
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInitiating photocatalytic degradation of organic pollutants under ultra-low light intensity via oxygen-centered organic radicals†
Yingge
He
a,
Yuyan
Huang
b,
Yu-Xin
Ye
*cd,
Yanchun
Deng
a,
Xin
Yang
 *a and
Gangfeng
Ouyang
*bcd
*a and
Gangfeng
Ouyang
*bcd
aSchool of Environmental Science and Engineering, Guangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, Sun Yat-sen University, Guangzhou, China. E-mail: yangx36@mail.sysu.edu.cn
bKey Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, LIFM, School of Chemistry, IGCME, Sun Yat-Sen University, Guangzhou, China. E-mail: cesoygf@mail.sysu.edu.cn
cSchool of Chemical Engineering and Technology, IGCME, Sun Yat-sen University, Zhuhai, China. E-mail: yeyuxin5@sysu.edu.cn
dSouthern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, China
First published on 22nd January 2025
Abstract
Photocatalysis is a promising method for in situ water pollution remediation but faces challenges due to the limited natural light intensity. Herein, we achieved highly-efficient photocatalytic removal of organic pollutants even under ultra-low light intensities of only 0.1 mW cm−2. This was accomplished by developing and effectively stabilizing novel reactive species, oxygen-centered organic radicals (OCORs), which have an impressive half-life of up to seven minutes in water. With lifetimes that are 8 to 11 orders of magnitude longer than for traditional transient radicals, OCORs can effectively wait for pollutants to diffuse, enabling them to remove organic pollutants through polymerization and degradation pathways. The mechanism behind the stability of OCORs lies in the enhanced electron-withdrawing ability of the electron acceptor and the extended conjugation of the catalyst, which effectively prevent back electron transfer. This study provides a theoretical foundation for practical applications of photochemistry in pollution remediation.
Introduction
Photocatalysis provides a promising avenue for in situ remediation of water pollution, as it allows for pollutant degradation by exclusively utilizing solar irradiation and a catalyst.1 This approach circumvents the need for additional energy sources or extensive infrastructure, thereby presenting a valuable opportunity for mitigating water pollution.2,3 Nevertheless, the low light intensity observed in natural water poses formidable challenges for the implementation of photocatalytic technology in situ for water remediation.4,5 In natural water bodies, it is challenging to reach a light intensity of 100 mW cm−2 that is usually required in the previous photocatalytic degradation systems, as it occurs at the water surface during a short period of direct sun irradiation. To achieve in situ photocatalytic remediation of water pollution under natural light, it is crucial to reduce the light intensity required for photocatalysis. Additionally, when photocatalysis occurs at lower light intensities, its scenarios of applications will be expanded. For instance, the scattered sun light indoor is sufficient for pollution remediation. In addition, even external light sources are used for pollution remediation, energy consumption will be largely reduced, which therefore makes photocatalysis viable for pollution remediation in various water environments.To attain effective and stable photocatalysis under low light conditions, the development of new oxidative species becomes imperative. The traditional transient oxidative species have lifetimes in the nanosecond to microsecond range in water6 and tend to undergo rapid quenching owing to the reactions with the surrounding environment before participating in the degradation of organic pollutants.7–9 Additionally, conventional oxidative species may indiscriminately oxidize the catalyst's surfaces, resulting in the deactivation of catalysts (Scheme 1a).10 Regarding these side reactions, most transient oxidative species are wasted, which results in the requirement of high light intensity to achieve high degradation efficiencies. Therefore, it is crucial to create novel active species with prolonged lifespans that slow down their reactions with the surrounding environment and avoid degrading the catalyst, thus enabling effective photocatalysis under low light intensity and bolstering the stability of photocatalysts as well.
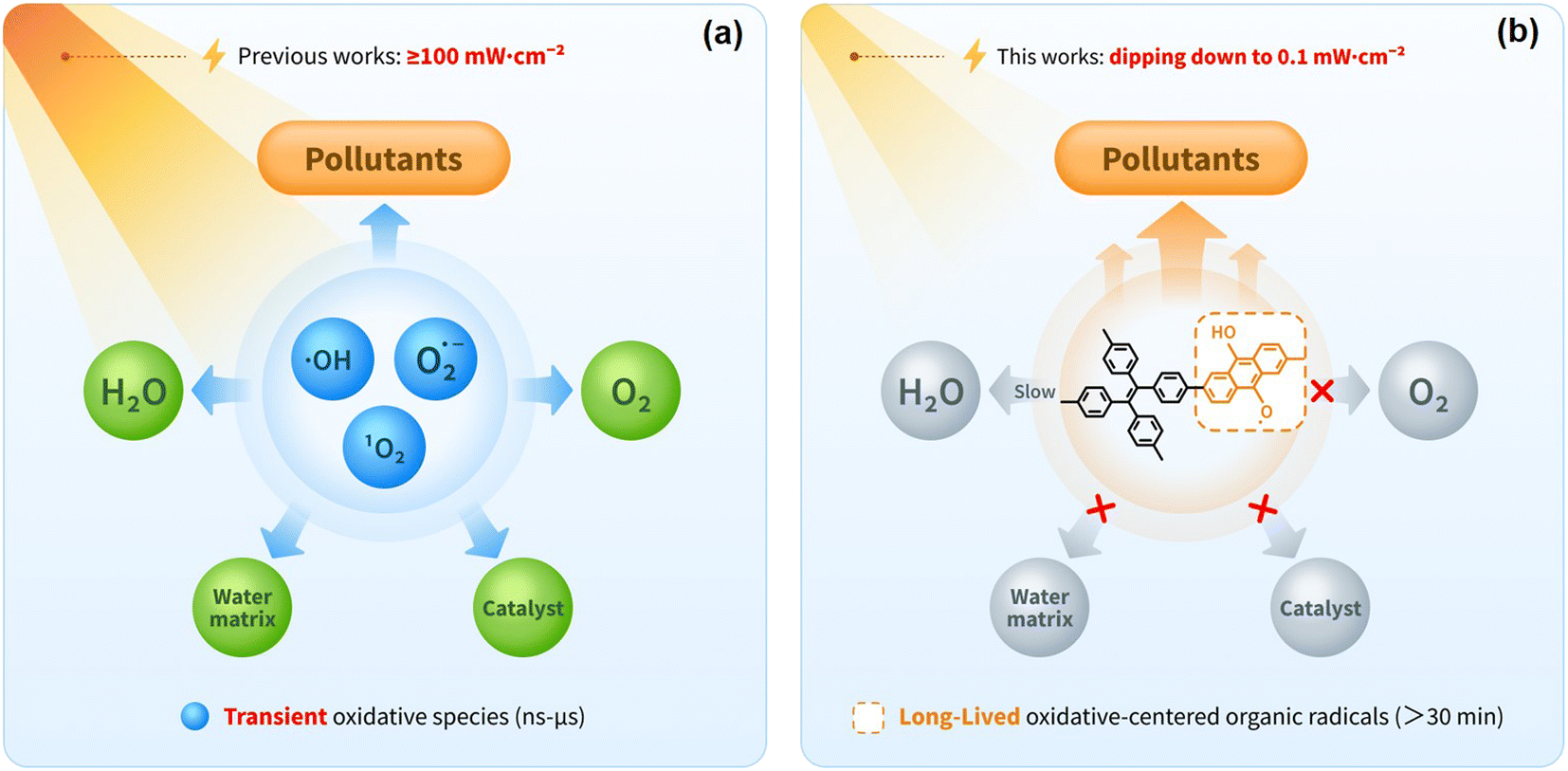 | ||
| Scheme 1 Comparison between traditional transient oxidative species (a) and long-lived oxygen-centered organic radicals (b). | ||
Drawing inspiration from natural aquatic environments where organic pollutants degrade, long-lived organic radicals (LLORs), particularly oxygen-centered organic radicals (OCORs) discovered within natural organic matter, exhibit remarkable resistance to water, with lifetimes extending up to several hours in aqueous environments.11–15 However, there is a lack of in-depth exploration regarding the relationship between the lifespans of natural LLORs and their molecular structures. More importantly, there are no reports on how to artificially generate LLORs for the degradation of pollutants using photoexcitation. Existing artificial simulation methods predominantly rely on redox reactions or thermal processes for LLOR generation.16 While it is common to utilize photons to generate traditional transient radicals for reactions, the direct and stable production of radical species through light would greatly advance pollutant control in natural water bodies and photochemical methodology.
