
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenaelectro-catalyzed C–H phosphorylation: ortho to para position-selectivity switch†
Xue-Ya
Gou‡
,
João C. A.
Oliveira‡
 ,
Shan
Chen
,
Simon L.
Homölle
,
Sven
Trienes
,
Tristan
von Münchow
,
Bo-Sheng
Zhang
and
Lutz
Ackermann
*
,
Shan
Chen
,
Simon L.
Homölle
,
Sven
Trienes
,
Tristan
von Münchow
,
Bo-Sheng
Zhang
and
Lutz
Ackermann
*
Wöhler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 29th November 2024
Abstract
The position-selective C–H bond activation of arenes has long been a challenging topic. Herein, we report an expedient ruthenium-electrocatalyzed site-selective ortho-C–H phosphorylation of arenes driven by electrochemical hydrogen evolution reaction (HER), avoiding stoichiometric amounts of chemical redox-waste products. This strategy paved the way to achieve unprecedented ruthenaelectro-catalyzed para-C–H phosphorylation with excellent levels of site-selectivity. This electrocatalytic approach was characterized by an ample substrate scope with a broad range of arenes containing N-heterocycles, as well as several aryl/alkylphosphine oxides were well tolerated. Moreover, late-stage C–H phosphorylation of medicinal relevant drugs could also be achieved. DFT mechanistic studies provided support for an unusual ruthenium(III/IV/II) regime for the ortho-C–H phosphorylation.
Introduction
Organophosphorus compounds have been widely employed in organic synthesis,1 medicinal chemistry,2 material sciences,3 as well as prominent ligands4 in catalytic reactions. The presence of a phosphoryl group in target molecules has often been reported to enhance their physical-chemical properties, such as their hydrophilicity, thereby improving their solubility and tolerance in biological systems.5 Compounds bearing phosphine oxide motifs have revealed potential in decreasing inflammation, reducing blood sugar, and even anti-HIV activity.6 Additionally, phosphorylated heteroarenes have been reported as the one of the main components in phosphorescent OLEDs, due to their excellent luminescence properties (Scheme 1a).7 In this context, the development of novel and efficient strategies for the construction of aromatic C–P bonds poses great significance.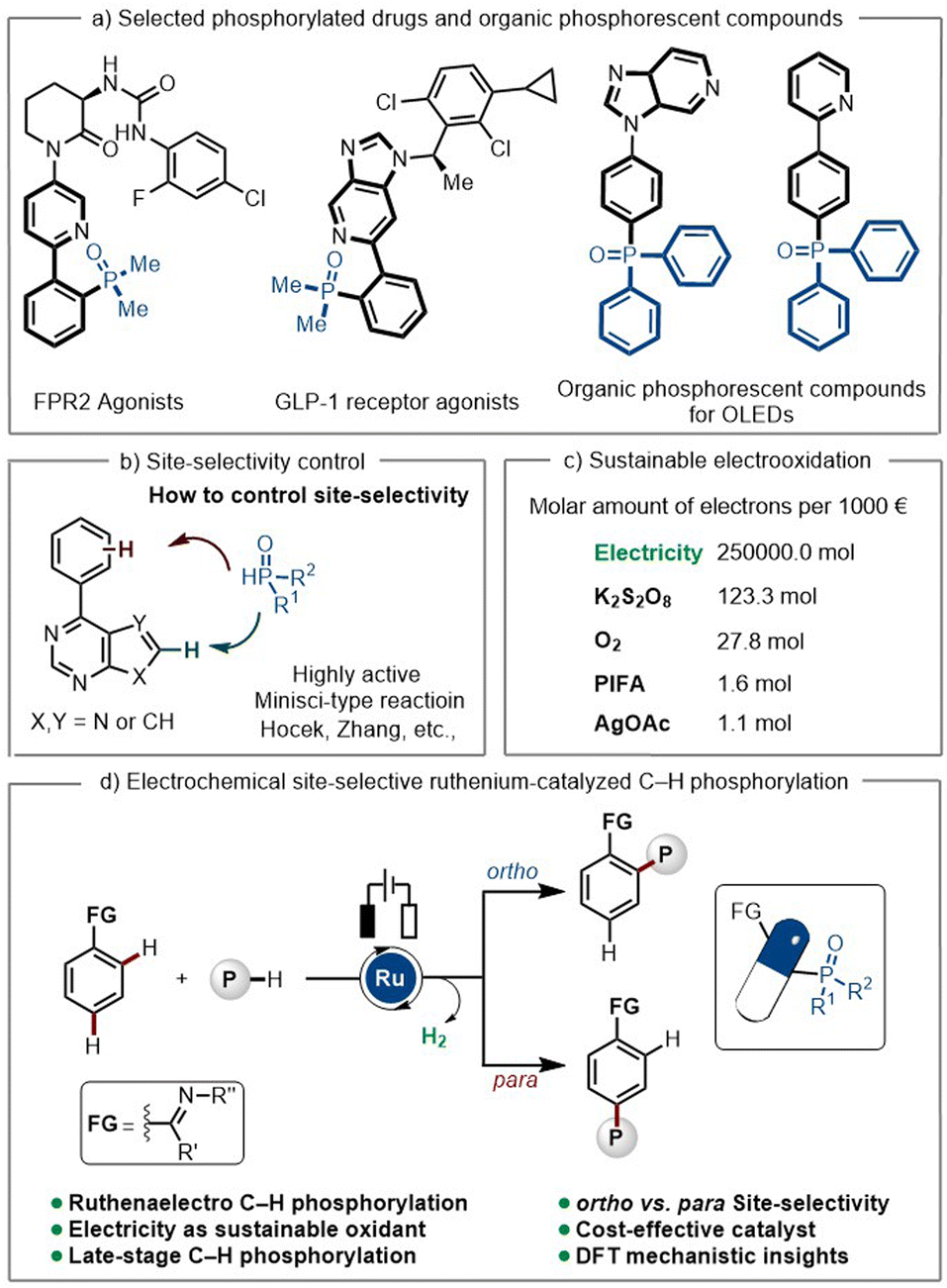 | ||
| Scheme 1 Electrochemical ruthenium-catalyzed site-selective C–H phosphorylation. | ||
Transition metal-catalyzed C–H activation has emerged as a powerful tool in modern synthesis.8 Direct C–H phosphorylation of arenes has proven to be challenging due to the strong coordination ability of the phosphine reagents, which often leads to catalyst deactivation. To overcome this drawback, distinct strategies have been developed. These involve the slow addition of the phosphine reagents,9 the use of masked phosphine reagents, which slowly release the active phosphine compounds10 or their sequential addition.11 However, these approaches involve the use of stoichiometric amounts of chemical oxidants, which strongly jeopardizes the sustainability of the overall approaches. On a different note, phosphine radical-based strategies for the synthesis of aryl phosphonyl compounds have been limited to electron-rich arenes, with poor position-selectivity.12 Furthermore, Minisci-type reactivity of nitrogen-containing heterocycles based on phosphine radicals is predominant,13 rendering phosphorylation at remote position difficult.
