
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico screening of P,N-ligands facilitates optimization of Au(III)-mediated S-arylation†
Joseph W.
Treacy
 ,
James A. R.
Tilden
,
Elaine Y.
Chao
,
Zihuan
Fu
,
Alexander M.
Spokoyny
*,
K. N.
Houk
* and
Heather D.
Maynard
*
,
James A. R.
Tilden
,
Elaine Y.
Chao
,
Zihuan
Fu
,
Alexander M.
Spokoyny
*,
K. N.
Houk
* and
Heather D.
Maynard
*
Department of Chemistry and Biochemistry and California NanoSystems Institute, University of California, Los Angeles, California 90095-1569, USA. E-mail: spokoyny@chem.ucla.edu; houk@chem.ucla.edu; maynard@chem.ucla.edu
First published on 16th January 2025
Abstract
Metal-mediated cysteine S-arylation is an emerging bioconjugation technique due to its high chemoselectivity, rapid kinetics, and aqueous compatibility. We have previously demonstrated that by altering the steric profile of the ligand and aryl groups of an Au(III) oxidative addition complex, one can modulate the kinetics of the bimolecular coordination and induce rate constants up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 s−1. To further enhance the rate of coordination, density functional theory (DFT) calculations were performed to investigate the steric properties of the P,N-ligated Au(III) oxidative addition complex as well as the thermodynamics of the S-arylation reaction. This allowed for the accelerated screening of 13 new Au(III) oxidative addition complexes. Three of the more sterically available, synthetically accessible P,N-ligands were synthesized, incorporated into Au(I) and Au(III) complexes, and their rates determined experimentally. The comprehensive mechanistic insights from the DFT calculations led to the development of new reagents with bimolecular coordination rate constants as fast as 20200 M−1 s−1. Further experimental characterization of these reagents' efficacy as S-arylation reagents led to a proposed switch in selectivity-determining step for the fastest reagent, which was further confirmed by profiling the reductive elimination kinetics. This work provides a concise workflow for the screening of metal-mediated cysteine S-arylation reagents and new fundamental insights into the coordination chemistry behavior of Au(III) systems.
600 M−1 s−1. To further enhance the rate of coordination, density functional theory (DFT) calculations were performed to investigate the steric properties of the P,N-ligated Au(III) oxidative addition complex as well as the thermodynamics of the S-arylation reaction. This allowed for the accelerated screening of 13 new Au(III) oxidative addition complexes. Three of the more sterically available, synthetically accessible P,N-ligands were synthesized, incorporated into Au(I) and Au(III) complexes, and their rates determined experimentally. The comprehensive mechanistic insights from the DFT calculations led to the development of new reagents with bimolecular coordination rate constants as fast as 20200 M−1 s−1. Further experimental characterization of these reagents' efficacy as S-arylation reagents led to a proposed switch in selectivity-determining step for the fastest reagent, which was further confirmed by profiling the reductive elimination kinetics. This work provides a concise workflow for the screening of metal-mediated cysteine S-arylation reagents and new fundamental insights into the coordination chemistry behavior of Au(III) systems.
Introduction
Effective bioconjugation reagents are chemoselective for specific residues, amenable to mild and aqueous reaction conditions, and able to perform reactions at low concentrations, which necessitates favorable reaction kinetics. Cysteine presents itself as a common target for site-selective bioconjugation due to its high nucleophilicity, relatively low abundance in the proteome, and ability to be incorporated into proteins, antibodies, and other biomolecules through molecular biology techniques.1–3 Commonly used cysteine modifications rely on addition to maleimides or other Michael acceptors. However, these linkages are susceptible to retro-Michael reactions under reducing conditions, leading to reversible modification of the biomolecule.4–9 Accordingly, cysteine S-arylation has emerged as a unique approach to produce C(sp2)–S bonds, which are less susceptible to reversibility.10 Many cysteine S-arylation approaches rely on nucleophilic aromatic substitution (SNAr), which necessitates electron withdrawing groups ortho or para to a leaving group.11–19 This limits the aryl substrate scope for potential modification. An alternative method uses metal-mediated or metal-catalyzed cysteine S-arylation, which allows for the ex situ or in situ formation of an oxidative addition complex (OAC).20–30 These OACs are then able to permit initial thiol coordination to the metal center and subsequent reductive elimination to form the stable, C(sp2)–S bond. The use of transition metals allows for the aryl scope to be broadly expanded to contain both electron donating and withdrawing substituents as well as different substitution patterns.The ex situ formation of the OAC can provide a set of bench stable reagents that can be used for subsequent biomolecule transformations without any additional precautions to preserve the metal reagent.31,32 By using thiophilic metal centers (e.g. Pd and Au), exquisite chemoselectivity for thiols and rapid kinetics are observed. Notably, the mechanism for the S-arylation proceeds via a two-step process of an initial coordination of the thiol to the metal center followed by reductive elimination to form the S-aryl bond. This stands in contrast to other cysteine modification mechanisms that rely on displacement of an alkyl halide or 1,4-addition into a Michael acceptor. Our groups have reported the use of P,N-ligated Au(III) OACs for the S-arylation of cysteine (Fig. 1),33–35 and recently, we have shown that stopped-flow/UV-vis spectroscopy is a powerful technique to characterize the rates of the two distinct elementary steps of the S-arylation. This allowed for the quantification of the bimolecular thiol coordination rate constant (kCo) for P,N-ligated Au(III) OACs with di-1-adamantylphosphine (PAd2) 1 and di-tert-butylphosphine (PtBu2) 2P-substituents as 2460 and 4220 M−1 s−1, respectively. The clear steric dependence on this coordination event led to the synthesis of a dicyclohexylphosphine-based (PCy2) P,N-ligated Au(III) OAC reagent 3 with an unprecedented kCo of 16600 M−1 s−1.35