Generating LLORs through light requires surmounting challenges in chemical design related to both photoproduction and stabilization factors. Two primary obstacles hinder the realization of this concept: (1) unpaired electrons produced upon photoirradiation are highly prone to ultrafast back-pairing relaxation and (2) there is a lack of structural characteristics to stabilize radicals in chemical systems, even if they are successfully formed.16 Moreover, LLORs face challenges not only in their formation but also in their susceptibility to quenching by oxygen or water.17,18 Compounding this complexity is the necessity to possess not only high generation efficiencies and high stabilities but also sufficient oxidative potentials to effectively degrade organic pollutants. Consequently, the development of a catalytic system that harnesses LLORs as oxidative species would represent a significant advancement in photocatalysis.
In this study, we have successfully developed a catalytic system that utilizes LLORs as the oxidizing species (Scheme 1b), enabling the effective degradation of organic pollutants under ultralow light intensities (0.1 mW cm−2), which is only 1/1000 of the light intensity commonly employed in the previous photocatalytic studies. The catalytic system presented remarkable resistance to interference and maintained stable performance over nine cycles. The generation of long-lived OCORs through photoexcitation was attributed to the interplay of electron-donating and redox electron-accepting conjugated polymers. This unique photocatalyst is characterized by a polymeric structure featuring alternating tetraphenylethylene, which acts as an electron donor with steric hindrance effects, and anthraquinone, exhibiting electron-withdrawing and redox properties as an electron acceptor. Furthermore, a suite of in situ characterization techniques was developed to investigate the properties of OCORs and gain detailed insights into the entire process of organic pollutant degradation mediated by OCORs. These groundbreaking findings provide valuable theoretical support for the practical application of photochemistry in environmental remediation.
Experimental
Synthesis of TPE-AQ and TPE-FN
In a 50 mL three-necked flask, 2,6-dibromoanthraquinone (183 mg, 0.5 mmol), [ethene-1,1,2,2-tetrayltetrakis(benzene-4,1-diyl)]tetraboronic acid (126.93 mg, 0.25 mmol), K2CO3 (100 mg, 0.72 mmol), Pd(PPh3)4 (12.5 mg, 0.01 mmol), and a mixed solvent of DMF/H2O (7.5 mL/1 mL, respectively) were added. The mixture was purged with argon gas for 30 minutes. Then, the system was stirred for 48 h at 150 °C. The mixture was centrifuged after cooling down to room temperature, and the sediment was washed with DMF, ethanol, and ultrapure water. After being vacuum dried at 80 °C, the finally obtained solid was ground into a powder for further use. The TPE-FN was synthesized via the same method as TPE-AQ, except for using 2,7-dibromo-9-fluorenone (169 mg, 0.5 mmol) instead of 2,6-dibromoanthraquinone.Transient absorption spectroscopy
A Helios femtosecond transient absorption spectrometer (Ultrafast Systems, LLC) with an 800 nm laser pulse was used to perform the ultrafast TAS investigation of catalysts. The laser pulse was separated into pump and probe pulses using a beam splitter. An optical parametric amplifier (OPerA Solo, Coherent) that can generate a wavelength-tunable laser pulse from 250 nm to 2500 nm was used to generate the pump laser at the excitation wavelength (400 nm), and the 400 nm laser intensity that arrived at the sample was 50 μW. The probe pulse was set up at particular time delays with a mechanical delay stage, which delayed it by an adjustable period relative to the pump pulse. The pump pulse spatially overlapped with the continuous probe pulse on the sample.The following steps were taken to prepare the sample: 10 mg of photocatalysts were dispersed in 50 mL of ultrapure water by ultrasonication for 30 minutes, the resulting suspension was then broken through a cell crusher, allowed to settle, and the supernatant was taken for testing.
Computational methods
All calculations were carried out using the Gaussian 09 D.01 program package.19 The geometry optimizations were performed at the ωb97xd/6-311g(d,p) level.20,21 Then, the optimized structures were used for single-point energy calculations with PBE0/ma-TZVP.22–24 Time-dependent density functional theory (TD-DFT) was carried out at the PBE0/6-31G(d,p) level of theory and applied to the investigation of the transfer direction of electrons.25–27Results and discussion
Synthesis and structure
The photocatalyst, named TPE-AQ, was synthesized by interconnecting electron-donating [ethene-1,1,2,2-tetrayltetrakis(benzene-4,1-diyl)]tetraboronic acid (TPE) and electron-accepting 2,6-dibromoanthraquinone (AQ) moieties via a one-step Suzuki reaction. Another photocatalyst, referred to as TPE-FN, was synthesized using 2,7-dibromo-9-fluorenone (FN) moieties as the electron acceptor for comparison (Fig. 1a). Both of these photocatalysts are insoluble in common organic solvents and water.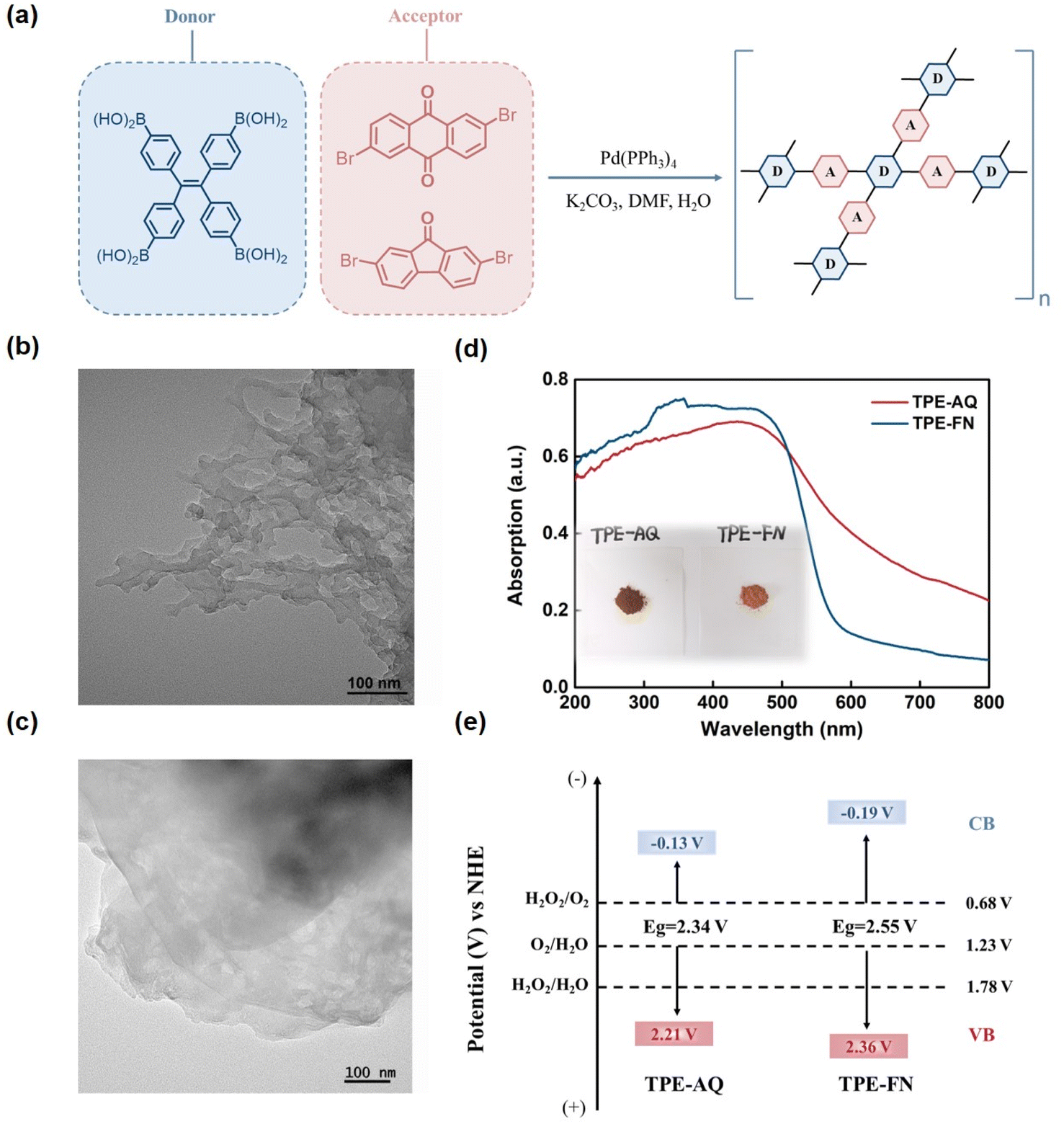 | ||
| Fig. 1 The structure of CPs. (a) molecular structures of CPs. (b and c) TEM images of TPE-AQ (b) and TPE-FN (c). (d) The UV-visible diffuse reflectance spectra of CPs. (e) The electronic structure of CPs. | ||