Ruthenium catalysis has surfaced as a uniquely versatile platform for proximal and distal bond functionalizations.14–16 Hence, we wondered whether position-selective ruthenium-catalyzed C–H phosphorylation would be viable in a position-selectivity-divergent manner by the judicious choice of the reaction conditions. The emergence of electrochemistry applied to organic synthesis has strongly revolutionized molecular synthesis by avoiding the use of chemical oxidants, leading to more sustainable and environmentally friendly synthetic routes,17 such as in transition metal-catalyzed C–H activation.18–23
Electrochemically24 driven ortho-C–H phosphorylation has solely been accomplished with expensive rhodium catalysts25 or through nickel catalysis, employing high-temperature conditions (110 °C, DG = 8-aminoquinoline).26 In sharp contrast, studies on metalla-electrocatalyzed C–H phosphorylations with versatile ruthenium catalysts have thus far proven elusive. Herein, we report a mild, electrochemically driven and cost-effective position-selective ruthenaelectro-catalyzed C–H phosphorylation with controlled position-switch from ortho to para. Moreover, phosphonyl units could be successfully introduced into relevant pharmaceutical compounds via late-stage C–H phosphorylation to access structurally diverse active compounds in a single step.
Results and discussion
Reaction optimization of ortho-C–H phosphorylation
We initiated our studies by exploring the envisioned ruthenium-electrocatalyzed ortho-C–H phosphorylation of arene 1a and diphenylphosphine oxide 2a using a graphite felt (GF) and a platinum (Pt) electrode as anode and cathode materials, respectively, in an undivided cell setup (Table 1). The use of [Ru(OAc)2(p-cymene)] as the catalyst, in the presence of HFIP as the solvent, led to the formation of the desired phosphorylated product 3a, with complete ortho-selectivity, in 73% isolated yield (entry 1). Several other solvents and bases were also considered, but significantly reduced efficacy was noted (entries 2–3). Furthermore, other transition metal catalysts, such as Co(OAc)2·4H2O, Cu(OAc)2 or Ni(DME)Cl2, gave unsatisfactory results (entry 4). Control experiments demonstrated that electricity is crucial for the C–H phosphorylation (entry 6).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 3ab(%) |
| a Reaction conditions A: undivided cell, GF anode, Pt cathode, 1a (0.3 mmol), 2a (4.0 equiv.), [Ru(OAc)2(p-cymene)] (10 mol%), NaOAc (4.0 equiv.), HFIP: 1,1,1,3,3,3-hexafluoropropan-2-ol (3.0 mL), air, 65 °C, 3.5 mA, 24 h. b Isolated yields. c Co(OAc)2·4H2O (10 mol%)/Cu(OAc)2 (10 mol%)/Ni(DME)Cl2 (10 mol%). | ||
| 1 | None | 73 |
| 2 | H2O/CH3CN/TFE as solvent | −/−/65 |
| 3 | Na2CO3/Na3PO4/NaOPiv as base | 62/66/53 |
| 4c | Co(OAc)2·4H2O/Cu(OAc)2/Ni(DME)Cl2 | Trace/0/0 |
| 5 | N2 instead of air | 72 |
| 6 | Without electricity | 0 |
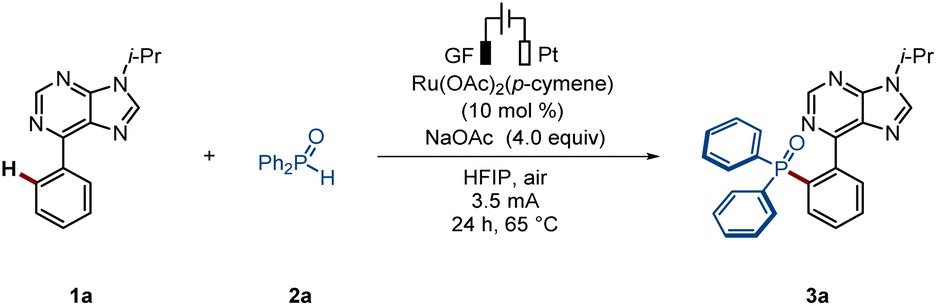
Substrate scope investigation for the ortho-C–H phosphorylation
With the optimized reaction conditions in hand, we first investigated the viable scope of the ruthenium-electrocatalyzed ortho-C–H phosphorylation (Scheme 2). First, various N-substituted arylpurines were probed, including bio-relevant 6-phenylpurine nucleosides, efficiently affording the desired products in good yields (3a–3d). Additionally, both electron-withdrawing groups (e.g., –F) and electron-donating groups (e.g., –Me) on the arenes were well tolerated (3e–3f). Second, a bipyridine-substituted arene was also tolerated, delivering solely the mono-phosphorylation product (3g). Moreover, oxime derivatives selectively delivered the desired product (3i). Third, various types of arenes were explored, featuring a wide range of oxazolinyl, pyrazyl or pyridyl substituents (3j–3n). Afterwards, the scope of phosphorus coupling partners was further investigated. Fourth, diphenylphosphine oxides bearing electron-donating and electron-withdrawing groups were shown to be suitable coupling partners leading to the formation of the expected products 3o–3s in good yields. Other pentavalent phosphine oxides, comprised of phenylalkyl phosphine oxides, dithienyl phosphine oxides, and commonly used dimethyl phosphine oxides, in phosphorylated drugs, have been demonstrated to be successful coupling partners (3t–3w).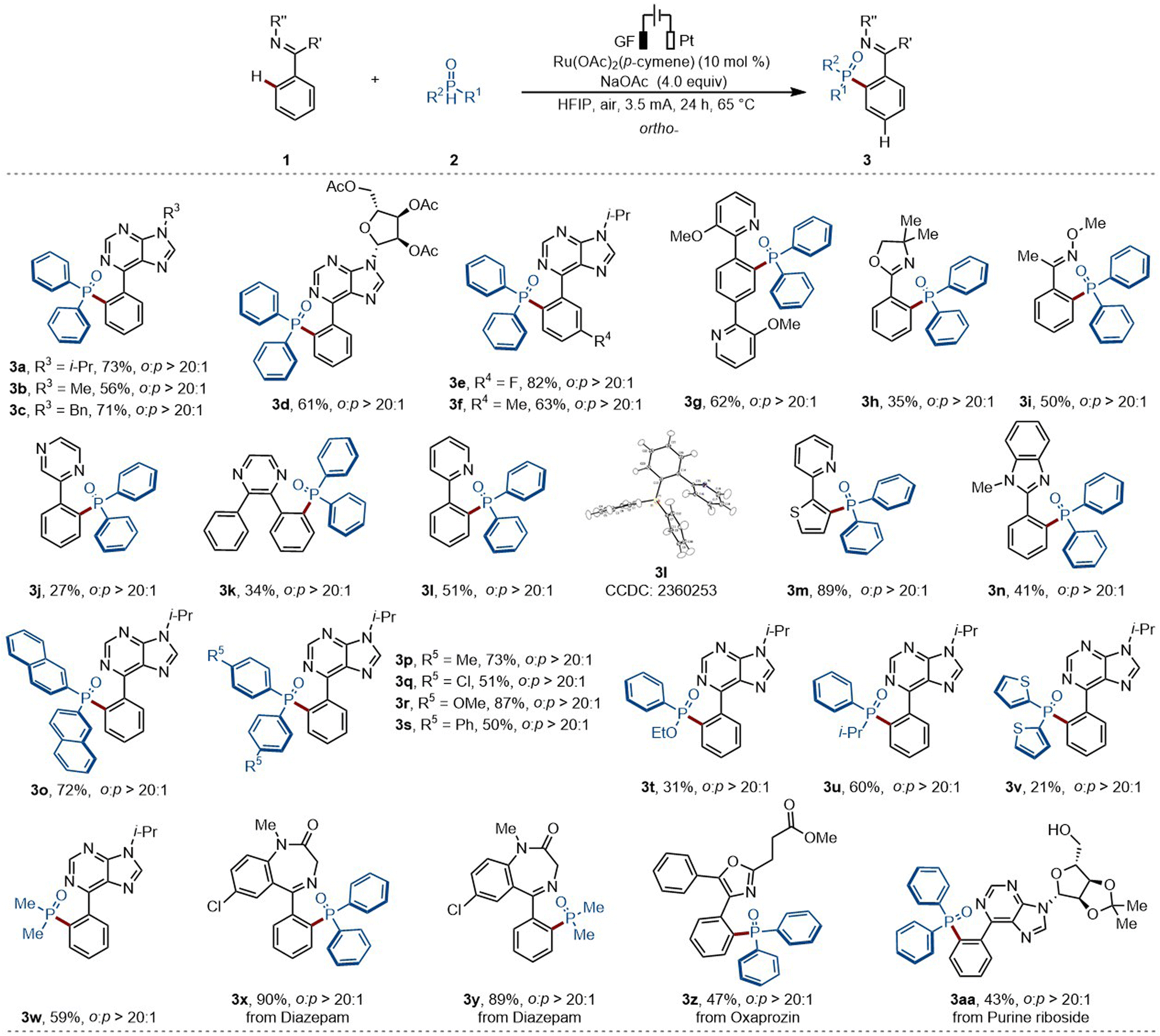 | ||
| Scheme 2 Scope of the ortho-selective C–H phosphorylation. | ||
Phosphorylation motifs can confide biological activity to drug molecules.5 In this context, the direct installation of phosphorylation units into target molecules, accounts for a more sustainable access to structural diversity, facilitating the expansion of the chemical space. Hence, we wondered whether our strategy could be exploited for late-stage C–H phosphorylations. To our delight, diazepam - a therapeutic drug used for acute tension and anxiety states - proved to be an amenable substrate, delivering the desired products 3x–3y in high yields. Additionally, oxaprozin and 6-phenylpurine riboside, delivered the desired products (3z–3aa) with excellent levels of position-selectivity.
Mechanistic studies of ortho-C–H phosphorylation
To gain insights into the reaction mechanism, a series of control experiments were conducted. The addition of equimolar amounts of TEMPO resulted in the inhibition of the ortho-C–H phosphorylation. However, given that TEMPO is a good reducing agent,27 such observations do not conclusively support the formation of a P-centered radical. Therefore, additional mechanistic experiments were conducted in the presence of representative radical scavengers, including 1,2-diphenylethylene and vinylcyclopropane 6, which is known to undergo ring opening in the presence of radial species. As a result, the ortho-phosphorylation product 3a was obtained in comparable isolated yields of 63% and 68%, respectively. These findings provide support for a non-radical pathway (Scheme 3a). Next, the addition of D2O as co-solvent resulted in a H/D scrambling at the ortho-position, suggesting a reversible ortho-C–H activation (Scheme 3b).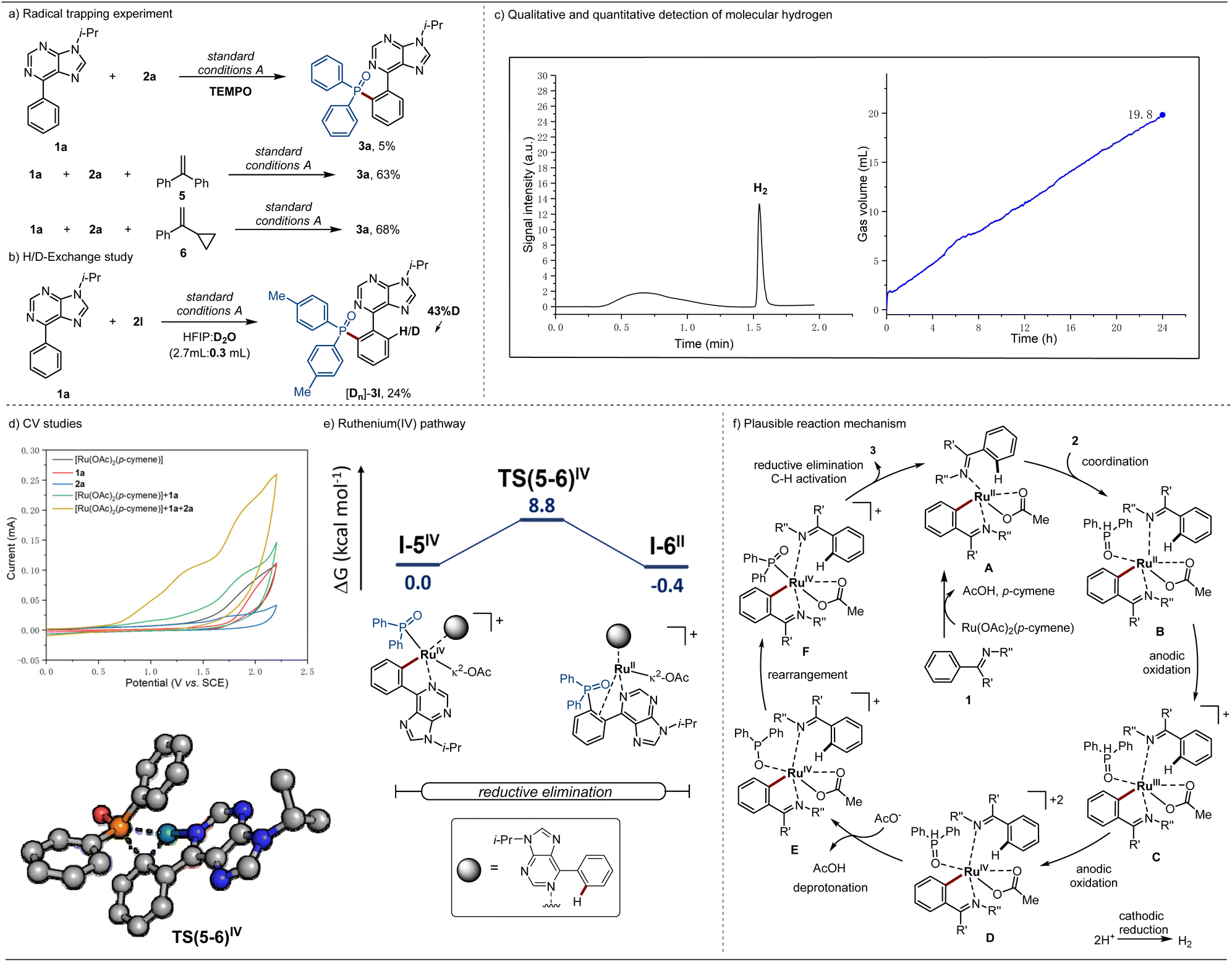 | ||
| Scheme 3 (a) Radical trapping experiment. (b) H/D-Exchange study. (c) Headspace GC analysis after catalysis (left side) and measurement of gas evolution during catalysis (right side). (d) Cyclic voltammetry studies. (e) Relative Gibbs free energies (ΔG338.15) are given in kcal mol−1 for the ruthenium-catalyzed ortho-C–H phosphorylation reductive elimination step at the PBE0-D4/def2-TZVPP-SMD(HFIP)//PBE0-D3BJ/def2-SVP level of theory. In the computed transition state structure, non-relevant hydrogens were omitted for clarity. (f) Plausible reaction mechanism for the ruthenaelectro-catalyzed ortho-C–H phosphorylation.27 | ||
In order to gain insights into the cathodic process, headspace gas chromatography was used and the formation of molecular hydrogen was probed. Thus, we monitored and quantified the formation of molecular hydrogen during the electrocatalytic reaction, which was determined to be 19.8 mL by the end of the electrocatalysis, translating into a faradaic efficiency of 59% (Scheme 3c and ESI Fig. S2, S3†). The result confirmed the hydrogen evolution reaction (HER) as the primary cathodic process, highlighting the unique potential of ruthenium electrocatalysis as a sustainable technology for organic synthesis.