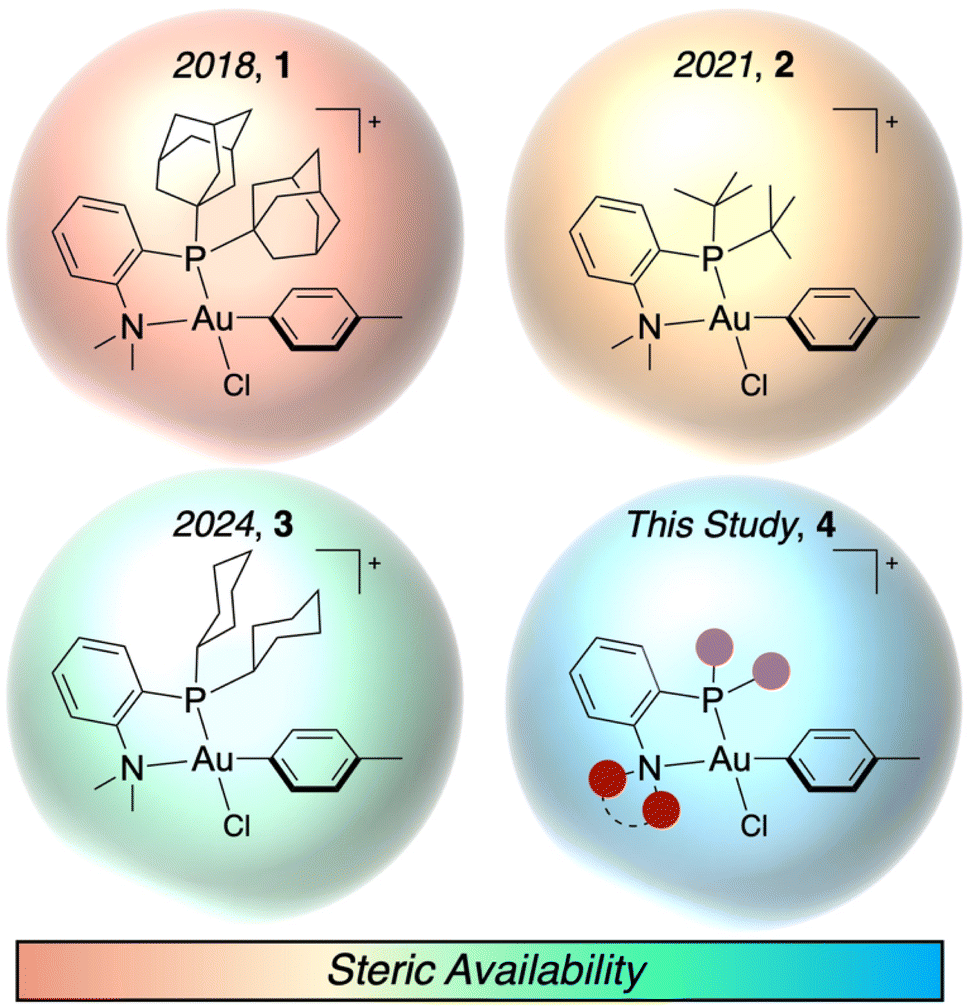 | ||
| Fig. 1 P,N-ligated Au(III) complexes developed for rapid and selective cysteine S-arylation. Purple and red circles represent sites for modification on the ligand scaffold. Counterion not included for clarity. | ||
Alternatively, by increasing the steric bulk of the aryl group, the coordination of the thiol to the Au(III) center was slowed down, which allowed for the chemoselective S-arylation of cysteine. By calculating the percent buried volume (% VBur) of the Au(III) OAC, a steric parameter was produced that could be used to predict the rate of coordination to the Au(III) center for both the ligand and aryl modifications. Herein, we demonstrate the extrapolation of % VBur as a screening metric for the expansion of Au(III) OACs for S-arylation. As the dialkylphosphine and amine could both be modified during the synthesis of the P,N-ligand, in silico screening was performed for eight dialkylphosphine substituents and five amine groups to examine their steric properties (Fig. 1, 4). Additionally, the free energy of reaction (ΔGRxn) as well as the free energy of activation for the reductive elimination (ΔG‡RE) were calculated to examine the thermodynamics of each P,N-ligated Au(III) OAC. This allowed for the rapid screening of 13 different Au(III) OACs. Three of these novel P,N-ligated Au(III) OACs were synthesized, then their kinetics and efficiency as S-arylation reagents were determined. This led to the generation of kCo up to 20200 M−1 s−1, improving upon the previously developed Au(III) OACs and demonstrating the computational and experimental synergy available in the investigation of metal-mediated bioconjugation reactions. These results also provide additional mechanistic insight into the coordination chemistry of Au(III) compounds.36–40
Results and discussion
Computational screening of P,N-Ligated Au(III) OACs
Previously, DFT calculations for the S-arylation of methanethiol were performed with NMe2-containing P,N-ligated Au(III) phenyl OACs with PAd2 (5), PtBu2 (6), and PCy2 (7) P-substituents.35 This reaction proceeds through a reversible coordination of the thiol to the Au(III) center of SM, generating Int1. Reductive elimination (TSRE) from Int1 irreversibly forms the C(sp2)–S bond (Int2), then product dissociation forms the free S-arylated product (PAr) in addition to the Au(I) chloride starting material (PAu, Fig. 2A).41 These calculations revealed that reagents 5–7 had a low ΔG‡RE and a thermodynamically favorable ΔGRxn. Additionally, by calculating the % VBur of the Au(III) OAC (SM) of 5–7, % VBur of 70.0, 69.8, and 67.1 were obtained, respectively.42 This steric parameter is in agreement with the experimentally determined kCo for each of these complexes wherein the more sterically available complexes (i.e. less % VBur) exhibited faster thiol coordination. We therefore hypothesized that the % VBur parameter could be used as a predictive metric for the rate of thiol coordination to the Au(III) center.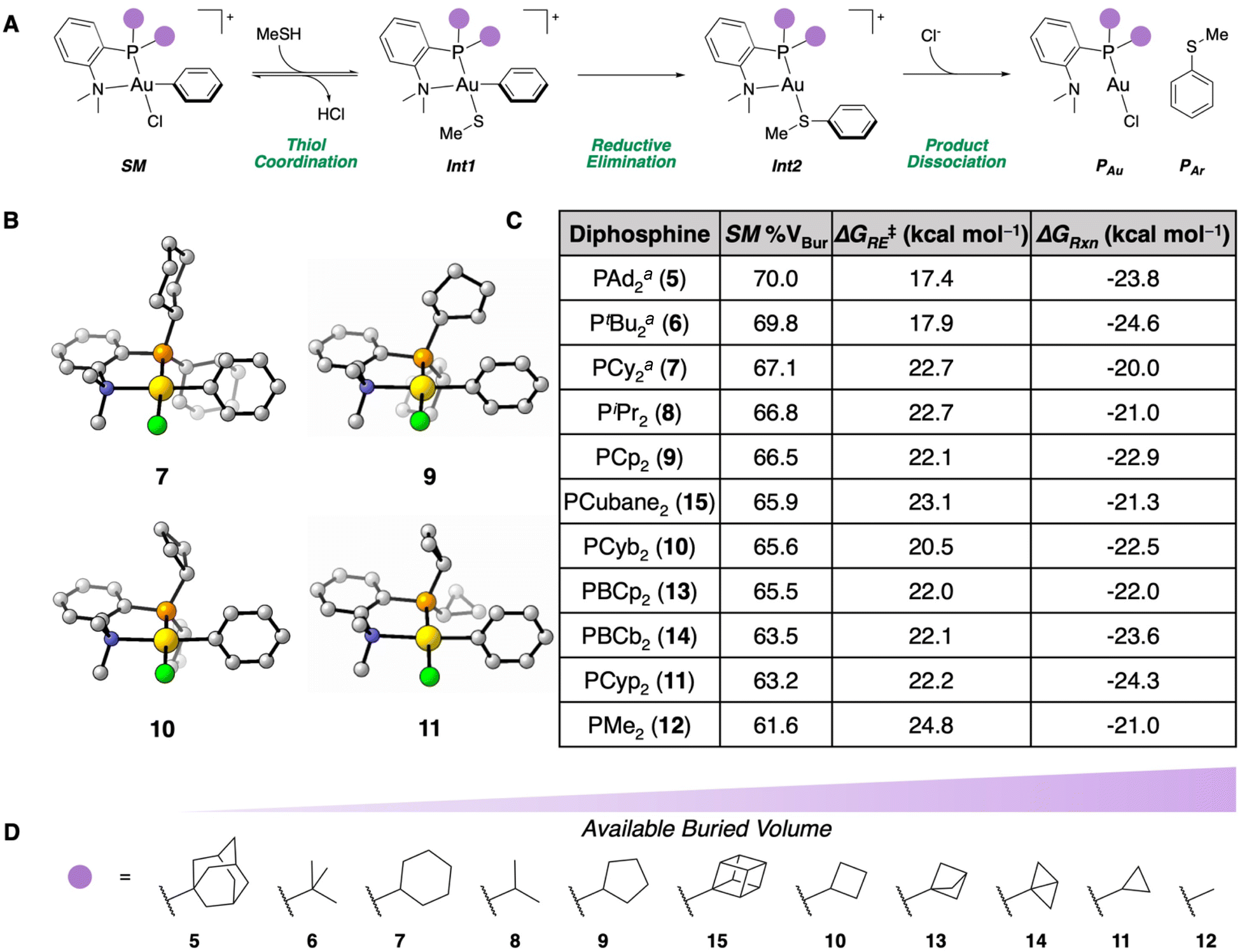 | ||
| Fig. 2 (A) Proposed mechanism for the S-arylation of methanethiol mediated by Au(III) OACs. (B) SM geometries calculated for reagents 7, 9, 10, and 11 at the ωB97X-D/6-311+G(d,p), SDD, CPCM(water)//B3LYP-D3/6-31G(d), LANL2DZ, CPCM(water) level of theory. (C) Table of % VBur, ΔG‡RE, and ΔGRxn for the different dialkylphosphine substituents examined. aThese parameters were calculated previously.35 (D) Schematic representation showing the dialkylphosphine variations in order of least to most available % VBur. | ||
To further enhance this rate of coordination, we sought to screen smaller dialkylphosphines to provide more sterically available P,N-ligated Au(III) OACs using NMe2 as the N-substituent. Accordingly, diisopropylphosphine-(PiPr2) (8) and dicyclopentylphosphine-based (PCp2) (9) P,N-ligated Au(III) SM geometries were calculated at the ωB97X-D/6-311+G(d,p), SDD, CPCM(water)//B3LYP-D3/6-31G(d), LANL2DZ, CPCM(water) level of theory. Complexes 8 and 9 had % VBur of 66.8 and 66.5, respectively. These values are very similar to the NMe2-PCy2-ligated OAC 7, so these ligands should affect a modest improvement on kCo. For 8 and 9, the comparable angles of the dialkylphosphine alkyl groups lead to similarly sized substituents compared to the cyclohexyl ring, which contributes to the small difference in % VBur compared to 7. To this end, smaller ring sizes on the dialkylphosphine fragment of the P,N-ligand were examined to affect larger changes in % VBur. Dicyclobutylphosphine-(PCyb2) (10) and dicylopropylphosphine-based (PCyp2) (11) P,N-ligated OACs were determined to have 65.6 and 63.2 % VBur, respectively. These values demonstrate that the smaller alkyl substituents induce more steric availability around the Au(III) center (Fig. 2B). To determine the lower limit of % VBur for these dialkylphosphine P,N-ligated Au(III) OACs, dimethylphosphine-based (PMe2) P,N-ligated SM (12) was examined and found to have 61.6 % VBur.
Inspired by recent work developing bicyclo[1.1.1]pentyl- and bicyclo[1.1.0]butyl-containing phosphines that are more strained and consequently smaller compared to their cycloalkyl counterparts,43,44 the % VBur for the di(bicyclo[1.1.1]pentyl)phosphine-(PBcp2) (13) and di(bicyclo[1.1.0]butyl)phosphine-based (PBcb2) (14) P,N-ligated OACs were computed. Compound 13 had a % VBur of 65.5 while 14 had a % VBur of 63.5. These dialkylphosphines provide a significant decrease in % VBur compared to P,N-ligated SM9 (PCp2) and 10 (PCyb2), which is in agreement with the additional strain in the bicyclic systems leading to smaller ring sizes and therefore less % VBur. The dicubylphosphine-based (PCubane2) P,N-ligated SM (15) was also examined, which could be employed due to its similarity to 6 (PtBu2). Complex 15 had a % VBur of 65.9, indicating that formation of the cyclic scaffold does provide more steric availability to the SM. Taken together, these eight new Au(III) OACs (8–15) provide significant changes in % VBur compared to previously examined reagents 5–7 (Fig. 2D).
We previously determined that while the coordination to the Au(III) center is important, the subsequent, irreversible reductive elimination to form the C(sp2)–S bond (PAr) remains a key step in the S-arylation.35 Accordingly, the ΔG‡RE and ΔGRxn were computed for each of the complexes. All of the reactions were calculated to be exergonic and irreversible. With the exception of the PMe2P-substituted OAC (12), the ΔG‡RE values are reasonable to undergo sufficiently fast reductive elimination at 25 °C (20.5–23.1 kcal mol−1), but the ΔG‡RE of 24.8 kcal mol−1 for 12 likely prevents it from performing an efficacious reductive elimination at ambient temperature (Fig. 2C).
While prior work from our group has focused on modifying the dialkylphosphine component of the P,N-ligand, there has been research varying the amino fragment of these P,N-ligands in Pd catalysis, most notably to morpholine and piperidine.45–49 Accordingly, these Au(III) complexes with morpholine (16) and piperidine (17) were calculated with a PAd2P-substituent (Fig. 3A), but there was a marginal difference in % VBur compared to the dimethylamino reagent (5, Fig. 3B). We hypothesized that by contracting the ring size, one could generate more sterically available Au(III) OACs. Pyrrolidine (18), azetidine (19), and aziridine (20) were consequently incorporated into the PAd2-based P,N-ligated Au(III) complex and determined to have % VBur of 69.5, 68.6, and 66.7 (Fig. 3B–D), respectively. These results are in line with the dialkylphosphine calculations (Fig. 2) wherein the five-membered ring is similar to the six-membered ring, but significant changes are observed in the four- and three-membered rings. To ensure that these amine modifications did not have significant effects on the thermodynamics of the reaction, the ΔG‡RE and ΔGRxn were computed for 16–20. Each reaction was calculated to be exergonic and irreversible with a low ΔG‡RE (12.2–18.8 kcal mol−1) (Fig. 3B). Notably, the ΔG‡RE for the piperidine-containing OAC (16) is significantly lower than the other complexes, potentially due to relaxation of the amine fragment from the twist-boat conformation in the S-coordinated Int1 to the chair conformation in the transition state (see ESI† for details). This complex may warrant further examination to determine its ability to mediate other, more challenging reductive eliminations,50 but the high % VBur indicates that it will likely not have a faster coordination than the other complexes examined herein.
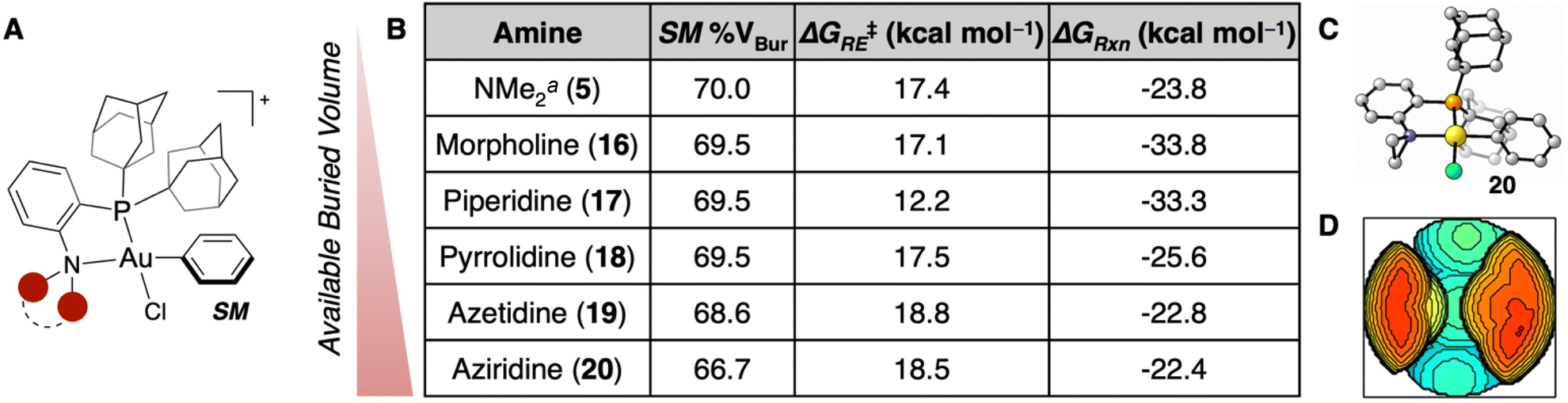 | ||
| Fig. 3 (A) Generalized geometry for the Au(III) SM being modified. (B) Table of SM % VBur, ΔG‡RE, and ΔGRxn for the different amine substituents examined. aThese parameters were calculated previously.35 (C) SM geometry for reagent 20 calculated at the ωB97X-D/6-311+G(d,p), SDD, CPCM(water)//B3LYP-D3/6-31G(d), LANL2DZ, CPCM(water) level of theory. (D) Buried volume plot for the SM of 20. | ||
Synthesis of P,N-ligands, Au(I) complexes, and Au(III) OACs
To validate these computational results, we sought to synthesize the compounds with both a lower % VBur than the NMe2-PCy2 ligated OAC (7) and a reasonable ΔG‡RE. Accordingly, the PCp2-, PCyb2-, and PCyp2-based P,N-ligands with NMe2 as the N-substituent were targeted due to their predicted rapid coordination and sufficiently fast reductive elimination. While the bicyclic systems 13 and 14 did have lower % VBur compared to their cyclic counterparts, they each had a similar % VBur to the more synthetically accessible PCyb2- (10) and PCyp2-based (11) systems. Due to this and their synthetic difficulty, 13 and 14 were not further examined. For the amine components, we wanted to synthesize the PAd2 ligand with azetidine and aziridine as the amino substituent. However, the phenyl aziridine complex polymerizes in water,51 which makes it an unsuitable reagent to perform bioconjugation reactions. We also wanted to examine if modifying both the P- and the N-substituents of the ligand simultaneously generated an additive effect of the two substitutions. To do this, the azetidine-PCy2 complex was targeted.The NMe2-PCp2 ligand 21 was synthesized by in situ formation of the aryl lithiate from 2-iodo-N,N-dimethylaniline and n-BuLi then substitution with the commercially available chlorodicyclopentylphosphine (see ESI† for details). Incomplete conversion to the desired P,N-ligand necessitated purification of the ligand by preparative high performance liquid chromatography (HPLC). Attempts to generate the NMe2-PCyb2 and NMe2-PCyp2 ligands relied on formation of the aryl lithiate and in situ generation of the chlorodialkylphosphine using phosphorus trichloride and the corresponding Grignard reagent. Purification of these reagents by preparative HPLC induced significant formation of the phosphine oxide, limiting the utility of these ligands. Other synthetic efforts to obtain these ligands involved formation of the phosphine oxide and subsequent reduction,52 as well as attempts to generate 2-(dichlorophosphaneyl)-N,N-dimethylaniline,53 which could then react with the cyclobutyl and cyclopropyl Grignard reagents. These routes provided low conversion to product and formation of multiply arylated byproducts, respectively.
However, the azetidine-PAd2 ligand (22) and azetidine-PCy2 ligand (23) could be readily accessed by Pd-catalyzed C(sp2)–P bond formation from the corresponding ortho-halo-substituted aniline species and the dialkylphosphine in good isolated yield (80% and 63% yield, respectively). With P,N-ligands 21–23 isolated (Fig. 4A), the Au(I) complexes (24–26) were generated using HAuCl4·3(H2O) and 2,2′-thiodiethanol as a water-soluble variant of the commercially available Au(I)Cl(SMe2) (Fig. 4B, see ESI† for details). The Au(III) OACs with a p-toluene aryl substituent (27–29) were synthesized using AgSbF6 as a halide scavenger and 4-iodotoluene to generate a model compound for kinetic investigations (Fig. 4C, see ESI† for details).
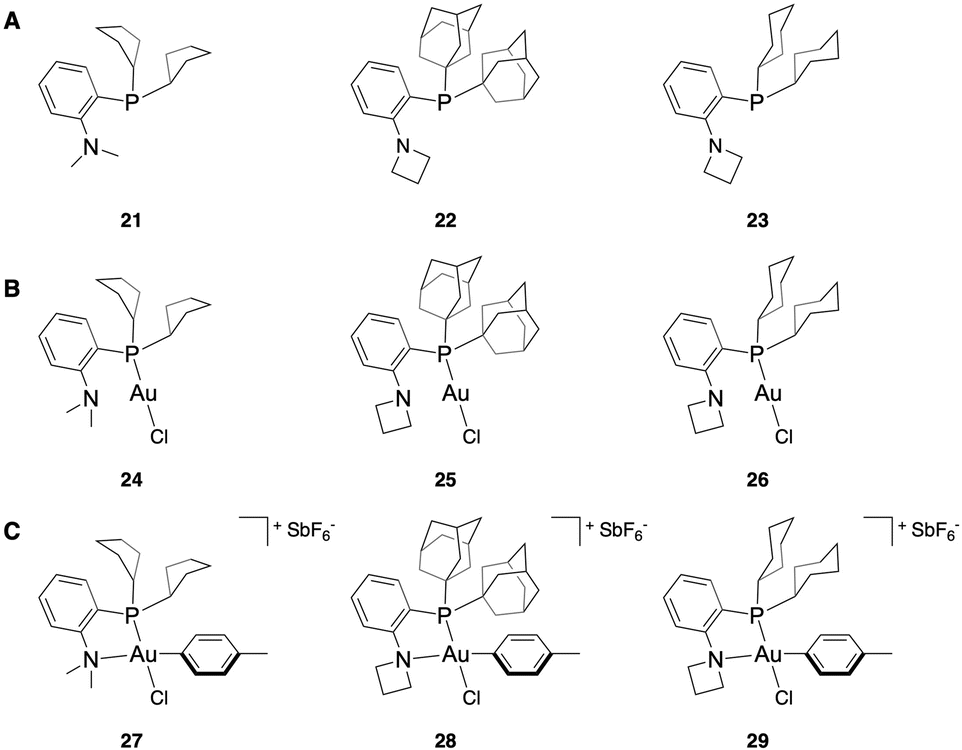 | ||
| Fig. 4 Structures of P,N-ligands (A), Au(I) complexes (B), and Au(III) OACs (C) synthesized. | ||
Thiol coordination rate monitoring
With the Au(III) OACs in hand, we wanted to test the kinetics of the thiol coordination to validate our computational predictions. To perform the coordination kinetics studies, stopped-flow/UV-vis spectroscopy experiments were performed at 5 μM of OAC with glutathione (GSH) as a model thiol under pseudo-first order conditions (500 μM–4 mM of GSH) at pH 7.4 and 20 °C (Fig. 5A, see ESI† for details). By monitoring the absorbance, one can determine the formation of Int1 and extrapolate the observed second-order rate constant, kCo, from the experimentally observed rate constant (kObs). Examining 27, a kCo of 18700 M−1 s−1 was calculated, which is faster than the NMe2-PCy2-ligated OAC 3 (16600 M−1 s−1). Also, the equilibrium of this complex is significantly shifted toward the S-coordinated intermediate (Int1) with a KEq (kCo/kDis) of 1530 M−1 (Fig. 5B and C).
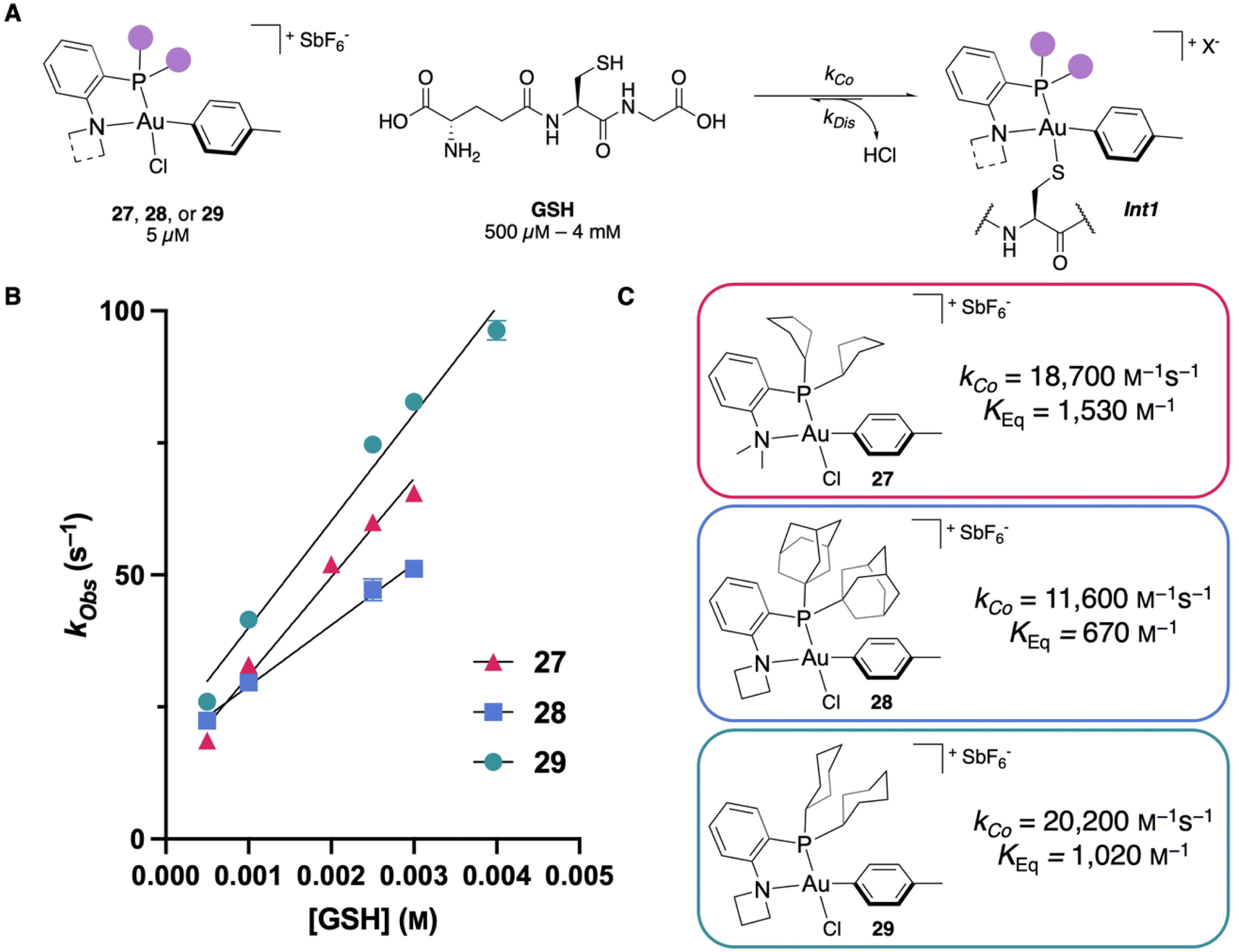 | ||
| Fig. 5 (A) Generalized scheme for coordination monitoring from Au(III) OACs. X− is used as a generalized counterion. Experiments were performed in 1:1 MeCN/100 mM Tris buffer at pH 7.4 and 20 °C. (B) Kinetic plots for coordination monitoring, n = 5 – some error bars are smaller than the markers. (C) Extracted kCo and KEq for reagents 27–29. | ||
Next, rate measurements were performed for 28 to determine if modifying the amine fragment was another viable strategy to affect the kCo. Kinetics experiments show that by utilizing the azetidine substituent, the kCo increases to 11600 M−1 s−1, which is nearly a five-fold increase compared to the NMe2-PAd2-ligated reagent 1 (2460 M−1 s−1). This increase in kCo may also be attributed to less electron donation from azetidine compared to NMe2, which would make the Au(III) center more electron poor and could accelerate the thiol coordination. By adding the P- and N-modifications together, we propose that 29 would provide the fastest kCo reported. The studies were performed and a kCo of 20200 M−1 s−1 was observed for 29 (Fig. 5B and C), which represents a 25% increase in kCo compared to 3 and is the fastest observed rate for S-coordination to an organometallic Au(III) compound to date. These results are all consistent with the % VBur calculations (Fig. 2C and 3B), validating the use of % VBur as a steric descriptor to predict the rate of bimolecular coordination for these Au(III) OACs.
S-Arylation competition experiments
With the kCo determined for these three OACs, as well as the equilibria showing preferential thermodynamic formation of Int1, we next wanted to determine if the bimolecular coordination elementary step was still the selectivity-determining step in the reaction by performing S-arylation competition experiments. These experiments were performed by incubating one equivalent of GSH with three equivalents of two different OACs. These OACs vary by one methylene unit, which allows us to determine by liquid chromatography-mass spectrometry (LC-MS) which Au(III) reagent mediates the S-arylation more effectively (Fig. 6A). The NMe2-PCy2-(30) and NMe2-PAd2-ligated (31) Au(III) OACs with a p-ethylbenzene aryl substituent were prepared accordingly.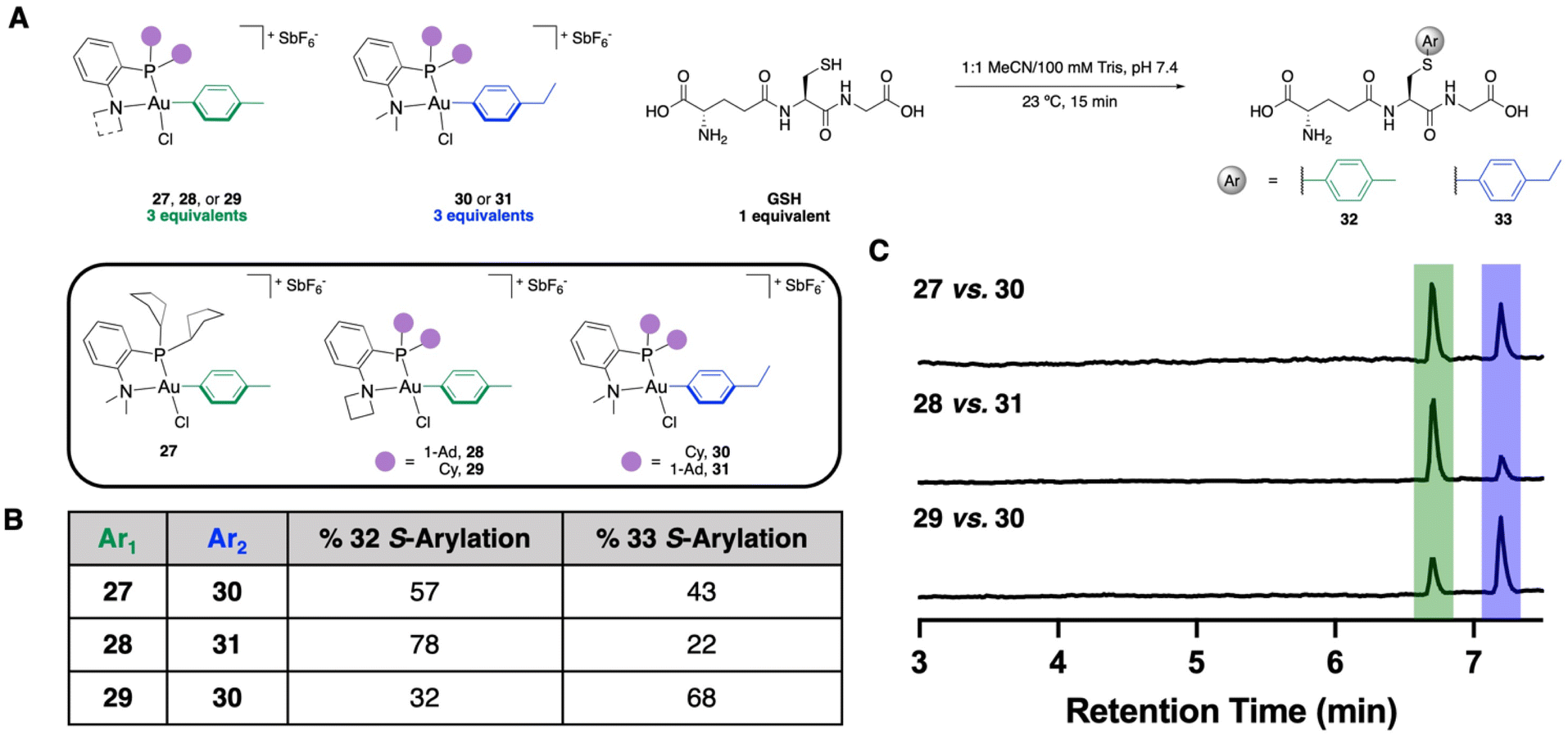 | ||
| Fig. 6 (A) Generalized scheme and reagents used for the S-arylation competition experiments. (B) Results of the S-arylation competition experiments, n = 3. See ESI† for further details. (C) Total ion chromatogram traces from a representative replicate from each LC-MS competition experiment. | ||
A competition experiment was performed between the NMe2-PCp2 (27) and NMe2-PCy2 (30) Au(III) OACs, which resulted in a 57:43 preference for the p-tolyl S-arylated product (32) over the p-ethylbenzene S-arylated product (33) (Fig. 6B and C). This preferential S-arylation from the NMe2-PCp2 OAC is in agreement with both the % VBur calculations as well as the kCo obtained from stopped-flow kinetics. To examine the amine fragments, competition experiments were performed using the PAd2-based P,N-ligated Au(III) OACs with azetidine (28) and dimethylamino (31) N-substitution. A 78:22 formation of 32:33 shows a preference for S-arylation from the azetidine-containing ligand (Fig. 6B and C), which is also in agreement with the % VBur calculations and the stopped-flow kinetics.
However, when the competition experiments were performed using the PCy2-based P,N-ligated Au(III) OACs using the azetidine (29) and dimethylamino (30) N-substituents, a 32:68 ratio for 32:33 was observed (Fig. 6B and C). This indicates preferential S-arylation from the dimethylamino-containing ligand 30. The % VBur calculations and the stopped-flow kinetics indicate that coordination is faster to 29, but we hypothesize that the reductive elimination is slower, allowing for a reversible coordination. This leads to a potential switch in the selectivity-determining step for this reaction and indicates that while the two ligand permutations are additive for the thiol coordination event, both elementary steps of the S-arylation need to be sufficiently fast to induce effective conjugation.
Reductive elimination rate monitoring
To examine the effects of the azetidine compounds on the reductive elimination, stopped-flow/UV-vis spectroscopy was performed using the conditions described vide supra (Fig. 7A, see ESI† for details). From Int1 of 28, a unimolecular rate constant was observed for the reductive elimination (kRE) of 2.66 s−1 (Fig. 7B), which is approximately three times slower than kRE of the NMe2-PAd2-ligated OAC 1 (Fig. 7D). This is also in line with the calculated ΔG‡RE for the phenyl OACs (Fig. 3B). Profiling the reductive elimination from Int1 of 29, a kRE of 0.133 s−1 was calculated (Fig. 7C), which is nearly ten times slower than the kRE for the NMe2-PCy2-ligated OAC 3 (Fig. 7D). These results confirm the proposed switch in selectivity-determining step for 29 as observed in the competition experiments. | ||
| Fig. 7 (A) Generalized scheme for reductive elimination monitoring from Int1. X− is used as a generalized counterion. Experiments were performed in 1:1 MeCN/100 mM Tris buffer at pH 7.4 and 20 °C. (B) Kinetic plot for the reductive elimination monitoring from 28-Int1, n = 5. (C) Kinetic plot for the reductive elimination monitoring from 29-Int1, n = 3 – some error bars are smaller than the markers. (D) kRE comparison for the amine and dialkylphosphine modifications. aThese parameters were determined previously.35 | ||
Altogether, this work demonstrates that % VBur can serve as a quickly calculated metric to screen the rate of bioconjugation to an Au(III) OAC, which will be valuable for the future screening of Au(III) and other metal complexes for bioconjugation. While the % VBur can be used to predict the rate of coordination to the Au(III) center, it can also be used to predict the rate of reductive elimination, though the trend is in the opposite direction (Fig. 8). These modifications that affect the steric bulk of the ligand may also modify the electronic properties of the complexes,54–56 which can impact both the rate of coordination to the metal center as well as the rate of reductive elimination.57–59 These factors should also be considered when designing new ligands for metal-catalyzed or metal-mediated transformations.
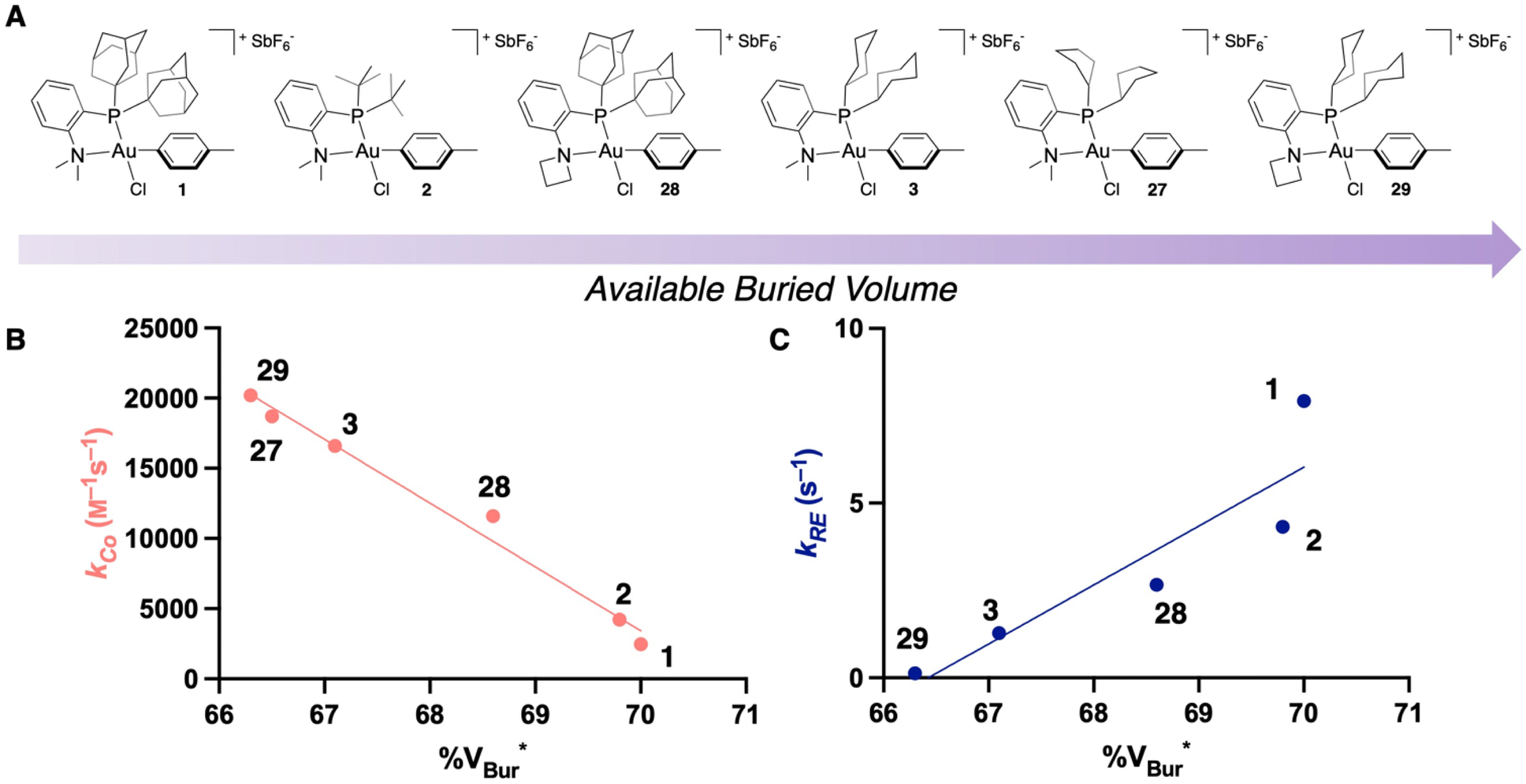 | ||
| Fig. 8 (A) Ordering of the experimentally characterized Au(III) OACs from this and previous studies in order of least to most available % VBur.35 (B) Plot of experimentally determined kCo against % VBur for the Au(III) OACs shown in Fig. 8A. *% VBur is for the phenyl OAC and the counterion is not included. (C) Plot of experimentally determined kRE against % VBur for the Au(III) OACs shown in Fig. 8A. *% VBur is for the phenyl OAC and the counterion is not included. | ||
We propose that this workflow should be amenable to other metal-mediated S-arylation reactions and provide a rapid way to screen the efficacy of these reagents. This work also provides fundamental mechanistic insight into the elementary steps of Au(III) chemistry,36,60–65 which is relatively understudied compared to Pd(II) and Pt(II) systems.
Conclusions
Thirteen Au(III) OACs were examined with eight different dialkylphosphines and five different amines in silico to determine their effects on both steps of the S-arylation reaction. Three novel Au(III) OACs were synthesized (27–29) and their bimolecular rate constants were determined to be between 11600–20,200 M−1 s−1. Finally, these OACs were examined in competition experiments to elucidate which of the two elementary steps of the S-arylation controls the selectivity, and a switch in the selectivity-determining step was confirmed experimentally for one of these reagents.
Data availability
All data are available free of charge in the ESI,† including experimental details, NMR spectra, characterization, kinetic data, and coordinates of computed structures. The raw data that support the findings of this study are available at https://github.com/jtreacy01/Ultrafaster/.Author contributions
Joseph W. Treacy: conceptualization, computation, synthesis, formal analysis, writing – original draft. James A. R. Tilden: formal analysis, writing – original draft. Elaine Y. Chao: computation, formal analysis. Zihuan Fu: synthesis. Alexander M. Spokoyny: conceptualization, supervision, writing – review and editing, funding acquisition. K. N. Houk: supervision, writing – review and editing, funding acquisition. Heather D. Maynard: supervision, writing – review and editing, funding acquisition. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare the following competing financial interest(s): A. M. S. and H. D. M. are co-inventors on several patent applications from UCLA associated with the Au(III) based bioconjugation technology.Acknowledgements
This work was funded in part by the NSF (CHE-2404202, CHE-2003946, and CHE-2153972) and the NIH (R01DK127908). A. M. S. thanks NIGMS (R35GM124746) for funding. J. W. T. thanks UCLA Dissertation Year Fellowship for support. Z. F. thanks National Institute of Health Chemistry-Biology Interface Training Grant (T32GM136614) for support. NMR spectrometers are supported in part by the S10 program of the NIH Office of Research Infrastructure Programs under grant S10OD028644. The stopped-flow/UV-vis spectrometer was supported by the NIH (1S10OD023584). This work used computational and storage services associated with the Hoffman2 Shared Cluster provided by the UCLA Institute for Digital Research and Education's Research Technology Group.References
- S. B. Gunnoo and A. Madder, Chemical Protein Modification through Cysteine, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed
.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed
- O. Boutureira and G. J. L. Bernardes, Advances in Chemical Protein Modification, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed
- A. D. Baldwin and K. L. Kiick, Tunable Degradation of Maleimide–Thiol Adducts in Reducing Environments, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed
- A. D. Baldwin and K. L. Kiick, Reversible maleimide–thiol adducts yield glutathione-sensitive poly(ethylene glycol)–heparin hydrogels, Polym. Chem., 2013, 4, 133–143 RSC
- E. H. Krenske, R. C. Petter and K. N. Houk, Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines, J. Org. Chem., 2016, 81, 11726–11733 CrossRef CAS PubMed
- S. Krishnan, R. M. Miller, B. Tian, R. D. Mullins, M. P. Jacobson and J. Taunton, Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis, J. Am. Chem. Soc., 2014, 136, 12624–12630 CrossRef CAS PubMed
- B. Shi and M. F. Greaney, Reversible Michael addition of thiols as a new tool for dynamic combinatorial chemistry, Chem. Commun., 2005, 886–888 RSC
- R. Walther, M. Park, N. Ashman, M. Welch, J. S. Carroll and D. R. Spring, Tuneable thiol exchange linkers for traceless drug release applications in prodrugs and ADCs, Chem. Commun., 2024, 60, 7025–7028 RSC
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Arylation Chemistry for Bioconjugation, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef CAS PubMed
- N. C. Price, M. Cohn and R. H. Schirmer, Fluorescent and spin label probes of the environments of the sulfhydryl groups of porcine muscle adenylate kinase, J. Biol. Chem., 1975, 250, 644–652 CrossRef CAS PubMed
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, π-Clamp-mediated cysteine conjugation, Nat. Chem., 2016, 8, 120–128 CrossRef CAS PubMed
- R. J. Taylor, M. Aguilar Rangel, M. B. Geeson, P. Sormanni, M. Vendruscolo and G. J. L. Bernardes, π-Clamp-Mediated Homo- and Heterodimerization of Single-Domain Antibodies via Site-Specific Homobifunctional Conjugation, J. Am. Chem. Soc., 2022, 144, 13026–13031 CrossRef CAS PubMed
- H. F. Motiwala, Y. H. Kuo, B. L. Stinger, B. A. Palfey and B. R. Martin, Tunable Heteroaromatic Sulfones Enhance in-Cell Cysteine Profiling, J. Am. Chem. Soc., 2020, 142, 1801–1810 CrossRef CAS PubMed
- K. C. Tang, S. M. Maddox, K. M. Backus and M. Raj, Tunable heteroaromatic azoline thioethers (HATs) for cysteine profiling, Chem. Sci., 2022, 13, 763–774 RSC
- B. M. Lipka, V. M. Betti, D. S. Honeycutt, D. L. Zelmanovich, M. Adamczyk, R. Wu, H. S. Blume, C. A. Mendina, J. M. Goldberg and F. Wang, Rapid Electrophilic Cysteine Arylation with Pyridinium Salts, Bioconjugate Chem., 2022, 33, 2189–2196 CrossRef CAS PubMed
- B. M. Lipka, D. S. Honeycutt, G. M. Bassett, T. N. Kowal, M. Adamczyk, Z. C. Cartnick, V. M. Betti, J. M. Goldberg and F. Wang, Ultra-rapid Electrophilic Cysteine Arylation, J. Am. Chem. Soc., 2023, 145, 23427–23432 CrossRef CAS PubMed
- C. Wan, Y. Zhang, J. Wang, Y. Xing, D. Yang, Q. Luo, J. Liu, Y. Ye, Z. Liu, F. Yin, R. Wang and Z. Li, Traceless Peptide and Protein Modification via Rational Tuning of Pyridiniums, J. Am. Chem. Soc., 2024, 146, 2624–2633 CrossRef CAS
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Organometallic palladium reagents for cysteine bioconjugation, Nature, 2015, 526, 687–691 CrossRef CAS PubMed
- J. A. R. Tilden, A. T. Lubben, S. B. Reeksting, G. Kociok-Köhn and C. G. Frost, Pd(II)-Mediated C−H Activation for Cysteine Bioconjugation, Chem.–Eur. J., 2022, 28, e202104385 CrossRef CAS PubMed
- S. Gukathasan, S. Parkin, E. P. Black and S. G. Awuah, Tuning Cyclometalated Gold(III) for Cysteine Arylation and Ligand-Directed Bioconjugation, Inorg. Chem., 2021, 60, 14582–14593 CrossRef CAS PubMed
- K. K.-Y. Kung, H.-M. Ko, J.-F. Cui, H.-C. Chong, Y.-C. Leung and M.-K. Wong, Cyclometalated gold(III) complexes for chemoselective cysteine modification via ligand controlled C–S bond-forming reductive elimination, Chem. Commun., 2014, 50, 11899–11902 RSC
- M. N. Wenzel, R. Bonsignore, S. R. Thomas, D. Bourissou, G. Barone and A. Casini, Cyclometalated Au-III Complexes for Cysteine Arylation in Zinc Finger Protein Domains: towards Controlled Reductive Elimination, Chem.–Eur. J., 2019, 25, 7628–7634 CrossRef CAS PubMed
- G. E. Mudd, S. J. Stanway, D. R. Witty, A. Thomas, S. Baldo, A. D. Bond, P. Beswick and A. Highton, Gold-Mediated Multiple Cysteine Arylation for the Construction of Highly Constrained Bicycle Peptides, Bioconjugate Chem., 2022, 33, 1441–1445 CrossRef CAS PubMed
- K. Hanaya, J. Ohata, M. K. Miller, A. E. Mangubat-Medina, M. J. Swierczynski, D. C. Yang, R. M. Rosenthal, B. V. Popp and Z. T. Ball, Rapid nickel(II)-promoted cysteine S-arylation with arylboronic acids, Chem. Commun., 2019, 55, 2841–2844 RSC
- L.-Z. Qin, H. Sun, X. Duan, S.-S. Zhu, J. Liu, M.-Y. Wu, X. Yuan, J.-K. Qiu and K. Guo, Visible-light-induced nickel-catalyzed selective S-arylation of peptides by exogenous-photosensitizer-free photocatalysis, Cell Rep. Phys. Sci., 2023, 4, 101292 CrossRef CAS
- V. Bacauanu, Z. N. Merz, Z. L. Hua and S. B. Lang, Nickel-Catalyzed Antibody Bioconjugation, J. Am. Chem. Soc., 2023, 145, 25842–25849 CrossRef CAS PubMed
- X. Chu, L. H. Shen, B. Li, P. Yang, C. Z. Du, X. Y. Wang, G. He, S. Messaoudi and G. Chen, Construction of Peptide Macrocycles via Palladium-Catalyzed Multiple S-Arylation: An Effective Strategy to Expand the Structural Diversity of Cross-Linkers, Org. Lett., 2021, 23, 8001–8006 CrossRef CAS PubMed
- P. Yang, X. Wang, B. Li, Y. Yang, J. Yue, Y. Suo, H. Tong, G. He, X. Lu and G. Chen, Streamlined construction of peptide macrocycles via palladium-catalyzed intramolecular S-arylation in solution and on DNA, Chem. Sci., 2021, 12, 5804–5810 RSC
- E. V. Vinogradova, Organometallic chemical biology: an organometallic approach to bioconjugation, Pure Appl. Chem., 2017, 89, 1619–1640 CrossRef CAS
- H. R. Montgomery, A. M. Spokoyny and H. D. Maynard, Organometallic Oxidative
Addition Complexes for S-Arylation of Free Cysteines, Bioconjugate Chem., 2024, 35, 883–889 CrossRef CAS PubMed
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, Organometallic Gold(III) Reagents for Cysteine Arylation, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS PubMed
- J. M. Stauber, A. L. Rheingold and A. M. Spokoyny, Gold(III) Aryl Complexes as Reagents for Constructing Hybrid Peptide-Based Assemblies via Cysteine S-Arylation, Inorg. Chem., 2021, 60, 5054–5062 CrossRef CAS PubMed
- E. A. Doud, J. A. R. Tilden, J. W. Treacy, E. Y. Chao, H. R. Montgomery, G. E. Kunkel, E. J. Olivares, N. Adhami, T. A. Kerr, Y. Chen, A. L. Rheingold, J. A. Loo, C. G. Frost, K. N. Houk, H. D. Maynard and A. M. Spokoyny, Ultrafast Au(III)-Mediated Arylation of Cysteine, J. Am. Chem. Soc., 2024, 146, 12365–12374 CrossRef CAS PubMed
- L. Rocchigiani and M. Bochmann, Recent Advances in Gold(III) Chemistry: Structure, Bonding, Reactivity, and Role in Homogeneous Catalysis, Chem. Rev., 2021, 121, 8364–8451 CrossRef CAS PubMed
- M. S. M. Holmsen, A. Nova and M. Tilset, Cyclometalated (N,C) Au(III) Complexes: The Impact of Trans Effects on Their Synthesis, Structure, and Reactivity, Acc. Chem. Res., 2023, 56, 3654–3664 CrossRef CAS PubMed
- K. Muratov, E. Zaripov, M. V. Berezovski and F. Gagosz, DFT-Enabled Development of Hemilabile (P∧N) Ligands for Gold(I/III) RedOx Catalysis: Application to the Thiotosylation of Aryl Iodides, J. Am. Chem. Soc., 2024, 146, 3660–3674 CrossRef CAS PubMed
- A. Genoux, M. Biedrzycki, E. Merino, E. Rivera-Chao, A. Linden and C. Nevado, Synthesis and Characterization of Bidentate (P^N)Gold(III) Fluoride Complexes: Reactivity Platforms for Reductive Elimination Studies, Angew. Chem., Int. Ed., 2021, 60, 4164–4168 CrossRef CAS PubMed
- J. Martín, E. Gómez-Bengoa, A. Genoux and C. Nevado, Synthesis of Cyclometalated Gold(III) Complexes via Catalytic Rhodium to Gold(III) Transmetalation, Angew. Chem., Int. Ed., 2022, 61, e202116755 CrossRef PubMed
- S.-L. Zhang and J.-J. Dong, Mechanism and chemoselectivity origins of bioconjugation of cysteine with Au(III)-aryl reagents, Org. Biomol. Chem., 2019, 17, 1245–1253 RSC
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed
- G. L. Perry and N. D. Schley, Tris(bicyclo[1.1.1]pentyl)phosphine: An Exceptionally Small Tri-tert-alkylphosphine and Its Bis-Ligated Pd(0) Complex, J. Am. Chem. Soc., 2023, 145, 7005–7010 CrossRef CAS PubMed
- G. L. Perry and N. D. Schley, Bis(bicyclo[1.1.1]pentyl)chlorophosphine as a Precursor for the Preparation of Bis(bicyclo[1.1.1]pentyl)phosphines, Org. Lett., 2024, 26, 6071–6075 CrossRef CAS PubMed
- S. M. Crawford, C. A. Wheaton, V. Mishra and M. Stradiotto, Probing the effect of donor-fragment substitution in Mor-DalPhos on palladium-catalyzed C–N and C–C cross-coupling reactivity, Can. J. Chem., 2018, 96, 578–586 CrossRef CAS
- M. A. MacLean, C. A. Wheaton and M. Stradiotto, Developing backbone-modified Mor-DalPhos ligand variants for use in palladium-catalyzed C–N and C–C cross-coupling, Can. J. Chem., 2018, 96, 712–721 CrossRef CAS
- B. J. Tardiff, R. McDonald, M. J. Ferguson and M. Stradiotto, Rational and Predictable Chemoselective Synthesis of Oligoamines via Buchwald–Hartwig Amination of (Hetero)Aryl Chlorides Employing Mor-DalPhos, J. Org. Chem., 2012, 77, 1056–1071 CrossRef CAS PubMed
- P. G. Alsabeh, R. J. Lundgren, R. McDonald, C. C. C. Johansson Seechurn, T. J. Colacot and M. Stradiotto, An Examination of the Palladium/Mor-DalPhos Catalyst System in the Context of Selective Ammonia Monoarylation at Room Temperature, Chem.–Eur. J., 2013, 19, 2131–2141 CrossRef CAS PubMed
- R. Lundgren, K. Hesp and M. Stradiotto, Design of New ‘DalPhos’ P,N-Ligands: Applications in Transition-Metal Catalysis, Synlett, 2011, 2011, 2443–2458 CrossRef
- A. J. Mallek, B. L. Pentelute and S. L. Buchwald, Selective N-Arylation of p -Aminophenylalanine in Unprotected Peptides with Organometallic Palladium Reagents, Angew. Chem., Int. Ed., 2021, 60, 16928–16931 CrossRef CAS PubMed
- H. W. Heine, B. L. Kapur, J. L. Bove, R. W. Greiner, K. H. Klinger and C. Mitch, The Synthesis of Some N-Arylethylenimines, J. Am. Chem. Soc., 1954, 76, 2503 CrossRef CAS
- Y. Li, L.-Q. Lu, S. Das, S. Pisiewicz, K. Junge and M. Beller, Highly Chemoselective Metal-Free Reduction of Phosphine Oxides to Phosphines, J. Am. Chem. Soc., 2012, 134, 18325–18329 CrossRef CAS PubMed
- J. W. M. MacMillan, R. T. McGuire, A. M. McMahon, T. S. Anderson, K. N. Robertson and M. Stradiotto, DalPhos on Demand: Facile Ligand Generation Enables New Ligand Discovery and Expedient Catalyst Screening, ACS Catal., 2024, 14, 4074–4081 CrossRef CAS
- C. A. Tolman, Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis, Chem. Rev., 1977, 77, 313–348 CrossRef CAS
- D. J. Durand and N. Fey, Computational Ligand Descriptors for Catalyst Design, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed
- T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’Addario, M. S. Sigman and A. Aspuru-Guzik, A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed
- J. F. Hartwig, Electronic Effects on Reductive Elimination To Form Carbon−Carbon and Carbon−Heteroatom Bonds from Palladium(II) Complexes, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS PubMed
- R. Martin and S. L. Buchwald, Palladium-Catalyzed Suzuki−Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed
- J. W. Treacy, E. Y. Chao, G. E. Kunkel, T. Louis-Goff, J. A. R. Tilden, A. M. Spokoyny, H. D. Maynard and K. N. Houk, Electronic Effects of Bidentate P,N -Ligands on the Elementary Steps of Au(I)/Au(III) Reactions Relevant to Cross-Coupling Chemistry, Org. Lett., 2024, 26, 10875–10879 CrossRef CAS PubMed
- M. S. Winston, W. J. Wolf and F. D. Toste, Halide-Dependent Mechanisms of Reductive Elimination from Gold(III), J. Am. Chem. Soc., 2015, 137, 7921–7928 CrossRef CAS PubMed
- A. Casini, In My Element : Gold, Chem.–Eur. J., 2019, 25, 12234 CrossRef CAS
- M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, Oxidative Addition, Transmetalation, and Reductive Elimination at a 2,2′-Bipyridyl-Ligated Gold Center, J. Am. Chem. Soc., 2018, 140, 4440–4445 CrossRef CAS PubMed
- D. Vesseur, S. Li, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, Ligand-Enabled Oxidative Fluorination of Gold(I) and Light-Induced Aryl–F Coupling at Gold(III), J. Am. Chem. Soc., 2024, 146, 11352–11363 CAS
- C. C. Chintawar, V. W. Bhoyare, M. V. Mane and N. T. Patil, Enantioselective Au(I)/Au(III) Redox Catalysis Enabled by Chiral (P,N)-Ligands, J. Am. Chem. Soc., 2022, 144, 7089–7095 CrossRef CAS PubMed
- A. Portugués, M. Á. Martínez-Nortes, D. Bautista, P. González-Herrero and J. Gil-Rubio, Reductive Elimination Reactions in Gold(III) Complexes Leading to C(sp3)–X (X = C, N, P, O, Halogen) Bond Formation: Inner-Sphere vs. SN2 Pathways, Inorg. Chem., 2023, 62, 1708–1718 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05920d |
| This journal is © The Royal Society of Chemistry 2025 |