The X-ray diffraction analysis confirmed that all CPs exhibited characteristics consistent with amorphous carbon (Fig. S1†). Furthermore, SEM and TEM images illustrated the porous and layered nature of the CPs (Fig. 1b, c and S2†). In the FT-IR spectrum, a distinct peak at 1590 cm−1 corresponded to the stretching vibration of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds within the aromatic structures, while peaks at 1671 cm−1 and 1716 cm−1 were attributed to the carbonyl groups in TPE-AQ and TPE-FN, respectively.28,29 These findings confirmed the preservation of the anthraquinone and fluorenone moieties (Fig. S3†).
C bonds within the aromatic structures, while peaks at 1671 cm−1 and 1716 cm−1 were attributed to the carbonyl groups in TPE-AQ and TPE-FN, respectively.28,29 These findings confirmed the preservation of the anthraquinone and fluorenone moieties (Fig. S3†).
The X-ray photoelectron spectroscopy (XPS) survey of the CPs revealed signals at approximately 285 eV and 531 eV, corresponding to C 1s and O 1s, respectively.30 In the high-resolution C 1s XPS spectra, the peak at 284.8 eV denoted sp2 carbon within the aromatic frameworks. Distinctive peaks at 287.32 eV and 287.41 eV indicated the presence of –CO bonds, confirming the existence of carbonyl groups in TPE-AQ and TPE-FN, respectively31 (Fig. S4†), consistent with the FT-IR spectra.
Solid-state 13C NMR spectra (Fig. S5†) revealed broad peaks in the range of 120 to 150 ppm, corresponding to carbon atoms in the aromatic rings. Notably, the peaks at 131.78 ppm and 144.22 ppm in the TPE-AQ spectrum were attributed to carbon atoms at the linking sites between the benzene ring and the AQ moiety, respectively.31,32 Additionally, peaks at 181.72 ppm and 191.87 ppm were observed, corresponding to the characteristic –CO carbon atoms of anthraquinone and fluorenone moieties, respectively.29,32
The N2 adsorption–desorption isotherms indicated a high Brunauer–Emmett–Teller (BET) surface area for TPE-AQ (564.124 m2 g−1) and TPE-FN (457.804 m2 g−1) (Fig. S6†). These substantial surface areas, combined with the porous structure of the CPs, facilitated efficient mass transfer, resulting in significantly enhanced adsorption capacity and photocatalytic activity.33
Energy band structures of CPs
In the UV-vis diffuse-reflectance spectra (Fig. 1d), strong absorption across the entire visible light spectrum was observed for the CPs. Mott–Schottky analysis was conducted at various frequencies (ranging from 1 to 2 kHz) to determine the conduction band minimum (CBM) of the CPs (Fig. S7†). These analyses revealed typical n-type plots, with flat-band potentials positioned 0.1 to 0.3 V more positively than the CBM.Specifically, the flat-band values for TPE-AQ and TPE-FN were found to be −0.64 and −0.70 V, respectively, concerning the Ag/AgCl electrode. Additionally, the CBM of TPE-AQ and TPE-FN was determined to be −0.13 and −0.19 eV, respectively, relative to the reversible hydrogen electrode. To further elucidate the electronic structure, Valence Band X-ray Photoelectron Spectroscopy (VBXPS) and Atomic Force Microscopy (AFM) were employed to estimate the valence band maximum (VBM) position of the CPs (Fig. S8 and S9†). The VBM position for TPE-AQ and TPE-FN was determined to be 2.48 and 2.77 eV relative to the Fermi level (EVBXPS), respectively. Additionally, AFM was utilized to calculate the work functions of TPE-AQ and TPE-FN (Φs), yielding values of 4.233 and 4.093 eV, respectively. By adding these two values, the VBM position of the CPs relative to the vacuum level was obtained. Furthermore, eqn (1) was applied to calculate the VBM position of the CPs referenced to the normal hydrogen electrode (ENHE). Therefore, the VBM position of TPE-AQ and TPE-FN was established to be 2.213 and 2.363 eV relative to the Normal Hydrogen Electrode (NHE).
Consequently, the calculated bandgaps for TPE-AQ and TPE-FN were determined to be 2.34 and 2.55 eV, respectively, as depicted in Fig. 1e.
| ENHE = Φs + EVBXPS − 4.5 | (1) |
Degradation performance
First, an in-depth investigation was conducted to examine solar irradiance levels under various environmental conditions and scenarios (Fig. S10†). Throughout the day, the solar irradiance reached a peak of over 100 mW cm−2 for no more than 4 hours in sunny conditions at the water surface. On cloudy days, for the majority of the time, it remained at about 15 mW cm−2. Underwater, light attenuation was even more pronounced. At a depth of 25 cm in natural water, it diminished to approximately 10 mW cm−2, and even the solar irradiance at the water surface reaches 98 mW cm−2. Indoor environments exhibited significantly lower light intensities, with daytime illumination ranging from a mere 0.1 mW cm−2.Utilizing these precise measurements of light intensity, the degradation performance of conjugated polymers (CPs) towards organic pollutants was evaluated under conventional indoor light sources, using slightly elevated concentrations of bisphenol A (BPA) in contaminated natural water as a template. The corresponding spectra and devices are presented in Fig. S11.† As illustrated in Fig. S13,† the elimination of BPA entails a combination of adsorption and degradation processes, which are facilitated by the porous structure of the CPs. To enhance the clarity of the data presentation, we offer the performance results in terms of both removal efficiency and degradation percentage.
As shown in Fig. 2a, TPE-AQ demonstrated effective degradation of BPA at an unprecedented low light intensity of 0.1 mW cm−2. It is worth emphasizing that this light intensity represents merely one-thousandth of the conventional light intensity (100 mW cm−2) commonly utilized in photocatalysis. This light intensity is significantly lower than that of any previously reported photocatalytic systems (Fig. 2b, the literature data are presented in Table S1†). The degradation rate of BPA accelerated with increasing light intensity, and at 2 mW cm−2 TPE-AQ accomplished an 86% degradation of BPA within a mere half-hour (Fig. S12†). It is noteworthy that under these light-intensity conditions, conventional photocatalysts failed to exhibit any degradation effect. Most commonly used photocatalysts such as titanium dioxide, graphitic carbon nitride, and bismuth vanadate, even with an extended reaction time of 3 hours at this light intensity, showed no degradation of BPA (Fig. 2c). In addition, even at an extremely low dosage of 0.02 g L−1, TPE-AQ maintained its effective BPA degradation capability (Fig. 2d), while conventional catalysts often require dosages well above 0.1 g L−1. This dosage represented the lowest reported to date, offering substantial cost savings on catalyst usage and establishing a solid foundation for its practical application in water treatment.
 | ||
| Fig. 2 The photocatalytic performance of TPE-AQ for pollutant degradation. (a) Photocatalytic performance of TPE-AQ under different light intensities. (b) Comparison of pollutant degradation percentage, illumination time and light intensity for TPE-AQ and other reported catalysts (the numbers in brackets represent the degradation percentage). (c) Photocatalytic performance of common photocatalysts under low-light intensity. (d) Photocatalytic performance of TPE-AQ with different material dosages. (e) The BPA degradation of TPE-AQ in different natural waters. (f) Recycling experiments using TPE-AQ. Reaction conditions: (a) [BPA] = 2 μM, [TPE-AQ] = 0.05 g L−1, [reaction time] = 5 h, and I0 = 0–2.0 mW cm−2; (c–f) [BPA] = 2 μM, [photocatalysts] = 0.05 g L−1 (d was 0.01–0.05 g L−1), [reaction time] = 3 h, and I0 = 2.0 mW cm−2. | ||
In practical water treatment applications, the ability to withstand interference is important. TPE-AQ demonstrated exceptional resistance to interference, allowing it to maintain its BPA degradation performance even in the presence of various anions or dissolved organic matter in water. Moreover, TPE-AQ remained effective over a wide pH range of 4 to 9 and exhibited consistent performance in both river water and high-salinity seawater (Fig. 2e and S14†).
Stability is a crucial factor influencing the cost-effectiveness of a catalyst. TPE-AQ exhibited remarkable stability over nine cycles, as evidenced by its consistent degradation performance without any noticeable decline. Throughout the recycling process, the composition and structure of TPE-AQ remained unchanged (Fig. 2f, S15 and S16†).
Furthermore, TPE-AQ showcased remarkable efficacy in degrading a wide range of emerging organic pollutants, including challenging substances like carbamazepine and microcystin, which are known for their persistence and resistance to degradation (Fig. S17†).
Oxidation species identification
Subsequently, we embarked on an exploration of the active species within TPE-AQ, aiming to unveil the underlying mechanisms responsible for its remarkable efficacy. In the quenching experiments (Fig. 3a and b), the degradation performance of BPA by TPE-AQ was significantly inhibited by 82% in the presence of the quenching agent deferoxamine mesylate salt (DFO) for OCORs.34,35 Furthermore, when DFO and the hole quencher ethylenediaminetetraacetic acid disodium salt (EDTA-2Na) coexisted, the degradation of BPA was almost completely suppressed. These observations provided compelling evidence that the OCORs served as the primary active species, while the hole also contributed to certain reactions. Conversely, common reactive species such as superoxide radicals, singlet oxygen, hydroxyl radicals, hydrogen peroxide and electrons showed no involvement in the reaction. When their corresponding scavengers, such as superoxide dismutase (SOD), L-histidine, tert-butyl alcohol (TBA), catalase, and silver nitrate, were individually introduced into the system, the performance remained unchanged. This conclusion was further supported by the 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethyl-4-piperidinol (TEMP) trapping experiments, which yielded no detectable signals of features in the electron paramagnetic resonance (EPR) spectra (Fig. S18†). Furthermore, even under non-photoexcited conditions, 15% of BPA could still be degraded, which supports the stability of OCORs, as will be discussed in more detail later. | ||
| Fig. 3 Oxidation species identification. (a and b) Photocatalytic performance of TPE-AQ under different quenching conditions. (c–f) EPR spectra of TPE-AQ under continuous irradiation in the absence (c and e) and presence (d and f) of BPA, and the highest point of EPR signal intensity (g = 2.0063) was taken as the ordinate (e and f). Reaction conditions: [BPA] = 2 μM, [TPE-AQ] = 0.05 g L−1, [reaction time] = 30 min, [EDTA-2Na] = [AgNO3] = [L-histidine] = [TBA] = [DFO] = 10 mM, [catalase] = [SOD] = 50 U mL−1, and I0 = 2.0 mW cm−2. | ||
In order to provide further evidence for the involvement of OCORs participating in BPA degradation, the concentration of OCORs was monitored using in situ EPR. The testing method for in situ EPR was as follows: a specified amount of material solution was transferred from a capillary tube to a quartz tube in the sample chamber. Subsequently, the BPA solution was added, and the material solution was continuously illuminated. The EPR signals were recorded at regular intervals (Fig. 3c–f). The EPR spectra of TPE-AQ exhibited a distinct peak at g = 2.0042, indicating the presence of oxygen-centered radicals.36,37 Remarkably, under illumination conditions, the concentration of OCORs exhibited a significant decrease with time upon the addition of BPA. Importantly, this decrease was significantly different from the observed increase in the EPR signal in the absence of BPA. This finding unequivocally demonstrated that the OCORs generated by TPE-AQ under illumination were capable of reacting with BPA, further supporting the pivotal role of OCORs in the degradation process.
Generation process and the mechanism of OCORs
Upon establishing OCORs as the active species, our investigation delved into the generation process of OCORs. The generation of OCORs involved a process previously detailed in our earlier publication.38 Upon light excitation of TPE-AQ, excitons were generated and subsequently undergo separation, leading to the generation of photogenerated charge carriers. Following this, photogenerated electrons from TPE underwent intramolecular transfer to AQ, initiating a reaction with the CO double bond in AQ, thus converting it into C–O· radicals (Fig. 4a). Through theoretical calculations, it was evident that the C–O· radicals exhibited a resonant distribution on oxygen, consistent with the g = 2.0042 value observed in the EPR spectra.
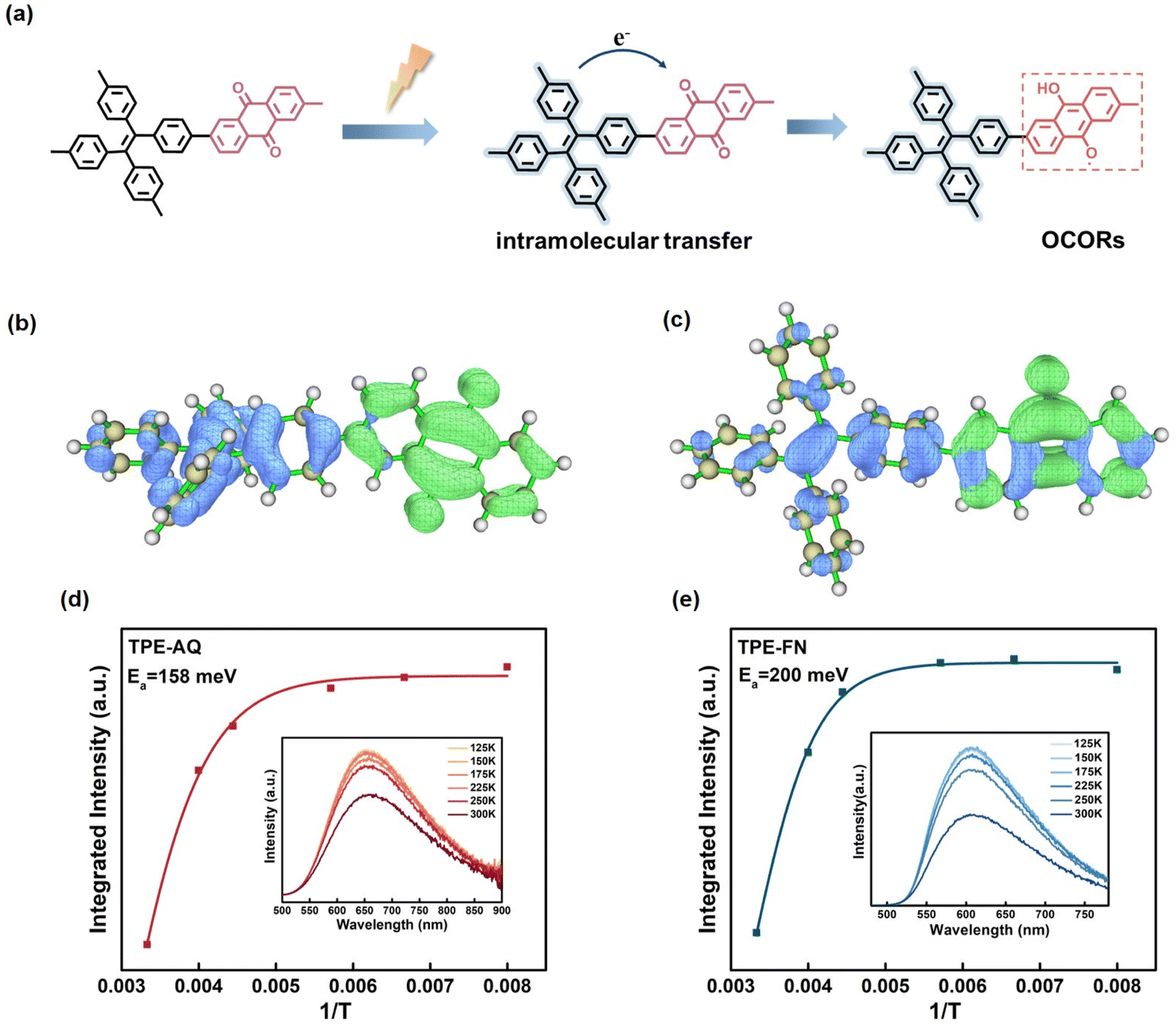 | ||
| Fig. 4 Generation process of OCORs. (a) Schematic diagram of oxygen-centered organic radical (OCOR) generation. (b and c) The analysis of the distribution of holes (blue) and electrons (green) for TPE-AQ (b) and TPE-FN (c) using TD-DFT calculations. (d and e) Temperature-dependent PL spectra of (d) TPE-AQ and (e)TPE-FN; inset: evolution of PL intensity as a function of temperature for (d) TPE-AQ and (e) TPE-FN. | ||
To elucidate the inherent mechanisms influencing the generation of OCORs, TPE-FN, a material structurally akin to TPE-AQ, was introduced. Both TPE-FN and TPE-AQ share analogous electron donors and polymeric structures, featuring a CO structure in their electron acceptors for electron storage. The distinctive dissimilarity stems from AQ's heightened electron-withdrawing capability, as illustrated in Fig. 4b, c and Table S3.† DFT calculations expose a more conspicuous delocalization effect in TPE-AQ compared to TPE-FN, as evidenced by their TD-DFT results. In the excited state of TPE-AQ, there is a substantial separation between electrons and holes, whereas in TPE-FN there is a partial overlap between electrons and holes in the FN moiety. The extent of electron delocalization in TPE-AQ and TPE-FN can also be characterized by the Electron Delocalization Index (EDI). The respective EDI values for TPE-AQ and TPE-FN are 8.20 and 8.78, with smaller EDI values indicating a higher degree of electron delocalization.
Firstly, in terms of absorbance, the strong delocalization effect resulting from AQ's powerful electron-withdrawing properties led to enhanced conjugation in TPE-AQ, resulting in stronger absorption in the visible light range compared to TPE-FN. Additionally, concerning exciton separation efficiency, TPE-AQ exhibited significantly lower fluorescence intensity (Fig. S19†), and both materials had notably low fluorescence quantum yields, measuring only 0.4% and 0.7% for TPE-AQ and TPE-FN, respectively. The effective exciton separation in TPE-AQ was further corroborated by its higher photocurrent intensity compared to TPE-FN (Fig. S20†).
The favorable exciton separation efficiency in TPE-AQ was attributed to the lower exciton activation energy (Ea) of TPE-AQ, which measured only 158 meV, in contrast to the 200 meV measured for TPE-FN (Fig. 4d and e). This reduction in Ea was a result of the stronger electron-withdrawing effect of AQ. The data collectively indicated that the robust electron-withdrawing efficiency of the electron acceptor significantly promoted the delocalization of the electron donor, thereby enhancing the material's absorbance and facilitating exciton separation through the reduction of Ea. Additionally, we carried out femtosecond transient absorption (fs-TA) measurements to shed light on the excitonic dissociation process within TPE-AQ and TPE-FN. This experimental pursuit involved the initial excitation of the conjugated polymer (CP) samples using a 400 nm pump pulse, followed by an examination of their behavior within the visible spectral region, as illustrated in Fig. 5a–d and S21.†
 | ||
| Fig. 5 Generation process and mechanism of OCORs. (a–c) The transient absorption spectra of TPE-AQ with different quenchers. TA values of CPs pumped at 400 nm for AgNO3 and EDTA-2Na were all 10 mM. (d) TA kinetic traces of TPE-AQ and TPE-FN in H2O probed at 634 nm and 615 nm, respectively. (e) Spectrogram of EPR signal intensity as a function of excitation light intensity. | ||
A pronounced and wide-ranging positive signal within the spectral range of 500 to 700 nm was detected for both TPE-AQ and TPE-FN. In order to elucidate the origin of this signal, either a hole sacrificial agent (EDTA-2Na) or an electron sacrificial agent (AgNO3) was introduced. It was consistently observed that the incorporation of sacrificial agents led to a reduction in the signal intensity. As these sacrificial agents facilitated the rapid removal of electrons or holes, they in turn expedited the separation of polarons, which comprised the bound electrons and holes. Consequently, we reasonably attributed the observed signals to the presence of polarons. Based on the aforementioned analysis, the lifetimes of polarons for TPE-AQ and TPE-FN were determined to be 997 ps and 904 ps, respectively. This signifies that both TPE-AQ and TPE-FN exhibit an equally swift rate of polaron dissociation.
Subsequently, the investigation focused on examining the influence of excitation wavelength and excitation light intensity on the concentration of OCORs generated through in situ EPR. The difference in EPR signal intensity (determined at the peak of EPR signal intensity, g = 2.0063) between samples exposed to different light intensities or excitation wavelengths for 60 seconds and the control group kept in darkness served as the ordinate. This difference was then plotted against light intensity or excitation wavelength. Remarkably, it was observed that, for the same duration of illumination, an increase in the EPR signal corresponded to higher light intensities (Fig. 5e). Moreover, the observed trend in the variation of EPR signal intensity with different excitation wavelengths was consistent with TPE-AQ's optical absorption spectrum (Fig. S22†). This once again underscores the connection between the concentration of OCORs generated and the material's inherent absorbance. The more excitons that were produced through photoexcitation, the higher the likelihood of achieving elevated OCOR concentration.
Stability mechanism for OCORs
Further investigation was conducted into the lifetime of OCORs. Due to the pioneering nature of artificially generated light-excited OCORs, a series of in situ methodologies were developed to characterize the lifetime of OCORs and elucidate the underlying factors influencing their stability. By employing EPR under ambient conditions (open air and no need for external light source excitation), the signal of OCORs in TPE-AQ remained detectable even after being placed for a year (Fig. 6a). Notably, while most radicals are readily reoxidized upon exposure to air,39–45 these OCORs exhibited stability under ambient conditions and required neither protective measures nor the low temperatures often necessary for stabilization. | ||
| Fig. 6 Stability mechanism for OCORs. (a) EPR spectra before and after TPE-AQ placement for one year. (b–d) The time-dependent in situ solid EPR spectra of TPE-AQ (b) and TPE-FN (c), and (d) the EPR spectrum and (b and c) the highest point of EPR signal intensity as the ordinate (g = 2.0063). (e) EPR spectra of TPE-AQ in water and acetonitrile before and after light irradiation. (f) TA kinetic traces of TPE-AQ and TPE-FN in H2O probed at 1110 nm and 972 nm, respectively. | ||
It's worth mentioning that due to the fact that OCORs in TPE-AQ can be excited even under very weak light conditions, the lack of significant changes in OCOR concentrations after one year of placement does not necessarily indicate a one-year lifetime. To more accurately determine the lifetime of OCORs, we employed in situ EPR and recorded the time it took for OCOR concentrations to recover from the photoexcited state to the baseline concentration after removing the light source. This time record better reflects the actual lifetime of OCORs, rather than just their concentration stability in the ambient environment. The analysis of Fig. 6b and S23a† confirmed the remarkable stability of OCORs in an air environment when in a solid powder form of TPE-AQ, as no significant decay was observed within 30 minutes after removing the light source. This observation is in line with our experimental findings, where OCORs returned to their initial state within 30 minutes in the case of TPE-FN, which possesses a less delocalization ability compared to TPE-AQ (Fig. 6c and S23b†). These results suggest a positive correlation between the electron-withdrawing capability of the acceptor group and the stability of OCORs. In this context, TPE-AQ, with its stronger delocalization effect compared to TPE-FN, exhibits the greater stability of OCORs. Concurrently, parallel investigations were extended to explore the behavior of small molecule monomers derived from TPE-AQ. It is noteworthy that our EPR analysis revealed a significantly reduced peak intensity for OCORs in these small molecules. Importantly, this decrease in peak intensity remained unchanged even under light excitation conditions, indicating an accelerated decay of OCORs in small molecules that goes undetected by non-transient EPR methods (Fig. 6d and S23c†). This observation underscores the crucial role of the extensive conjugation provided by polymeric structures in stabilizing OCORs.
Furthermore, it is essential to highlight that the fluorescence emission signal of small molecules was significantly higher than that of polymers (Fig. S19†). This disparity in fluorescence signals further substantiates that the inadequate dissociation of excitons in small molecules contributes to the observed differences. It emphasizes the importance of polymer structures in both the formation and stability of OCORs. These findings demonstrated the distinct behavior of small molecule monomers compared to polymer radicals, where the unpaired electrons in the assembled dimers are readily recombined or quenched in the presence of air.
Since electron and hole recombination through back electron transfer is one of the primary recombination mechanisms for organic radicals, it is essential to delve deeper into the impact of hole reactions and their lifetimes on the longevity of OCORs. To explore this further, TPE-AQ was separately dispersed in two different solvents: water and acetonitrile (Fig. 6e). The results unveiled distinct behaviors of OCORs in these solvents. Water exhibited a unique capacity to interact with the holes in TPE-AQ, leading to a significant enhancement of OCORs under illumination. In contrast, acetonitrile, even at an equivalent concentration, displayed a substantially lower OCOR signal, which remained relatively unchanged under light exposure. This disparity strongly suggests that water serves as a key electron donor to the holes, thereby enhancing the stability of OCORs through avoiding the back electron transfer.
To corroborate this hypothesis, we employed transient absorption spectroscopy, shedding light on the lifetimes of these holes. As illustrated in Fig. S24,† the positive signal within the 850 to 1300 nm range was specifically attributed to trapped holes, as demonstrated by their rapid decay in the presence of the hole scavenger EDTA-2Na. In Fig. 6f, it was observed that the lifetime of TPE-AQ's holes in water was relatively short, measuring at 5 ns. This lifetime contrasted significantly with the lifetimes of OCORs in water, as elaborated in the following section, which reached half-lives of 7 minutes. These findings suggest that water is the primary entity that readily interacts with the holes, significantly contributing to the stabilization of OCORs. Moreover, investigation was continued to determine whether the lifetime of holes affects the longevity of OCORs. The decay curves for the trapped hole of the two CPs revealed quite different lifetimes. The lifetime of TPE-AQ was 5 ns, which was two times longer than that of TPE-FN, measuring at 2 ns. The significantly longer hole lifetime observed in TPE-AQ provides further evidence of the enhanced charge separation facilitated by its long-lived OCORs, contributing to the prolonged lifetime of holes. The difference in hole lifetimes underscores the distinct advantages of TPE-AQ over TPE-FN in terms of efficient charge separation and prolonged hole lifetime, which are crucial for facilitating the efficient oxidation of organic pollutants.
OCOR quenching
Given their single-electron characteristics, OCORs exhibited the capability to either donate an electron to oxygen, resulting in the generation of superoxide radicals, or accept an electron from water, leading to the formation of hydroxyl radicals. The fact that TPE-AQ's OCORs remained stable in ambient air suggested its limited reactivity or sluggish reaction rate with oxygen. When TPE-AQ was dispersed in water, the changes in OCOR peak intensity were monitored after the light source was removed, revealing a gradual decline in concentration, indicative of its potential reactivity with water (Fig. 7a and S25†). This decline was particularly noticeable in the first 7 minutes, during which there was a relatively rapid decrease, reaching half of the overall change in signal intensity. Subsequently, there was a gradual decline observed. The EPR signal peak had returned to its pre-excitation level and persisted for a duration of 30 minutes. | ||
| Fig. 7 Quenching and reaction of OCORs. (a) The time-dependent in situ EPR spectra of TPE-AQ in H2O. (b) CV curves of TPE-AQ on glassy carbon electrodes in 0.1 M KCl solution at a sweep rate of 50 mV s−1. (c) H/D kinetic isotope effect for phenol degradation in the TPE-AQ system. (d) The Gibbs free energy (ΔE) of OCORs converted to anthrahydroquinone under different conditions. (e) The photocatalytic performance of TPE-AQ for H2O2 production with or without BPA. (f) Photocatalytic degradation of BPA by TPE-AQ by purging with O2 or Ar. Reaction conditions: (c) [phenol] = [phenol-D6] = 2 μM, [TPE-AQ] = 0.05 g L−1, and I0 = 2.0 mW cm−2; (e and f) [BPA] = 2 μM and [TPE-AQ] = 0.05 g L−1, I0 = 2.0 mW cm−2, and [reaction time] = 60 min (e) and 30 min (f). | ||
To elucidate this phenomenon, we employed an in situ cyclic voltammetry (CV) technique to identify the distinctive peak associated with OCORs (Fig. 7b). It is important to note that the attribution of OCORs' characteristic peak in CV required satisfying two criteria. First, the signal associated with the peak increased with prolonged exposure to light and persisted even after the removal of the light source, effectively eliminating the possibility of hole characteristic peaks. Secondly, a comparison with experimental data is crucial to exclude the oxygen peak.
The CV curve of TPE-AQ revealed a distinct oxidation peak at 1.5 VNHE, with no apparent reduction peak being observed. This demonstrated its preference for gaining electrons to oxidize water, rather than losing electrons to reduce oxygen. Importantly, the observed oxidation potential, slightly exceeding that of the water-to-oxygen pair (1.23 VNHE), offers evidence that the quenching of OCORs primarily stemmed from a slow reaction with water. The relatively slow reaction rate could be attributed to the limited occurrence of overpotentials.
OCOR reaction
The investigation into the reaction pathways between OCORs and organic pollutants persisted, and a more detailed analysis of experimental outcomes was conducted. In the in situ EPR analysis, TPE-AQ was subjected to light exposure, followed by the removal of the light source. During this phase, the concentration of OCORs exhibited a gradual decline over a 30-minute period, without fully returning to its pre-excitation levels. This observation underscored the impressive stability of OCORs and substantiated their photo-induced generation.Subsequent to the light source removal and the introduction of phenol into the system, a marked reduction in OCOR concentration was evident, and it remained significantly below the pre-excitation levels (Fig. S26†). This finding indicates that the reaction between OCORs and phenol occurred spontaneously, devoid of the need for external energy, such as light. This light-independent reaction signifies an intrinsic and spontaneous process.
Based on the aforementioned discussion, it was observed that both water and BPA possess the capability to quench OCORs by providing electrons or protons to OCORs. In order to determine the origin of the electrons and protons involved in the conversion of OCORs to anthrahydroquinone moieties, experiments were conducted to compare the reaction rates of fully deuterated phenol and regular phenol (Fig. 7c and S27†). A small kinetic isotope effect (KIE) value of 1.32 was observed, which excluded the hydrogen abstraction pathway and provided support for the idea that substrate oxidation is driven by electron transfer or proton-coupled electron transfer.46 The study also employed theoretical calculations to investigate the process. It was found that BPA served as the source of electrons and protons, and that this reaction had a lower Gibbs free energy change compared to the other two cases (Fig. 7d and Table S6†). Together with the results of the kinetic isotope effect experiment, these findings suggest that proton-coupled electron transfer between BPA and OCORs is responsible for substrate oxidation.
The preference of OCORs for acquiring electrons from BPA, driven by the superior electron-donating capacity of BPA, allows OCORs to be highly selective in their utilization for the removal of pollutants (Fig. 8). This oxidative process led to the formation of eliminated compounds through mineralization or polymerization pathways, resulting in a remarkable removal efficiency of total organic carbon (TOC) of 71% (Fig. S28, S29 and Table S7†). Additionally, 29% of the BPA was degraded into smaller molecules, and simulation calculations through the ECOSAR program of EPI Suite have indicated a significant reduction in the toxicity of these degraded small molecules. Nevertheless, the toxicity of the polymerization products increased, and they exhibited a propensity to adsorb onto the material's surface, preventing their release into water (Table S8†). Details of the degradation products and their pathways are extensively documented in the ESI.†
 | ||
| Fig. 8 The proposed mechanism of photocatalytic BPA degradation. Proposed mechanism of photocatalytic BPA degradation and the cycle of anthraquinone (AQ), long-lived oxygen-centered organic radicals (OCORs) and anthrahydroquinone. | ||
Subsequent to this, anthrahydroquinone moieties underwent oxidation by oxygen, reverting to their AQ state while concurrently generating hydrogen peroxide as a beneficial byproduct (Fig. 7e and 8). Given the stronger electron-donating propensity of BPA relative to H2O, the presence of BPA expedited the production of hydrogen peroxide. This deduction found support in the comparative analysis of the degradation of organic pollutants in both argon and air atmospheres (Fig. 7f and S30†). In the absence of oxygen, the degradation percentage of BPA remained at 54%, signifying that anthrahydroquinone could not be effectively restored to its AQ state, thereby impeding the sustained generation of OCORs.
Conclusion
Our study presents a groundbreaking catalytic system that utilizes photo-excited OCORs for ultra-low light intensity photocatalysis. This work also systematically analyzed the working principle of low-light intensity photocatalysis, demonstrating that the long lifespan of OCORs generated by light excitation in water is a key factor for effectively degrading organic pollutants under low-light conditions. It further revealed that OCORs can promote generation and stability by enhancing the electron-withdrawing ability of electron acceptors and expanding the conjugation of catalysts. Additionally, it characterized in detail the quenching process of this radical and the reaction process for removing organic pollutants. The adaptability of our system to practical environments using conventional light sources further expands its potential applications. This not only reduces energy consumption but also provides sustainable solutions for pollutant degradation and environmental remediation in various settings. Overall, this study represents a significant advancement in the field of photocatalysis and offers promising prospects for addressing energy and environmental challenges.Data availability
The data that support the findings of this study are included in the main text and ESI.† Additional data are available from the corresponding authors upon reasonable request.Author contributions
Y. He, X. Y. and Y. Y. co-proposed the idea and designed the experiments. Y. He performed the experiments. Y. Huang performed the DFT calculations. Y. He and Y. D. performed the electron paramagnetic resonance measurements and analyzed the data. Y. He and Y. Y. co-analyzed the results. Y. He, X. Y., Y. Y. and G. O. participated in writing the manuscript. X. Y., Y. Y. and G. O. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22176225, 22206209, and 22336007) and the Natural Science Foundation of Guangdong Province (2022A1515011953).Notes and references
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Environmental applications of semiconductor photocatalysis, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- S. Ghosh, N. A. Kouame, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, Conducting polymer nanostructures for photocatalysis under visible light, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- J. Lv, J. Xie, A. G. A. Mohamed, X. Zhang, Y. Feng, L. Jiao, E. Zhou, D. Yuan and Y. Wang, Solar utilization beyond photosynthesis, Nat. Rev. Chem, 2023, 7, 91–105 CrossRef PubMed.
- Y. Lin, S. N. Lai and J. M. Wu, Simultaneous piezoelectrocatalytic hydrogen-evolution and degradation of water pollutants by quartz microrods@Few-Layered MoS2 hierarchical heterostructures, Adv. Mater., 2020, 32, e2002875 CrossRef PubMed.
- W. H. Lee, C. W. Lee, G. D. Cha, B. H. Lee, J. H. Jeong, H. Park, J. Heo, M. S. Bootharaju, S. H. Sunwoo, J. H. Kim, K. H. Ahn, D. H. Kim and T. Hyeon, Floatable photocatalytic hydrogel nanocomposites for large-scale solar hydrogen production, Nat. Nanotechnol., 2023, 18, 754–762 CrossRef CAS PubMed.
- C. Zhao, J. Chen, R. Zhong, D. S. Chen, J. Shi and J. Song, Oxidative-species-selective materials for diagnostic and therapeutic applications, Angew. Chem., Int. Ed., 2021, 60, 9804–9827 CrossRef CAS PubMed.
- X. Yang, F. L. Rosario-Ortiz, Y. Lei, Y. Pan, X. Lei and P. Westerhoff, Multiple roles of dissolved organic matter in advanced oxidation processes, Environ. Sci. Technol., 2022, 56, 11111–11131 CrossRef CAS PubMed.
- T. Liu, S. Xiao, N. Li, J. Chen, X. Zhou, Y. Qian, C. H. Huang and Y. Zhang, Water decontamination via nonradical process by nanoconfined Fenton-like catalysts, Nat. Commun., 2023, 14, 2881 CrossRef CAS PubMed.
- U. von Gunten, Oxidation processes in water treatment: are we on track?, Environ. Sci. Technol., 2018, 52, 5062–5075 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Cocatalysts for selective photoreduction of CO2 into solar fuels, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- A. Paul, R. Stösser, A. Zehl, E. Zwirnmann, R. D. Vogt and C. E. W. Steinberg, Nature and abundance of organic radicals in natural organic matter: effect of pH and irradiation, Environ. Sci. Technol., 2006, 40, 5897–5903 CrossRef CAS PubMed.
- L. Klüpfel, A. Piepenbrock, A. Kappler and M. Sander, Humic substances as fully regenerable electron acceptors in recurrently anoxic environments, Nat. Geosci., 2014, 7, 195–200 CrossRef.
- D. T. Scott, D. M. McKnight, E. L. Blunt-Harris, S. E. Kolesar and D. R. Lovley, Quinone moieties act as electron acceptors in the reduction of humic substances by humics-reducing microorganisms, Environ. Sci. Technol., 1998, 32, 2984–2989 CrossRef CAS.
- N. Walpen, G. J. Getzinger, M. H. Schroth and M. Sander, Electron-donating phenolic and electron-accepting quinone moieties in peat dissolved organic matter: quantities and redox transformations in the context of peat biogeochemistry, Environ. Sci. Technol., 2018, 52, 5236–5245 CrossRef CAS PubMed.
- C. M. Sharpless, M. Aeschbacher, S. E. Page, J. Wenk, M. Sander and K. McNeill, Photooxidation-induced changes in optical, electrochemical, and photochemical properties of humic substances, Environ. Sci. Technol., 2014, 48, 2688–2696 CrossRef CAS PubMed.
- X. Chen, W. Zhao, G. Baryshnikov, M. L. Steigerwald, J. Gu, Y. Zhou, H. Ågren, Q. Zou, W. Chen and L. Zhu, Engineering stable radicals using photochromic triggers, Nat. Commun., 2020, 11, 945 CrossRef CAS PubMed.
- L. Li, Y. Su, Y. Ji and P. Wang, A long-lived water-soluble phenazine radical cation, J. Am. Chem. Soc., 2023, 145, 5778–5785 CrossRef CAS PubMed.
- S. Zhang, L. Ma, W. Ma, L. Chen, K. Gao, S. Yu, M. Zhang, L. Zhang and G. He, Selenoviologen-appendant metallacycles with highly stable radical cations and long-lived charge separation states for electrochromism and photocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202209054 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, M. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. E. Gomperts, O. Stratmann, A. J. Yazyev, R. Austin, C. Cammi, J. W. Pomelli, R. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Organic wastewater treatment by a single-atom catalyst and electrolytically produced H2O2, Nat Sustainability, 2021, 4, 233–241 CrossRef PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. a basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, Perspectives on basis sets beautiful: seasonal plantings of diffuse basis functions, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Minimally augmented karlsruhe basis sets, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. a polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent molecular-orbital methods. IX. an extended Gaussian-type basis for molecular-orbital studies of organic molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, An insoluble benzoquinone-based organic cathode for use in rechargeable lithium-ion batteries, Angew. Chem., Int. Ed., 2017, 56, 12561–12565 CrossRef CAS PubMed.
- V. S. Mothika, P. Sutar, P. Verma, S. Das, S. K. Pati and T. K. Maji, Regulating charge-transfer in conjugated microporous polymers for photocatalytic hydrogen evolution, Chem.–Eur. J., 2019, 25, 3867–3874 CrossRef CAS PubMed.
- X. Li, Z. Li and Y. Yang, Tetraphenylethylene-interweaving conjugated macrocycle polymer materials as two-photon fluorescence sensors for metal ions and organic molecules, Adv. Mater., 2018, 30, 1800177 CrossRef PubMed.
- L. Luo, W. Ma, P. Dong, X. Huang, C. Yan, C. Han, P. Zheng, C. Zhang and J.-X. Jiang, Synthetic control of electronic property and porosity in anthraquinone-based conjugated polymer cathodes for high-rate and long-cycle-life Na-organic batteries, ACS Nano, 2022, 9, 14590–14599 CrossRef PubMed.
- X. Xu, R. Sa, W. Huang, Y. Sui, W. Chen, G. Zhou, X. Li, Y. Li and H. Zhong, Conjugated organic polymers with anthraquinone redox centers for efficient photocatalytic hydrogen peroxide production from water and oxygen under visible light irradiation without any additives, ACS Catal., 2022, 12, 12954–12963 CrossRef CAS.
- Z. Luo, X. Chen, Y. Hu, X. Chen, W. Lin, X. Wu and X. Wang, Side-chain molecular engineering of triazole-based donor-acceptor polymeric photocatalysts with strong electron push-pull interactions, Angew. Chem., Int. Ed., 2023, e202304875, DOI:10.1002/anie.202304875.
- Y. Zhou, Y. Wu, Y. Lei, Y. Pan, S. Cheng, G. Ouyang and X. Yang, Redox-active moieties in dissolved organic matter accelerate the degradation of nitroimidazoles in SO4·-based oxidation, Environ. Sci. Technol., 2021, 55, 14844–14853 CrossRef CAS PubMed.
- B. Zhu, R. Har-El, N. Kitrossky and M. Chevion, New modes of action of desferrioxamine: scavenging of semiquinone radical and stimulation of hydrolysis of tetrachlorohydroquinone, Free Radic. Biol. Med., 1998, 24, 360–369 CrossRef CAS PubMed.
- J. Dou, Y. Tang, Z. Lu, G. He, J. Xu and Y. He, Neglected but efficient electron utilization driven by biochar-coactivated phenols and peroxydisulfate: polyphenol accumulation rather than mineralization, Environ. Sci. Technol., 2023, 57, 5703–5713 CrossRef CAS PubMed.
- J. Chen, Y. Wang, F. Wang and Y. Li, Photo-induced switching of CO2 hydrogenation pathway towards CH3OH production over Pt@UiO-66-NH2(Co), Angew. Chem., Int. Ed., 2023, 62, e202218115 CrossRef CAS PubMed.
- Y. Ye, J. Pan, Y. Shen, M. Shen, H. Yan, J. He, X. Yang, F. Zhu, J. Xu, J. He and G. Ouyang, A solar-to-chemical conversion efficiency up to 0.26% achieved in ambient conditions, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2115666118 CrossRef CAS PubMed.
- R. Kang, R. Miao, Y. Qi, X. Chang, C. Shang, L. Wang and Y. Fang, Tuning the formation of reductive species of perylene-bisimide derivatives in DMF via aggregation matter, Chem. Commun., 2017, 53, 10018–10021 RSC.
- S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, Extraordinary stability of naphthalenediimide radical ion and its ultra-electron-deficient precursor: strategic role of the phosphonium group, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed.
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, Excited doublet states of electrochemically generated aromatic imide and diimide radical anions, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS.
- D. Schmidt, D. Bialas and F. Würthner, Ambient stable zwitterionic perylene bisimide-centered radical, Angew. Chem., Int. Ed., 2015, 54, 3611–3614 CrossRef CAS PubMed.
- E. Shirman, A. Ustinov, N. Ben-Shitrit, H. Weissman, M. A. Iron, R. Cohen and B. Rybtchinski, Stable aromatic dianion in water, J. Phys. Chem. B, 2008, 112, 8855–8858 CrossRef CAS PubMed.
- M. A. Iron, R. Cohen and B. Rybtchinski, On the unexpected stability of the dianion of perylene diimide in water-a computational study, J. Phys. Chem. A, 2011, 115, 2047–2056 CrossRef CAS PubMed.
- V. V. Roznyatovskiy, D. M. Gardner, S. W. Eaton and M. R. Wasielewski, Radical anions of trifluoromethylated perylene and naphthalene imide and diimide electron acceptors, Org. Lett., 2014, 16, 696–699 CrossRef CAS PubMed.
- J. Wang, K. Hou, Y. Wen, H. Liu, H. Wang, K. Chakarawet, M. Gong and X. Yang, Interlayer structure manipulation of iron oxychloride by potassium cation intercalation to steer H2O2 activation pathway, J. Am. Chem. Soc., 2022, 144, 4294–4299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06339b |
| This journal is © The Royal Society of Chemistry 2025 |