Additionally, cyclic voltammetric (CV) experiments were conducted to assess the redox potential of the substrates as well as the catalyst (Scheme 3d). For the ortho-C–H phosphorylation, the mixture of phenyl-9H-purine (1a), diphenylphosphine oxide (2a), and the catalyst [Ru(OAc)2(p-cymene)] exhibited oxidation peaks at Ep/2 = 0.95 V and Ep/2 = 1.26 V vs. SCE. These findings are indicative of a ruthenacycle generated after C–H activation being coordinated by SPO 2a.
The catalyst mode of action for the ruthenium-electrocatalyzed ortho-C–H phosphorylation was further investigated through DFT calculations at the PBE0-D4/def2-TZVPP-SMD(HFIP)//PBE0-D3BJ/def2-SVP level of theory (Scheme 3e, Fig. S8 and S9, in the ESI†).28 Upon phosphine coordination two single electron oxidation steps take place, leading to the formation of the ruthenium(IV) intermediate I–1IV (Fig. S8†). Such is consistent with the CV studies, where two oxidation peaks were observed in the presence of phosphine. Subsequently, a facile phosphine deprotonation takes place (TS(2-3)) with an energy barrier of 6.8 kcal mol−1 giving rise to intermediate I-3, which after rearrangement originates a more exergonic intermediate I-5. The latter undergoes reductive elimination through transition state TS(5-6) with an energy barrier of 8.8 kcal mol−1. Additionally, an alternative pathway for reductive elimination under ruthenium(III) was also investigated (Fig. S9†). The latter has proven to be energetically disfavored, not only by the prohibitive calculated barrier of 40.3 kcal mol−1 but also by the formation of an endergonic ruthenium(I) intermediate I-6I. Such observations provide support for a ruthenium(III/IV/II) regime.
Based on our experimental and computational mechanistic studies, a plausible reaction mechanism is depicted in Scheme 3f. The ortho-phosphorylation commences with the ortho-C–H activation, forming the ruthenacycle complex A. Subsequently, ligand exchange occurs, leading to a more easily oxidized intermediate B, which after two single electron oxidation steps originates the ruthenium(IV) intermediate D. Then, deprotonation gives rise to intermediate E, which rearranges to form intermediate F. Finally, F undergoes reductive elimination to yield the desired product 3 and to regenerate the active ruthenium(II) catalyst A.
Reaction optimization of para-C–H phosphorylation
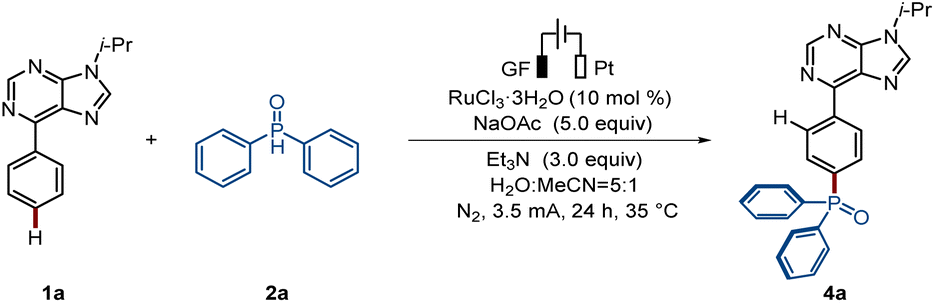
Next, we focused our attention on the envisioned switch towards ruthenaelectro-catalyzed para-C–H phosphorylation. We began our studies with arene 1a and diphenylphosphine oxide 2a using graphite felt (GF) and platinum (Pt) electrodes as anode and cathode materials, respectively, in an undivided cell setup (Table 2). When RuCl3·3H2O was used as a catalyst in the presence of MeCN as solvent a minor amount of para-C–H phosphorylated product was obtained. Upon further experimentation, the para-phosphorylated product 4a was selectively obtained in an isolated yield of 82% in the presence of RuCl3·3H2O, triethylamine, and a solvent mixture of MeCN and water (Entry 1). Notably, only unconverted substrate 1a was accounted for in the mass balance here, with no side product, from possible phosphorylation at the purine C–H bond, being detected. Other tested solvents and bases did not improve the efficacy of the reaction (entries 3–4). Moreover, the electricity was shown to be essential for the catalysis to take place (entry 6).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 4ab (%) |
a Reaction conditions B: undivided cell, GF anode, Pt cathode, 1a (0.3 mmol), 2a (4.0 equiv.), [RuCl3·3H2O] (10 mol%), NaOAc (5.0 equiv.), Et3N (3.0 equiv.), MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = (0.5:2.5 mL), N2, 35 °C, 3.5 mA, 24 h.
b Isolated yields.
c PPh3 (10 mol%), 2,2′-bipyridine (10 mol%). :H2O = (0.5:2.5 mL), N2, 35 °C, 3.5 mA, 24 h.
b Isolated yields.
c PPh3 (10 mol%), 2,2′-bipyridine (10 mol%).
|
||
| 1 | None | 82 |
| 2 | Under air | 66 |
| 3 | H2O/CH3CN/TFE as solvent | 25/12/— |
| 4 | Na2CO3/Na3PO4/NaOPiv as base | 78/55/23 |
| 5c | PPh3/2,2′-bipyridine as ligand | 55/42 |
| 6 | Without electricity | 0 |
Substrate scope investigation for the para-C–H phosphorylation
With the optimized reaction conditions in hand, we directed our attention to the viable substrate scope of the ruthenium-electrocatalyzed para-C–H phosphorylation (Scheme 4a). Several N-substituted 6-phenylpurines were explored, enabling the formation of the corresponding desired products in good yields with exclusive para-selectivity (4a–4d). Furthermore, both electron-withdrawing groups (–F) and electron-donating (–Me) on the arenes were well tolerated (4e–4f). Diphenylphosphine oxide compounds bearing different substituents (–Cl, –Me, –OMe, –OCF3) were also well tolerated (4g–4k). Noticeably, the 1-phenylpyrazole yielded the expected single para-C–H phosphorylation product, with no phosphorylation occurring at the pyrazole ring (4m). Arenes bearing different substituents, such as pyrimidyl, pyridyl, or tetrazolyl groups, demonstrated higher efficacy, in the absence of triethylamine, with excellent para-selectivity (4o–4q). Moreover, our approach could be successfully applied to the late-stage C–H phosphorylation of 6-phenylpurine riboside, delivering the desired product (4r) with excellent position-selectivity.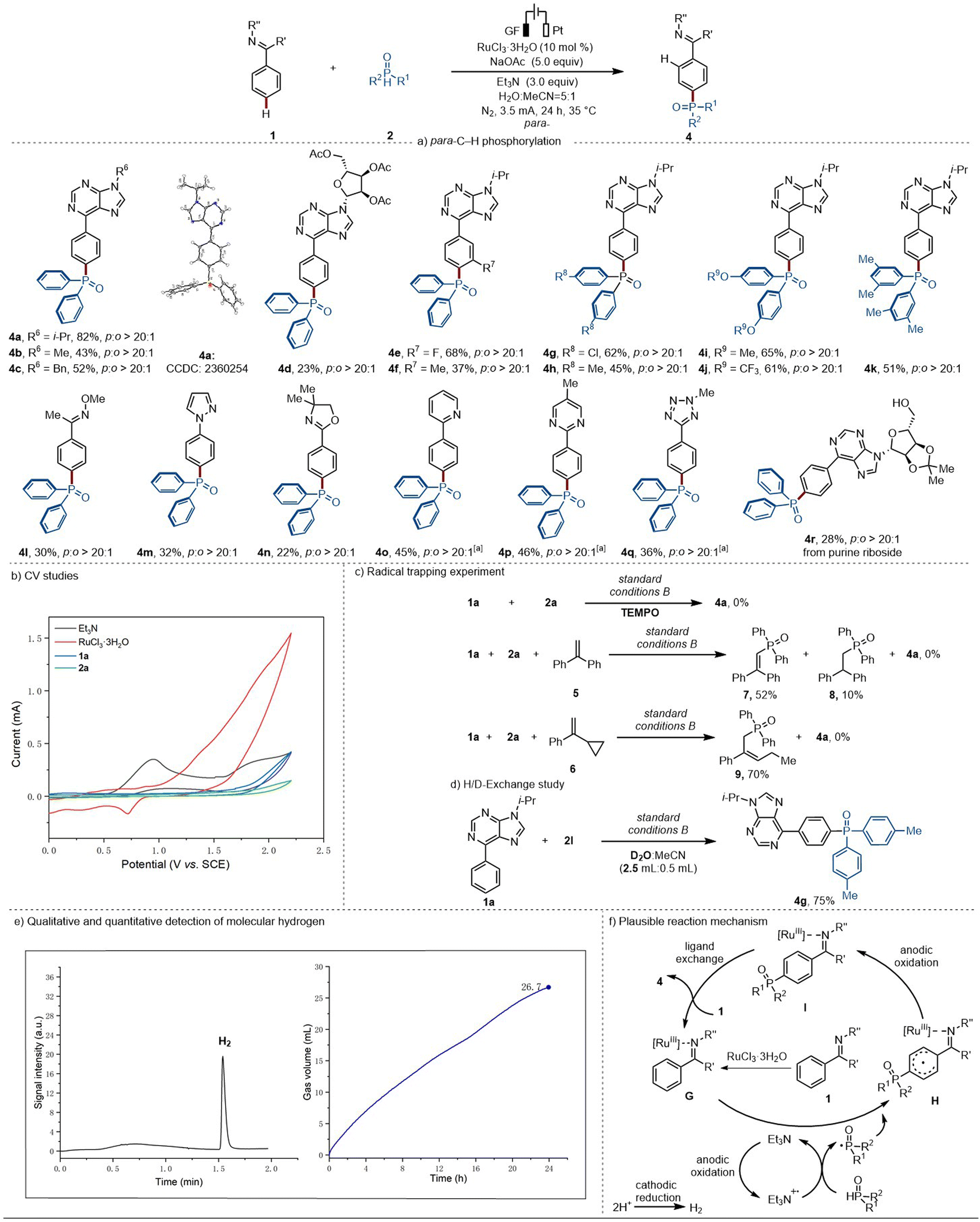 | ||
| Scheme 4 (a) Scope of the ruthenium-electrocatalyzed para-selective C–H phosphorylation, ano Et3N. (b) Cyclic Voltammograms. (c) Radical trapping experiment. Yield ratio calculated by phosphorus NMR from a mixture of 7 and 8. (d) H/D-exchange study. (e) Headspace GC analysis after catalysis (left side) and measurement of gas evolution during catalysis (right side). (f) Plausible reaction mechanism involved in the ruthenaelectro-catalyzed of para-C–H phosphorylation. | ||
Mechanistic studies for the para-C–H phosphorylation
To gain mechanistic insights into the ruthenium-electrocatalyzed para-C–H phosphorylation, cyclic voltammetry (CV) experiments were conducted to access the redox potential of the substrates as well as the catalyst (Scheme 4b). The Et3N features an oxidation peak at Ep/2 = 0.91 V vs. SCE, which is much lower than the one obtained for 1a, 2a, and RuCl3·3H2O, indicating that Et3N is more susceptible to be oxidized.Additionally, a series of control experiments were performed. The addition of TEMPO under otherwise identical reaction conditions inhibited the para-C–H phosphorylation. Moreover, upon the addition of 1,2-diphenylethylene, the corresponding radical intermediates 7 and 8 were trapped. When vinylcyclopropane 6 was added, the compound resulting from the ring opening (9) could be isolated in 70% yield. These findings provide strong support for the involvement of a phosphorus-centered radical in the para-phosphorylation (Scheme 4c). However, when D2O was added to the para-phosphorylation reaction, no H/D-scrambling was observed, suggesting that an ortho-C–H cycloruthenation is not relevant for the para-reaction pathway (Scheme 4d).
Additionally, we monitored and quantified the formation of molecular hydrogen during the electrocatalytic reaction, which was determined to be 26.7 mL by the end of the reaction time, translating into a faradaic efficiency of 68% (Scheme 4e and ESI Fig. S4, S5†). This provides support for the hydrogen evolution reaction (HER) to be the primary cathodic process.
Based on our experimental mechanistic studies, a plausible reaction mechanism is depicted in Scheme 4f. For the para-phosphorylation pathway, the nitrogen-containing heteroarene first coordinates to the ruthenium(III) catalyst to form intermediate G. Then, Et3N·+ may react with H-phosphonate or H-phosphine oxide to give rise to a phosphorus-centered radical. Subsequently, the electrophilic phosphine radical will attack at the para-position of the arene via a charge transfer-directed approach,29 followed by oxidative aromatization to generate the para-phosphorylated product. In the meanwhile, at the cathode, protons are reduced to generate molecular hydrogen by HER.
Conclusions
In conclusion, we have devised a position-selectivity switch for electrochemical ruthenium-catalyzed C–H phosphorylations enabled by hydrogen evolution reaction (HER). Thereby, we achieved selective ortho- and even para-C–H phosphorylation. The robustness of the ruthena-electrocatalysis was reflected by a wide substrate scope including various sensitive electrophilic functional groups. Our strategy thereby enabled challenging late-stage phosphorylations of biorelevant pharmaceuticals. Experimental and computational mechanistic studies provided strong support for an unusual ruthenium(III/IV/II) manifold for the ruthenaelectro-catalyzed proximal phosphorylation.Data availability
All data associated with this study are available in the article and ESI.†Author contributions
Conceptualization, L. A.; methodology, X.-Y. G.; investigation, X.-Y. G.; DFT calculation, J. C. A. O.; cyclic voltammetry studies, S. L. H.; headspace GC analysis and measurement, S .T.; HRMS studies, T. v. M.; writing – original Draft, X.-Y. G., B.-S. Z. and J. C. A. O.; writing – review & editing, X.-Y. G., J. C. A. O., and S. C.; funding acquisition, L. A.; resources, L. A.; supervision, L. A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the ERC Advanced grant no. 101021358, the DFG (Gottfried Wilhelm Leibniz award to L. A., SPP2363). We thank Dr Christopher Golz (University of Göttingen) for the assistance with the X-ray diffraction analysis,30 Dr Holm Frauendorf for HRMS analysis and Dr Michael John for NMR spectroscopy.Notes and references
- (a) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Review on Modern Advances of Chemical Methods for the Introduction of a Phosphonic Acid Group, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS; (b) Y.-Y. Zhu, T. Zhang, L. Zhou and S.-D. Yang, Concise synthesis of N-phosphorylated amides through three-component reactions, Green Chem., 2021, 23, 9417–9421 RSC.
- T. D. Baguley, H.-C. Xu, M. Chatterjee, A. C. Nairn, P. J. Lombroso and J. A. Ellman, Substrate-Based Fragment Identification for the Development of Selective, Nonpeptidic Inhibitors of Striatal-Enriched Protein Tyrosine Phosphatase, J. Med. Chem., 2013, 56, 7636–7650 CrossRef CAS.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Conventional and Unconventional Metal–Organic Frameworks Based on Phosphonate Ligands: MOFs and UMOFs, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS.
- (a) J. Holz, H. Jiao, M. Gandelman and A. Börner, About the Inversion Barriers of P-Chirogenic Triaryl-Substituted Phosphanes, Eur. J. Org Chem., 2018, 2984–2994 CrossRef CAS; (b) Y.-N. Ma, S.-X. Li and S.-D. Yang, New Approaches for Biaryl-Based Phosphine Ligand Synthesis via P═O Directed C–H Functionalizations, Acc. Chem. Res., 2017, 50, 1480–1492 CrossRef CAS PubMed.
- C. A. Tian and C. C. Chiu, Importance of Hydrophilic Groups on Modulating the Structural, Mechanical, and Interfacial Properties of Bilayers: A Comparative Molecular Dynamics Study of Phosphatidylcholine and Ion Pair Amphiphile Membranes, Int. J. Mol. Sci., 2018, 19, 1552–1571 CrossRef PubMed.
- P. S. Shirude, A. K. K. Chattopadhyay, K. Ellen and N. R. Wurtz, Biaryl dialkyl phosphine oxide Fpr2 agonists, WO2020257161A1, 2020.
- (a) J. Zhao, Z. Feng, D. Zhong, X. Yang, Y. Wu, G. Zhou and Z. Wu, Cyclometalated Platinum Complexes with Aggregation-Induced Phosphorescence Emission Behavior and Highly Efficient Electroluminescent Ability, Chem. Mater., 2018, 30, 929–946 CrossRef CAS; (b) G. Sarada, A. Maheshwaran, W. Cho, T. Lee, S. H. Han, J. Y. Lee and S.-H. Jin, Pure blue phosphorescence by new N-heterocyclic carbene-based Ir(III) complexes for organic light-emitting diode application, Dyes Pigm., 2018, 150, 1–8 CrossRef CAS.
- (a) J. H. Docherty, T. M. Lister, G. McArthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Transition-Metal-Catalyzed C–H Bond Activation for the Formation of C–C Bonds in Complex Molecules, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS; (b) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, C–H activation, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS; (c) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Arene diversification through distal C(sp2)−H functionalization, Science, 2021, 372, eabd5992 CrossRef CAS PubMed; (d) R. R. Karimov and J. F. Hartwig, Transition-Metal-Catalyzed Selective Functionalization of C(sp3)−H Bonds in Natural Products, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS; (e) Z. Zhang, K. Tanaka and J.-Q. Yu, Remote site-selective C–H activation directed by a catalytic bifunctional template, Nature, 2017, 543, 538–542 CrossRef CAS; (f) H. M. L. Davies, J. Du Bois and J.-Q. Yu, C–H Functionalization in organic synthesis, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC; (g) L. Ackermann, Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- C.-G. Feng, M. Ye, K.-J. Xiao, S. Li and J.-Q. Yu, Pd(II)-Catalyzed Phosphorylation of Aryl C–H Bonds, J. Am. Chem. Soc., 2013, 135, 9322–9325 CrossRef CAS.
- (a) M. Min, D. Kang, S. Jung and S. Hong, Rhodium-Catalyzed Direct C–H Phosphorylation of (Hetero)arenes Suitable for Late-Stage Functionalization, Adv. Synth. Catal., 2016, 358, 1296–1301 CrossRef CAS; (b) C. Li, T. Yano, N. Ishida and M. Murakami, Pyridine-Directed Palladium-Catalyzed Phosphonation of C(sp2)-H Bonds, Angew. Chem., Int. Ed., 2013, 52, 9801–9804 CrossRef CAS.
- S. Wang, R. Guo, G. Wang, S.-Y. Chen and X.-Q. Yu, Copper-catalyzed phosphorylation of sp2 C–H bonds, Chem. Commun., 2014, 50, 12718–12721 RSC.
- (a) O. Berger and J.-L. Montchamp, Manganese-Catalyzed and Mediated Synthesis of Arylphosphinates and Related Compounds, J. Org. Chem., 2019, 84, 9239–9256 CrossRef CAS PubMed; (b) L. Niu, J. Liu, H. Yi, S. Wang, X.-A. Liang, A. K. Singh, C.-W. Chiang and A. Lei, Visible-Light-Induced External Oxidant-Free Oxidative Phosphonylation of C(sp2)–H Bonds, ACS Catal., 2017, 7, 7412–7416 CrossRef CAS; (c) O. Berger and J.-L. Montchamp, Manganese-Mediated Homolytic Aromatic Substitution with Phosphinylidenes, Chem. Rec., 2017, 17, 1203–1212 CrossRef CAS.
- (a) N. Sabat, L. Poštová Slavětínská, B. Klepetářová and M. Hocek, C–H Phosphonation of Pyrrolopyrimidines: Synthesis of Substituted 7- and 9-Deazapurine-8-phosphonate Derivatives, J. Org. Chem., 2016, 81, 9507–9514 CrossRef CAS PubMed; (b) C.-B. Xiang, Y.-J. Bian, X.-R. Mao and Z.-Z. Huang, Coupling Reactions of Heteroarenes with Phosphites under Silver Catalysis, J. Org. Chem., 2012, 77, 7706–7710 CrossRef CAS PubMed.
- (a) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Recent advances in ruthenium-based olefin metathesis, Chem. Soc. Rev., 2018, 47, 4510–4544 Search PubMed; (b) L. Ackermann, Carboxylate-Assisted Ruthenium-Catalyzed Alkyne Annulations by C–H/Het–H Bond Functionalizations, Acc. Chem. Res., 2014, 47, 281–295 Search PubMed; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (d) L. Ackermann and R. J. C. A. Vicente, Ruthenium-catalyzed direct arylations through C–H bond cleavages, Top. Curr. Chem., 2010, 292, 211–229 CrossRef CAS; (e) R. Noyori and S. Hashiguchi, Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- (a) G.-W. Wang, M. Wheatley, M. Simonetti, D. M. Cannas and I. Larrosa, Cyclometalated Ruthenium Catalyst Enables Ortho-Selective C–H Alkylation with Secondary Alkyl Bromides, Chem, 2020, 6, 1459–1468 CrossRef CAS; (b) W.-T. Fan, Y. Li, D. Wang, S.-J. Ji and Y. Zhao, Iron-Catalyzed Highly para-Selective Difluoromethylation of Arenes, J. Am. Chem. Soc., 2020, 142, 20524–20530 CrossRef CAS; (c) X.-G. Wang, Y. Li, H.-C. Liu, B.-S. Zhang, X.-Y. Gou, Q. Wang, J.-W. Ma and Y.-M. Liang, Three-Component Ruthenium-Catalyzed Direct Meta-Selective C–H Activation of Arenes: A New Approach to the Alkylarylation of Alkenes, J. Am. Chem. Soc., 2019, 141, 13914–13922 CrossRef CAS; (d) M. Simonetti, D. M. Cannas, X. Just-Baringo, I. J. Vitorica-Yrezabal and I. Larrosa, Cyclometallated ruthenium catalyst enables late-stage directed arylation of pharmaceuticals, Nat. Chem., 2018, 10, 724–731 CrossRef CAS PubMed; (e) G. Li, D. Li, J. Zhang, D.-Q. Shi and Y. Zhao, Ligand-Enabled Regioselectivity in the Oxidative Cross-coupling of Arenes with Toluenes and Cycloalkanes Using Ruthenium Catalysts: Tuning the Site-Selectivity from the ortho to meta Positions, ACS Catal., 2017, 7, 4138–4143 CrossRef CAS; (f) J. A. Leitch and C. G. Frost, Ruthenium-catalysed σ-activation for remote meta-selective C–H functionalisation, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC; (g) C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Ruthenium-Catalyzed meta-Selective C-H Bromination, Angew. Chem., Int. Ed., 2015, 54, 11677–11680 CrossRef CAS PubMed.
- (a) L. Guillemard, L. Ackermann and M. J. Johansson, Late-stage meta-C–H alkylation of pharmaceuticals to modulate biological properties and expedite molecular optimisation in a single step, Nat. Commun., 2024, 15, 3349–3358 CrossRef CAS PubMed; (b) K. Korvorapun, M. Moselage, J. Struwe, T. Rogge, A. M. Messinis and L. Ackermann, Regiodivergent C−H and Decarboxylative C−C Alkylation by Ruthenium Catalysis: ortho versus meta Position-Selectivity, Angew. Chem., Int. Ed., 2020, 59, 18795–18803 CrossRef CAS; (c) P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Visible-Light-Enabled Ruthenium-Catalyzed meta-C−H Alkylation at Room Temperature, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS; (d) J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Ruthenium(II)-catalysed remote C–H alkylations as a versatile platform to meta-decorated arenes, Nat. Commun., 2017, 8, 15430 CrossRef; (e) N. Hofmann and L. Ackermann, meta-Selective C–H Bond Alkylation with Secondary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS; (f) L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Ruthenium-Catalyzed Regioselective Direct Alkylation of Arenes with Unactivated Alkyl Halides through C-H Bond Cleavage, Angew. Chem., Int. Ed., 2009, 48, 6045–6048 CrossRef CAS.
- (a) M. Y. Gao and C. J. I. J. O. C. Gosmini, Transition Metal-Catalyzed Electroreductive Cross-Couplings for C− C Bond Formation, Isr. J. Chem., 2024, 64, e202300074 CrossRef CAS; (b) L. Ackermann, R. C. D. Brown, P. Enders, P. Fang, A. A. Folgueiras-Amador, R. Francke, J. Galczynski, C. Gosmini, J. W. Hodgson, Z. W. Hou, H. Huang, Z. Huang, S. Inagi, K. Kuciński, M. Kuriyama, K. Lam, T. H. Lambert, M. C. Leech, A. J. J. Lennox, Z. Lin, R. D. Little, L. Massignan, T. S. Mei, T. H. Meyer, K. D. Moeller, O. Onomura, A. Prudlik, Z. Ruan, A. Scheremetjew, P. Schiltz, M. Selt, E. Villani, S. R. Waldvogel, Z. H. Wang, T. Wu, Y. K. Xing, H. C. Xu and K. Yamamoto, Electrochemistry in Organic Synthesis, 2022, DOI:10.1055/b000000126; (c) J. C. Siu, N. Fu and S. Lin, Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS; (d) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis?, Chem, 2020, 6, 2484–2496 CrossRef CAS; (e) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, A Survival Guide for the “Electro-curious”, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (f) Y. Yuan and A. Lei, Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed; (g) P. Xiong and H.-C. Xu, Chemistry with Electrochemically Generated N-Centered Radicals, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (h) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed; (i) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- (a) E. M. Alvarez, G. Stewart, M. Ullah, R. Lalisse, O. Gutierrez and C. A. Malapit, Site-Selective Electrochemical Arene C–H Amination, J. Am. Chem. Soc., 2024, 146, 3591–3597 CrossRef CAS PubMed; (b) Y. Gao, B. Zhang, J. He and P. S. Baran, Ni-Electrocatalytic Enantioselective Doubly Decarboxylative C(sp3)–C(sp3) Cross-Coupling, J. Am. Chem. Soc., 2023, 145, 11518–11523 CrossRef CAS; (c) T. S.-B. Lou, Y. Kawamata, T. Ewing, G. A. Correa-Otero, M. R. Collins and P. S. Baran, Scalable, Chemoselective Nickel Electrocatalytic Sulfinylation of Aryl Halides with SO2, Angew. Chem., Int. Ed., 2022, 61, e202208080 CrossRef CAS PubMed; (d) M. Saito, Y. Kawamata, M. Meanwell, R. Navratil, D. Chiodi, E. Carlson, P. Hu, L. Chen, S. Udyavara, C. Kingston, M. Tanwar, S. Tyagi, B. P. McKillican, M. G. Gichinga, M. A. Schmidt, M. D. Eastgate, M. Lamberto, C. He, T. Tang, C. A. Malapit, M. S. Sigman, S. D. Minteer, M. Neurock and P. S. Baran, N-Ammonium Ylide Mediators for Electrochemical C–H Oxidation, J. Am. Chem. Soc., 2021, 143, 7859–7867 CrossRef CAS.
- (a) Z.-W. Hou, H. Yan, J. Song and H.-C. Xu, Photoelectrocatalytic C–H amination of arenes, Green Chem., 2023, 25, 7959–7962 RSC; (b) N. Chen and H.-C. Xu, Electrochemically Driven Radical Reactions: From Direct Electrolysis to Molecular Catalysis, Chem. Rec., 2021, 21, 2306–2319 CrossRef CAS; (c) P. Xu, P.-Y. Chen and H.-C. Xu, Scalable Photoelectrochemical Dehydrogenative Cross-Coupling of Heteroarenes with Aliphatic C−H Bonds, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed; (d) X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Electrophotocatalytic Decarboxylative C−H Functionalization of Heteroarenes, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed; (e) F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, Ruthenium-Catalyzed Electrochemical Dehydrogenative Alkyne Annulation, ACS Catal., 2018, 8, 3820–3824 CrossRef CAS.
- (a) Y. Yuan, J. Yang and A. Lei, Recent advances in electrochemical oxidative cross-coupling with hydrogen evolution involving radicals, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC; (b) X. Hu, L. Nie, G. Zhang and A. Lei, Electrochemical Oxidative [4+2] Annulation for the π-Extension of Unfunctionalized Heterobiaryl Compounds, Angew. Chem., Int. Ed., 2020, 59, 15238–15243 CrossRef CAS; (c) X. Hu, G. Zhang, L. Nie, T. Kong and A. Lei, Electrochemical oxidation induced intermolecular aromatic C-H imidation, Nat. Commun., 2019, 10, 5467–5476 CrossRef PubMed; (d) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, Electrochemical-Oxidation-Induced Site-Selective Intramolecular C(sp3)–H Amination, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS.
- (a) Y.-K. Xing, Z.-H. Wang, P. Fang, C. Ma and T.-S. Mei, Divergent synthesis of aryl amines and dihydroquinazolinones via electrochemistry-enabled rhodium-catalyzed C–H functionalization, Sci. China:Chem., 2023, 66, 2863–2870 CrossRef CAS; (b) D. Liu, Z.-R. Liu, Z.-H. Wang, C. Ma, S. Herbert, H. Schirok and T.-S. Mei, Paired electrolysis-enabled nickel-catalyzed enantioselective reductive cross-coupling between α-chloroesters and aryl bromides, Nat. Commun., 2022, 13, 7318–7326 CrossRef CAS; (c) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS.
- (a) T. Michiyuki, I. Maksso and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C−H Arylation Polymerization at Ambient Temperature, Angew. Chem., Int. Ed., 2024, e202400845 CAS; (b) Z. Lin, J. C. A. Oliveira, A. Scheremetjew and L. Ackermann, Palladium-Catalyzed Electrooxidative Double C–H Arylation, J. Am. Chem. Soc., 2024, 146, 228–239 CrossRef CAS PubMed; (c) Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Electrochemical Late-Stage Functionalization, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS; (d) T. von Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Enantioselective electrochemical cobalt-catalyzed aryl C–H activation reactions, Science, 2023, 379, 1036–1042 CrossRef PubMed; (e) Y. Li, S. Dana and L. Ackermann, Recent advances in organic electrochemical functionalizations for specialty chemicals, Curr. Opin. Electrochem., 2023, 40, 101312 CrossRef CAS; (f) Y. Wang, H. Simon, X. Chen, Z. Lin, S. Chen and L. Ackermann, Distal Ruthenaelectro-Catalyzed meta-C−H Bromination with Aqueous HBr, Angew. Chem., Int. Ed., 2022, 61, e202201595 CrossRef CAS; (g) X. Tan, X. Hou, T. Rogge and L. Ackermann, Ruthenaelectro-Catalyzed Domino Three-Component Alkyne Annulation for Expedient Isoquinoline Assembly, Angew. Chem., Int. Ed., 2021, 60, 4619–4624 CrossRef CAS; (h) L. Ackermann, Metalla-electrocatalyzed C–H Activation by Earth-Abundant 3d Metals and Beyond, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- (a) J. Qi, J. Xu, H. T. Ang, B. Wang, N. K. Gupta, S. R. Dubbaka, P. O'Neill, X. Mao, Y. Lum and J. Wu, Electrophotochemical Synthesis Facilitated Trifluoromethylation of Arenes Using Trifluoroacetic Acid, J. Am. Chem. Soc., 2023, 145, 24965–24971 CAS; (b) H. Huang, K. A. Steiniger and T. H. Lambert, Electrophotocatalysis: Combining Light and Electricity to Catalyze Reactions, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed; (c) S. Jin, J. Kim, D. Kim, J.-W. Park and S. Chang, Electrolytic C–H Oxygenation via Oxidatively Induced Reductive Elimination in Rh Catalysis, ACS Catal., 2021, 11, 6590–6595 CrossRef CAS; (d) M.-J. Luo, M. Hu, R.-J. Song, D.-L. He and J.-H. Li, Ruthenium(ii)-catalyzed electrooxidative [4+2] annulation of benzylic alcohols with internal alkynes: entry to isocoumarins, Chem. Commun., 2019, 55, 1124–1127 RSC.
- (a) S. Wang, Q. Xue, Z. Guan, Y. Ye and A. Lei, ACS Catal., 2021, 11, 4295–4300 CrossRef CAS; (b) Y. Kurimoto, J. Yamashita, K. Mitsudo, E. Sato and S. Suga, Org. Lett., 2021, 23, 3120–3124 CrossRef CAS PubMed; (c) K.-J. Li, Y.-Y. Jiang, K. Xu, C.-C. Zeng and B.-G. Sun, Green Chem., 2019, 21, 4412–4421 RSC.
- Z.-J. Wu, F. Su, W. Lin, J. Song, T.-B. Wen, H.-J. Zhang and H.-C. Xu, Scalable Rhodium(III)-Catalyzed Aryl C−H Phosphorylation Enabled by Anodic Oxidation Induced Reductive Elimination, Angew. Chem., Int. Ed., 2019, 58, 16770–16774 CrossRef CAS.
- S.-K. Zhang, A. Del Vecchio, R. Kuniyil, A. M. Messinis, Z. Lin and L. Ackermann, Electrocatalytic C–H phosphorylation through nickel(III/IV/II) catalysis, Chem, 2021, 7, 1379–1392 CAS.
- X.-Y. Qian, S.-Q. Li, J. Song and H.-C. Xu, TEMPO-Catalyzed Electrochemical C–H Thiolation: Synthesis of Benzothiazoles and Thiazolopyridines from Thioamides, ACS Catal., 2017, 7, 2730–2734 CrossRef CAS.
- For detailed information see ESI.†.
- G. B. Boursalian, W. S. Ham, A. R. Mazzotti and T. Ritter, Charge-transfer-directed radical substitution enables para-selective C–H functionalization, Nat. Chem., 2016, 8, 810–815 CrossRef CAS.
- For X-ray single crystal diffraction data: (a) CCDC 2360253: Experimental Crystal Structure Determination, 2024, DOI: 10.5517/ccdc.csd.cc2k7162; ; (b) CCDC 2360254: Experimental Crystal Structure Determination, 2024, DOI: 10.5517/ccdc.csd.cc2k7173..
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06219a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |