently, Pd(IV) D undergoes C–H insertion to generate intermediate E and t-BuOH. Finally, reductive elimination of species E produces the ortho-methylation product and regenerates Pd(OAc)2.
2.1.1.2 Cobalt catalysis. In 2019, Cai and co-workers reported the successful ortho-methylation of anilides by using a cobalt/silver catalyst system in the presence of DTBP (2 equiv.). The reaction produced up to 80% yield and a total of 16 methylated anilide derivatives were reported (Scheme 7A).21 It was found that the arenes containing directing groups such as amides, ketones and esters also gave good yields of the methylated products. Interestingly, this rection method did not work with 2-phenylpyridine compounds at all (Scheme 7B). Although the Pd(OAc)2/DCP system reported by Cai in 2017 allowed the ortho-methylation of benzamides, only two examples were provided (Schemes 5 and 7). In the previous examples, the ortho-methylation of benzamide compounds varied depending on the nature of the N-substituent.
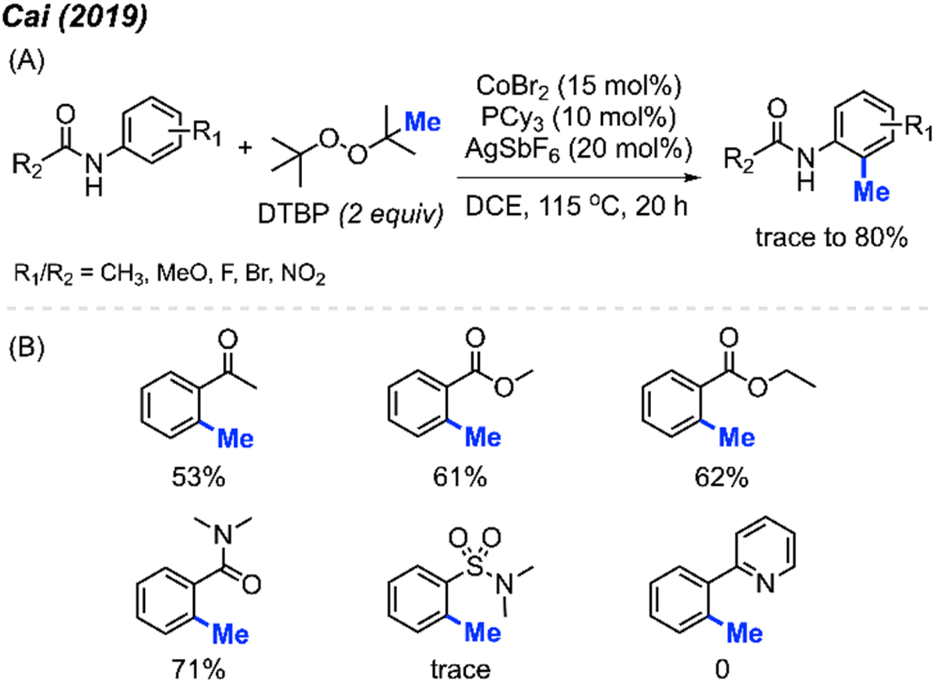 | ||
| Scheme 7 Cobalt-catalysed ortho-methylation of anilides and arenes using DCP as a methyl source. (A) Optimised reaction conditions, (B) reaction yields with different directing groups. | ||
In 2016, Lu and co-workers reported an ortho-methylation of benzamide bearing a 2-pyridinylisopropyl (PIP) directing group, which was catalysed by 10 mol% Co(acac)2 with DCP (3 equiv.) (Scheme 8A).22 A total of 17 substrates were reported, and the isolated yields ranged between 65–87%. When the meta-position was occupied, only the mono-methylated product was obtained, which is in agreement with Cheng's observation20 (Scheme 6). The authors also performed an intermolecular competition experiment, showing that an electron-withdrawing group (–CF3) on the aromatic ring favours substrate methylation compared to an electron-donating group (–OMe) (Scheme 8B). Notably, the authors successfully methylated heteroaryl derivatives (thiophenes) and included one example of a cyclic α,β-unsaturated amide.
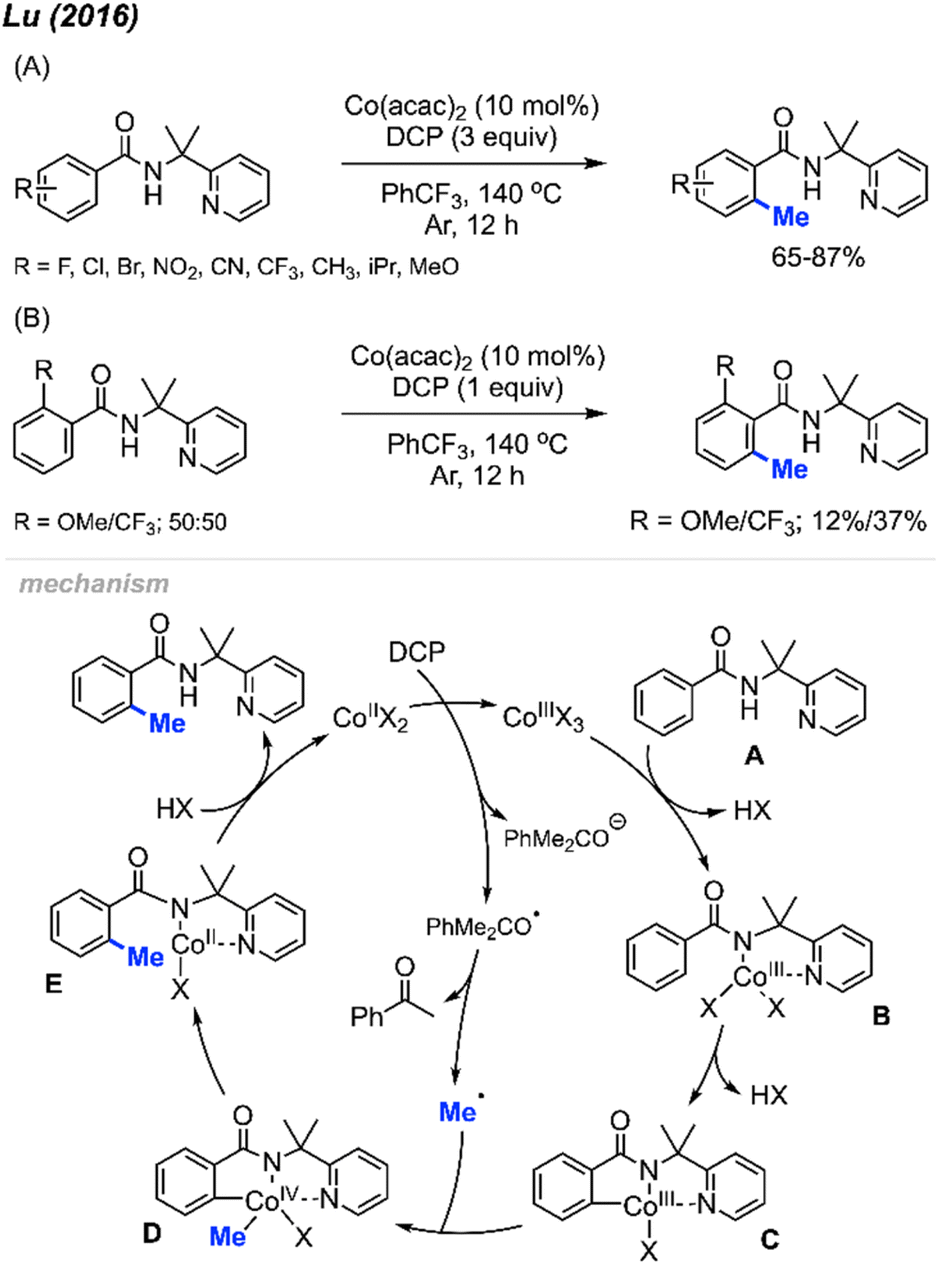 | ||
| Scheme 8 Cobalt-catalysed and directing group facilitated ortho-methylation of benzamide using DCP as a methyl source. (A) Optimised reaction conditions, (B) intermolecular competition experiment. | ||
A possible mechanism is shown in Scheme 8. Upon heating, DCP undergoes a single electron transfer (SET) process with Co(II), which forms the Co(III) species along with one radical and one anionic alkoxy fragments of DCP. The Co(III) species further coordinates to the benzamide molecule A by ligand exchange to form intermediate B. Then intermediate B undergoes C–H insertion to generate Co(III) species C. β-Methyl elimination within the DCP alkoxy radical fragment generates a methyl radical, which then reacts with intermediate C to yield species D by oxidative addition. Finally, reductive elimination within species D and subsequent protonation yields the final product and regenerates the Co(II) catalyst.
2.1.1.3 Nickel catalysis. In the same year (2016), Chatani and co-workers reported the ortho-methylation of benzamides containing 8-aminoquinoline as a directing group. Products were isolated in 44–86% yields in the presence of 10 mol% NiCl2(PCy3)2, DCP (2 equiv.) and Na2CO3 (3 equiv.) using tert-butylbenzene as the solvent (Scheme 9).23 The scope included 30 benzamide derivatives with various functional groups. The authors found that meta-substituted benzamides favour methylation at the less sterically hindered C(sp2)–H bond, which agrees with the discovery by both Cheng20 (Scheme 6) and Lu22 (Scheme 8). Through a competition experiment, the authors demonstrated that electron withdrawing groups on the aromatic ring promote the methylation reaction compared to electron donating groups, which aligns with Lu's22 investigation in 2016 (Scheme 8).
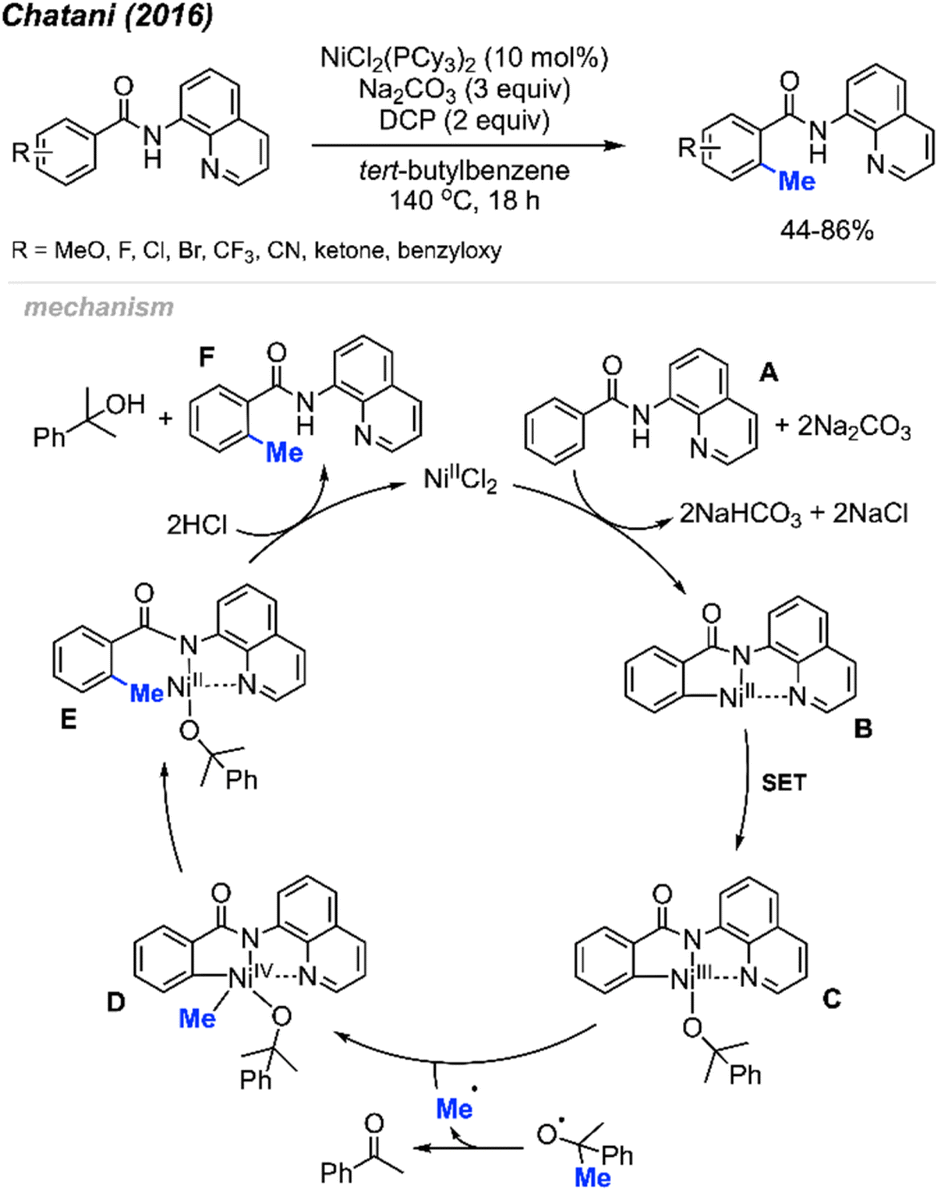 | ||
| Scheme 9 Nickel-catalysed ortho-methylation of benzamides containing 8-aminoquinoline directing groups and using DCP as a methyl source. | ||
The authors performed detailed mechanistic studies and proposed a Ni(II)/Ni(IV) catalytic cycle (Scheme 9). First, the benzamide A reacts with the Ni(II) species with the help of Na2CO3 base to form intermediate B. Then, a SET process between DCP and Ni(II) B generates the Ni(III) specie C and an alkoxy radical fragment. The alkoxy radical will decompose to yield acetophenone and a methyl radical which will react with species C to form Ni(IV) D. Finally, intermediate D undergoes reductive elimination to give intermediate E, and subsequent protonation yields the target product F and regenerates the Ni(II) catalytic species.
In 2019, Chen and co-workers reported an alternative catalytic system for benzamide methylation, with 20 examples being reported.24 They found that benzamide compounds can be ortho-methylated in the presence of Ni(OAc)2·4H2O (20 mol%) and DTBP (4 equiv.) in CH3CN at 140 °C for 12 hours, in 44–81% yields (Scheme 10A). Compared to the Chatani23 method (Scheme 9), this reaction eliminates the need for added base and generates acetone as a by-product which can more readily be removed compared to acetophenone.
 | ||
| Scheme 10 Nickel-catalysed methylation of benzamides using DTBP as a methyl course. (A) Optimised reaction conditions, (B) replacing quinoline with naphthalene, (C) replacing amide with carboxylic ester. | ||
In agreement with previous observations, the authors also found that meta-substituted benzamides encouraged methylation at the less sterically hindered site, and that electron-withdrawn groups on the aromatic ring are favoured for this reaction. Furthermore, the authors demonstrated that both the quinoline and amide components were necessary functionalities within the directing group. If the quinoline is replaced with a naphthalene, or if the amide is replaced with a carboxylic ester, no product is observed in either scenario (Scheme 10B and C).
2.1.2.1 Photocatalysis. In 2014, DiRocco and co-workers reported the late-stage photoredox catalysed methylation of biologically active heterocycles (Scheme 11).25 Six-membered aromatic heterocycles (e.g. pyridines, pyrazines, and pyrimidines) and some electron-rich five-membered aromatic heterocycles (e.g. imidazoles) reacted with peroxides efficiently. Notably, this reaction proceeds in the presence of various functionalities such as basic amines, alcohols, amides, and esters without the need for protecting groups. The author also found that DTBP and TBHP did not work for this reaction, with tert-butylperacetate (TBPA) being the best methylation reagent.
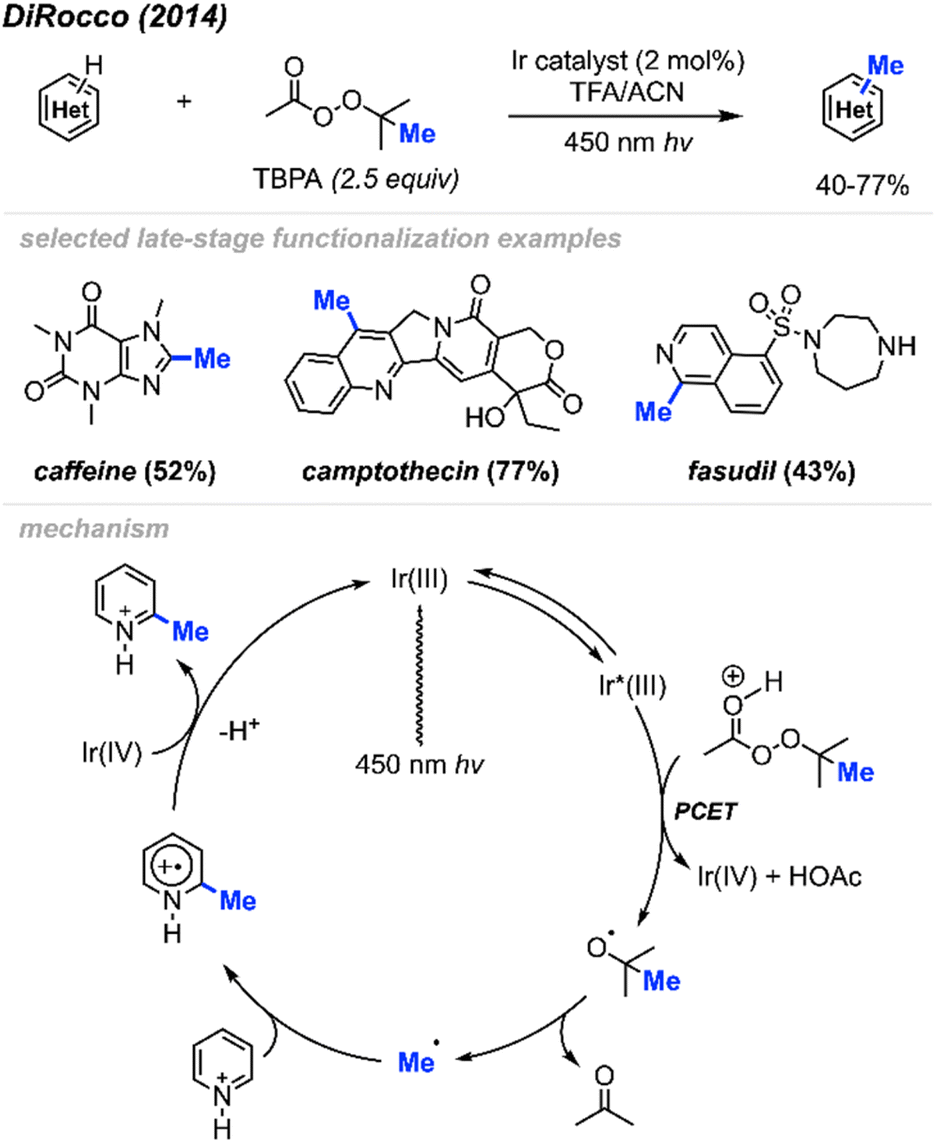 | ||
| Scheme 11 Bio-active heterocycle methylation through photoredox catalysis using TBPA. | ||
The proposed catalytic cycle is shown in Scheme 11. A proton-coupled electron transfer (PCET) between the excited Ir(III)* photoredox catalyst and TBPA under acidic conditions generates a tert-butoxy radical, which subsequently decomposes via β-scission, yielding a methyl radical and acetone. The methyl radical then undergoes addition to the protonated heterocycle, resulting in an amino-radical cation, which is then oxidised by Ir(IV), yielding the desired product and regenerating the initial Ir(III) photocatalyst.
2.1.2.2 Metal-free, thermal reactions. In 2015, Li and co-workers reported that DCP can be used for methylation of pyridine N-oxides at the 2-position under inert atmosphere at 120 °C in the absence of a metal catalyst.26 The authors provided 13 examples containing various functionalities (Cl, NO2, and OCH3), with yields ranging from 54–84% (Scheme 12A). It is noted that earlier studies show that methyl radicals from high temperature decomposition of peroxides can generate trace and mixture of methylation products with heterocycles.27,28 Not only were pyridine N-oxides effective substrates under these conditions, methylation of quinoline N-oxide compounds and isoquinoline N-oxide also gave good yields. Furthermore, the authors performed a gram scale reaction, and were able to obtain a 68% isolated yield (Scheme 12B). Similar to our work in 2008, this pyridine N-oxide methylation method suffers from the generation of both mono- and di-methylated products, which can complicate purifications. This study significantly extended the radical methylation via a peroxide that did not require transition metal catalysis.
 | ||
| Scheme 12 Methylation of pyridine N-oxide derivatives with DCP. (A) Optimised reaction conditions, (B) gram scale reaction. | ||
The reaction mechanism (Scheme 12) begins with DCP decomposition under heat, generating two molecules of 2-phenylpropanoxy radical, which can further undergo β-scission to form the methyl radical and acetophenone. The methyl radical then attacks the N-oxide at the 2-position, forming an intermediate amino-radical cation. With the help of another DCP derived radical, hydrogen abstraction occurs to restore the aromatic ring.
In 2017, Zhang and co-workers discovered that metal-free conditions can be used to methylate pyrimidinones and pyridinones in acetic acid with DCP (3 equiv.) at 120 °C.29 14 examples of pyrimidinones were provided with 63–96% yields (Scheme 13A) and 18 examples of pyridinones were generated in 22–79% yields (Scheme 13B). For both pyrimidinone and pyridinones compounds, it was observed that aromatic substituents improved yields compared to non-aryl groups and that compounds lacking at least one aryl substituent were unreactive. This observation can be attributed to the ability of aromatic groups to better stabilise the intermediate radical formation. Notably, there is one example of the C-3 methylation of indole, albeit at a low yield of 22%.
 | ||
| Scheme 13 Metal-free methylation of pyrimidinones and pyridinones using DCP. (A) Optimised reaction conditions for pyrimidinones, (B) optimised reaction conditions for pyridinones, (C) gram scale reaction. | ||
The authors also performed a 50 g scale reaction, with 70% isolated yield being achieved (Scheme 13C), which indicated its utility in organic synthesis. The proposed mechanism is in-line with previously reported radical mechanisms (Scheme 12). First, homolysis followed by β-scission of DCP generates the methyl radical and acetophenone by-product. This is followed by selective addition of the methyl radical at the 5-position, and subsequent re-aromatisation to generate the final product.
By using the exact same reaction conditions as Zhang29et al. (Scheme 13), Cai and co-workers reported the direct methylation of azole compounds in up to 75% yields, with 5 examples being reported (Scheme 14).30 This work further highlighted the versatility of peroxide-mediated methylation and broadened the scope of valuable heterocycles that it can be applied to.
 | ||
| Scheme 14 Metal-free methylation of azoles using DCP. | ||
In 2020, Lin and co-workers discovered a third application of the same metal-free DCP-mediated methylation of imidazo[1,2-a]pyridine compounds at the C-5 position (Scheme 15A).31 A total of 11 imidazopyridine compounds were methylated, with yields ranging from trace amounts to 74%. Both electron-donating groups (methyl, methoxy) and electron-withdrawing groups (halogen) were tolerated, however, no specific trends in reactivity were observed based on substituent variation.
 | ||
| Scheme 15 Metal-free methylation of imidazo[1,2-a]pyridines and quinoxalin-2(1H)-ones using either DCP or TBHP. (A) Optimised reaction conditions for imidazo[1,2-a]pyridine, (B) optimised reaction conditions for quinoxaline-2(1H)-one, (C) gram scale reaction. | ||
Although previous investigations into the use of tert-butyl hydroperoxide (TBHP) as a methylating reagent were not successful, the authors found that TBHP works excellently for the C-3 methylation of quinoxaline-2(1H)-one compounds. Under the same metal-free conditions, TBHP was shown to be the most suitable reagent compared to the commonly used DCP and DTBP (Scheme 15B). 15 examples were provided in trace-85% yields. A gram scale reaction gave 61% isolated yield (Scheme 15C).
In the same year, Xia and co-workers also developed a method for C-3 methylation of quinoxaline-2(1H)-one compounds (Scheme 16).32 In this case, the iodine-mediated methylation was achieved at a lower temperature of 80 °C in air, using TBHP as the methyl source. The authors report a large scope with yields ranging from 28–74%, with good functional group tolerance including aliphatic chains, esters, alkenes, alkynes, methoxy, nitro and halogens.
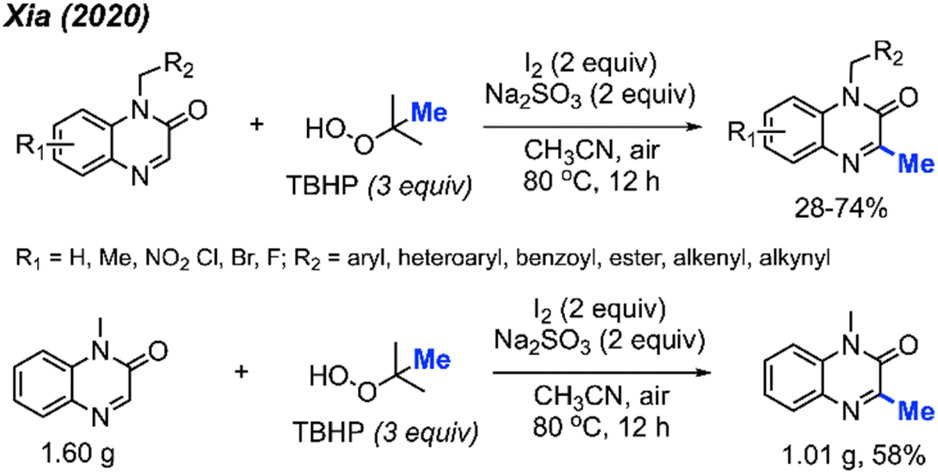 | ||
| Scheme 16 Metal-free methylation quinoxaline-2(1H)-ones using TBHP. | ||
2.1.2.3 Copper catalysis. In 2016, Zou and co-workers reported the methylation of coumarins catalysed by Cu(I) using DTBP as the methylation reagent (Scheme 17).33 The authors methylated 17 variously functionalised coumarins, which provided 50–68% yields. Higher yields were observed when electron-withdrawing groups were present on the phenyl ring compared to electron-donating groups, which likely increased the electrophilicity at the 3-position. Interestingly, the lower yields observed with electron-donating groups on the phenyl ring can be improved with the addition of an electron-donating groups at the 4-position, likely due to the compensatory stabilisation of the benzyl radical intermediate.
 | ||
| Scheme 17 Copper-catalysed regioselective methylation of coumarins. | ||
The authors proposed a mechanism (Scheme 17) that begins with Cu(I)-mediated cleavage of DTBP to generate a tert-butoxyl radical, which then undergoes β-scission to generate the methyl radical and a molecule of acetone. Selective addition of the methyl radical to the 3-position of coumarin forms the benzyl radical intermediate A, which is then oxidised by Cu(II) to yield the carbocation B. Finally, deprotonation of intermediate B produces product C.
2.1.2.4 Iron catalysis. Again in 2016, Zhou and co-workers reported an alternative method for C-3 methylation of coumarins, catalysed by FeCl2·4H2O (10 mol%) in the presence of DABCO (10 mol%) base and DTBP as a methyl radical source.34 This is the second example of coumarin methylation by DTBP, generating yields from 60–72% for 7 coumarin derivatives (Scheme 18).
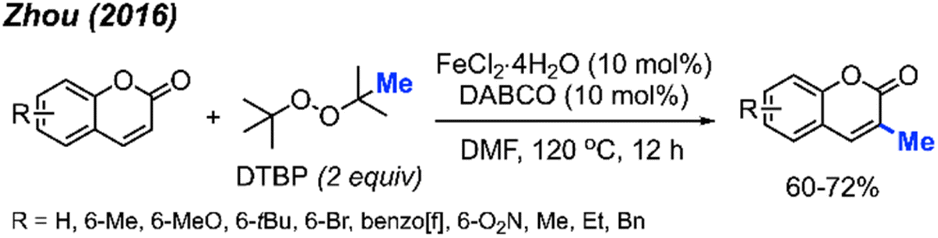 | ||
| Scheme 18 Iron-catalysed C-3 methylation of coumarins using DTBP. | ||
In the same year, Patel and co-workers discovered that tert-butyl peroxybenzoate (TBPB) functions as a reagent for the C-3 methylation of flavones when catalysed by Fe(acac)3 (Scheme 19).35 Although their publication focused on the use of TBPB as an intermediate oxidant for C-3 cyclo-alkylation and amidation, 5 examples of flavone methylation were provided using TBPB as the methyl source. Despite moderate to low yields, these examples highlight the ability of peroxide-derived methyl radicals to functionalise electron-rich sites of heterocycles.
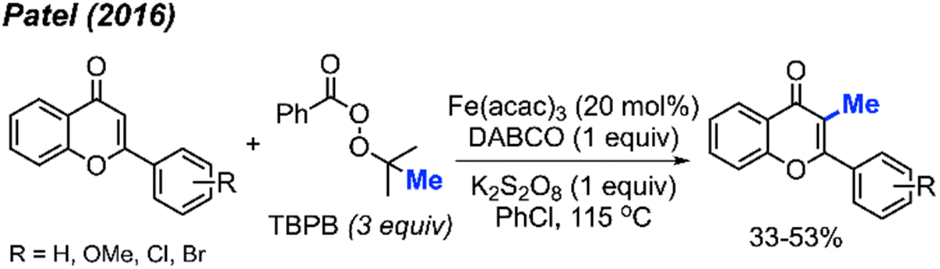 | ||
| Scheme 19 Iron-catalysed C-3 methylation of flavones with TBPB. | ||
The first report of a direct alkenyl C(sp2)–H methylation was published by Bao and co-workers in 2017, where they demonstrated an iron-catalysed methylation of vinyl arenes (Scheme 20).36 Using 2.5–5 mol% Fe(OTf)3 in dioxane at 80 °C, the authors achieved yields ranging from 57–84% using a variety of methyl-containing peroxides. Functional group tolerance included methyl groups, halogens, carboxylic acid, and dihydroxyborate.
 | ||
| Scheme 20 Iron-catalysed direct C–H methylation of vinyl arenes. | ||
The authors proposed the mechanism shown in Scheme 20, starting with the initial reduction of Fe(OTf)3 to Fe(II). The heat along with Fe(II) then facilitates the decomposition of the peroxide to form the methyl radical, a carboxylate and acetone byproduct. The methyl radical will then react at the terminal end of the alkene, forming the more stable benzylic radical that is subsequently oxidised to the carbocation by Fe(III) thereby reforming the Fe(II). A final deprotonation of the substrate by the carboxylate allows for the reformation of the double bond to form the final product.
In 2023, Luo and co-workers discovered that the methylation of enamide compounds using DCP can be copper- or iron-catalysed (Scheme 21).37 Both Cu(OAc)2·2H2O (5 mol%) and FeCl2 (5 mol%) can catalyse the direct β-C(sp2)–H methylation of enamides in 50–85% yields, with only E-isomer formation. It was found that the α-C(sp2) position needed to be aromatic in order to stabilise the intermediate radical generated, since aliphatic substitution at this position did not work under these conditions. Various functional groups were tolerated on the α-aryl moiety, where electron-donating groups generated slightly higher yields compared to electron-withdrawing groups.
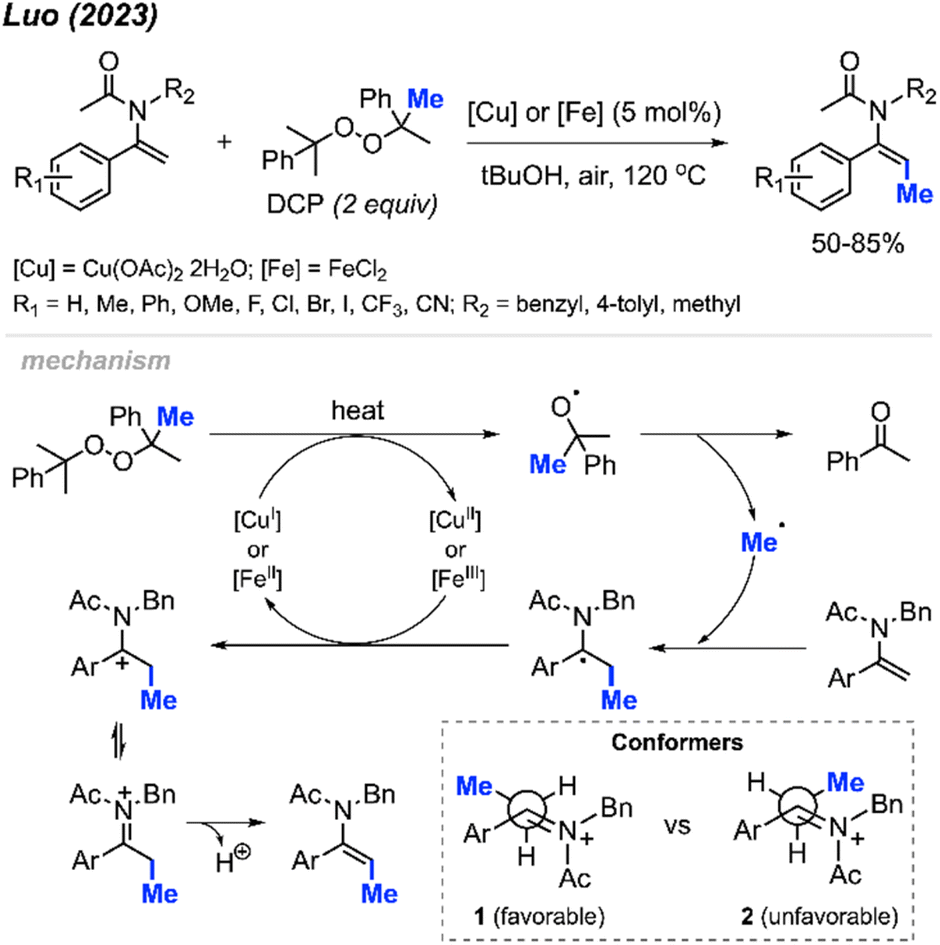 | ||
| Scheme 21 Copper- or iron-catalysed methylation of enamides using DCP. | ||
The authors proposed a mechanism to explain the exclusive E-isomer product formation (Scheme 21). As with previous mechanisms, homolytic cleavage of DCP followed by β-scission generates a methyl radical and acetophenone by-product. Regioselective addition of the methyl radical at the β-alkene position is favoured by the more stable radical intermediate formed. Either Cu(II) or Fe(II) then oxidises the radical to form a cationic intermediate or iminium ion. A look at the possible conformers shows that conformer 1 is sterically more favourable than conformer 2. Finally, deprotonation of conformer 1 generates the E-configured enamide product.
In 2014, Yu and co-workers reported that tert-butyl peroxybenzoate (TBPB) could promote α-methylation of β-keto ester compounds (Scheme 22).38 They found that Cu(BF4)2 (5 mol%) with TBPB (2 equiv.) in PhCl as a solvent were the best conditions for methylation of 18 examples in 38–78% yields. Notably, α-methylation of dimedone was achieved at 49% isolated yield using this method, which hints at the versatility of this method.
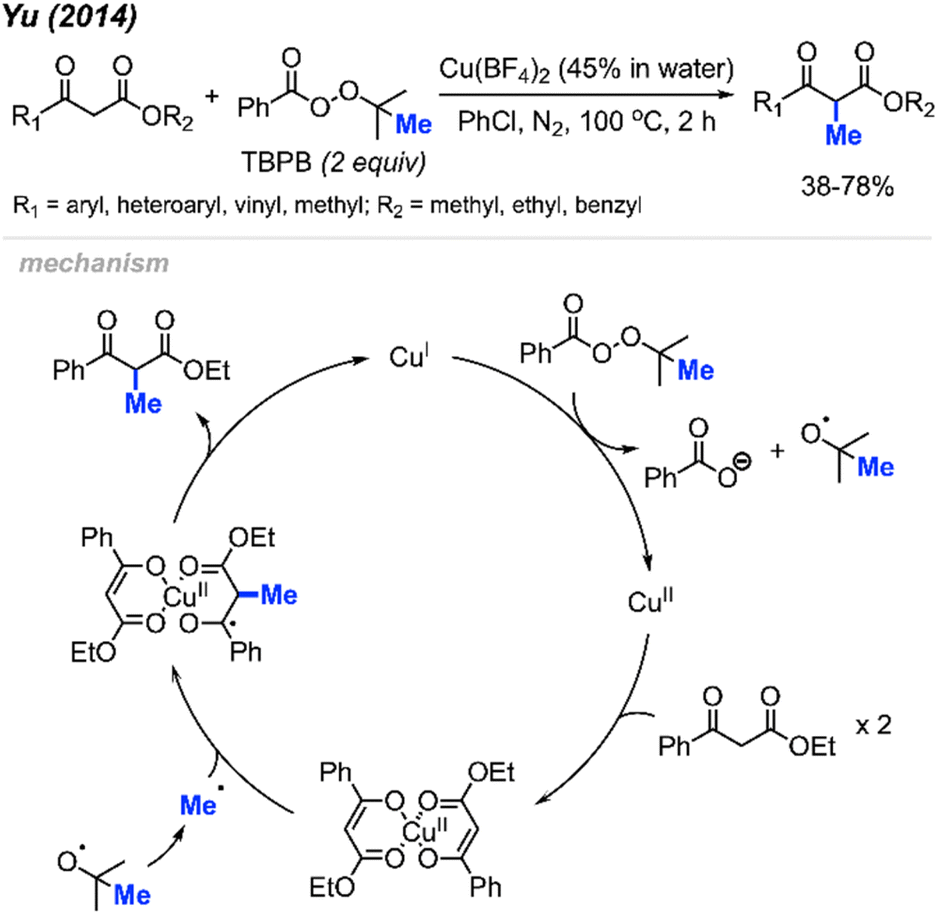 | ||
| Scheme 22 Copper-catalysed methylation of β-keto esters using TBPB. | ||
The authors proposed the mechanism shown in Scheme 22. At 100 °C, TBPB undergoes homolytic cleavage followed by β-scission to form the active methyl radical and one equivalent of acetone. Concurrently, the 1,3-dicarbonyl compound coordinates with Cu(II) to produce the copper-enolate intermediate. This intermediate reacts with the methyl radical to form the α-methylated 1,3-dicarbonyl product and Cu(I), which oxidises to regenerate Cu(II) by TBPB.
In 2017, Zou and co-workers found that CuCl (10 mol%) with TBPD (3 equiv.) in HOAc as a solvent can methylate 1,3-diketone compounds at the methylene position when heated to 120 °C (Scheme 23).39 The authors reported 32 product examples with yields ranging from 29–90%. Under these reaction conditions, methylation of dimedone was achieved in 68% yield, much better than the method in Scheme 22.
 | ||
| Scheme 23 Copper-catalysed methylation of 1,3-diketones using TBPB. | ||
Aromatic and aliphatic substituted as well as cyclic 1,3-diketones all reacted with TBPD to give moderate to good yields. The authors found that electronic effects are important in the reaction: 1,3-diketones with electron-withdrawing halogens and nitro groups on the phenyl rings gave lower yields, while diketones with electron-donating groups such as methoxy on the phenyl ring achieved higher yields. Various heteroaryl (pyridine, quinoline, pyrrole, thiophene, and furan) substituted 1,3-diketone also generated moderate to good yields. The aromatic-aliphatic asymmetric diketones also worked very well, however, ethyl benzoylacetate was not reactive under these conditions, which contrasts with the previously reported method by Yu et al. Notably, the pyridine and quinoline substituted diketones afforded demethylation as minor by-products. In the case of pyrrole and furan, N-methylation of pyrrole and 5-methylation of furan in 19% and 29% yields respectively were obtained. The reaction was also successfully scaled up to 50 grams, yielding 60% isolated product (Scheme 23).
In 2021, Stahl and colleagues introduced a single-step process for methylating C(sp3)–H bonds employing nickel photocatalysis under visible light conditions (Scheme 24).40 Employing either NiCl2(DME)[tButpy] or NiCl2(DME)[TPA] (4 mol%) and an Ir–F photocatalyst (1 mol%), they devised four sets of conditions for methylating different substrate classes. These conditions involved using either DTBP or DCP (6 equiv.) as the peroxide methyl source while varying the acid additives (0.5 equiv.) and solvents. The authors obtained 30 substrates in low to moderate yields ranging from 28–61%, including 5 instances of late-stage functionalisation. The reaction generally lacked enantioselectivity and, in certain cases, exhibited less site-selectivity, resulting in a mixture of diastereomeric and regioisomeric ratios. The remaining mass balance primarily comprised incomplete conversion as well as di- and multi-methylation. Depending on the set of conditions, several reactive functional groups were tolerated, including aryl and alkyl chlorides, aryl boronates and bromides, acidic O–H and N–H bonds, silyl-protected alcohols, quaternary alcohols, esters, benzisoxazole, lactam, ketones, primary sulfonamides, trifluoroacetamide, and carboxylic acid.
 | ||
| Scheme 24 Nickel-mediated C(sp3)–H methylation under various conditions. | ||
The authors further demonstrated that chemoselectivity can be controlled either by protonating or Boc-protecting basic amines with activated α-amino C(sp3)–H sites in the presence of competing benzylic reactive sites. Under innate selectivity, α-N-Boc C(sp3)–H sites preferentially undergo methylation in the presence of benzylic hydrogens. However, under modified conditions with a Brønsted acid present, the unprotected basic amine will protonate, thereby deactivating the α-amino C(sp3)–H sites. This causes a shift in chemoselectivity that favours benzylic C(sp3)–H methylation. Several examples were provided, yielding products in low to moderate yields ranging from 21–51%, including compounds such as safinamide, trimethoxybenzamide, cinacalcet, and homo-phenylalanine.
In the proposed mechanism (Scheme 24), the peroxide serves a dual role: it acts both as a source of the methyl radical through β-scission and facilitates hydrogen atom transfer (HAT) to generate the carbon radical necessary for cross-coupling. Initially, the methyl radical produced via β-scission coordinates with a Ni(I) species, forming a Ni(II)–Me intermediate. Subsequently, this methyl radical on the Ni(II) species reacts with the carbon radical, generated through HAT, to produce the desired product and regenerate the Ni(I) catalyst. Interestingly, despite the slightly acidic conditions, Minisci-type methyl addition to pyridine-containing substrates did not compete with C(sp3)–H methylation. By tuning specific reaction parameters tailored to different substrate classes, the authors demonstrated the potential of achieving a balance between the rates of hydrogen atom transfer (HAT) and methyl-radical formation.
In 2023, Gong and co-workers reported the Fe(III)-catalysed aliphatic C(sp3)–H methylation of glycine derivatives and peptides (Scheme 25).41 In the presence of DTBP, various N-aryl glycine esters and N-aryl glycine amides including some modified peptides can be selectively methylated, and 40–88% yields were achieved for 50 examples.
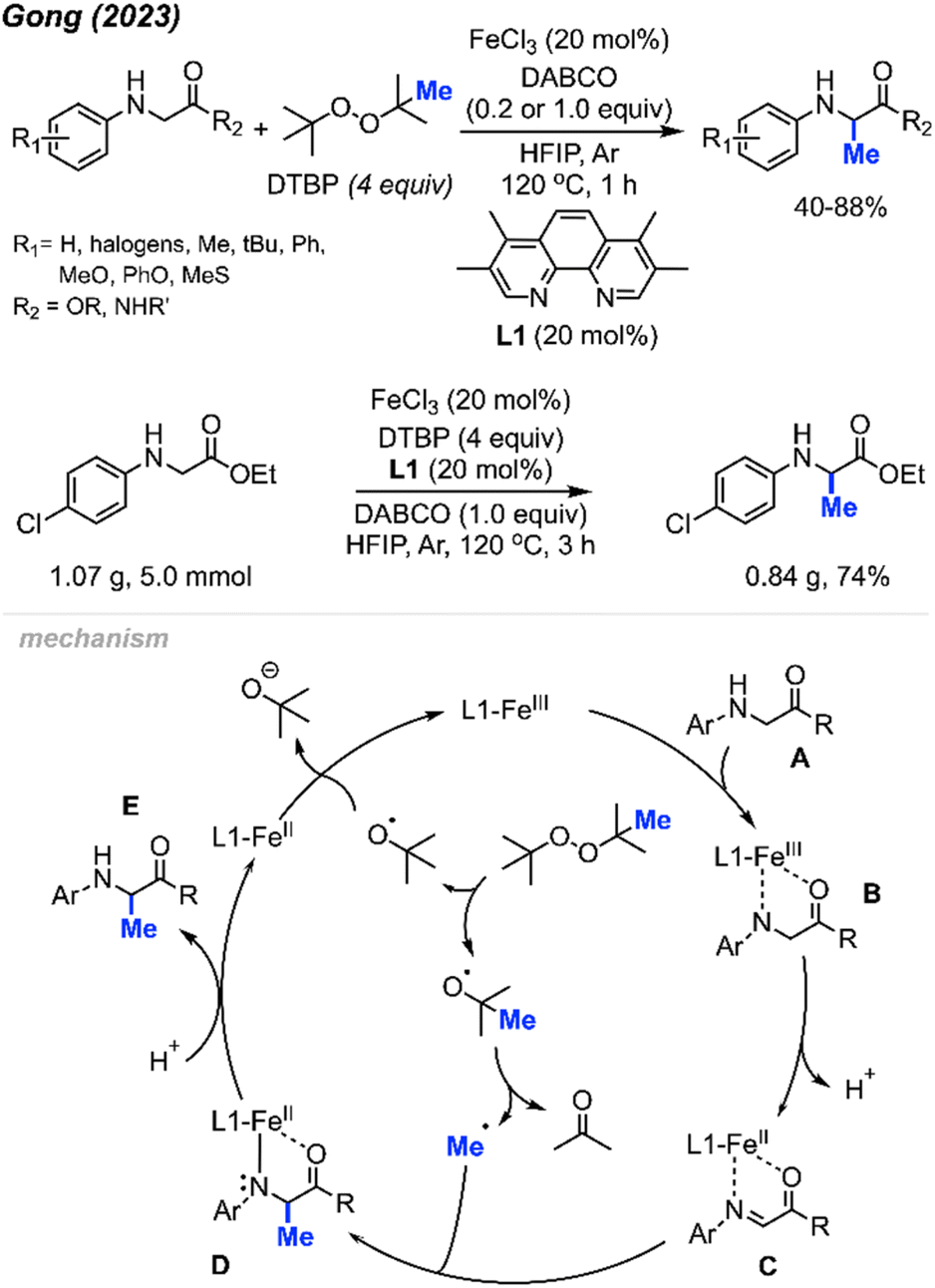 | ||
| Scheme 25 Iron-catalysed methylation of glycine and peptide derivatives. Scale-up of iron-catalysed methylation of glycine derivatives. | ||
Using HFIP as the solvent drastically decreased the reaction time to 1 hour. The authors also examined the effects of N-substituents on glycine derivatives, and mono N-aryl substitution was found to be necessary. Several mono- and di-substituted glycine derivatives including N-Ph2, N-PhMe, N-HBoc, N-HFmoc, N-HCbz, and N-HMe did not work for this transformation. Showcasing the peroxide-mediated methylation of biologically relevant targets remains underexplored and has a wide application potential within pharmaceutical chemistry. To demonstrate the synthetic utility, the authors performed a gram scale reaction that yielded 74% isolated product within 3 hours under standard conditions (Scheme 25).
The proposed mechanism of this reaction involved an Fe(III)/Fe(II) catalytic cycle (Scheme 25). Firstly, N-aryl glycine substrate A undergoes ligand exchange with FeCl3 to form intermediate B. Subsequent intramolecular electron transfer and further deprotonation generates Fe(II)–imine complex C. Concurrently, DTBP undergoes thermal homolysis to release the active tert-butoxy radical, which generates the methyl radical through β-scission. The methyl radical reacts with species C to form a coordinated complex D, which is protonated to produce the final product E. The released Fe(II) intermediate is then oxidised by DTBP to regenerate the Fe(III) catalyst.
2.2 Methylation of carboxylic acids
Methyl esters can be synthesised using various methods, including the Fisher esterification, the Steglich esterification, direct reaction of an acyl chloride with alcohols, and electrophilic methylation of carboxylic acid under basic conditions. Direct methylation of carboxylic acids by peroxides provides another option to obtain methyl ester derivatives. Until now, only two papers have been published regarding methyl esterification of carboxylic acids via peroxides.In 2013, Mao and co-workers reported that benzoic acid derivatives can be used to synthesise their corresponding benzoyl methyl esters via copper-catalysed cleavage of tert-butyl hydroperoxide (TBHP),42 providing 26 example reactions (Scheme 26). Benzoic acids with electron-withdrawing (nitro, cyano, benzoyl) or electron-donating groups (methoxy, methyl, tert-butyl) on the phenyl ring were all effective. Carboxylic acids on heterocyclic compounds such as indazole, thiophene, and furan were well tolerated, generating moderate to good yields.
 | ||
| Scheme 26 Copper-catalysed methyl esterification of carboxylic acids. | ||
In the same year, Chen and co-workers reported another methylation of carboxylic acids using peroxide catalysed by CuCl (Scheme 27).43 Both DCP and DTBP are good methylation reagents in the presence of CuCl (10 mol%), and 15 reaction examples were provided with 30–85% isolated yields. The authors found that alkyl acids gave higher yields than benzoic acids. Electron-withdrawn group at the para-position in benzoic acid strongly decreased the yields.
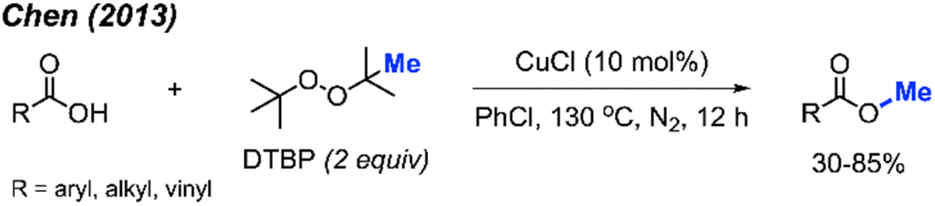 | ||
| Scheme 27 Copper-catalysed methyl esterification of carboxylic acids using DTBP. | ||
2.3 S-Methylation of sulfur containing compounds
Aryl methyl sulfides are a prevalent structural motif in bioactive natural products and therapeutic agents, while also serving as versatile synthetic intermediates for sulfoxides, sulfones, thiols and arenes. The synthesis of aryl methyl sulfides typically involves a coupling reaction between aryl halides and dimethyldisulfide, the reduction of methyl sulfoxides, or heteroatom-facilitated lithiation of aromatic carbon–hydrogen bonds followed by electrophilic substitution with dimethyldisulfide.In 2018, Wu and co-workers reported a simple synthesis of aryl methyl sulfides using peroxides (Scheme 28).44 This reaction was run in neutral conditions, in absence of metal catalysts. Various functional groups (alkyl, halogen, methoxy, nitro, and heteroaryl) were tolerated in moderate to good yields. This represents a facile and environmentally friendly alternative to the traditional synthesis of aryl methyl sulfides.
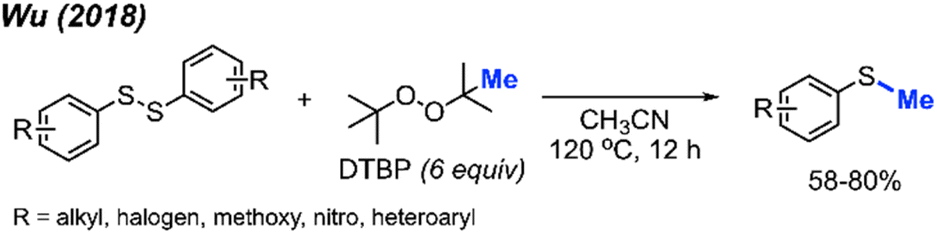 | ||
| Scheme 28 Metal-free sulfur methylation of diaryl disulfides with TDBP. | ||
In 2018, Yuan and co-workers demonstrated a new method for synthesizing aryl methyl sulfones using sodium sulfinates and DTBP in water (Scheme 29).45 Under this metal-free system, products were obtained in 61–93% yields with good functional group tolerance, including halogens, ketones, thiophene, alkyl and methoxy groups. The authors suggest that the DTBP plays a dual role, first as an oxidant to generate the sulfinate radical and secondly to form the methyl radical that can react with the sulfinate radical.
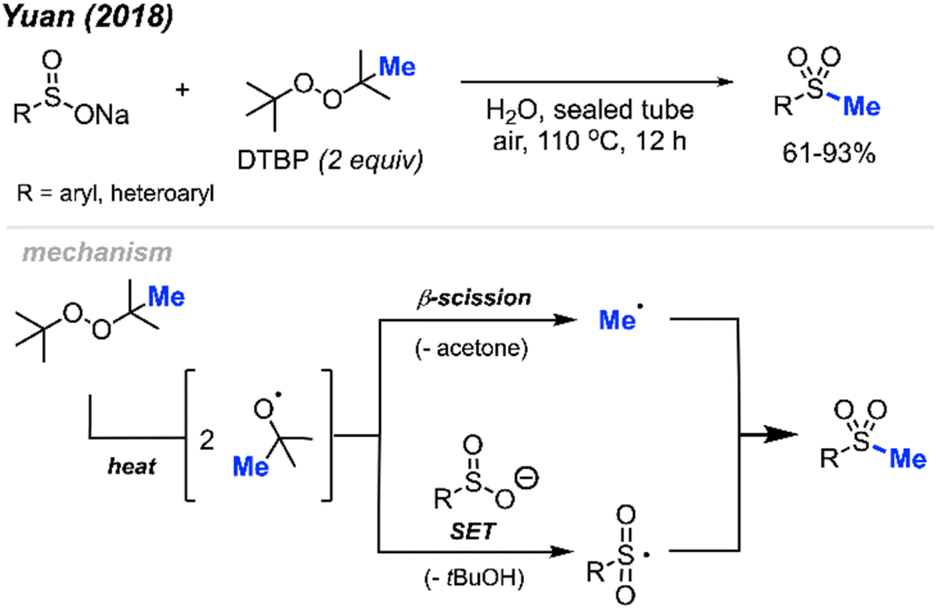 | ||
| Scheme 29 Metal-free sulfur methylation of diaryl disulfides with TDBP. | ||
In 2019, Wu and co-workers reported the direct C(sp2)–H methylsulfonylation of styrene derivatives using 1.0 equivalent of FeCl3 and 50 mol% quinoline under nitrogen at 130 °C (Scheme 30).46 DTBP was used as the methyl source and sodium metabisulfite (2.0 equiv.) was used to introduce the sulfone component. The product yields ranged from 61–93%, with functional group tolerance including halogens, aliphatic, and trifluoromethyl groups and one example with a vinyl thiophene methylsulfonylation. In the case of allylbenzene, prop-1-en-2-ylbenzene, allylbenzene and chalcone, no product was observed. This suggests that both the steric hindrance and electronics play a key role in this transformation.
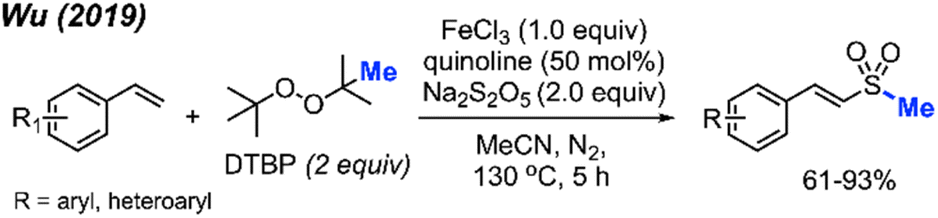 | ||
| Scheme 30 Iron-promoted C(sp2)–H methylsulfonylation of alkenes using DTBP. | ||
2.4 Methylation of N–H bonds
The investigations of N–H bond methylation by peroxides are focused on amides (sulfonamides and methanamides), phosphonamides, phosphinamides, sulfoximines, and tetrazole compounds. Amides are widely present in nature and pharmaceutical compounds. The synthesis of amides commonly involves nucleophilic substitution of primary amides with alkyl halides under basic conditions, the reaction of acyl chlorides with amines, or a Steglich amide formation. Again, direct methylation of amide by peroxides provides another avenue for the amide synthesis.In 2013, Chen reported the N-methylation of various aromatic primary amides by DCP (2 equiv.) catalysed by CuCl (10 mol%) at 130 °C in PhCl as the solvent (Scheme 31).43 Both electron-donating groups (methyl, tert-butyl, methoxy) and electron-withdrawing groups (halogen, nitro, trifluoromethyl) on the phenyl ring worked well under these conditions. Heteroaryl amides (thiophene and furan) as well as cinnamamide were also successfully methylated. Secondary amides can be further N-methylated under the same conditions. Both aromatic amides and aliphatic amides afforded good yields (38–91%), including lactams.
 | ||
| Scheme 31 Copper-catalysed N-methylation of primary and secondary amides with DCP. | ||
In 2017, Cai reported a nickel-catalysed N-methylation of amides using peroxides (Scheme 32).19 Both amides and sulfonamides containing various substituents including methyl, methoxy, halogens, and nitro provided good yields under these reaction conditions. A total of 20 examples afforded 53–76% yields. Notably, N-methylation of maleimide, phthalimide, lactams, nicotinamide and indole were all successful. Lactams showed higher reactivity than linear alkyl amides.
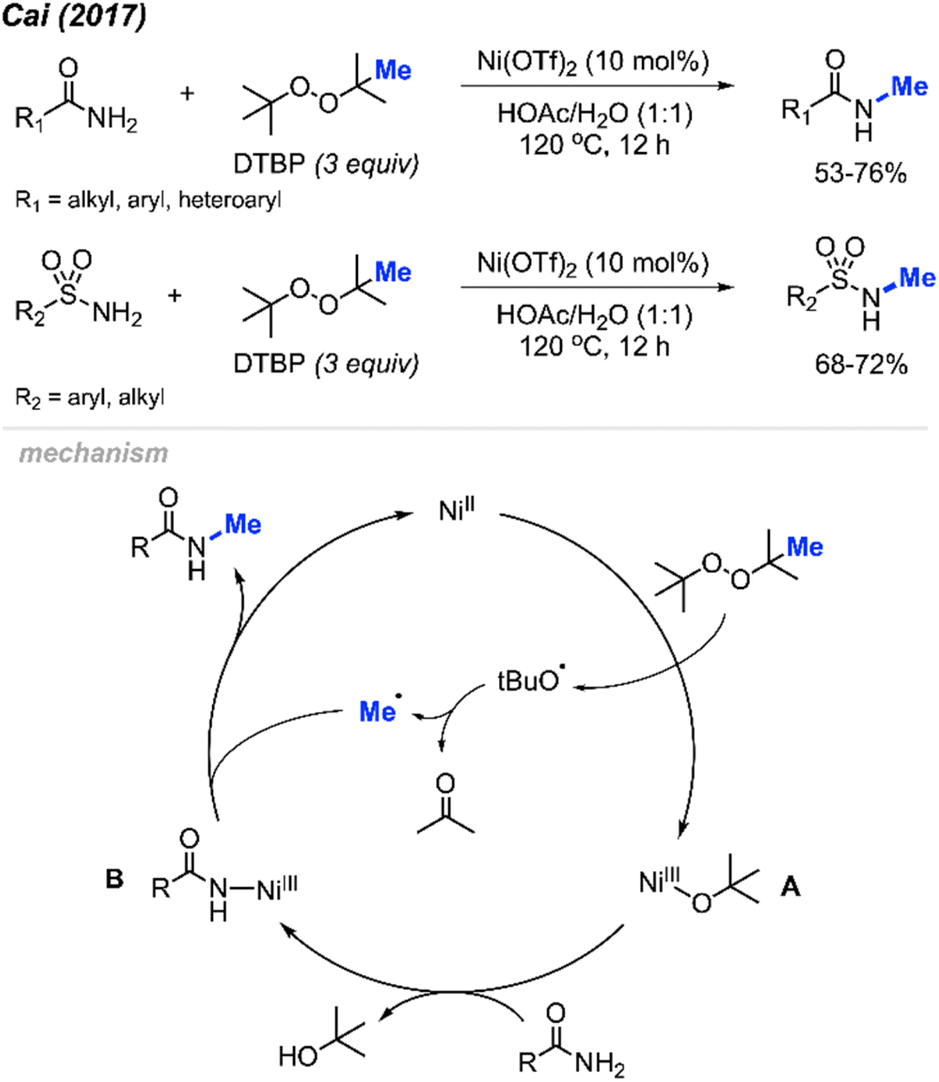 | ||
| Scheme 32 Nickel-catalysed N-methylation of amide and sulfonamides using DTBP. | ||
In 2021, Zhao and co-workers showed that methylation of sulfonamides can also be achieved with copper catalysis (Scheme 33).47 In this reaction, 10 mol% Cu(acac)2 is used in combination with DCP as the methyl source in acetone at 120 °C to obtain 21 product in yields ranging from 62–90%. Notably, the reaction demonstrated broad functional group tolerance, including halogens, methoxy, alkyl, trifluoromethyl, nitro, cyano, pyridine, quinoline, and thiophene. The authors also provided one example to show that the reaction is applicable to benzylamide.
 | ||
| Scheme 33 Copper-catalysed N-methylation of sulfonamides using DCP. | ||
Each of the N-methylation reactions undergo similar mechanisms (Scheme 32). Highlighted in Scheme 32 is the proposed mechanism from Cai and co-workers19. Initially, thermal homolysis of DTBP produces two alkoxy radicals, which then undergo β-scission to form the methyl radical and the corresponding ketone. Concurrently, DTBP oxidises Ni(II) to the Ni(III) alkoxide intermediate A. Metathesis of this intermediate with the amide substrate generates intermediate B and releases tBuOH. Intermediate B reacts with the methyl radical to form the N-methylated product while regenerating the Ni(II) catalyst.
Phosphonamides and phosphinamides are typical amino-fused organophosphorus compounds widely used in pharmaceutical and pesticide applications. An and co-workers first developed the N-methylation of phosphonamides and phosphinamides by peroxides via Cu-catalysis in 2017 (Scheme 34).48 DCP proved to be the best N-methylation reagent for these substrates, with 13 reaction examples presented. This is a novel and effective synthetic method toward N-methyl phosphonamides and phosphinamides.
 | ||
| Scheme 34 Copper-catalysed N-methylation of phosphonamides and phosphinamides. | ||
In 2015, Yu reported the N-methylation of sulfoximines by DTBP catalysed by Cu(OAc)2 in DMSO at 110 °C (Scheme 35).49 A total of 15 reaction examples were provided with 36–87% isolated yields. Various functional groups (methyl, halogen, phenyl, methoxy, acetyl, and ester) on the phenyl ring were tolerated during the reaction. S,S-Diarylsulfoximines gave higher yields than S-aryl-S-alkyl sulfoximines. This reaction was conducted under neutral conditions without the use of acids and bases.
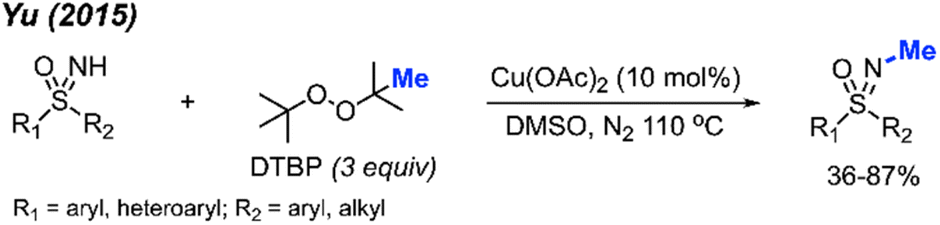 | ||
| Scheme 35 Copper-catalysed N-methylation of sulfoximines with DTBP. | ||
Tetrazole-based N-heterocyclic compounds are widely used in biological, pharmaceutical, and material sciences. Tetrazoles have been used in agricultural applications including ligands herbicides and fungicides. In 2020, Patel and co-workers developed a direct N-methylation method of aryl tetrazoles using peroxides catalysed by Bu4NI (Scheme 36).50 In this metal-free reaction, TBHP was found to be the best methylation reagent. The authors were also able to methylate 1-methyl-1H-benzo[d][1,2,3]triazole and 2-methylbenzo[d]isothiazol-3(2H)-one 1,1-dioxide in 42% and 68% respectively. Notably, the reaction worked well in the presence of ketones on the substrate; however, aldehydes tended to undergo side reactions with peroxide radicals. Overall, electron-withdrawing groups on the aryl ring afforded slightly higher yields than electron-donating groups.
 | ||
| Scheme 36 Metal-free N-methylation of aryl tetrazoles using TBHP. | ||
3 Methylation and other functionalisation
3.1 Methylative difunctionalisation of alkenes
In 2018, Zhu and co-workers were able to achieve methylative difunctionalisation of alkenyl C(sp2)–H bonds using copper catalysis (Scheme 37).51 Two sets of conditions were devised, one in which the solvent acts as the nucleophile (Scheme 37A) and the second in which the nucleophile is LiN3 (Scheme 37B). Both conditions afforded good overall yields, with similar functional group tolerance including methyl, methoxy, and halogens. In the case of the 1,2-azido methylation, the reaction also tolerated cyano, nitro, and trifluoromethoxy groups.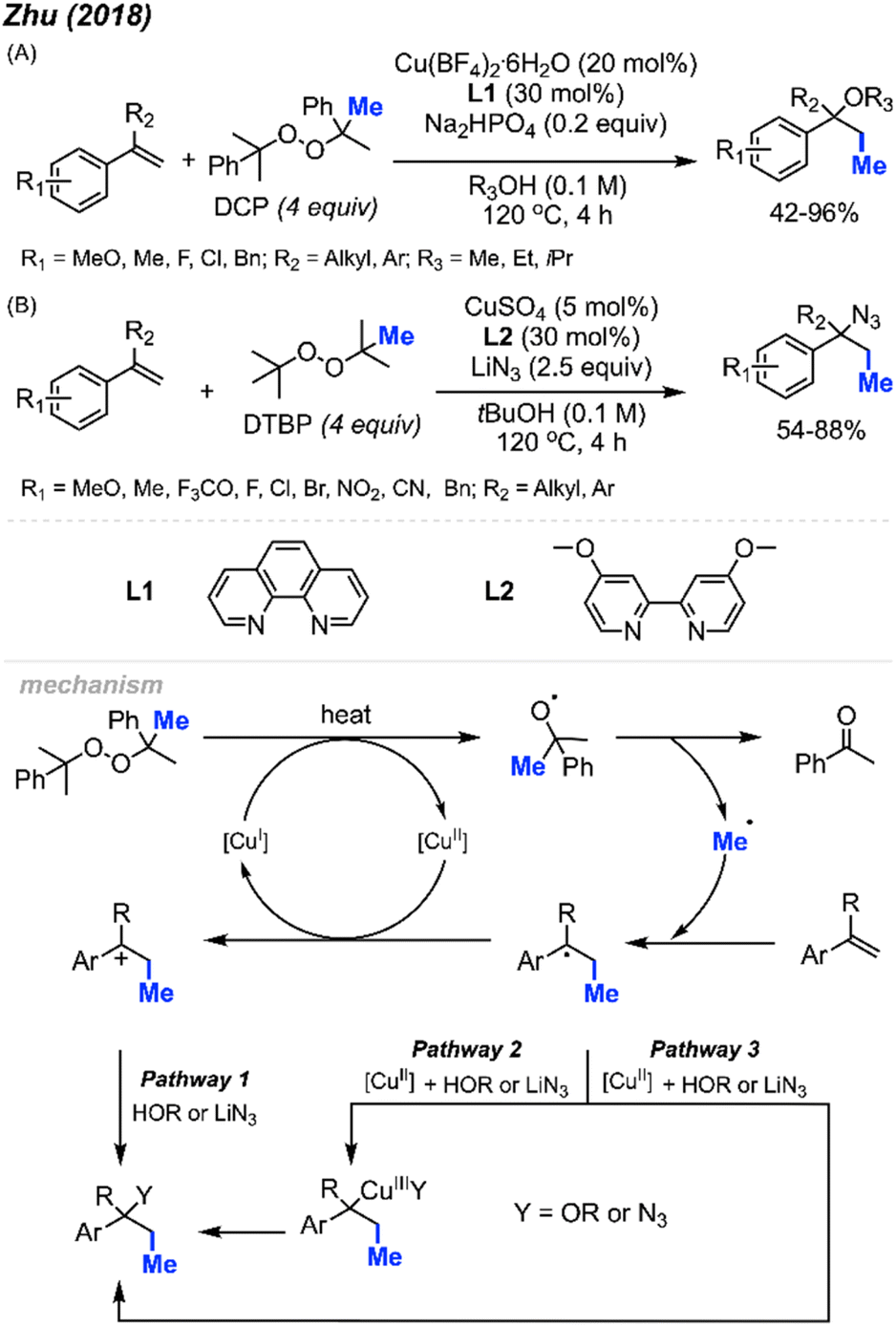 | ||
| Scheme 37 Copper-catalysed methylative difunctionalisation of alkenes. (A) Optimised reaction conditions using the solvent as the nucleophile, (B) optimised reaction conditions using LiN3 as the nucleophile. | ||
The mechanism (Scheme 37) is similar to that of Bao and co-coworkers.36 First, the homolytic cleavage of the peroxide is facilitated by Cu(I) and heat. Subsequent β-scission of the alkoxy radical generates the methyl radical, which reacts at the terminal end of the alkene to generate the more stable benzylic radical. The radical can then be oxidised by Cu(II) to form the carbocation that can undergo nucleophilic attack (Pathway 1). Alternatively, the nucleophilic addition to the benzylic radical intermediate can either be achieved via a radical rebound to generated a Cu(III) intermediate with the nucleophile and substrate which is then followed by reductive elimination (Pathway 2) or through a Cu-centred redox transfer process (Pathway 3).
More recently in 2023, Wu, Zhu, and co-workers were similarly able to achieve methylative difunctionalisation of alkenes using iron catalysis (Scheme 38).52 This was achieved with 20 mol% FeSO4 at an elevated temperature of 130 °C under nitrogen, using DTBP as a methyl source. In this case, the authors demonstrated the use of anisoles as nucleophiles for C–N bond formation, as well as pyrrole and indole nucleophiles that afforded C–C bond formation.
 | ||
| Scheme 38 Iron-catalysed intermolecular 1,2-difunctionalisation of alkenes using DTBP. | ||
The authors proposed the mechanism shown in Scheme 32. Initially, Fe(II) and heat promote the homolytic cleavage of DTBP, followed by β-scission to form tert-butoxide, acetone and the methyl radical. Upon addition of the methyl radical to the terminal end of the alkene, two possible pathways are proposed. Both pathways lead to an Fe(IV) intermediate containing the substrate and nucleophile, where subsequent reductive elimination afford the final product and regenerates Fe(II).
3.2 Decarboxylative methylation
In 2014, Mao and co-workers discovered the peroxide mediated decarbonylative methylation of cinnamic acid derivatives (Scheme 39).53 The reaction was achieved using FeCl3 catalyst and DTBP, where 12 reaction examples provided 20–53% yields with retained double bond configuration. Notably, β-substituted substrates such as β-phenyl cinnamic acid were tolerated.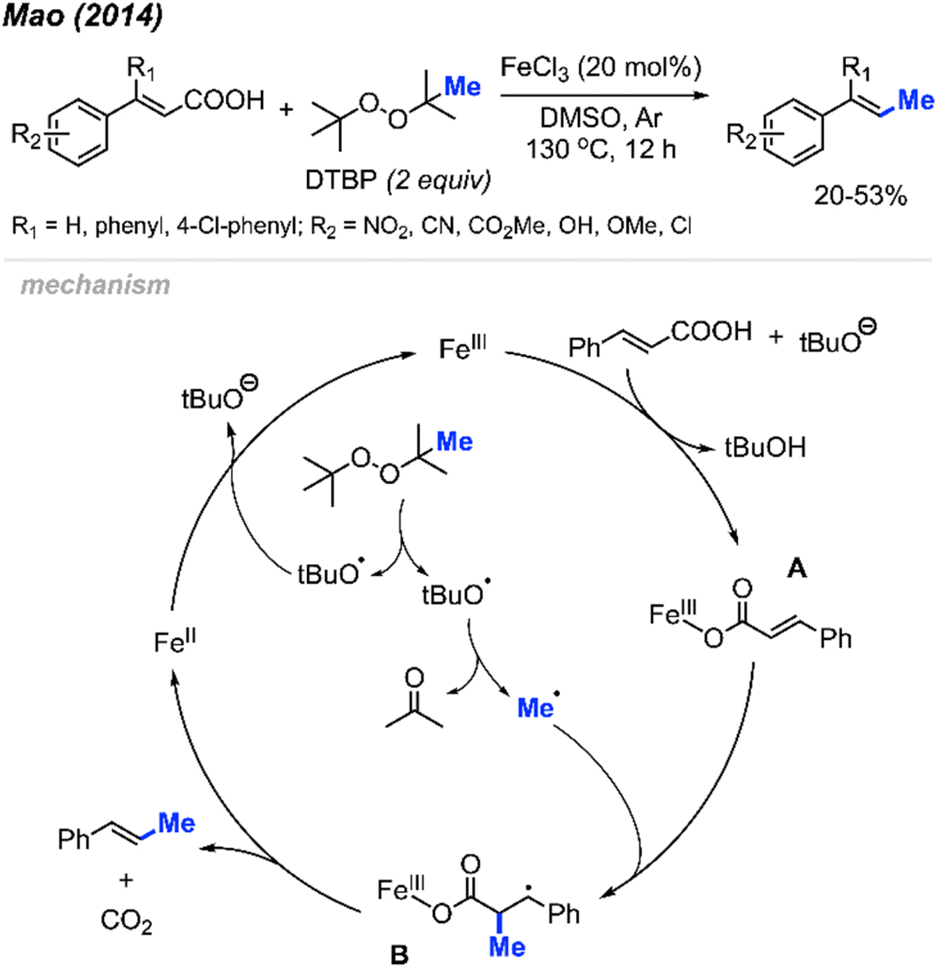 | ||
| Scheme 39 Iron-catalysed decarboxylative methylation of cinnamic acids. | ||
Scheme 39 illustrates the proposed mechanism. The catalytic cycle initiates with the formation of a methyl radical released from a tert-butoxy radical. This radical subsequently adds to the α-position of the double bond on the ferric cinnamate intermediate A, formed through the reaction between cinnamic acid and ferric chloride, yielding intermediate B. Elimination of CO2 and Fe(II) from intermediate B results in the final product. The regeneration of Fe(III) occurs as Fe(II) undergoes oxidation by DTBP.
3.3 Methylation and desulfonylation
In 2016, Li and co-workers reported a peroxide-promoted methylation/1,4-aryl migration/desulfonylation cascades of N-(arylsulfonyl)acrylamides, resulting in the formation of 2,2-disubstituted-N-arylbutanamides without the need for any metal catalyst (Scheme 40).54 The study presented 21 example reactions, showcasing yields ranging from 30–75%. Various peroxides, including DTBP, DCP, TBHP, and TBPB, exhibited reactivity in these conditions, with DTBP emerging as the most effective methylation reagent. The degree of reactivity was observed to vary with the position of the N-aryl R2 substituent, following the order: para > meta > ortho. This research introduces a facile and straightforward synthetic route for 2,2-disubstituted-N-arylbutanamides featuring an all-carbon quaternary stereocenter. | ||
| Scheme 40 Oxidative 1,2-arylmethylation cascades of N-(arylsulfonyl)acryl-amides with DTBP. | ||
The authors proposed a possible mechanism outlined in Scheme 40. DTBP undergoes facile decomposition, yielding a methyl radical and acetone. Addition of the methyl radical across the alkene of the N-(arylsulfonyl)acrylamide A starting material produces the radical intermediate B. An intramolecular 5-ipso-cyclisation of intermediate B generates intermediate C, which subsequently undergoes desulfonylation to transform into amidyl radical species D. Finally, hydrogen abstraction by intermediate D leads to the formation of the desired product E.
In the same year, Yu and co-workers reported a similar investigation, finding that Fe2(SO4)3 could serve as a catalyst for the arylmethylation of sulfonyl acrylamides in the presence of DTBP (Scheme 41A).55 A total of 22 reaction examples were presented, achieving up to 90% yields. Notably, the authors observed a substantial substituent effect on the terminal alkene of the starting material. When a single methyl substituent was present at the terminal alkene, the reaction yield decreased to 41%. Interestingly, employing a di-methylated terminal alkene resulted in no observable reaction (Scheme 41B).
 | ||
| Scheme 41 Iron-catalysed oxidative 1,2-arylmethylation cascades of N-(arylsulfonyl)acrylamides with DTBP. (A) Optimised reaction conditions, (B) product not observed when using di-methylated terminal alkenes. | ||
3.4 Oxidation and methylation
In 2013, Mao and co-workers reported that benzyl alcohols and aldehydes can be oxidised and subsequently O-methylated in a one-pot synthesis of the corresponding benzoyl methyl esters using TBHP when catalysed by Cu(II)-quinolate (Scheme 42).42 For these two reactions, both electron-withdrawing groups and electron-donating groups on the phenyl rings provided good yields. For the benzyl alcohol reactions, K3PO4 (2 equiv.) was used as base to assist the oxidation process. Tetrabutylammonium iodide (TBAI) was used as an additive since it was observed to accelerate the methyl esterification process.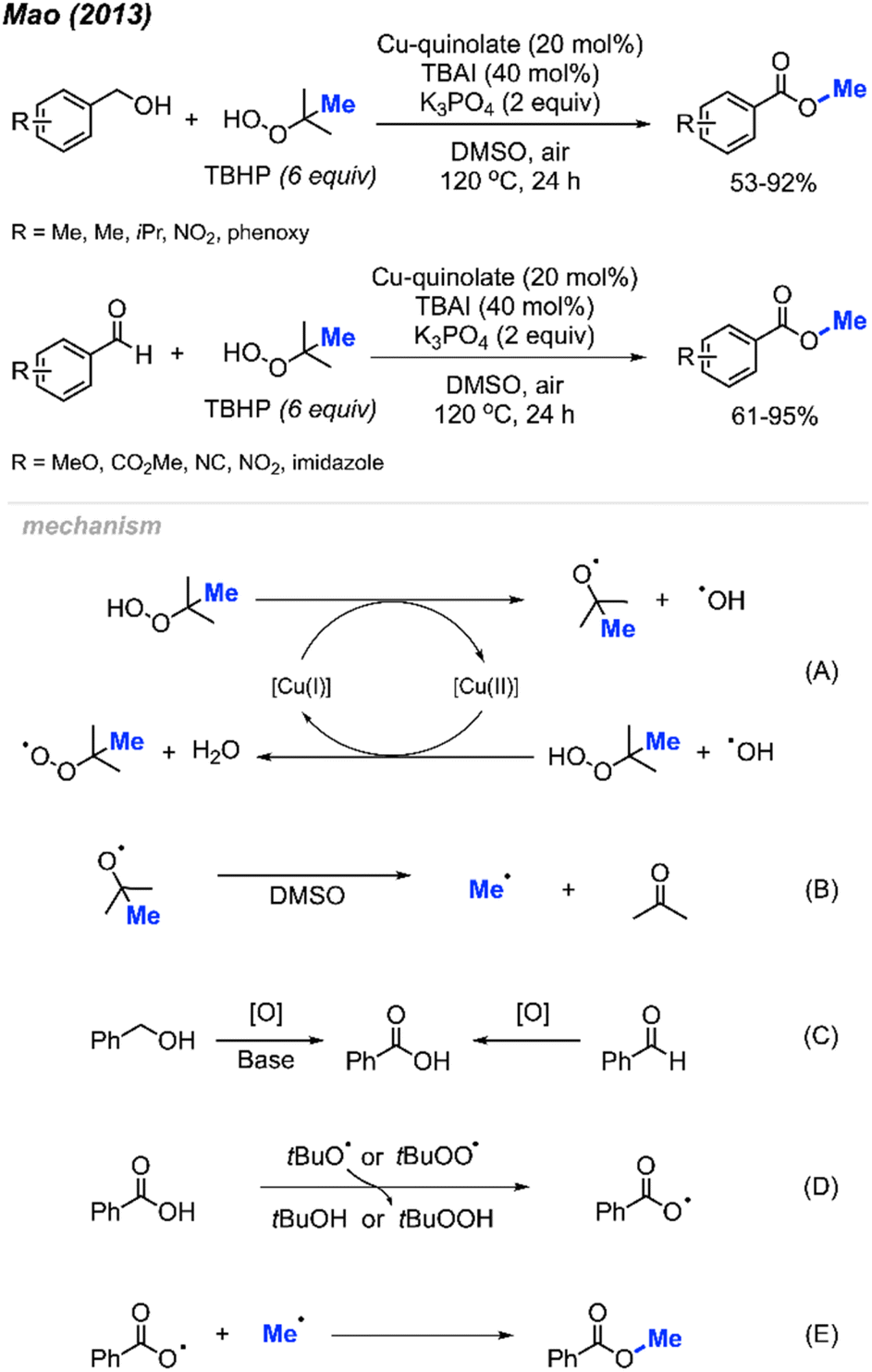 | ||
| Scheme 42 Copper-catalysed methyl esterification of benzyl alcohols and benzaldehydes. | ||
In 2014, Li and co-workers demonstrated the copper-catalysed methyl esterification of benzaldehydes and benzoic alcohols under similar conditions (Scheme 43).56 In this case, a lower catalyst loading of 10 mol% CuF2 was used with a one to one mixture of DMSO and water as the solvent. Reaction yields ranged from 44–90%, with functional group tolerance including halogens, cyano, methoxy, nitro, methyl, and hydroxyl groups. Notably, thiophene-2-carbaldehyde was successfully esterified at 90% yield.
 | ||
| Scheme 43 Copper-catalysed methyl esterification of benzyl alcohols and benzaldehydes. | ||
Both sets of authors proposed similar reaction mechanisms, as shown in Scheme 42. First, cleavage of TBHP to produce a tert-butoxy radical and hydroxide anion is facilitated by the coupled oxidation of Cu(I) to Cu(II). The tert-butoxy radical further decomposes to yield the methyl radical and acetone. The Cu(II) subsequently facilitates the conversion of another TBHP and a hydroxide anion into a tert-butylperoxy radical and water, thereby reducing back to Cu(I). Concurrently, the benzylic alcohol or aryl aldehyde is oxidised by the peroxide to the corresponding carboxylic acid with the help of base for the former. Hydrogen atom abstraction of the resulting carboxylic acid by the tert-butoxy or tert-butylperoxy radical then generates the acyloxy radical. Finally, the acyloxy radical reacts with the methyl radical to form the desired methyl ester.
In 2018, Jia and co-workers reported the peroxide facilitated methylation and oxidation of aromatic isocyanides to produce N-arylacetamide compounds (Scheme 44).57 This was achieved in the presence of FeCp2 (10 mol%) catalyst and DTBP, affording moderate to good yields (31–81%). Aromatic isocyanides containing various functional groups were well tolerated; however, aliphatic isocyanides such as tert-butyl, cyclohexyl, n-butyl, and benzyl isocyanides were not successful. As shown in Scheme 44, the authors propose a mechanism in which metal-assisted cleavage of DTBP generates a methyl radical. This radical then reacts with the isocyanide to form intermediate A, which reacts with oxygen to produce intermediate B. Homolytic cleavage of the peroxide then forms intermediate C, which undergoes hydrogen abstraction to yield the final product.
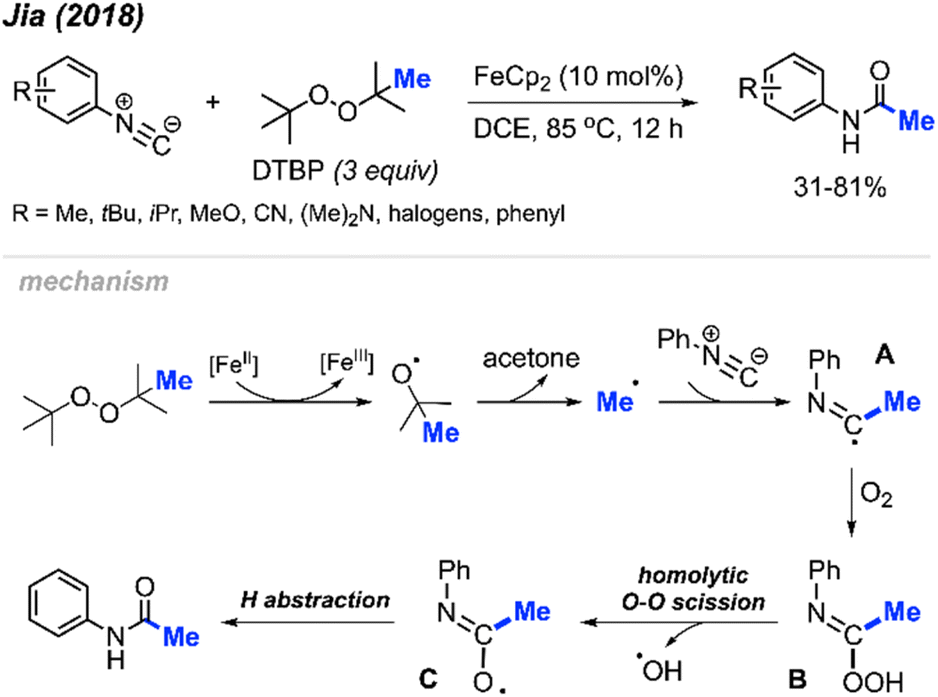 | ||
| Scheme 44 Iron-catalysed methylation and oxidation of aryl isocyanides. | ||
3.5 Methylation and cyclisation
Initially, Cheng and co-workers observed that FeCl2 facilitates the carbomethylation of arylacrylamides using DTBP, resulting in the synthesis of 3-ethyl-3-substituted indolin-2-one in moderate to good yields (Scheme 45).58 The reaction exhibited tolerance towards various functional groups, such as cyano, nitro, ethoxy carbonyl, halogen, and trifluoromethyl groups. The researchers provided approximately 30 examples of successful reactions.
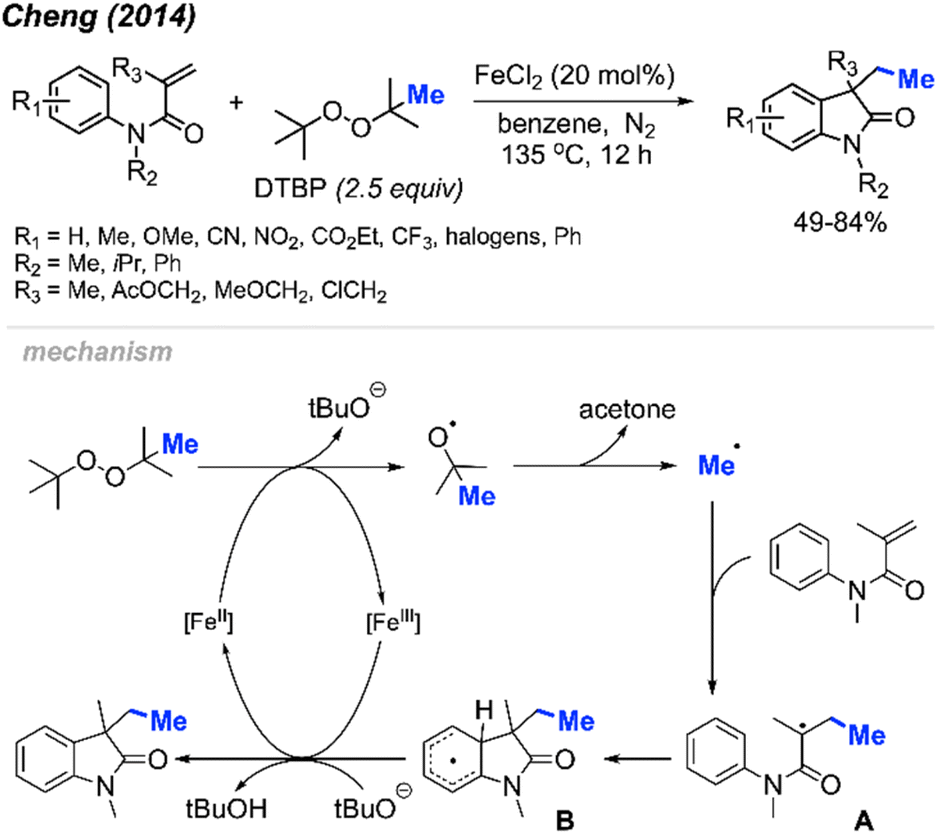 | ||
| Scheme 45 Iron-catalysed sequential difunctionalisation of alkenes with DTBP. | ||
The reaction mechanism is outlined in Scheme 45. Initially, the reaction between DTBP with Fe(II) produces tert-butoxy radicals, leading to the generation of acetone, a tert-butoxy anion, and a methyl radical. The methyl radical then reacts with the starting material, resulting in the formation of intermediate A. Subsequently, intermediate A undergoes intramolecular cyclisation with the phenyl ring, yielding intermediate B. The Fe(III) then oxidises intermediate B to form an intermediate cation, where subsequent deprotonation by the tert-butoxy anion restores aromaticity to generate the final product.
Li and co-workers presented a study with analogous findings (Scheme 46).59 The authors screened various peroxides, including TBHP, DTBP, DCP, and cumyl hydroperoxide. Among these, DCP emerged as the most effective reagent for the methylation/cyclisation reaction. Various functional groups demonstrated good compatibility under the optimised conditions. The reactivity order of substituents on the phenyl ring was observed to be para > meta > ortho. The authors also determined that the R2 substituent on the nitrogen atom cannot be a hydrogen, since the free NH acrylamide exhibited no reactivity under these conditions. The study presented 16 examples, showcasing yields ranging from 46–66%.
 | ||
| Scheme 46 Sequential difunctionalisation of alkenes with DCP. | ||
In the same year, Liu and co-workers reported the third example for the methylation/cyclisation reaction promoted by DCP (Scheme 47).60 The authors opted for CuBr as the catalyst, and, notably, hydroxyl groups and halogens are resilient under these reaction conditions. A total of 12 examples were presented containing various functional groups, yielding 51–89% yields.
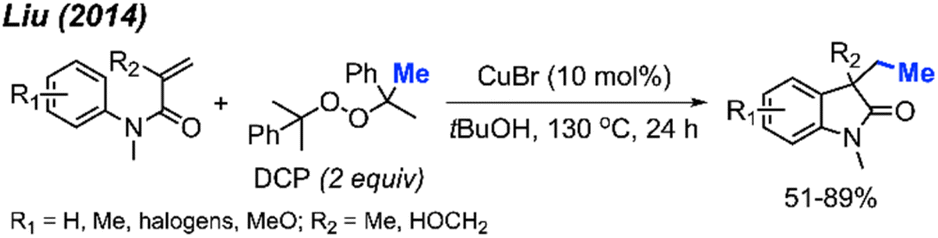 | ||
| Scheme 47 Sequential difunctionalisation of alkenes with DCP. | ||
In 2018, Yuan and co-workers showed that it was possible to achieve a metal-free methylative cyclisation of N-alkyl-N-methacryloylbenzamides to generate methylated isoquinoline-1,3-diones (Scheme 48).61 The reaction was performed in chlorobenzene at 120 °C, using DCP as the methyl source. The overall reaction yields ranged from 35–83%, with a total of 17 examples. Tolerance to halogens, trifluoromethyl, chloromethyl, methyl, and tert-butyl functional groups was observed. It was also observed that product formation was favoured with electron-donating groups and hindered in the presence of electron-withdrawing groups. In the case of p-methoxy substitution, a mixture of products was obtained that reflected reactions occurring at both the ortho- and meta-positions, likely due to increased reactivity of the benzene ring.
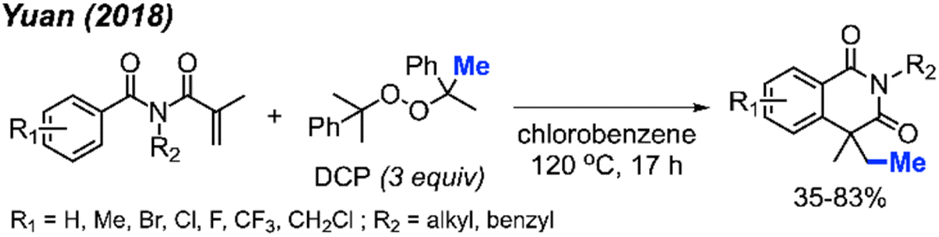 | ||
| Scheme 48 Metal-free sequential difunctionalisation of alkenes with DCP. | ||
In 2018, Zhu and co-workers utilised the difunctionalisation of styrenes to achieve methylative cycloetherification, lactonisation, and cycloamination (Scheme 49).51 In each case, the copper pre-catalyst and conditions were tuned to facilitate the specific cyclisation, with DTBP used as the methyl source in all cases. The product yields across the three cyclisation reactions ranged from 52–82%, with functional groups tolerance towards halogens, methoxy, methyl, iso-propyl, and cyano groups. Notably, a regioselective methylative lactonisation of (E)-4-phenylbut-3-enoic acid to afford the 4,5-trans-disubstituted γ-lactone as one diastereomer in a 41% yield was demonstrated. In this case, the mechanism follows that of the methylative difunctionalisation in Scheme 30, with the nucleophile directly on the starting substrate to facilitate cyclisation.
 | ||
| Scheme 49 Copper-catalysed methylative cycloetherification, lactonisation, and cycloamination using DTBP. (A) Optimised reaction conditions for methylative cycloetherification, (B) optimised reaction conditions for methylative lactonisation, (C) optimised reaction conditions for methylative cycloamination. | ||
In 2021, Pan and co-workers extended their exploration, introducing a novel peroxide-driven methylation/cyclisation method for the synthesis of benzimidazo[2,1-a]isoquinoline-6(5H)-ones. Starting from N-methyacryl-2-phenylbenzimidazoles and DTBP, this approach yielded the corresponding products in good yields, all achieved without the need for any metal catalyst (Scheme 50).62 The study showcased 30 reaction examples, demonstrating yields ranging from 28–84%. This research not only broadens the synthetic toolkit but also offers a new strategy for constructing benzimidazo-fused polycyclic scaffolds.
 | ||
| Scheme 50 Synthesis of benzimidazo[2,1-a]isoquinoline-6(5H)-ones promotred by DTBP. | ||
In 2019, Li and co-workers reported the cascade methylation and cyclisation of ortho-cyanoaryacrylamindes with DCP (Scheme 51).63 The reaction exhibited efficacy even in the absence of a transition metal catalyst, although higher yields were achieved in the presence of 5 mol% CuF2.
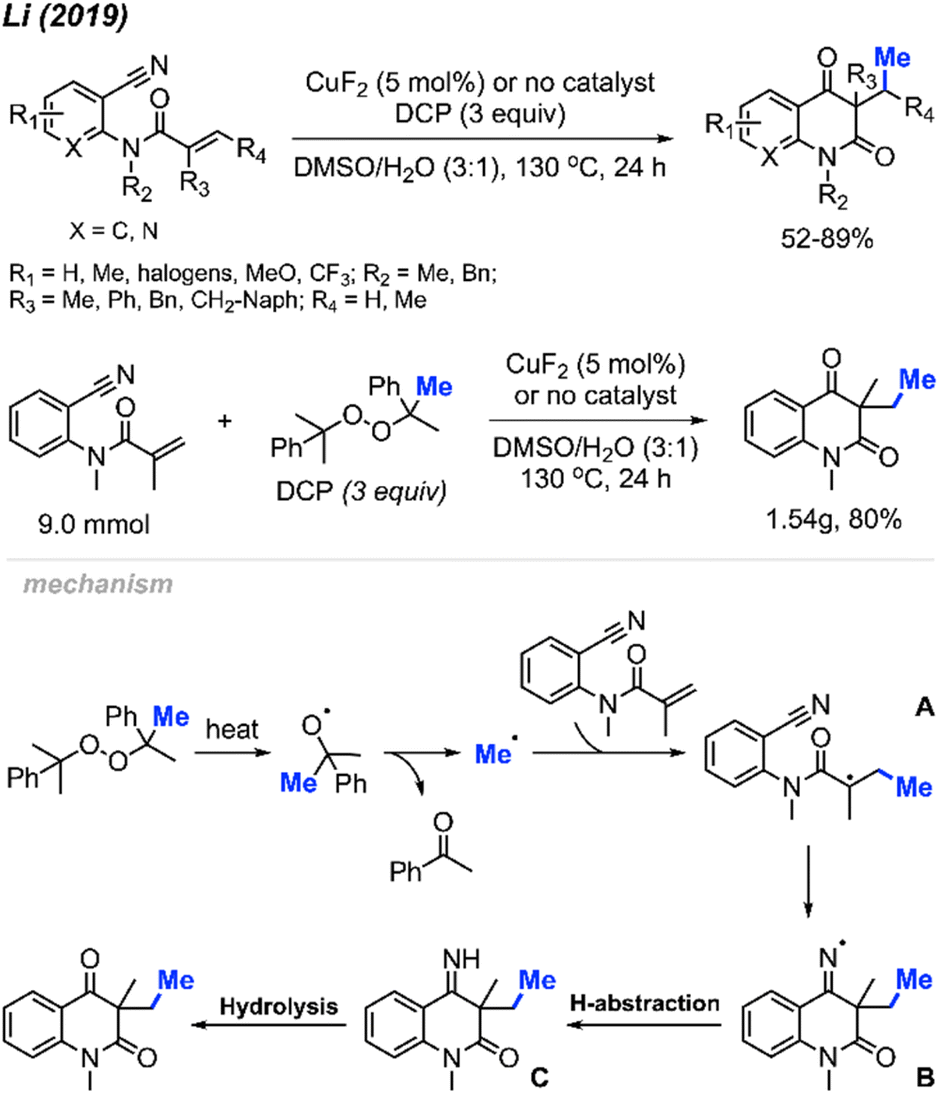 | ||
| Scheme 51 Copper-catalysed sequential difunctionalisation of ortho-cyanoarylacrylamides with DCP. | ||
This transformation provides a simple and straightforward method for synthesizing methylated quinoline-2,4(1H,3H)-dione compounds. Notably, a gram-scale reaction resulted in an 80% isolated yield, showcasing the application of the methylation/cyclisation procedure towards organic synthetic applications without sacrificing efficiency.
The reaction mechanism is illustrated in Scheme 51. Thermal homolytic cleavage of DCP generates the cumyloxyl radical, which then undergoes β-scission to yield the methyl radical and acetophenone. Subsequently, the methyl radical reacts with the acrylamide alkene to afford intermediate A. The radical intermediate A attacks the cyano group, leading to the generation of imine radical B. This imine radical then facilitates H-abstraction, resulting in the formation of imine C. Hydrolysis of imine C ultimately yields the final product. The presence of CuF2 was thought to either enhance the acrylamide alkene reactivity, or stabilise the radicals generated during the reaction.
Peroxides exhibit dual functionality by not only facilitating the methylation and cyclisation of N-arylacrylamides but also directing the methylation and intramolecular aromatisation of isonitriles. In 2014, Cheng and co-workers first reported the conversion of isonitrile into 6-methylphenanthridine derivatives utilizing DTBP and an Fe(II) catalyst (Scheme 52).64 The reaction was tolerant of various functional groups, including fluoro, chloro, acetyl, methoxycarbonyl, cyano, and trifluoromethyl groups. Interestingly, no clear substituent effect was observed to impact the reaction efficiency.
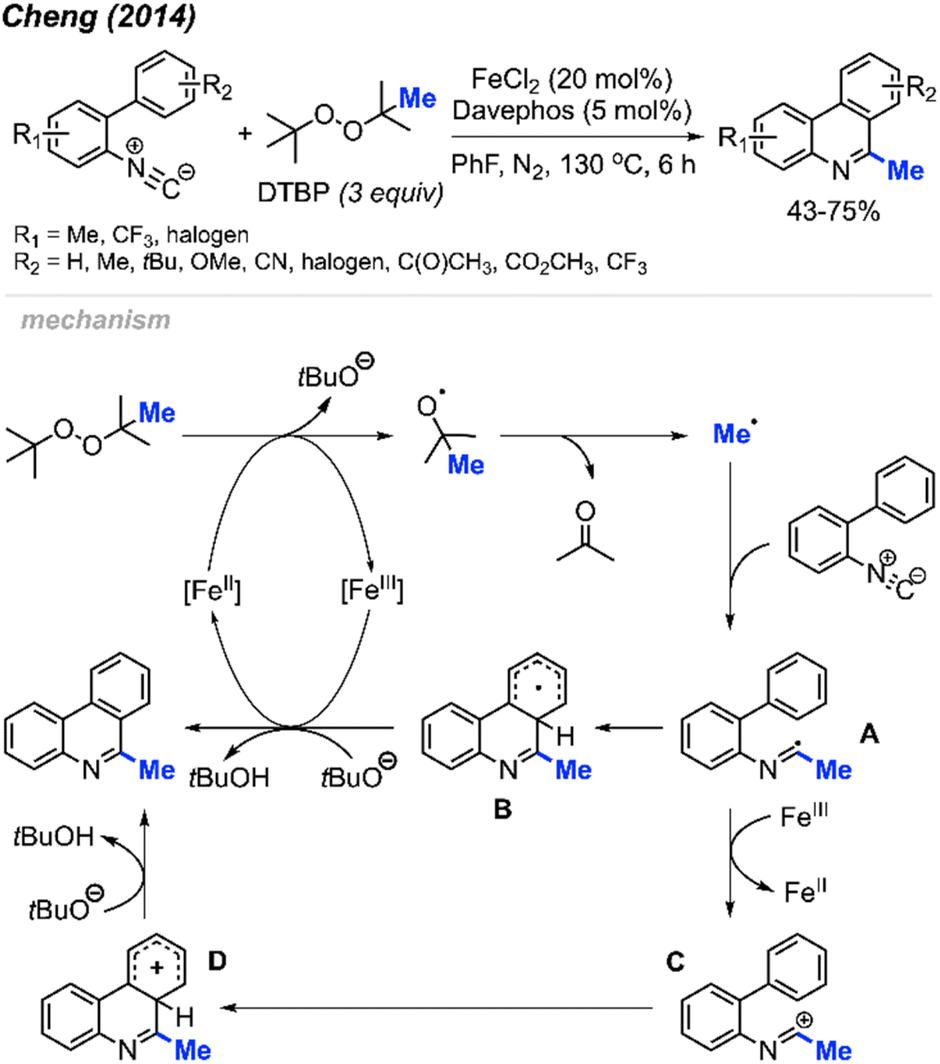 | ||
| Scheme 52 Iron-catalysed methylation and aromatisation of isonitrile promoted by DTBP. | ||
The proposed reaction mechanism is illustrated in Scheme 52. Initially, Fe(II)-assisted homolytic cleavage of DTBP generates the tert-butoxy radical, which then decomposes to the methyl radical and acetone. The methyl radical subsequently adds to the initial isonitrile substrate, forming intermediate A. Intramolecular cyclisation of intermediate A leads to intermediate B. Finally, Fe(III) oxidises intermediate B to form an intermediate cation, which subsequently get deprotonated by tert-butoxide to form the final product. An alternative pathway involves single electron transfer (SET) between Fe(III) and intermediate A, yielding cationic intermediate C. Subsequent aromatic electrophilic substitution (SEAr) produces intermediate D, which undergoes proton loss to form the final product.
Almost at the same time, Liu and co-workers reported a comparable study, revealing that DCP could promote the metal-free transformation of isocyanide into 6-methylphenanthridine compounds (Scheme 53).60 In this case, potassium fluoride was found to efficiently promote the reaction, although it was also functional in the absence of the KF promoter in lower yields.
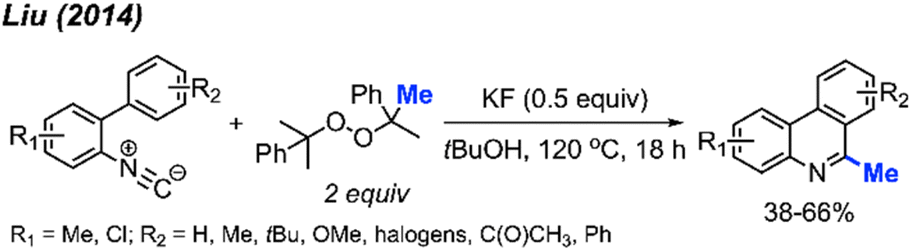 | ||
| Scheme 53 Methylation and aromatisation of isonitrile promoted by DCP. | ||
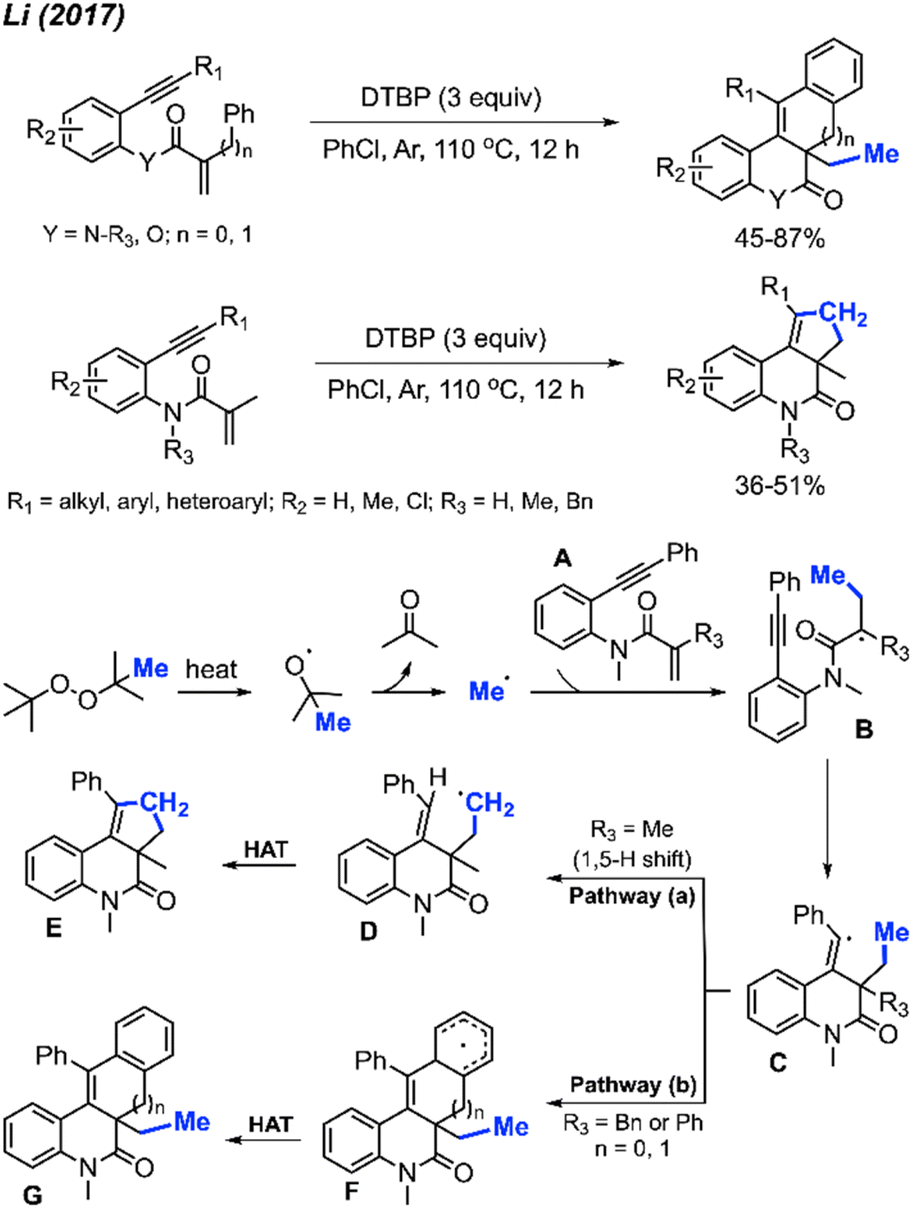 | ||
| Scheme 54 Metal-free annulation cascades of 1,7-enynes using DTBP. | ||
Several peroxides, including DTBP, TBHP, TBPH, and DCP, exhibited efficacy in this reaction, with DTBP identified as the optimal methylation reagent. Among the solvents tested, PhCl proved to be the most suitable. The proposed reaction mechanism is depicted in Scheme 54. Under heat, DTBP generates a methyl radical and acetone. The methyl radical adds to the alkene on the starting material A to produce intermediate B. Subsequently, the radical intermediate B undergoes intramolecular cyclisation, forming the vinyl radical intermediate C. The fate of intermediate C diverges based on the nature of the substituents on the starting material. Pathway (a) involves an intramolecular 1,5-hydrogen shift, yielding the new alkyl radical intermediate D when R3 is a methyl group. The process concludes with a hydrogen atom transfer (HAT) of intermediate D, facilitated by an oxidant, to produce the final product E. In contrast, Pathway (b) entails a direct intramolecular cyclisation with the aryl ring, leading to the aryl radical intermediate F when R3 is a benzyl or phenyl group. Intermediate F proceeds through HAT to produce final product G.
In 2018, Li and co-workers demonstrated another example of a metal-free bicyclisation to access methyl substituted polyheterocycles (Scheme 55).66 The authors provided 8 examples with yields ranging from 46–78%, including methoxy, fluoro and extended aryl groups. A variety of N-methyl groups were also tolerated, including methyl and benzyl groups, however, the secondary amide was not tolerated. When R1 was a meta-substituted methyl group, a mixture of two regioselective products was obtained. The reaction follows a similar mechanism to that show in Scheme 54.
 | ||
| Scheme 55 Metal-free radical bicyclisation of 2-cyanoaryl acrylamides with TBPB. | ||
In 2023, Li and co-workers extended the exploration of their metal-free ortho-cyanoaryl acrylamide radical bicyclisation reaction, introducing a new method towards benzo[h]naphtho[1,8-bc][1,6]naphthyridine scaffold synthesis (Scheme 56).67 A total of 22 example compounds were successfully synthesised in 20–78% yields. Tolerance of various functional groups was observed, including methyl, methoxy, methyl ester, halogens and trifluoromethyl groups on the phenyl ring. Moreover, pyridine, thiophene, and alkyne-substituted phenylacrylamides also resulted in satisfactory yields. Notably, the authors also found that acrylamides with N-benzyl or a free N–H were not suitable for this transformation.
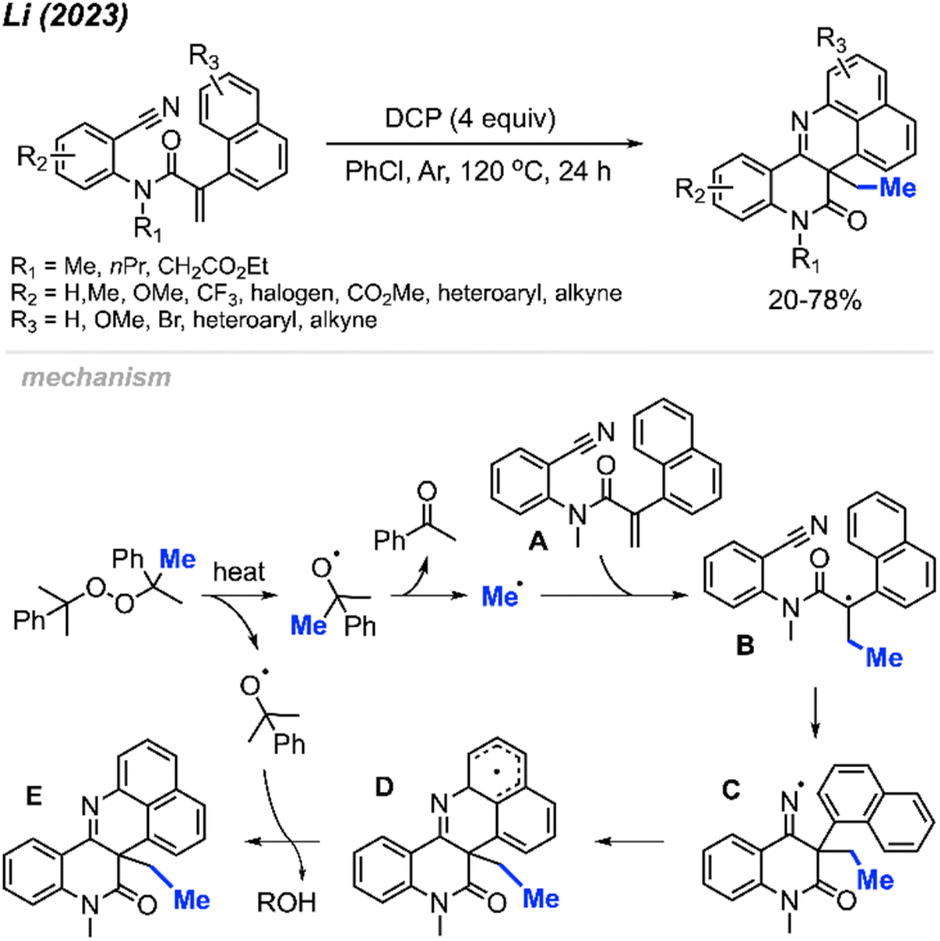 | ||
| Scheme 56 Metal-free radical bicyclisation of 2-cyanoaryl acrylamides with DCP. | ||
The reaction mechanism is depicted in Scheme 56. When heated, homolytic cleavage of DCP releases cumyloxyl radicals, leading to acetophenone and the methyl radical formation through β-scission. The methyl radical then adds to the alkene of acrylamide A, forming the carbon-centred radical intermediate B. Subsequent intramolecular cyclisation of intermediate B results in the formation of the imine radical C, which further cyclises with the naphthyl ring to produce intermediate D. Finally, the oxidative aromatisation of intermediate D culminates in the formation of the final polyheterocycle product.
3.6 Methylene formation via intermediate methylation
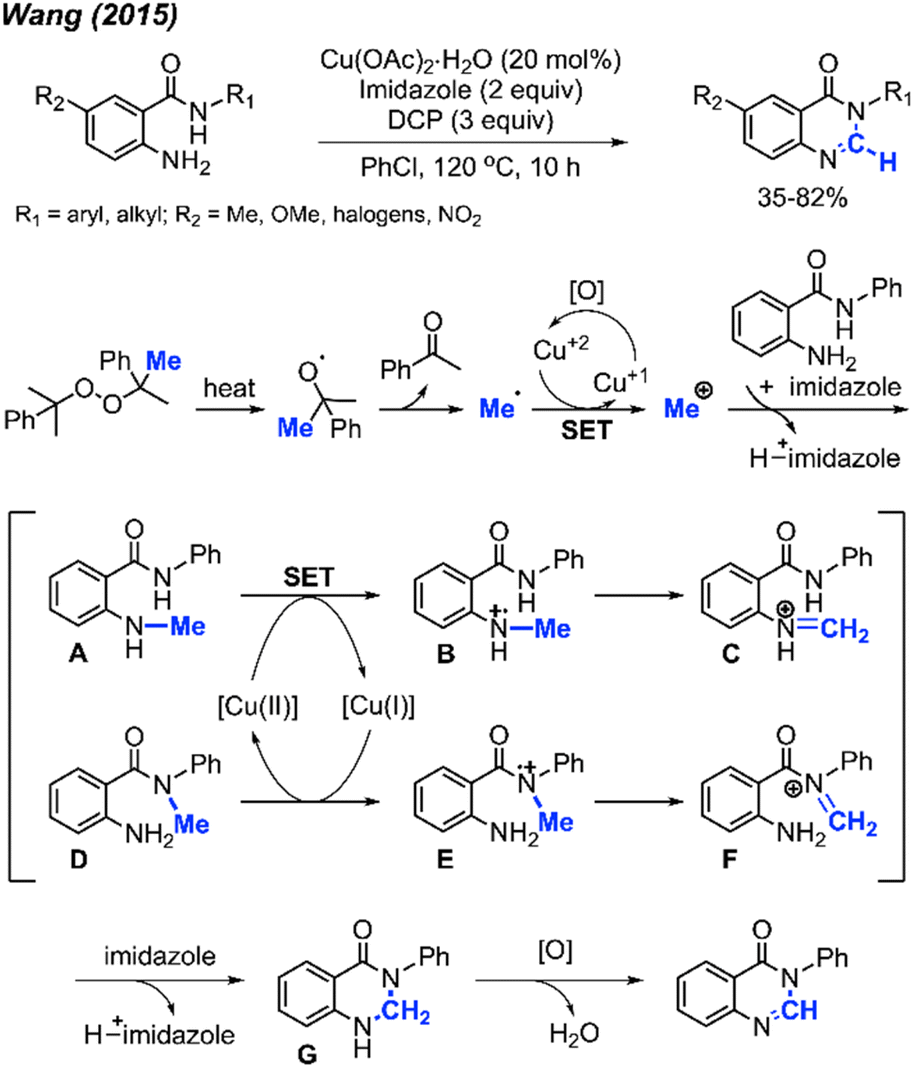 | ||
| Scheme 57 Copper-catalysed methylation and cyclisation towards the synthesis of quinazolinones promoted by DCP. | ||
Scheme 57 depicts the proposed reaction mechanism. The process initiates with the thermal decomposition of DCP, yielding two cumyloxyl radicals. A cumyloxyl radical further decomposes to form a methyl radical and acetophenone. The methyl radical is then converted into a methyl cation through a single electron transfer (SET) facilitated by Cu(II). Subsequently, deprotonation of the starting material by imidazole base followed by its nucleophilic attack on the methyl cation affords either intermediate A or D. In the presence of Cu(II), intermediate B can be derived from intermediate A. Following a hydrogen radical abstraction, intermediate C is formed. The amination product G is generated through an intramolecular nucleophilic attack within intermediate C, followed by deprotonation. Finally, intermediate G is oxidised to yield the final quinazolinone product.
Later in 2016, Liu and co-workers extended their previous work by coordinating a domino reaction to achieve methylation, aromatisation, and perfluorinated alcohol functionalisation under metal-free conditions (Scheme 58).69 The reaction was achieved at an elevated temperature of 130 °C using DCP as a methyl source. The authors demonstrated 17 examples in yields ranging from 22–58%, with functional groups including methyl, chloro, methoxy, and tert-butyl groups. A variety of polyfluorinated alcohols were also well tolerated. The reaction initially follows the same mechanism as Scheme 52. This is followed by phenanthridine isomerisation to afford the active enamine, which can subsequently react with the polyfluorinated alcohols to produce the final product.
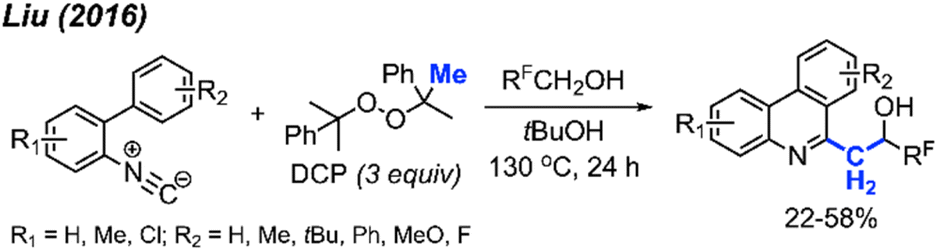 | ||
| Scheme 58 Metal-free methylation, aromatisation, and perfluoronated alcohol functionalisation cascade promoted by DCP. | ||
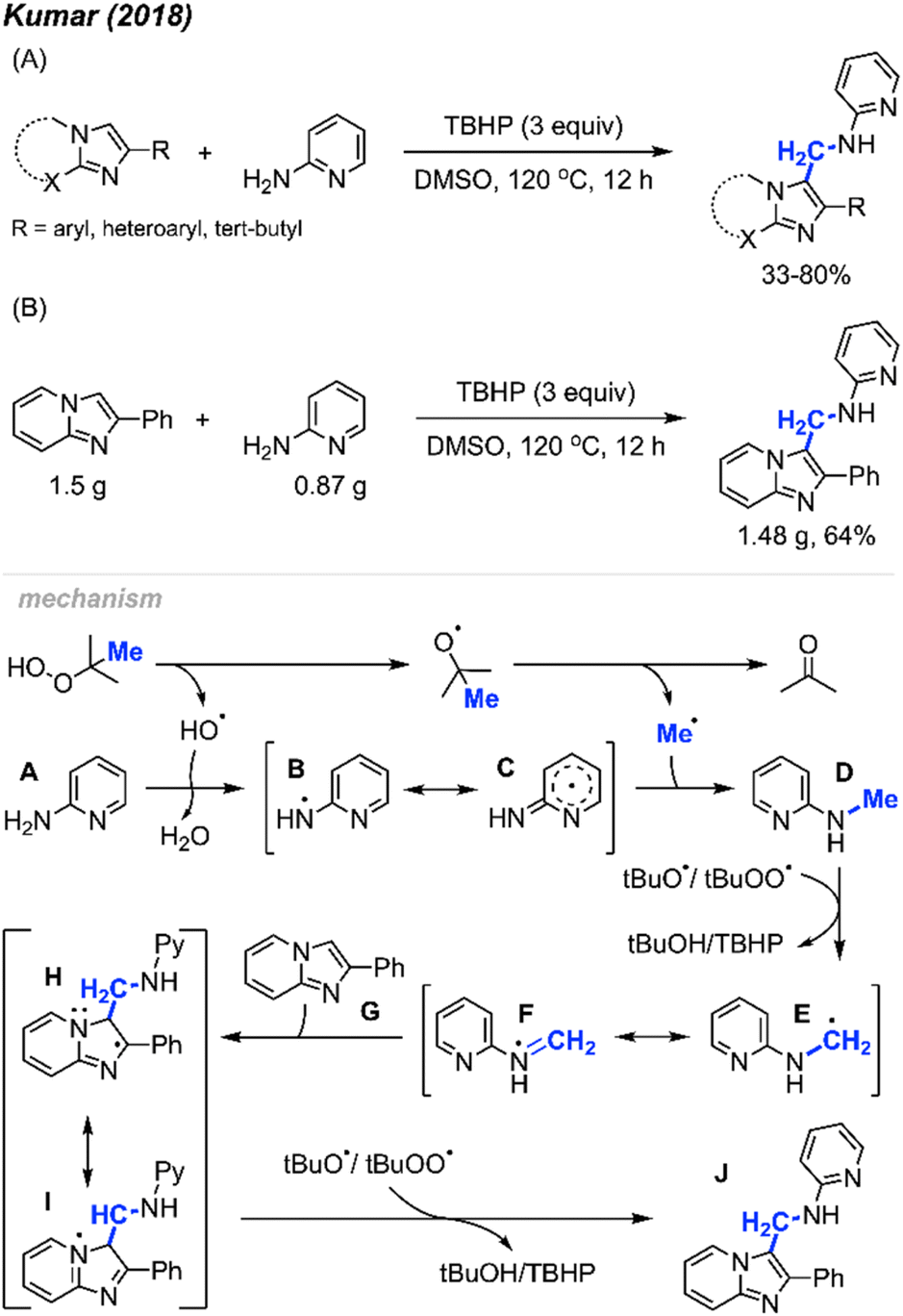 | ||
| Scheme 59 Metal-free synthesis of amino-methylated imidazo-heterocycles. (A) Optimised reaction conditions, (B) gram scale reaction. | ||
The proposed reaction mechanism is illustrated in Scheme 59. TBHP undergoes homolytic cleavage, generating a tert-butoxy radical that then produces the methyl radical through β-scission. Subsequently, either a tert-butoxy, hydroxy, or tert-butylperoxy intermediate radical abstracts a hydrogen radical from 2-aminopyridine A, resulting in the formation of the aminyl radical intermediate B, which is stabilised by the pyridine ring. Intermediate B then reacts with the methyl radical to form intermediate D. Hydrogen atom abstraction from intermediate D yields the radical species E. The radical addition of E to the starting substrate G generates intermediate H, which, upon hydrogen abstraction, affords the desired product J.
3.7 Methylation and alkynylation of terminal alkenes
In 2021, Xiang and co-workers reported an oxidative, metal-free method for alkene methylation/alkynylation of 1,4-enyn-3-ols using DCP as the methyl source (Scheme 60).71 The 1,4-enyn-3-ol derivatives are reactive in PhCF3 at 120 °C, resulting in the synthesis of challenging quaternary-carbon containing but-3-yn-1-one products. DCP and DTBP were comparably effective as methylation reagents. The authors also found that the degree of reactivity varied with position of the R1 substituent on the alkynyl–aryl ring, following the sequence: para > meta > ortho. This study serves as another illustrative example of peroxides' utility as a methylation reagent within organic synthesis applications. | ||
| Scheme 60 Metal-free methylation/alkynylation of terminal alkenes using DCP. | ||
The authors proposed the mechanism depicted in Scheme 60. DCP thermally decomposes to generate a methyl radical and acetophenone. The methyl radical reacts with the starting material A, leading to the formation of radical intermediate B. This is followed by an unconventional, “anti-Baldwin” 3-exo-dig cyclisation and alkynyl migration, involving homolysis of the carbon–carbon bond, generating the hydroxyalkyl radical intermediate D. Single electron oxidation of intermediate D by a cumyloxyl radical produces the carbocation intermediate E. Finally, deprotonation of intermediate E yields the desired product F.
4 Conclusions
This review highlights significant advancements in the utilisation of peroxides as methylating reagents. Many of these innovative reactions operate without the need for transition metal catalysts, resulting in cleaner reactions, simpler workups, and enhanced suitability for late-stage functionalisation of biological compounds. Tandem processes have also been discovered, paving the way for more concise synthetic routes to generate useful products. Many of these reactions exhibit broad tolerance for various functionalities, encompassing both electron-donating and withdrawing groups. Particularly noteworthy is the inert nature of halogen substituents in this chemistry, providing an opportunity to use them as potential handles for further functionalisation. Insofar, the use of methyl-containing peroxides has emerged as a versatile pathway for the methylation of value-added products.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Daliah Farajat and Yuhua Zhang prepared the draft, which was revised and edited by Professor Chao-Jun Li.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the funding from the National Sciences & Engineering Research Council (NSERC), Canada Research Chair Foundation, Canada Foundation for Innovations, the FRQNT Centre for Green Chemistry and Catalysis (CGCC), and the McGill University Chemistry Department.References
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- R. E. Williams, J. Chem. Ed., 2010, 87, 1348 CrossRef.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- S. Sun and J. Fu, Bioorg. Med. Chem. Lett., 2018, 28, 3283–3289 CrossRef CAS.
- Y. Chen, Chem.–Eur. J., 2019, 25, 3405–3439 CrossRef CAS PubMed.
- G. Yan, A. J. Borah, L. Wang and M. Yang, Adv. Synth. Catal., 2015, 357, 1333–1350 CrossRef CAS.
- Z. Li and C.-J. Li, J. Am. Chem. Soc., 2004, 126, 11810–11811 CrossRef CAS PubMed.
- Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 6968–6969 CrossRef CAS.
- Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 3672–3673 CrossRef CAS.
- Z. Li and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 56–57 CrossRef CAS PubMed.
- Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242–4243 CrossRef CAS.
- Y. Zhang and C. J. Li, Angew. Chem., 2006, 118, 1983–1986 CrossRef.
- Y. Zhang, J. Feng and C.-J. Li, J. Am. Chem. Soc., 2008, 130, 2900–2901 CrossRef CAS.
- A. K. Sharma, D. Roy and R. B. Sunoj, Dalton Trans., 2014, 43, 10183–10201 RSC.
- Q. Dai, Y. Jiang, J.-T. Yu and J. Cheng, Synthesis, 2016, 48, 329–339 CrossRef CAS.
- J. Würmel and J. M. Simmie, J. Phys. Chem. A, 2024, 128, 8672–8678 CrossRef.
- R. D. Bach and H. B. Schlegel, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRef CAS PubMed.
- Z.-l. Li and C. Cai, Org. Chem. Front., 2017, 4, 2207–2210 RSC.
- W. Lv, S. Wen, J. Liu and G. Cheng, J. Org. Chem., 2019, 84, 9786–9791 CrossRef CAS PubMed.
- Z.-l. Li, P.-Y. Wu and C. Cai, Org. Chem. Front., 2019, 6, 2043–2047 RSC.
- Q. Li, Y. Li, W. Hu, R. Hu, G. Li and H. Lu, Chem.–Eur. J., 2016, 22, 12286–12289 CrossRef CAS PubMed.
- T. Kubo and N. Chatani, Org. Lett., 2016, 18, 1698–1701 CrossRef CAS PubMed.
- D. Liu, L. Yu, Y. Yu, Z. Xia, Z. Song, L. Liao, Z. Tan and X. Chen, Eur. J. Org Chem., 2019, 2019, 6930–6934 CrossRef CAS.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS.
- G. Li, S. Yang, B. Lv, Q. Han, X. Ma, K. Sun, Z. Wang, F. Zhao, Y. Lv and H. Wu, Org. Biomol. Chem., 2015, 13, 11184–11188 RSC.
- M. Araki, M. Maeda and Y. Kawazoe, Tetrahedron, 1976, 32, 337–340 CrossRef CAS.
- M. F. Zady and J. L. Wong, J. Am. Chem. Soc., 1977, 99, 5096–5101 CrossRef CAS.
- P.-Z. Zhang, J.-A. Li, L. Zhang, A. Shoberu, J.-P. Zou and W. Zhang, Green Chem., 2017, 19, 919–923 RSC.
- Z.-L. Li, L.-K. Jin and C. Cai, Org. Chem. Front., 2017, 4, 2039–2043 RSC.
- S. Jin, H. Yao, S. Lin, X. You, Y. Yang and Z. Yan, Org. Biomol. Chem., 2020, 18, 205–210 RSC.
- X. Rong, L. Jin, Y. Gu, G. Liang and Q. Xia, Asian J. Org. Chem., 2020, 9, 185–188 CrossRef CAS.
- H. Zhuang, R. Zeng and J. Zou, Chin. J. Chem., 2016, 34, 368–372 CrossRef CAS.
- W. Zhao, L. Xu, Y. Ding, B. Niu, P. Xie, Z. Bian, D. Zhang and A. Zhou, Eur. J. Org Chem., 2016, 2016, 325–330 CrossRef CAS.
- B. A. Mir, A. Banerjee, S. K. Santra, S. Rajamanickam and B. K. Patel, Adv. Synth. Catal., 2016, 358, 3471–3476 CrossRef CAS.
- N. Zhu, J. Zhao and H. Bao, Chem. Sci., 2017, 8, 2081–2085 RSC.
- F. Zhang, H. Liu, X.-J. Jia, L. Li, Y. Liang, X. Luo and H. Luo, New J. Chem., 2023, 47, 17060–17065 RSC.
- S. Guo, Q. Wang, Y. Jiang and J.-T. Yu, J. Org. Chem., 2014, 79, 11285–11289 CrossRef CAS PubMed.
- Z.-H. Zhou, C.-K. Li, S.-F. Zhou, A. Shoberu and J.-P. Zou, Tetrahedron, 2017, 73, 2740–2746 CrossRef CAS.
- A. Vasilopoulos, S. W. Krska and S. S. Stahl, Science, 2021, 372, 398–403 CrossRef CAS PubMed.
- S. Song, X. Cheng, S. Cheng, Y.-M. Lin and L. Gong, Chem.–Eur. J., 2023, 29, e202203404 CrossRef CAS.
- Y. Zhu, H. Yan, L. Lu, D. Liu, G. Rong and J. Mao, J. Org. Chem., 2013, 78, 9898–9905 CrossRef CAS PubMed.
- Q. Xia, X. Liu, Y. Zhang, C. Chen and W. Chen, Org. Lett., 2013, 15, 3326–3329 CrossRef CAS.
- X. Wu and Y. Wang, Tetrahedron Lett., 2018, 59, 1240–1243 CrossRef CAS.
- J. Lai and G. Yuan, Tetrahedron Lett., 2018, 59, 524–527 CrossRef CAS.
- F.-S. He, X. Gong, P. Rojsitthisak and J. Wu, J. Org. Chem., 2019, 84, 13159–13163 CrossRef CAS PubMed.
- S. Che, Q. Zhu, Z. Luo, Y. Lian and Z. Zhao, Synth. Commun., 2021, 51, 935–942 CAS.
- J. Xiao, Q. Su, W. Dong, Z. Peng, Y. Zhang and D. An, J. Org. Chem., 2017, 82, 9497–9504 CrossRef CAS.
- F. Teng, J. Cheng and J.-T. Yu, Org. Biomol. Chem., 2015, 13, 9934–9937 RSC.
- S. Rajamanickam, C. Sah, B. A. Mir, S. Ghosh, G. Sethi, V. Yadav, S. Venkataramani and B. K. Patel, J. Org. Chem., 2020, 85, 2118–2141 CrossRef CAS.
- X. Bao, T. Yokoe, T. M. Ha, Q. Wang and J. Zhu, Nat. Commun., 2018, 9, 3725 CrossRef.
- F. Zhu, J. Xue and X.-F. Wu, Tetrahedron, 2023, 149, 133735 CrossRef CAS.
- G. Rong, D. Liu, L. Lu, H. Yan, Y. Zheng, J. Chen and J. Mao, Tetrahedron, 2014, 70, 5033–5037 CrossRef CAS.
- F.-L. Tan, R.-J. Song, M. Hu and J.-H. Li, Org. Lett., 2016, 18, 3198–3201 CrossRef CAS.
- J.-T. Yu, W. Hu, H. Peng and J. Cheng, Tetrahedron Lett., 2016, 57, 4109–4112 CrossRef CAS.
- P. Li, J. Zhao, R. Lang, C. Xia and F. Li, Tetrahedron Lett., 2014, 55, 390–393 CrossRef CAS.
- X. Zhang, Z. Liu, Y. Gao, F. Li, Y. Tian, C. Li, X. Jia and J. Li, Adv. Synth. Catal., 2018, 360, 272–277 CrossRef CAS.
- Q. Dai, J. Yu, Y. Jiang, S. Guo, H. Yang and J. Cheng, Chem. Commun., 2014, 50, 3865–3867 RSC.
- J.-H. Fan, M.-B. Zhou, Y. Liu, W.-T. Wei, X.-H. Ouyang, R.-J. Song and J.-H. Li, Synlett, 2014, 25, 657–660 CrossRef CAS.
- Z. Xu, C. Yan and Z.-Q. Liu, Org. Lett., 2014, 16, 5670–5673 CrossRef CAS.
- Y.-N. Niu, X.-F. Xia and Y. Yuan, Synlett, 2018, 29, 617–620 CrossRef CAS.
- C. Pan, C. Yuan and J.-T. Yu, Org. Biomol. Chem., 2021, 19, 619–626 RSC.
- T. Yang, W.-J. Xia, B. Zhou, Y. Xin, Y. Shen and Y.-M. Li, Eur. J. Org Chem., 2019, 2019, 5749–5755 CrossRef CAS.
- Q. Dai, J.-T. Yu, X. Feng, Y. Jiang, H. Yang and J. Cheng, Adv. Synth. Catal., 2014, 356, 3341–3346 CrossRef CAS.
- F.-L. Tan, M. Hu, R.-J. Song and J.-H. Li, Adv. Synth. Catal., 2017, 359, 3602–3610 CrossRef CAS.
- J.-Q. Shang, S.-S. Wang, H. Fu, Y. Li, T. Yang and Y.-M. Li, Org. Chem. Front., 2018, 5, 1945–1949 RSC.
- T. G. Fan, X. L. Ding, B. X. Sun, Y. J. Hou, B. Jiang, X. N. Wang and Y. M. Li, Adv. Synth. Catal., 2023, 365, 579–583 CrossRef CAS.
- Y. Bao, Y. Yan, K. Xu, J. Su, Z. Zha and Z. Wang, J. Org. Chem., 2015, 80, 4736–4742 CrossRef CAS PubMed.
- Z. Xu, Z. Hang and Z.-Q. Liu, Org. Lett., 2016, 18, 4470–4473 CrossRef CAS PubMed.
- O. P. S. Patel, N. K. Nandwana, A. K. Sah and A. Kumar, Org. Biomol. Chem., 2018, 16, 8620–8628 RSC.
- Y.-Q. Qin, D. Chen, L. Liu, J.-J. Zhang, X.-J. Peng, Y.-Y. Luo, W. Deng and J. Xiang, Synthesis, 2021, 53, 4700–4708 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |