
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A silicon analogue of a fused bicyclic borirene derivative†
Si Jia Isabel
Phang‡
a,
Zheng-Feng
Zhang‡
b,
Chi-Shiun
Wu
 b,
Zhen Xuan
Wong
a,
Ming-Der
Su
*bc and
Cheuk-Wai
So
*a
b,
Zhen Xuan
Wong
a,
Ming-Der
Su
*bc and
Cheuk-Wai
So
*a
aSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore 637371, Singapore. E-mail: CWSo@ntu.edu.sg
bDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan
cDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 27th January 2025
Abstract
The replacement of all carbon atoms in aromatic rings with main-group elements to afford inorganic ring systems is highly desirable due to their distinct aromatic character. However, fused polycyclic main-group element rings are rare and the feasibility of aromaticity in such compounds has yet to be explored. To explore aromaticity in fused polycyclic main-group element rings, a stable di-silicon analogue of fused bicyclic borirene, namely bicyclo[1.1.0]-2,4-diborylenyldisil-1(3)-ene 5 was synthesized from an N-phosphinoamidinato chlorosilylene 3. Compound 5 consists of a bridgehead Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double bond bonded with two bridging borons resulting in an unsaturated fused bicyclic skeleton. The bridgehead SiSi σ- and π-electrons and bridging Si–B σ-electrons are stabilized by both σ- and π-aromatic delocalization on the Si2B2 fused bicyclic ring.
Si double bond bonded with two bridging borons resulting in an unsaturated fused bicyclic skeleton. The bridgehead SiSi σ- and π-electrons and bridging Si–B σ-electrons are stabilized by both σ- and π-aromatic delocalization on the Si2B2 fused bicyclic ring.
Introduction
Aromaticity is a key concept in understanding the electronic properties and reactivity of aromatic compounds in chemistry.1 Aside from the typical cyclic carbon based aromatic compounds such as benzene, aromaticity has been successfully extended to other main-group elements such as boron, silicon and phosphorus.2 Aromatic compounds with cyclic delocalized (4n + 2) electrons are important synthons in organic chemistry; therefore it is highly desirable to replace all carbon atoms in aromatic rings with main-group elements to afford inorganic ring systems that retain aromaticity.2 The diverse characteristics of main group elements compared to carbon give rise to distinct aromatic character which allows such rings to function as versatile building blocks for complex, yet highly functional molecules. However, the synthesis of such aromatic rings containing these main group elements is a formidable challenge due to the lack of methodology. To date, only a few stable examples have been reported and most examples consist of highly charged aromatic rings,2–8 such as triboracyclopropenyl dianion I,3 cyclopentagallene dianion II,4 cyclotrigermenium cation III,5 cyclotrisilenylium cation IV,6 and tetrasilacyclobutadiene dication V.7 Neutral aromatic rings are relatively rare;9–16 tetrasilacyclobutane-1,3-diyl VI,10 tetrasilacyclobutadiene VII,11 disilaborirene VIII,12 and cyclo-2,4-dibora-1,3-disilabutane-1,3-dilyl IX13,14 are such examples, and they were only synthesized recently (Scheme 1). | ||
| Scheme 1 Aromatic main-group element rings. | ||
It is noteworthy that compounds I–IX are all mono-cyclic aromatic rings, and are representative main group element analogues of aromatic carbon molecules, such as cyclobutane-1,3-diyl and the cyclopropenyl cation. Based on fused polycyclic aromatic hydrocarbons such as naphthalene, it is plausible to extend the concept of aromaticity to fused polycyclic inorganic rings, which have yet to be discovered.
Among fused polycyclic aromatic rings, bicyclic borirene X is the smallest neutral fused aromatic ring.17 It features bicyclic 2π-aromatic delocalization, resulting from the overlapping of the bridgehead CC π orbital with two empty p orbitals on the bridging boron center. Compared to typical alkenes such as ethylene and propylene, high strain energy owing to the exceptionally strained bridgehead CC double bond in the bicyclic scaffold, arising from an inversion of the conformation about the CC double bond, is expected. The isolation of such a ring remains unknown experimentally. Thus, in an effort to isolate a more stable analogue of bicyclic borirene X, we replaced the carbon atoms in the ring with silicon to offer more chemical adaptability and reduced ring strain due to the larger atomic size of silicon. We further used a bidentate ligand bonded with the boron atoms to provide enhanced kinetic stabilization. Herein, we report a phosphinoamidinate-stabilized bicyclo[1.1.0]-2,4-diborylenyldisil-1(3)-ene, where the bridgehead SiSi σ- and π-electrons and bridging Si–B σ-electrons are aromatically delocalized on the Si2B2 fused bicyclic ring, accounting for the stability of the fused bicyclic borirene ring structure.
Results and discussion
Synthesis and characterization
The N-phosphinoamidinato dichlorosilane 2 (Fig. S27†) was prepared and reacted with LiN(SiMe3)2·Et2O in toluene at room temperature for 16 h to form N-phosphinoamidinato chlorosilylene 3 (Scheme 2), which was isolated as a yellow crystalline solid (yield: 61%). The N-phosphinoamidinate ligand is bonded with the silylene center in an N,P-chelate fashion. Its 31P{1H} NMR spectrum shows a singlet at 67.8 ppm with a pair of satellite signals (JSi–P = 186.5 Hz), which is upfield shifted compared with that of (phosphine)(amido)bromosilylene (74.7 ppm).18 The 29Si{1H} NMR spectrum shows a doublet at 8.0 ppm (JSi–P = 186.6 Hz), which is intermediate between that of (phosphine)(amido)bromosilylene (−18.4 ppm)18 and amidinato chlorosilylene (14.6 ppm).19 The X-ray crystal structure shows the silicon center adopting a trigonal pyramidal geometry (Fig. 1a, sum of the bond angles: 280.0°), indicating the presence of a Si lone pair of electrons. | ||
| Scheme 2 Synthesis of compound 5 (yields reported are all isolated yields). | ||
 | ||
| Fig. 1 X-ray crystal structures of 3 and 4 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. (a) Selected bond lengths (Å) and angles (deg.) of 3: Si1–N1 1.862(3), Si1–P1 2.3685(13), Si1–Cl1 2.1770(14), P1–Si1–N1 81.03(9), P1–Si1–Cl1 99.06(5), N1–Si1–Cl1 99.89(10). (b) Selected bond lengths (Å) and angles (deg.) of 4: Si1–B1 2.021(5), Si1–N1 1.787(3), Si1–I1 2.4356(11), Si1–Cl1 2.0845(15), B1–I2 2.243(5), B1–I3 2.260(5), B1–P1 2.009(5), N1–Si1–B1 112.94(17), Si1–B1–P1 101.1(2), B1–P1–N2 110.17(19). | ||
Compound 3 underwent oxidative addition with BI3 in toluene at 65 °C for 16 h to afford the N-phosphinoamidinate-bridged borylsilane 4 (Scheme 2), which was isolated as a colorless crystalline solid (yield: 55%). The 29Si{1H} NMR signal of 4 (−8.8 ppm) is broad due to quadrupolar coupling with the B nucleus. The 31P{1H} and 11B{1H} NMR signals are found at 26.6 ppm and −45.6 ppm, respectively. X-ray crystallography shows a six-membered ring, where the ligand is an N,P-chelate, bridged between the Si–B bond (Fig. 1b). The Si1–B1 bond (2.021(5) Å) is typical of a single bond. A similar B–Cl bond oxidative addition was observed in the reactivity of a disilicon(I) compound.20
Compound 4 was reacted with excess KC8 in THF at room temperature for 3 h to afford N-phosphinoamidinato bicyclo[1.1.0]-2,4-diborylenyldisil-1(3)-ene 5, which was isolated as a reddish-orange crystalline solid (yield: 10%). Compound 5 is considered a bridging boron analogue of the bicyclo[1.1.0]tetrasil-1(3)-ene reported by Iwamoto et al.,21–25 but the presence of the bridging N-phosphinoamidinato boron moieties in compound 5 induces some degree of difference in electronic properties compared with the bicyclo[1.1.0]tetrasil-1(3)-ene, as indicated by NMR spectroscopy and X-ray crystallography (Scheme 3). The 31P{1H} NMR signal (40.9 ppm) is broad and the 11B{1H} NMR signal (30.5 ppm) is in the low-field region, even though the boron centers in compound 5 are four-coordinate. It is upfield shifted in comparison with that of the endocyclic boron center in VIII (50.1 ppm).12 The 29Si{1H} NMR resonance (233.0 ppm) is intermediate between cyclotrisilenylium cations (284.6–288.1 ppm)6 and bicyclo[1.1.0]tetrasil-1(3)-ene (217 ppm).21 The broad 29Si{1H} NMR signal of compound 5 is also downfield shifted in comparison with those of disilenes (50–155 ppm)26 and VIII (112.2 ppm). The molecular structure of 5 obtained by X-ray crystallography shows the planar fused bicyclo-Si2B2 ring being orthogonal to two N-phosphinoamidinate ligands (Fig. 2). It is expected that the exceptionally strained bridgehead SiSi double bond in the bicyclic scaffold distorts the structural parameters. The Si–B (2.034(4), 2.048(3) Å) and Si1–Si1A (2.3583(19) Å) bond lengths are longer than those in compound VIII (Si–B: 1.911(7)–1.952(3); Si–Si: 2.133(2)– 2.1469(11) Å).12 The Si1–Si1A bond is intermediate between those in disilenes (2.14–2.29 Å)26 and bicyclo[1.1.0]tetrasil-1(3)-ene (2.4873(10) Å).21 The Si–Si bond length in compound 5 is comparable with the Si–Si single bond length (ca. 2.3 Å). The UV-vis spectrum of compound 5 in THF (dark orange solution) shows an absorption band at λmax = 412 nm corresponding to the  (HOMO−2 → LUMO) and
(HOMO−2 → LUMO) and  (HOMO → LUMO+2) with equal contribution, and an intense absorption band at 474 nm corresponding to the
(HOMO → LUMO+2) with equal contribution, and an intense absorption band at 474 nm corresponding to the 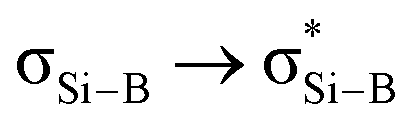 (HOMO−1 → LUMO).
(HOMO−1 → LUMO).
 | ||
| Scheme 3 Comparison of NMR signals (in red) and SiSi bond lengths (in purple) between bicyclo[1.1.0]tetrasil-1(3)-ene and compound 5. | ||
 | ||
| Fig. 2 X-ray crystal structure of 5 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg.): Si1–Si1A 2.3583(19), B1–Si1 2.034(4), B1–Si1A 2.048(3), B1–P1 1.970(3), B1–N1 1.550(4), B1–Si1–Si1A 54.98(10), B1–Si1–B1A 109.41(12), P1–B1–N1 95.34(18). | ||
The downfield 11B{1H} and 29Si{1H} NMR signals together with long SiSi double bond length indicate that the bridgehead SiSi σ and π electrons in compound 5 could delocalize in the fused bicyclic scaffold. However, the multinuclear NMR signals and the distorted structural parameters of the Si2B2 ring cannot definitively confirm the presence of aromaticity. 1H NMR spectroscopy is a widely used experimental tool to assess whether a molecule is aromatic, although determining the degree of aromaticity remains a challenge.27 In the case of compound 5, there are no 1H NMR signals for the Si2B2 ring. Therefore, the degree of electron delocalization as σ- and π-aromaticity can only be evaluated through DFT calculations, which is discussed later in the manuscript.
The reaction of 4 with KC8 was performed in a less polar solvent, namely toluene, at room temperature (Scheme 2). The reaction was traced by 31P{1H} and 11B{1H} NMR spectroscopy after stirring for 16 h, where a mixture of 1,3-dibora-2,4-disilacyclobutanes 6a and 6b was observed (31P{1H} NMR: 102.4 and 108.4 ppm; 11B{1H} NMR: −11.3 and −34.8 ppm). They were isolated as a co-crystalline colorless solid (Fig. 3, yield: 10%) from the reaction mixture. The formation of 6a and 6b indicates that halogen scrambling occurred during the reduction. X-ray crystallography of 6a shows that the N-phosphinoamidinate ligands are in a P,N-chelate fashion, bridging across the B–Si bonds (Fig. 3). The B–Si bond lengths (2.033(10) and 2.025(11) Å) are comparable to those of 5. The Si1–N1 (1.804(7) Å) and P1–B1 bonds (1.979(10) Å) are typical single bonds. The C–N bond lengths (C1–N1: 1.416(10) and C1–N2: 1.281(10) Å) are unequal, showing that the exocyclic N2 atom is an imine moiety. When the reaction of 4 with KC8 in toluene was performed for 3 days, compound 5 was observed, indicating that compounds 6a and 6b are intermediates for the formation of compound 5. To support this, a mixture of compounds 6a and 6b were reacted with excess KC8 in toluene for 16 h, leading to the formation of compound 5.
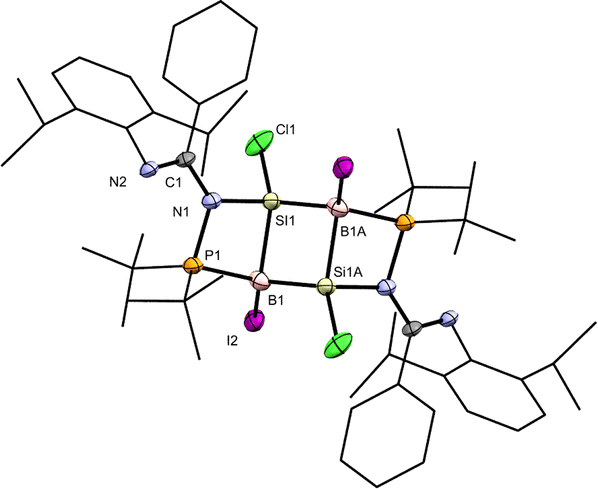 | ||
| Fig. 3 X-ray crystal structure of 6a with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg.): Si1–B1 2.033(10), Si1–B1A 2.025(11), Si1–N1 1.804(7), B1–P1 1.979(10), P1–N1 1.716(7), C1–N1 1.416(10), C1–N2 1.281(10), B1A-Si1-B1 95.6(4), Si1–B1–Si1A 84.4(4), N1–Si1–B1 86.2(4), Si1–B1–P1 83.8(4), B1–P1–N1 90.3(4), P1–N1–Si1 99.2(3). | ||
From compounds 6a and 6b, it is plausible that the reduction of compound 4 with KC8 proceeds through the formation of compounds 6a and 6b first, followed by further reduction to form a diboradisilacyclobutadiene intermediate, which is anti-aromatic (Scheme S1†). Subsequently, it undergoes rearrangement by coordinating the N-phosphinoamidinate ligands with the boron centers to form the bridgehead SiSi double bond and bridging boron in compound 5.
The electronic properties of the bridgehead Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) Si double bond
Si double bond
DFT calculations (M06-2X/def2-TZVP) were performed to elucidate the electronic structure of 5 (Fig. 4). Calculations revealed a singlet ground state of 5 with a singlet-triplet energy gap of 17.4 kcal mol−1. First, the molecular orbital analysis shows that the highest occupied molecular orbital (HOMO) is a π orbital delocalized over the entire fused bicyclic Si2B2 ring. It can be considered as the overlapping of pπ orbitals at the Si atoms with the B–N and B–P σ* orbitals, having pseudo π symmetry at the boron atoms. A total occupancy value of 0.59 e in B–N and B–P σ* orbitals from the NBO analysis reaffirms their participation in delocalization. The NBO analysis does not show a Si–Si π orbital, but instead shows two Si pπ orbitals with an occupancy value of 0.76 e. These NBO data suggest that the Si–Si π bond (total electron occupancy = 0.76 + 0.76 = 1.52 e) is weak due to electron delocalization (0.59 e) with the B–N and B–P σ* orbitals. In comparison, Cui et al. reported tetrasilacyclobutane-1,3-diyl VI,10 where 2π electrons are aromatically delocalized through the σ* orbitals with pseudo π symmetry on the four-coordinate silicon centers. The HOMO−2 represents the σ-interaction between two bridgehead silicon atoms, which significantly extends over the fused bicyclic Si2B2 ring. The HOMO−1, HOMO−12, HOMO−19 and HOMO−77 show the presence of four B–Si σ orbitals. Second, the Wiberg bond index (WBI) between the bridgehead silicon atoms is 1.01, indicating weak SiSi double bond character.
 | ||
| Fig. 4 Molecular orbitals of 5 at the M06-2X/Def2-TZVP level of theory. | ||
To understand the nature of the Si–Si σ-orbital, the electron localization function (ELF) was calculated (Fig. 5a). The ELF plot of the fused bicyclic Si2B2 ring reveals four Si–B bond critical points (blue dots) and one Si2B2 ring critical point (orange dot). Notably, a weak σ-interaction is observed between the two bridgehead Si atom as reflected by the small ELF values (in green). It is because some of the Si–Si σ-electrons are delocalized with the B–Si σ-electrons (annular red region). Upon the removal of σ electrons from HOMO−2 (Fig. 5b), the distribution of energy density is altered, revealing a nearly non-existent σ bond between the two bridgehead Si atoms (in blue). This confirms the presence of a weak Si–Si σ bond which cannot be completely disregarded.
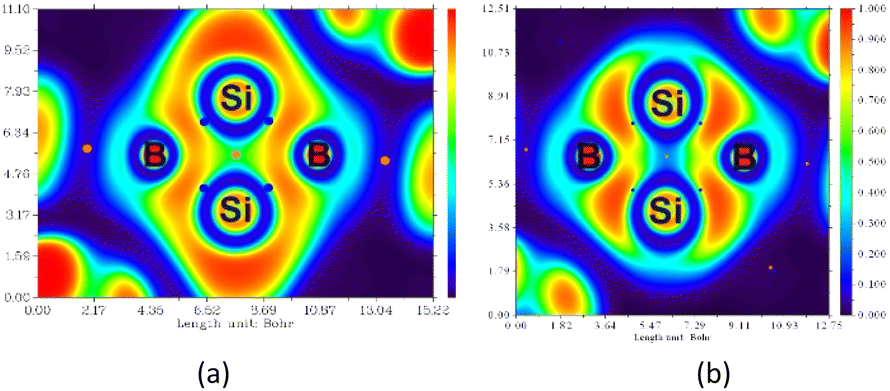 | ||
| Fig. 5 (a) ELF map of the fused bicyclic Si2B2 ring in 5; red and blue represent the strong and weak degree of electron localization of the core electrons, respectively. (b) Removing electrons from the σ-bonding HOMO−2 results in an almost non-existent σ bond between the two bridgehead Si atoms (ELF ≈ 0.01). | ||
A quantum theory of atoms in molecules (QTAIM) analysis was conducted to quantify the Si–Si σ-interaction. QTAIM revealed the formation of 4 Si–B σ bonds, as seen from the bond critical point (BCP: blue dot 1–4, Fig. 6a) along each Si–B bond path. The mean Laplacian distribution ∇2ρ(rc) and energy density H(r) of the Si–B bonds at the BCP are 0.009 e a−5 and –0.060 hartree a−3, respectively. It should be noted that the π-electron density at the ring critical point (RCP) becomes zero, which is attributed to the RCP being located precisely on the nodal plane of the π orbital. As such, the values of ∇2ρ(rc) (0.008 e a−5) and H(r) (−0.032 hartree a−3) observed at the Si2B2 RCP (orange dot 5) indicate the presence of a distinct and weak σ bond between the two Si atoms. Upon the removal of σ electrons from HOMO−2 (Fig. 6b), the H(r) of the RCP reduced from −0.032 hartree a−3 to −0.017 hartree a−3, suggesting that HOMO−2 contributes to the weak σ bond between the two bridgehead Si atoms. These results are in line with ELF calculations. The feasibility of weak σ interaction in the strained and saturated fused bicyclic ring system has also been illustrated by Foroutan-Nejad's theoretical calculations.28
 | ||
| Fig. 6 (a) Laplacian distribution of electron energy of the fused bicyclic B2Si2 ring. Positive and negative areas are shown by crimson and blue lines, representing electron depletion and accumulation, respectively. (b) By removing electrons from the σ-bonding HOMO−2, the σ-electron density concentrations within the fused bicyclic B2Si2 σ skeleton are altered. | ||
Electronic delocalization in bicyclo[1.1.0]-2,4-diborylenyldisil-1(3)-ene 5
Recently, Driess et al. showed that a highly strained all-germanium spiro molecule containing one GeGe double bond in each spiro ring, namely spiro[2.2]pentagerma-1,3-diene, exhibits both σ and 2π electronic delocalization in order to consolidate the pentagerma-spiro skeleton and to reduce the magnitude of the ring strain, respectively.29 In addition, Cui et al. pointed out that Iwamoto's bicyclo[1.1.0]tetrasil-1(3)-ene21 possesses both σ and π delocalization through DFT calculations.10 We reported a highly strained all-silicon spiro molecule containing two bent trisilicon-allene skeletons in each spiro ring, namely spiro[3.3]heptasila-2,6-diylidone possessing both σ and π electronic delocalization to enhance the stability of the entire molecule.30 In this context, it is anticipated that the stability of the fused bicyclic borirene skeleton in compound 5 could arise from some degree of σ and π electronic delocalization, as indicated by the HOMO and ELF calculations.
To understand the overall σ and π electronic delocalization in the entire Si2B2 ring, adaptive natural density partitioning (AdNDP) analysis was performed to elucidate the electronic structure of 5 in terms of the classical Lewis elements (lone-pairs and two-center-two-electron bonds) and delocalized n-center two-electron (nc-2e) bonds (Fig. 7) in order to identify possible delocalized electron-pair bonding.31 A simplified truncated model 5-H was used since all the bonding features of interest are located at the Si2B2 core in 5, which is preserved and clearly presented in the model. First, a 2-center-2-electron π-bond with occupation number (ON) = 1.41 e was found between two Si atoms (Fig. 7, top left). Such low occupancy indicates that the Si–Si π-bond is delocalized over the Si2B2 bicyclic ring, supporting the presence of 2π-aromaticity. Second, a 4-center-2-electron σ bond with ON = 1.99 e is found between two silicon atoms (Fig. 7, top right). In addition, four 4-center-2-electron σ bonds with ON = 1.88–1.99 e are observed at the Si2B2 ring (Fig. 7, bottom). These five 4-center-2-electron σ bonds indicate the presence of σ-aromatization,32 which is also illustrated by the contour plot (annular red region) in ELF calculations (Fig. 5a).
 | ||
| Fig. 7 Adaptive natural population density (AdNDP) analysis of a simplified truncated model 5-H, where substituents (Dipp, Ph, and tBu) in compound 5 are substituted with hydrogen atoms for clarity. | ||
Anisotropy of the induced current density (ACID) was calculated to elucidate the σ- and π-aromaticity. The current density vectors plotted onto the ACID isosurface indicate a strong diatropic ring current in the π system above and underneath the ring, showing π-aromaticity in the Si2B2 ring (Fig. 8a). It is supported by the strong diatropic ring current in the HOMO (Fig. 8c), where the Si–Si π bond exhibits 2π-aromatic delocalization. Besides π delocalization, the current density vectors plotted onto the ACID isosurface indicate a diatropic ring current in the molecular plane mainly localized inside the four-membered ring (Fig. 8a). This is supported by the diatropic ring current in HOMO-1 (Fig. 8b), which illustrates the presence of σ-tangential aromatic delocalization33 among the Si–B bonds. It is noteworthy that the diatropic ring current is restricted to the periphery. A similar observation can be found in naphthalene.34 In addition, the positive natural population analysis (NPA) charge on the bridgehead Si atoms (0.36e) and the negative NPA charge on the B atoms (−0.55e) suggest that the electron density in the ring flows from the silicon to boron atoms. The highly negative Nucleus Independent Chemical Shift (NICS) values at the center of each three-membered ring [NICS(0): −38.7, NICS(1): −13.8, NICS(1)zz: −22.4 ppm] and at the center of the whole Si2B2 four-membered ring [NICS(0): −38.5, NICS(1): −22.1, NICS(1)zz: −34.0 ppm] indicate considerable inorganic aromaticity in the fused bicyclic 2π-Si2B2 ring.
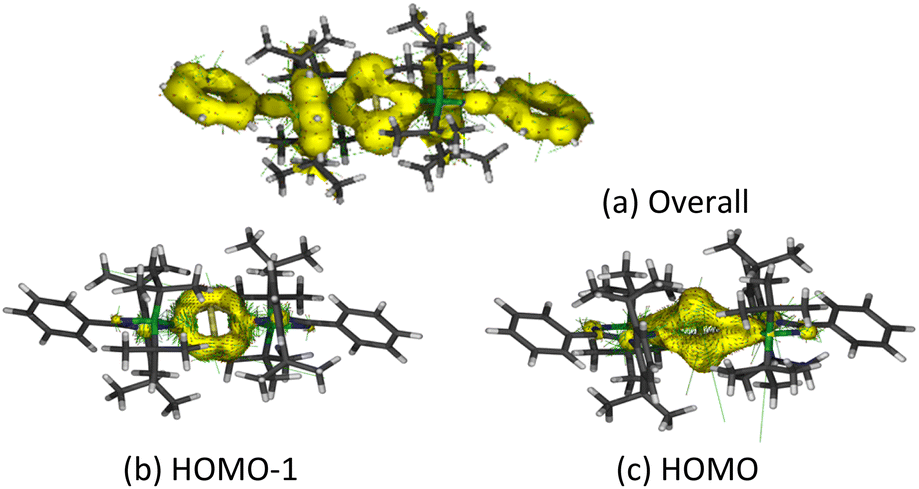 | ||
| Fig. 8 Anisotropy of the current induced density (ACID) of 5. (a) Overall, (b) HOMO-1, (c) HOMO. | ||
To further quantify the aromaticity of the B2Si2 four-membered ring in compound 5, the electron density of delocalized bonds (EDDB) was employed. The EDDB value for compound 5 is 1.901 e. The EDDB value comprises the EDDBσ component (1.193 e) and the EDDBπ component (0.708 e) within the B2Si2 four-membered ring of compound 5. These results illustrate that compound 5 possesses both σ- and π-aromaticity in the Si2B2 ring of compound 5. The EDDBπ component (0.708 e) is comparable with that of VI and related 2π-aromatic Si4 ring analogues (0.586–0.979 e, CAM-B3LYP/def2-TZVP).10 Other ring systems containing both σ and π aromatic delocalization have been reported by Saito and Berndt.35
5-NMe2 with two -NMe2 substituted tricoordinate boron centers was used as a model molecule (Fig. 9), and its NICS and EDDB calculations were performed for comparison with compound 5. Similar to compound 5, the 5-NMe2 model possesses both σ- and π-aromatic delocalization. The HOMO of the 5-NMe2 model is a π orbital delocalized over the entire fused bicyclic Si2B2 ring. It can be considered as the overlapping of pπ orbitals on the Si atoms with the B–N π* orbitals. The HOMO-1 of the 5-NMe2 model represents the σ-interaction between two bridgehead silicon atoms, which significantly extends over the fused bicyclic Si2B2 ring. The NICS value at the center of the Si2B2 ring [NICS(0): −37.5, NICS(1): −23.2, NICS(1)zz: −35.8 ppm], the EDDBσ value (2.071 e) and the EDDBπ value (1.396 e) illustrate the σ- and π-aromaticity in the Si2B2 ring of the 5-NMe2 model. The higher EDDBπ value of 5-NMe2 is attributed to the empty p orbital on the boron center which allows for more effective overlapping with the SiSi π orbital. The lower EDDBπ value in compound 5 is due to the less effective overlapping of pπ orbitals on the Si atoms with the B–N and B–P σ* orbitals, having pseudo π symmetry at the four-coordinate boron atoms.
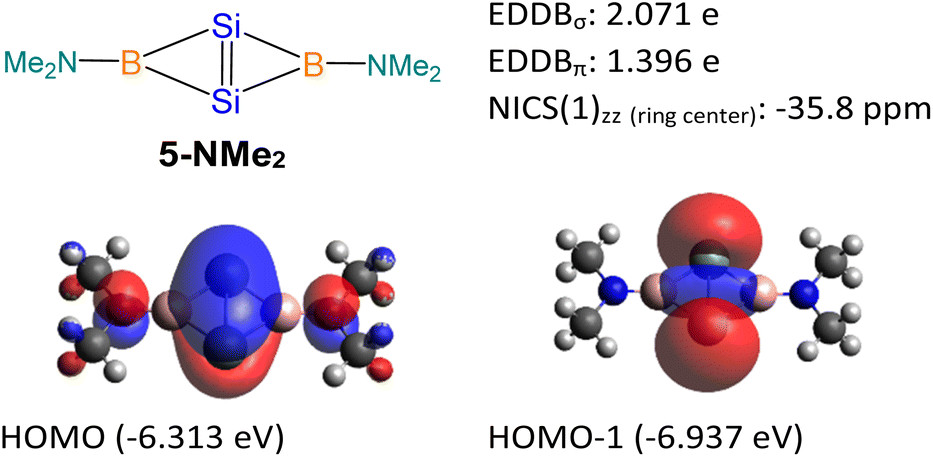 | ||
| Fig. 9 Theoretical data of 5-NMe2. | ||
Reactivity of bicyclo[1.1.0]-2,4-diborylenyldisil-1(3)-ene 5
The lack of reactivity of compound 5 with small molecules (examples: H2 and CO), unsaturated molecules (example: p-tolyl isocyanate) and transition metal complexes (examples: [Ir(cod)Cl]2, PdCl2, and Pt(PtBu3)2), can be attributed to the electronic delocalization of 5 which enhances the stability of the SiSi and Si–B bonds. Reactivity with the chlorinating agent (C2Cl6) led to the oxidation of compound 5 resulting in a mixture of compounds including compound 7 and di-tert-butylchlorophosphine (Scheme 4 and Fig. 10). Reaction with protic reagents (examples: MeOH, NH3BH3 and H2O), and boron substrates (BI3 and B(OH)3) leads to the hydrolysis of compound 5 to form the free N-phosphinoamidine ligand (tracked by in situ31P{1H} NMR), indicating the decomposition of 5.
 | ||
| Scheme 4 Synthesis of compound 7. | ||
 | ||
| Fig. 10 X-ray crystal structure of 7 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg.): Si1–Si2 2.4114(16), Si1–B1 2.080(6), Cl2–Si1 2.1008(16), Cl3–Si1 2.0837(15), B1–N1 1.550(6), B1–Cl1 1.900(5), Cl4–Si2 2.1043(16), Cl5–Si2 2.0738(15), B2–N3 1.546(6), B2–Cl6 1.898(5), B2–P2 2.025(5), B2–Si2 2.085(5), N1–B1–Cl1 109.9(3), Cl1–B1–P1 109.0(3), N1–B1–P1 95.5(3), N1–B1–Si1 128.6(3), Cl1–B1–Si1 92.6(2), P1–B1–Si1 120.6(2), N3–B2–Cl6 109.4(3), Cl3–Si1–Cl2 100.88(6). | ||
Conclusions
In conclusion, the first base-stabilized di-silicon analogue of fused bicyclic borirene, namely bicyclo[1.1.0]-2,4-diborylenyldisil-1(3)-ene 5 was synthesized. It consists of a bridgehead SiSi double bond bonded with two bridging N-phosphinoamidinato boron moieties. The bridgehead SiSi σ- and π-electrons and bridging Si–B σ-electrons are both σ- and π–aromatically delocalized at the fused Si2B2 bicyclic ring. Other main-group element analogues of fused bicyclic borirene are currently under investigation.36
Data availability
The data supporting this article have been included as part of the ESI.† Deposition numbers 2266104 (for 2), 2266105 (for 3), 2266106 (for 4), 2266107 (for 5), 2266108 (for 6) and 2380825 (for 7) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fach Informationszentrum Karlsruhe Access Structures service.Author contributions
S. J. I. Phang performed experiments. Z.-F. Zhang, C.-S. Wu, Z. X. Wong, and M.-D. Su conducted theoretical calculations. C.-W. So prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by A*STAR MTC Individual Research Grants (M21K2c0117) and the Ministry of Education Singapore AcRF Tier 1 (RG72/21). M.-D. Su acknowledges the National Center for High-Performance Computing of Taiwan for providing generous amounts of computing time, and the Ministry of Science and Technology of Taiwan for the financial support.Notes and references
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023, 14, 5569–5576 RSC.
- K. Ota and R. Kinjo, Chem. Soc. Rev., 2021, 50, 10594–10673 RSC.
- T. Kupfer, H. Braunschweig and K. Radacki, Angew. Chem., Int. Ed., 2015, 54, 15084–15088 CrossRef CAS PubMed.
- O. Kysliak, S. H. F. Schreiner, N. Grabicki, P. Liebing, F. Weigend, O. Dumele and R. Kretschmer, Angew. Chem., Int. Ed., 2022, 61, e202206963 CrossRef CAS PubMed.
- A. Sekiguchi, M. Tsukamoto and M. Ichinohe, Science, 1997, 275, 60–61 CrossRef CAS PubMed.
- M. Ichinohe, M. Igarashi, K. Sanuki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 9978–9979 CrossRef CAS PubMed.
- X. Sun, T. Simler, R. Yadav, R. Köppe and P. W. Roesky, J. Am. Chem. Soc., 2019, 141, 14987–14990 CrossRef CAS PubMed.
- M. Fischer, S. Nees, T. Kupfer, J. T. Goettel, H. Braunschweig and C. Hering-Junghans, J. Am. Chem. Soc., 2021, 143, 4106–4111 CrossRef CAS PubMed.
- S. K. Sarkar, R. Chaliha, M. M. Siddiqui, S. Banerjee, A. Münch, R. Herbst-Irmer, D. Stalke, E. D. Jemmis and H. W. Roesky, Angew. Chem., Int. Ed., 2020, 59, 23015–23019 CrossRef CAS PubMed.
- Y. Li, S. Dong, J. Guo, Y. Ding, J. Zhang, J. Zhu and C. Cui, J. Am. Chem. Soc., 2023, 145, 21159–21164 CrossRef CAS PubMed.
- S.-H. Zhang, H.-W. Xi, K. H. Lim and C.-W. So, Angew. Chem., Int. Ed., 2013, 52, 12364–12367 CrossRef CAS PubMed.
- M. Tian, J. Zhang, L. Guo and C. Cui, Chem. Sci., 2021, 12, 14635–14640 RSC.
- S. K. Kushvaha, P. Kallenbach, S. S. Rohman, M. K. Pandey, Z. Hendi, F. Rüttger, R. Herbst-Irmer, D. Stalke, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2023, 145, 25523–25527 CrossRef CAS PubMed.
- J. Fan, L. Yue, C. Liu, B. Rao, G. Zhou, A. Li and B. Su, J. Am. Chem. Soc., 2024, 146, 39–44 CrossRef CAS PubMed.
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 668–672 CrossRef CAS PubMed.
- H. Xu, S. Saebo and C. U. Pittman, Struct. Chem., 2003, 14, 325–335 CrossRef CAS.
- A. Rosas-Sánchez, I. Alvarado-Beltran, A. Baceiredo, D. Hashizume, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 4814–4818 CrossRef PubMed.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- S. Khoo, Y.-L. Shan, M.-C. Yang, Y. Li, M.-D. Su and C.-W. So, Inorg. Chem., 2018, 57, 5879–5887 CrossRef CAS PubMed.
- T. Iwamoto, T. Abe, K. Sugimoto, D. Hashizume, H. Matsui, R. Kishi, M. Nakano and S. Ishida, Angew. Chem., Int. Ed., 2019, 58, 4371–4375 CrossRef CAS PubMed.
- T. Nukazawa, T. Kosai, S. Honda, S. Ishida and T. Iwamoto, Dalton Trans., 2019, 48, 10874–10880 RSC.
- T. Nukazawa and T. Iwamoto, Chem. Commun., 2021, 57, 9692–9695 RSC.
- T. Nukazawa and T. Iwamoto, Organometallics, 2021, 40, 3511–3515 CrossRef CAS.
- T. Nukazawa and T. Iwamoto, J. Am. Chem. Soc., 2020, 142, 9920–9924 CrossRef CAS PubMed.
- M. Weidenbruch in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Chichester, U.K., 2001, vol. 3, p 391 Search PubMed.
- (a) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed; (b) R. H. Mitchell, Chem. Rev., 2001, 101, 1301–1316 CrossRef CAS PubMed.
- C. Foroutan-Nejad, Nat. Commun., 2021, 12, 4037 CrossRef CAS PubMed.
- Y. Guo, Z. Xia, J. Liu, J. Yu, S. Yao, W. Shi, K. Hu, S. Chen, Y. Wang, A. Li, M. Driess and W. Wang, J. Am. Chem. Soc., 2019, 141, 19252–19256 CrossRef CAS PubMed.
- M. Y.-S. Wee, S. Quek, C.-S. Wu, M.-D. Su and C.-W. So, J. Am. Chem. Soc., 2024, 146, 14410–14415 CrossRef CAS PubMed.
- G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623 CrossRef CAS PubMed.
- I. A. Popov, A. A. Starikova, D. V. Steglenko and A. I. Boldyrev, Chem.–Eur. J., 2018, 24, 292–305 CrossRef CAS PubMed.
- A. P. Sergeeva, B. B. Averkiev and A. I. Boldyrev, in Metal-Metal Bonding, ed. G. Parkin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 275–305 Search PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- Double aromaticity systems: (a) S. Furukawa, M. Fujita, Y. Kanatomi, M. Minoura, M. Hatanaka, K. Morokuma, K. Ishimura and M. Saito, Commun. Chem., 2018, 1, 60 CrossRef; (b) M. Unverzagt, G. Subramanian, M. Hofmann, P. v. R. Schleyer, S. Berger, K. Harms, W. Massa and A. Berndt, Angew Chem. Int. Ed. Engl., 1997, 36, 1469–1472 CrossRef.
- Z.-F. Zhang, C.-W. So and M.-D. Su, Dalton Trans., 2024, 53, 14866–14874 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, complete characterization data, computational details, and NMR spectra. CCDC 2266104–2266108 and 2380825 for 2–7. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05867d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |