
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA lesion-gated dumbbell nanodevices enable on-demand activation of the cGAS-STING pathway for enhancing cancer immunotherapy†
Mei-Ling
Zhao
a,
Yan-Mei
Lei
ab,
Jing-Yi
Tang
a,
Wen
Li
a,
Xin-Yu
Cao
a,
Wen-Bin
Liang
 a,
Ruo
Yuan
a,
Chaoyong
Yang
bc and
Ying
Zhuo
*a
a,
Ruo
Yuan
a,
Chaoyong
Yang
bc and
Ying
Zhuo
*a
aKey Laboratory of Luminescence Analysis and Molecular Sensing (Southwest University), Ministry of Education, Institute of Developmental Biology and Regenerative Medicine, College of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, P. R. China. E-mail: yingzhuo@swu.edu.cn
bInstitute of Molecular Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 2002127, P. R. China
cThe MOE Key Laboratory of Spectrochemical Analysis and Instrumentation, Department of Chemical Biology, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China
First published on 23rd December 2024
Abstract
Utilizing the cGAS-STING pathway to combat immune evasion is one of the most promising strategies for enhancing cancer immunotherapy. However, current techniques for activating the cGAS-STING pathway often face a dilemma, mainly due to the balance between efficacy and safety. Here, we develop a uracil base lesion-gated dumbbell DNA nanodevice (UBLE) that allows on-demand activation and termination of the cGAS-STING pathway in tumor cells, thereby enhancing cancer immunotherapy. The UBLE integrates two deoxyuridines (dU) in the stem for DNA lesion recognition, two locked complementary primer sequences (primers A and B) for DNA self-assembly, and a Förster resonance energy transfer pair (Cy3 and Cy5) attached to the loop for activation assessment. Upon the orthogonal recognition of tumor-specific repair indicators (UDG and APE1), the UBLE undergoes a conformational change to create massive nicked double-stranded DNA (dsDNA) units. These units self-assemble to generate long fluorescent dsDNA structures, permitting selective evaluation and on-demand activation of the cGAS-STING pathway. Furthermore, we demonstrate that the UBLE can effectively activate the cGAS-STING pathway in tumor cells, enhancing NK cell-targeted cancer immunotherapy. This work develops a DNA lesion-gated strategy for on-demand activation and termination of the cGAS-STING pathway, affording an innovative avenue for enhancing cancer immunotherapy.
Introduction
Cancer immunotherapy is a revolutionary clinical technology that holds immense promise in combating tumors by offering systemic treatment, long-lasting outcomes, and fewer side effects.1–4 Until now, classical anti-tumor immunity technologies, such as immune checkpoint blockade therapy and chimeric antigen receptor T-cell immunotherapy, have made tremendous advancements in cancer treatment.5–7 However, these technologies still face the challenge of immune escape, leading to low efficiency in cancer immunotherapy.8–11 Numerous recent studies have demonstrated the potential of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway in combating immune escape.12–16 Upon activating the cGAS-STING pathway, it induces the secretion of type I interferons (IFNs) to promote antigen presentation and initiate immune cell infiltration, thereby improving the effectiveness of cancer immunotherapy.17–19 Therefore, the on-demand activation of the cGAS-STING pathway is a promising avenue for enhancing cancer immunotherapy.To date, several types of agonists, including organic chemotherapeutic agents (such as cisplatin and carboplatin),20,21 biomolecule agonists (e.g., cyclic dinucleotides and their derivatives),22,23 and inorganic metal ions (like Mn2+ and Zn2+),24–26 have been developed to activate the cGAS-STING pathway for cancer immunotherapy. However, these agonists often suffer from low metabolic stability and unpredictable toxicity, which limit their clinical applicability.27,28 Recent studies have shown that long double-stranded (dsDNA) can be recognized by cGAS for activating the cGAS-STING pathway in a sequence-independent but length-dependent manner.29–31 For example, exogenous DNA circles and DNA hairpins have been used to assemble long dsDNA structures (>45 bp) that serve as agonists for activating the cGAS-STING pathway.32,33 Despite the encouraging progress of these DNA agonists in cancer immunotherapy, current techniques for activating the cGAS-STING pathway often face a dilemma, mainly due to the balance between efficacy and safety. On one hand, effective stimulation of the cGAS-STING pathway enhances anti-tumor immunity by promoting the production of IFNs and other pro-inflammatory cytokines. On the other hand, excessive activation of this pathway might heighten the risk of various health complications, including inflammation, autoimmune disorders, and chronic diseases. Therefore, there is an urgent need to develop new DNA agonists that allow on-demand activation and termination of the cGAS-STING pathway.
Logic-gated DNA nanodevices have been widely used in molecule sensing, disease diagnosis, and cancer therapy, benefiting from their excellent programmability, high flexibility, and controllable recognition.34–39 Recent studies have demonstrated that multiple-gated operations enable successive or simultaneous responses to multiple stimuli, showing higher specificity and stronger anti-interference ability than a single input.40–45 Thus, utilizing multiple-gated nanodevices to activate the cGAS-STING pathway may be a powerful approach for enhancing the specificity of cancer treatments. It is well-documented that the DNA lesion-driven base excision repair pathway remains highly active during tumorigenesis and progression, due to the overexpression of various repair enzymes that repair damaged bases in tumor cells.46–48 For instance, uracil DNA glycosylase (UDG) can recognize damaged uracil bases, leading to the production of abasic (AP) sites, which are then cleaved by apurinic/apyrimidinic endonuclease 1 (APE1) to initiate downstream repairs for tumor survival.49–52 Recently, our group demonstrated that APE1 serves as an indicator for evaluating NK cell-targeted cancer immunotherapy through granzyme A-induced inactivation of APE1.53 Therefore, APE1 could act as a modulator for on-demand activation and termination of the cGAS-STING pathway during NK cell-targeted cancer immunotherapy, which has not yet been explored.
Herein, we develop a uracil base lesion-gated dumbbell DNA nanodevice (UBLE) that allows on-demand activation and termination of the cGAS-STING pathway in tumor cells and effectively enhances cancer immunotherapy (Fig. 1). The UBLE integrates two deoxyuridines (dU) in the stem for lesion recognition, two locked complementary primer sequences (primers A and B) for DNA self-assembly, and a Förster resonance energy transfer (FRET) pair (Cy3 and Cy5) attached to the loop for activation assessment. The UBLE exhibits three notable advantages in cancer immunotherapy compared to the existing DNA-based activation strategies. Firstly, the UBLE utilizes the orthogonal recognition of UDG and APE1 in tumor cells to create massive nicked dsDNA units, offering a highly specific avenue to activate the cGAS-STING pathway. Secondly, the nicked dsDNA units self-assemble into long fluorescent dsDNA structures, enabling specific imaging and selective evaluation of the activation of the cGAS-STING pathway in tumor cells. Thirdly, the UBLE with an enclosed structural feature shows superior biostability in tumor cells, ensuring its functional integrity for precise and effective cancer immunotherapy. Finally, we demonstrate that the UBLE is capable of effectively activating the cGAS-STING pathway in tumor cells, thereby enhancing NK cell-targeted cancer immunotherapy. This work offers an innovative DNA lesion-gated avenue for on-demand activation and termination of the cGAS-STING pathway and effectively enhances cancer immunotherapy.
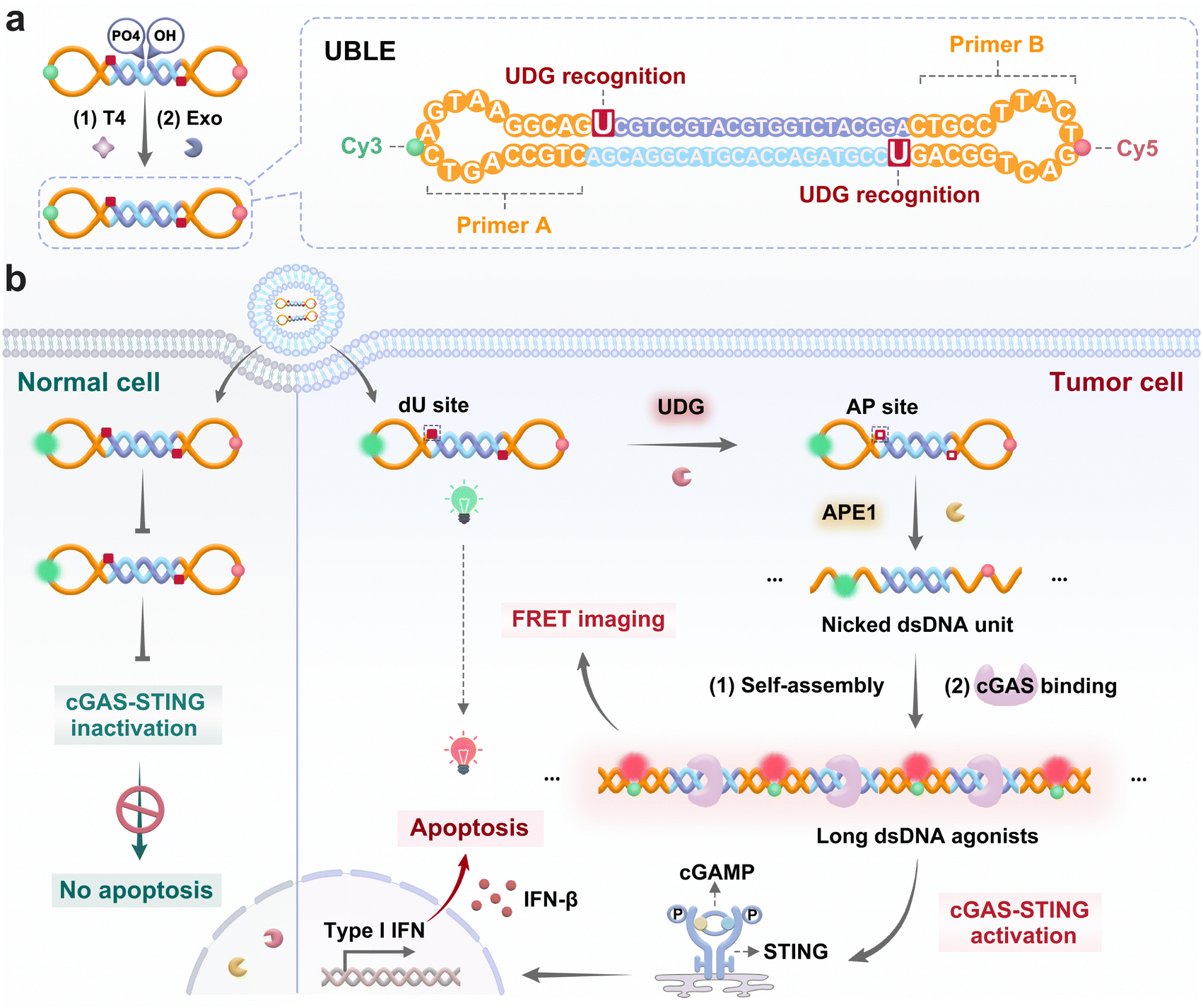 | ||
| Fig. 1 Schematic illustration of UBLE for on-demand activation of the cGAS-STING pathway and enhancement of cancer immunotherapy. (a) Design of UBLE with DNA lesion recognition and activation assessment. (b) Operating mechanism of UBLE for activating the cGAS-STING pathway in tumor cells and enhancing cancer immunotherapy. | ||
Results and discussion
Design and operation principle of UBLE
In this work, we have designed a DNA lesion-gated nanodevice (i.e., UBLE) for on-demand activation of the cGAS-STING pathway in tumor cells and the effective enhancement of cancer immunotherapy (Fig. 1). The UBLE integrates three functional domains, including two artificial dU sites in the stem for DNA lesion recognition, two locked complementary primer sequences (primers A and B) for DNA self-assembly, and a FRET pair (Cy3 and Cy5) attached to the loop for activation assessment (Fig. 1a). When the UBLE is recognized and cleaved orthogonally by the tumor-specific indicators UDG and APE1 (Fig. 1b), it undergoes a conformational change, outputting massive nicked dsDNA units. Subsequently, these units self-assemble through the hybridization of the exposed primers, generating long fluorescent dsDNA structures. Notably, the long dsDNA can be directly recognized by cGAS to activate the cGAS-STING pathway, producing abundant IFNs (i.e., IFN-β) and enhancing cancer immunotherapy. As a result, the UBLE provides a reliable avenue for the on-demand activation of the cGAS-STING pathway and effective enhancement of cancer immunotherapy.Preparation and characterization of the UBLE
The UBLE was constructed through three main steps (Fig. 2a). First, phosphorylated DNA (p-DNA) was annealed to form a middle-opened dumbbell nanodevice (MOD). The MOD was then ligated with T4 DNA ligase (T4) to generate an enclosed configuration. Finally, a mixture of exonuclease I and exonuclease III (Exo I/III) was added to degrade any unconnected MOD or other by-products, resulting in a pure UBLE. The formation of the UBLE was confirmed through native polyacrylamide gel electrophoresis (PAGE) analysis (Fig. 2b). After annealing the p-DNA, two distinct bands appeared in lane 1, representing the MOD and the hybrid dsDNA by-products. When T4 was added, no new bands appeared following the ligation reaction (lane 2). Notably, upon the addition of Exo I/III to the T4-treated MOD, only a clean and distinct band was observed in lane 3, confirming the successful formation of a pure UBLE with an enclosed configuration. In contrast, when T4 was absent, the addition of Exo I/III led to the complete degradation of the MOD and the hybrid dsDNA by-products (lane 4). These results validate that the formation of the UBLE is dependent on the co-presence of T4 and Exo I/III.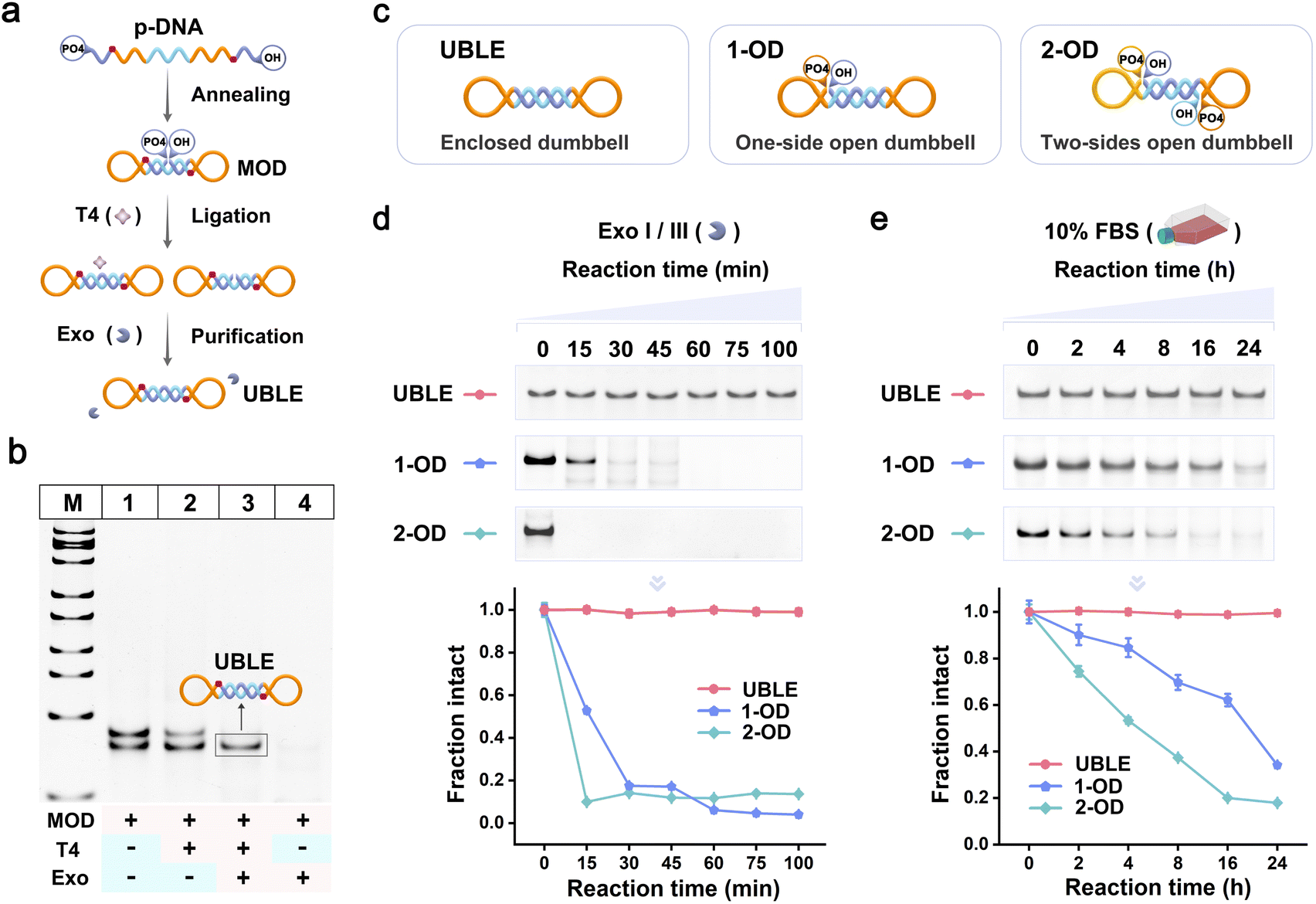 | ||
| Fig. 2 Preparation and biostability characterization of the UBLE. (a) Schematic illustration of the formation of the UBLE through ligation by T4 and purification with Exo I/III. (b) PAGE analysis of the formation of the UBLE. Lane 1, MOD; lane 2, MOD + T4; lane 3, MOD + T4 + Exo I/III (equivalent to UBLE); lane 4, MOD + Exo I/III. (c) Schematic design of the UBLE (without open sides) and two dumbbell probes including 1-OD (with one open side) and 2-OD (with two open sides). (d) and (e) Resistance comparison of the UBLE, 1-OD, and 2-OD treated with Exo I/III (d) and 10% FBS (e) at different time points, respectively. | ||
To assess the structural stability of the UBLE, the degradation rates of the UBLE were measured in Exo I/III and 10% fetal bovine serum (FBS). As a comparison, we designed alternative dumbbell probes, 1-OD and 2-OD, with one and two open sides, respectively (Fig. 2c) (detailed sequences in Fig. S1 and Table S1†). First, we added the UBLE, 1-OD, and 2-OD into Exo I/III for different time points at 37 °C, and then analyzed the treated samples using PAGE. As shown in Fig. 2d, most 1-OD and 2-OD were degraded within 30 min, while the UBLE exhibited great structural integrity even after 100 min. The quantitative analysis demonstrated that the UBLE exhibited an extremely lower degradation rate, approximately 653-fold and 1820-fold lower than 1-OD and 2-OD, respectively (Fig. S2†). We further evaluated the nuclease resistance of the UBLE in the biological environment. After incubation with 10% FBS for different time points at 37 °C, the UBLE remained stable with almost no discernible degradation after 24 h, while 1-OD and 2-OD exhibited an obvious degradation trend (Fig. 2e). Quantitative analysis showed that the UBLE had a degradation time 95-fold and 271-fold longer compared to 1-OD and 2-OD, respectively (Fig. S3†). Additionally, we also carried out a comparison evaluation of the UBLE towards phosphorylation-modified hairpin and traditional hairpin (Fig. S4 and S5†), further confirming the superior biostability of the UBLE. These results suggest that the enclosed configuration confers significantly enhanced nuclease resistance, indicating minimal impact from nucleases and the biological environment on the UBLE.
On-demand activation of the UBLE by DNA lesion repair enzymes
DNA lesion repair enzymes such as UDG and APE1 are widely recognized as being overexpressed in the occurrence and progression of tumor cells. Therefore, we chose UDG and APE1 as tumor-specific indicators to activate the UBLE, triggering the DNA lesion-gated long dsDNA self-assembly to stimulate the cGAS-STING pathway (Fig. 3a). Initially, we performed PAGE analysis and FRET-based assays to investigate the orthogonal recognition of UDG and APE1 on UBLE activation. PAGE analysis showed no new bands when individual UDG or APE1 was added to the UBLE (lanes 2 and 3) compared to the UBLE alone (lane 1), demonstrating that neither UDG nor APE1 could activate the UBLE (Fig. 3b). However, upon the addition of both UDG and APE1, a distinct ladder-like band emerged (lane 4), indicating the successful formation of long dsDNA structures. We further evaluated the orthogonal recognition of UDG and APE1 on the UBLE using FRET-based assays (Fig. 3c). As shown in Fig. 3d, a strong FRET signal for dsDNA (i.e., Fcy5/Fcy3) was detected when UDG and APE1 were co-present. In contrast, weak signals were observed when either UDG or APE1 was added alone, or in the absence of both enzymes. This result demonstrated that the orthogonal recognition of UDG and APE1 could activate the UBLE to form long fluorescent dsDNA. Additionally, as shown in Fig. 3e, a significant kinetic increase was observed when the UBLE was incubated with UDG and APE1, further confirming the orthogonal recognition of UDG and APE1 in triggering DNA lesion-gated long dsDNA self-assembly. Moreover, to validate the essential role of DNA lesions in UBLE activation, we designed a control probe (nBLE, lacking dU sites) for identical FRET-based assays. The nBLE remained inactive in the co-presence of UDG and APE1, demonstrating the necessity of DNA lesion-gated recognition for UBLE activation (Fig. S6†). These results confirm that the orthogonal recognition of UDG and APE1 allows on-demand activation of the UBLE and triggers DNA lesion-gated self-assembly for long dsDNA formation.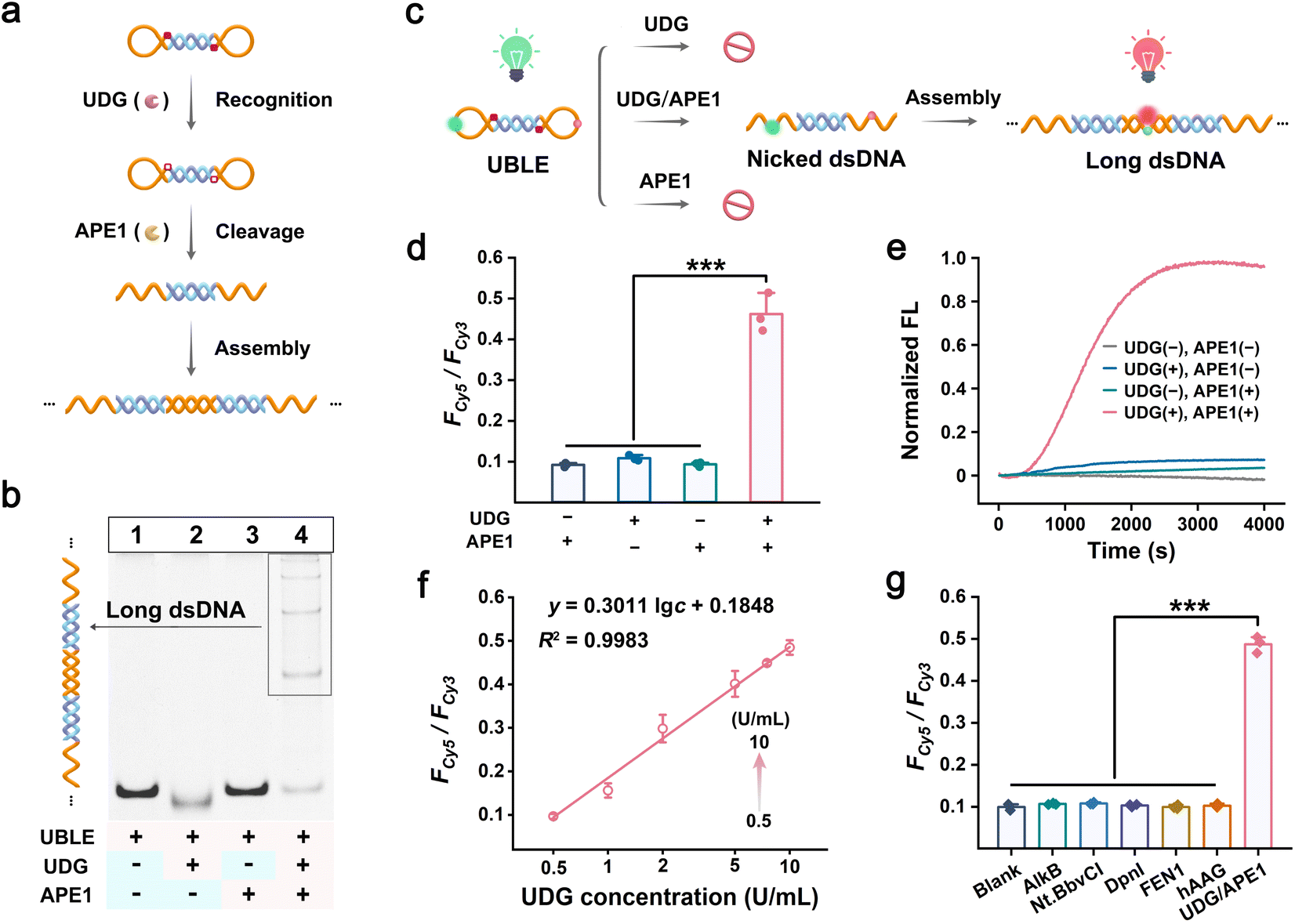 | ||
| Fig. 3 On-demand activation of the UBLE by lesion repair enzymes of UDG and APE1. (a) Schematic illustrating the orthogonal recognition of UDG and APE1 on UBLE to trigger long dsDNA self-assembly. (b) PAGE analysis of long dsDNA formation by DNA lesion-gated self-assembly of the UBLE. Lane 1, UBLE; Lane 2, UBLE + UDG; lane 3, UBLE + APE1; lane 4, UBLE + UDG + APE1. (c) Schematic illustration of the FRET response to DNA lesion-gated long dsDNA formation. (d) and (e) FRET response (d) and kinetic analysis (e) of the UBLE in the presence of UDG and APE1 alone or together. (f) Calibration curves for quantifying different concentrations of UDG on the UBLE (where the concentration of APE1 is consistent with UDG concentration). (g) FRET response analysis of the UBLE in response to various interfering enzymes. Data are represented as mean ± SD (n = 3). ***p < 0.001 by two-tailed Student's t-test. | ||
We next evaluated the response of the UBLE by co-incubating it with varying concentrations of UDG. The FRET signals for dsDNA progressively increased within the concentration of UDG from 0 to 50 U mL−1 and reached a plateau at 10 U mL−1 (Fig. S7†). Calibration analysis showed a gradual enhancement of the FRET signals for dsDNA in a linear range of 0.5–10 U mL−1 and with a detection limit (LOD) of 0.46 U mL−1 (Fig. 3f). In addition, we investigated the effect of the UDG inhibitor (uracil glycosylase inhibitor, UGI) on UDG by incubating it with different concentrations of UGI. With the increased concentrations of UGI, it was observed that the FRET signals for dsDNA gradually decreased (Fig. S8†), confirming the key role of repair enzymes in UBLE activation. We also evaluated the specificity of the UBLE by testing the FRET response in the presence of other interfering enzymes. As shown in Fig. 3g, the FRET signals for dsDNA were significantly higher in the co-presence of UDG and APE1 compared to those with other interfering enzymes. This result indicated that the UBLE can strongly ensure high specificity in indicator recognition. Collectively, these results confirm that the UBLE can be activated to form long fluorescent dsDNA via the orthogonal recognition of UDG and APE1, demonstrating its potential for the on-demand activation of the cGAS-STING pathway in tumor cells.
Validation of DNA lesion-gated self-assembly of the UBLE
To validate the capacity of UBLE for long dsDNA self-assembly in tumor cells, we performed a comparison analysis of the UBLE and nBLE in human cervical cancer cell line (HeLa) and human normal hepatocytes (LO2) cells. Fig. 4a illustrates the operating procedures, including probe preparation, sample transfection, and FRET imaging. As shown in Fig. 4b, fluorescence imaging results showed that only UBLE-incubated HeLa cells exhibited strong FRET signals for dsDNA. In contrast, no significant signal was observed in nBLE-incubated HeLa cells, nor in LO2 cells incubated with either UBLE or nBLE. Quantitative analysis confirmed the significant difference (p < 0.001) after incubation with the UBLE (Fig. 4c). These results indicate that overexpressed UDG and APE1 in HeLa cells can effectively activate the UBLE for long fluorescent dsDNA formation.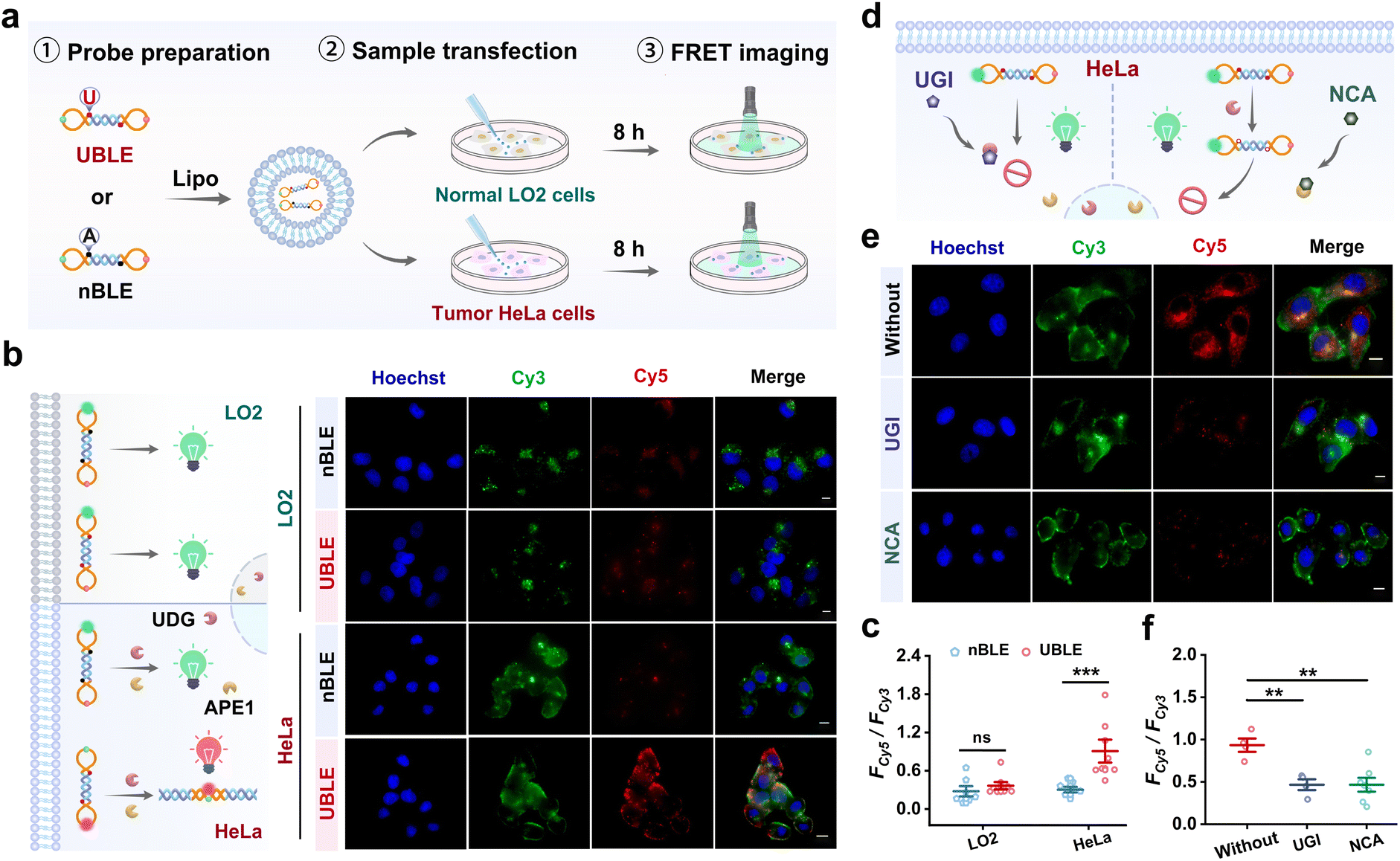 | ||
| Fig. 4 Validation of DNA lesion-gated self-assembly of the UBLE in tumor cells. (a) Operating procedures of the UBLE for UDG and APE1 imaging analysis. (b) and (c) Fluorescence imaging (b) and quantitative analysis (c) of UDG and APE1 in LO2 and HeLa cells after incubation with the UBLE or nBLE, respectively. (d) Schematic illustration of the inhibitor analysis in HeLa cells. (e) and (f) Fluorescence imaging (e) and quantitative analysis (f) of HeLa cells treated with UGI and NCA before incubation with the UBLE. Scale bars: 10 μm. Data are represented as mean ± SD. ns, not significant, **p < 0.01 and ***p < 0.001 by two-tailed Student's t-test. | ||
To further assess the role of UDG and APE1 in UBLE activation, we then selected the UDG inhibitor (UGI) and APE1 inhibitor (7-nitroindole-2-carboxylic acid, NCA) to pre-treat HeLa cells (Fig. 4d). Both fluorescence imaging (Fig. 4e) and quantitative analysis (Fig. 4f) showed that the FRET signals for dsDNA in HeLa cells pre-treated with UGI or NCA were significantly reduced when compared to untreated HeLa cells, indicating the necessity of UDG and APE1 for activating the UBLE for long dsDNA formation. Additionally, we also assessed the inhibition effect by transfecting UDG and APE1 into LO2 cells for comparisons. Further analysis demonstrated an obvious signal difference (p < 0.001) in LO2 cells compared to those pre-treated with UGI or NCA (Fig. S9†). These findings demonstrate that the presence of UDG and APE1 is the necessary condition to trigger lesion-gated self-assembly of long fluorescent dsDNA, providing a selective approach to activate the cGAS-STING pathway in tumor cells.
Evaluation of UBLE activation for enhancing cancer immunotherapy
To evaluate whether the UBLE can effectively activate the cGAS-STING pathway and enhance cancer immunotherapy, we performed comparative analyses by monitoring the changes in HeLa cells after incubation with the UBLE or nBLE. Before this analysis, the cell viability effects of UBLE and nBLE on LO2 and HeLa cells were assessed by using the cell counting kit-8 (CCK-8) assay. Notably, the UBLE caused a significant decrease in HeLa cell viability with increasing UBLE concentration while no such effect was observed in UBLE-incubated LO2 cells (Fig. 5a and b). Meanwhile, as the concentration of nBLE increased in LO2 and HeLa cells, the cell viability remained at a relatively stable level. These results indicated that the UBLE could selectively induce HeLa cell death. Furthermore, calcein acetoxymethyl ester (Calcein-AM) and propidium iodide (PI) staining assays were employed to verify the cell death visually. As shown in Fig. 5c, only UBLE-incubated HeLa cells exhibited extensive red fluorescence, further confirming the capability of UBLE to induce tumor cell death.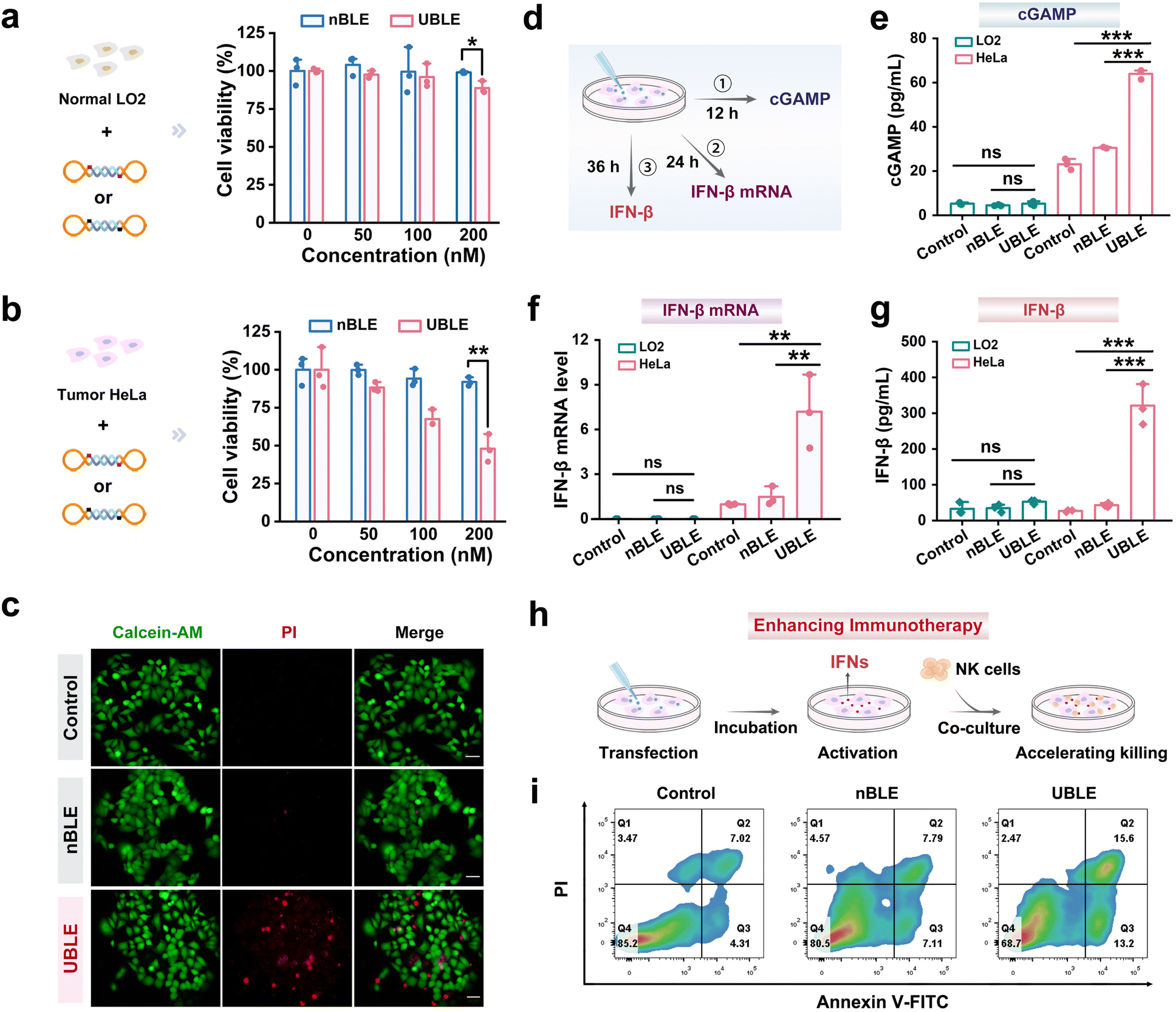 | ||
| Fig. 5 Evaluation of the UBLE to activate the cGAS-STING pathway and enhance cancer immunotherapy. (a) and (b) Cell viability analysis of LO2 (a) and HeLa (b) cells after incubation with different concentrations of UBLE or nBLE. (c) Calcein-AM/PI staining analysis of HeLa cells after incubation with the UBLE or nBLE (dead cells marked in red and live cells marked in green). Scale bars: 50 μm. (d) Schematic illustrating the sample collection of key markers (i.e., cGAMP and IFN-β mRNA) in the cGAS-STING pathway and a pivotal cytokine (i.e., IFN-β) in modulating anti-tumor immune response from HeLa cells after incubation with the UBLE or nBLE at different times. (e) ELISA analysis of the abundance of cGAMP in LO2 and HeLa cells. (f) RT-qPCR analysis of the expression levels of IFN-β mRNA in LO2 and HeLa cells. (g) ELISA analysis of the production of IFN-β by LO2 and HeLa cells. (h) Schematic illustration of a co-culture system of HeLa cells and NK cells without treatment (control), and with UBLE or nBLE treatment. (i) Flow cytometry analysis of HeLa cell apoptosis with Annexin V-FITC/PI staining in the co-culture system derived from (h). Data are represented as mean ± SD (n = 3). ns, not significant, *p < 0.05, **p < 0.01 and ***p < 0.001 by two-tailed Student's t-test. | ||
We next compared the quantity of key markers including cGAMP, IFN-β mRNA, and IFN-β at different times after transfecting the UBLE into HeLa cells (Fig. 5d). To evaluate the potential of the UBLE in activating the cGAS-STING pathway, we quantified the production of cGAMP—a critical intermediate in this pathway—and the mRNA expression levels of downstream cytokines using enzyme-linked immunosorbent assay (ELISA) and real-time quantitative polymerase chain reaction (RT-qPCR) analysis, respectively. Interestingly, the treatment with the UBLE led to a marked increase in cGAMP abundance and significant upregulation of IFN-β mRNA expression levels in HeLa cells, but no detectable changes were observed in LO2 cells or nBLE-treated cells (Fig. 5e and f). Additionally, RT-qPCR analysis also demonstrated the significant upregulation of other pro-inflammatory cytokines-related mRNAs such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) after treatment with the UBLE in HeLa cells (Fig. S10†). These results reveal that the UBLE can effectively activate the cGAS-STING pathway in tumor cells.
To further assess the impact of UBLE-induced activation of the cGAS-STING pathway on cancer immunotherapy, we first measured the production of IFN-β (a pivotal cytokine in modulating the anti-tumor immune response) using ELISA. As shown in Fig. 5g, the ELISA results revealed a significant elevation in IFN-β production in HeLa cells following UBLE treatment, whereas the UBLE-treated LO2 cells exhibited no detectable increase in IFN-β production, indicating the robust production of IFN-β after the on-demand activation of the cGAS-STING pathway. Most notably, we further established a co-culture system of tumor cells (HeLa) and immune cells (natural killer, NK cells). HeLa cells were pre-treated with the UBLE for 20 h, followed by co-culture with NK cells at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 for 8 h (Fig. 5h). Flow cytometry analysis revealed an obvious increase in apoptosis (Q2 + Q3) within the UBLE-treated co-culture system, approximately 2.54-fold higher than that observed with NK cell-mediated cancer immunotherapy alone (Fig. 5i), indicating the significant effect of UBLE in enhancing cancer immunotherapy against tumor cells. Taken together, these findings demonstrate that the UBLE allows on-demand activation of the cGAS-STING pathway and effectively enhances cancer immunotherapy.
:5 for 8 h (Fig. 5h). Flow cytometry analysis revealed an obvious increase in apoptosis (Q2 + Q3) within the UBLE-treated co-culture system, approximately 2.54-fold higher than that observed with NK cell-mediated cancer immunotherapy alone (Fig. 5i), indicating the significant effect of UBLE in enhancing cancer immunotherapy against tumor cells. Taken together, these findings demonstrate that the UBLE allows on-demand activation of the cGAS-STING pathway and effectively enhances cancer immunotherapy.
Conclusions
In summary, we developed a DNA lesion-gated nanodevice (UBLE) that allows on-demand activation and termination of the cGAS-STING pathway in tumor cells and effectively enhances cancer immunotherapy. Compared to the existing DNA-based activation strategies, we have demonstrated that the UBLE exhibits three notable advancements in this work. Firstly, the DNA lesion-gated activation strategy affords an innovative and high-specificity avenue to activate the cGAS-STING pathway in tumor cells. Secondly, the UBLE-triggered long fluorescent dsDNA enables specific imaging evaluation and on-demand activation of the cGAS-STING pathway. Thirdly, the enclosed structure of the UBLE ensures functional integrity for precise and effective cancer immunotherapy. Owing to the high tumor-specificity and superior nuclease-resistance of the UBLE, this work developed a precise and reliable approach to selectively activate the cGAS-STING pathway and effectively enhance cancer immunotherapy.Data availability
All the data supporting the findings of this study are available within the article and its ESI† and can be obtained from the corresponding author upon reasonable request.Author contributions
Mei-Ling Zhao: conceptualization; data curation; formal analysis; investigation; writing – original draft; writing – review & editing. Yan-Mei Lei: review & editing; supervision. Jing-Yi Tang: investigation. Wen Li: investigation. Xin-Yu Cao: investigation. Wen-Bin Liang: review & editing; supervision. Ruo Yuan: review & editing; supervision. Chaoyong Yang: review & editing; supervision. Ying Zhuo: review & editing; supervision; project administration; funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the NNSF of China (22374123) and the Fundamental Research Funds for the Central Universities (SWU-XJLJ202303), China. We are grateful to Dr Yan Li (Analytical and Testing Center, Southwest University) for assistance in imaging observations.References
- I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS PubMed.
- M. S. Goldberg, Nat. Rev. Cancer, 2019, 19, 587–602 CrossRef CAS.
- W. Jiang, Y. Wang, J. A. Wargo, F. F. Lang and B. Y. S. Kim, Nat. Nanotechnol., 2020, 16, 6–15 CrossRef PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef CAS.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- N. N. Shah and T. J. Fry, Nat. Rev. Clin. Oncol., 2019, 16, 372–385 CAS.
- M. Saxena, S. H. van der Burg, C. J. M. Melief and N. Bhardwaj, Nat. Rev. Cancer, 2021, 21, 360–378 CrossRef CAS.
- L. Cao, H. Tian, M. Fang, Z. Xu, D. Tang, J. Chen, J. Yin, H. Xiao, K. Shang, H. Han and X. Li, Biomaterials, 2022, 290, 121856 CrossRef CAS PubMed.
- K. Wang, Y. Li, X. Wang, Z. Zhang, L. Cao, X. Fan, B. Wan, F. Liu, X. Zhang, Z. He, Y. Zhou, D. Wang, J. Sun and X. Chen, Nat. Commun., 2023, 14, 2950 CrossRef CAS PubMed.
- H. Lei, Q. Li, G. Li, T. Wang, X. Lv, Z. Pei, X. Gao, N. Yang, F. Gong, Y. Yang, G. Hou, M. Chen, J. Ji, Z. Liu and L. Cheng, Bioact. Mater., 2024, 31, 53–62 CAS.
- S. Yang, J. Wu, Z. Wang, Y. Cheng, R. Zhang, C. Yao and D. Yang, Angew. Chem., Int. Ed., 2024, 63, e20231907 Search PubMed.
- Q. Chen, L. Sun and Z. J. Chen, Nat. Immunol., 2016, 17, 1142–1149 CrossRef CAS.
- J. Kwon and S. F. Bakhoum, Cancer Discovery, 2020, 10, 26–39 CrossRef CAS.
- A. Decout, J. D. Katz, S. Venkatraman and A. Ablasser, Nat. Rev. Immunol., 2021, 21, 548–569 CrossRef CAS PubMed.
- N. Samson and A. Ablasser, Nat. Cancer, 2022, 3, 1452–1463 CrossRef CAS PubMed.
- F. Ding, J. Liu, K. Ai, C. Xu, X. Mao, Z. Liu and H. Xiao, Adv. Mater., 2023, 36, 2306419 CrossRef.
- E. C. Carroll, L. Jin, A. Mori, N. Munoz-Wolf, E. Oleszycka, H. B. T. Andersen, C. Cunningham, P. Hertzog, K. A. Fitzgerald, A. G. Bowie and E. C. Lavelle, Immunity, 2016, 44, 597–608 CrossRef CAS PubMed.
- E. Duong, T. B. Fessenden, E. Lutz, T. Dinter, L. Yim, S. Blatt, A. Bhutkar, K. D. Wittrup and S. Spranger, Immunity, 2022, 55, 308–323 CrossRef CAS PubMed.
- Y. Feng, G. Wang, W. Li, J. Yan, X. Yu, H. Tian, B. Li and Y. Dai, Adv. Healthcare Mater., 2023, 13, 2302811 CrossRef PubMed.
- Y. Y. Ling, X. Y. Xia, L. Hao, W. J. Wang, H. Zhang, L. Y. Liu, W. Liu, Z. Y. Li, C. P. Tan and Z. W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210988 CrossRef CAS PubMed.
- L. Zhou, Q. Xu, L. Huang, J. Jin, X. Zuo, Q. Zhang, L. Ye, S. Zhu, P. Zhan, J. Ren, T. Lv and Y. Song, Cancer Lett., 2021, 500, 163–171 CrossRef CAS.
- Y. Liu, W. N. Crowe, L. Wang, Y. Lu, W. J. Petty, A. A. Habib and D. Zhao, Nat. Commun., 2019, 10, 5108 CrossRef CAS.
- D. Shae, K. W. Becker, P. Christov, D. S. Yun, A. K. R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D. B. Johnson, J. M. Balko and J. T. Wilson, Nat. Nanotechnol., 2019, 14, 269–278 CrossRef CAS PubMed.
- Y. Huang, G. Qin, T. Cui, C. Zhao, J. Ren and X. Qu, Nat. Commun., 2023, 14, 4647 CrossRef CAS PubMed.
- X. Wang, Y. Liu, C. Xue, Y. Hu, Y. Zhao, K. Cai, M. Li and Z. Luo, Nat. Commun., 2022, 13, 5658 CrossRef PubMed.
- H. Tian, G. H. Wang, W. Sang, L. S. Xie, Z. Zhang, W. X. Li, J. Yan, Y. Tian, J. Li, B. Li and Y. L. Dai, Nano Today, 2022, 43, 101405 CrossRef CAS.
- L. Zhang, K. Shang, X. Li, M. Shen, S. Lu, D. Tang, H. Han and Y. Yu, Adv. Funct. Mater., 2022, 32, 2204589 CrossRef CAS.
- Y. Xing, A. Peng, J. Yang, Z. Cheng, Y. Yue, F. Liu, F. Li, Y. Liu and Q. Liu, Adv. Sci., 2024, 11, 2309583 CrossRef CAS PubMed.
- L. Andreeva, B. Hiller, D. Kostrewa, C. Lässig, C. C. de Oliveira Mann, D. Jan Drexler, A. Maiser, M. Gaidt, H. Leonhardt, V. Hornung and K. P. Hopfner, Nature, 2017, 549, 394–398 CrossRef CAS.
- M. Du and Z. J. Chen, Science, 2018, 361, 704–709 CrossRef CAS PubMed.
- L. Wang, S. Li, K. Wang, N. Wang, Q. Liu, Z. Sun, L. Wang, L. Wang, Q. Liu, C. Song, C. Liu and Q. Yang, Nat. Commun., 2022, 13, 7107 CrossRef CAS.
- X. Xu, H. Fan, Y. Yang, S. Yao, W. Yu, Z. Guo and W. Tan, Angew. Chem., Int. Ed., 2023, 62, e202303010 CrossRef CAS PubMed.
- F. Yu, X. Li, J. Zhao, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202305837 CrossRef CAS PubMed.
- W. Liu, N. Liao, Y. M. Lei, W. B. Liang, X. Yang, R. Yuan, C. Y. Yang and Y. Zhuo, Adv. Sci., 2024, 2401253 CrossRef CAS.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS PubMed.
- T. Song, A. Eshra, S. Shah, H. Bui, D. Fu, M. Yang, R. Mokhtar and J. Reif, Nat. Nanotechnol., 2019, 14, 1075–1081 CrossRef CAS PubMed.
- S. Chen, Z. Xu, W. Yang, X. Lin, J. Li, J. Li and H. Yang, Angew. Chem., Int. Ed., 2019, 58, 18186–18190 CrossRef CAS PubMed.
- D. Wang, S. Li, Z. Zhao, X. Zhang and W. Tan, Angew. Chem., Int. Ed., 2021, 60, 15816–15820 CrossRef CAS PubMed.
- Y. Zhang, W. Chen, Y. Fang, X. Zhang, Y. Liu and H. Ju, J. Am. Chem. Soc., 2021, 143, 15233–15242 CrossRef CAS PubMed.
- Z. Fan, J. Zhao, X. Chai and L. Li, Angew. Chem., Int. Ed., 2021, 60, 14887–14891 CrossRef CAS PubMed.
- Z. Xiang, J. Zhao, J. Qu, J. Song and L. Li, Angew. Chem., Int. Ed., 2021, 61, e202111836 CrossRef.
- Y. Shang, L. Zhu, Y. Xiao, S. Du, R. Ji, B. Li, J. Chen, S. Deng and K. Ren, Adv. Funct. Mater., 2023, 34, 2311722 CrossRef.
- Y. Wu, J. Huang, H. He, M. Wang, G. Yin, L. Qi, X. He, H. H. Wang and K. Wang, Nano Lett., 2023, 23, 1801–1809 CrossRef CAS.
- Y. Guo, J. Ren and E. Wang, Nano Today, 2022, 44, 101476 CrossRef CAS.
- M. Bai, F. Chen, X. W. Cao, Y. Zhao, J. Xue, X. Yu, C. H. Fan and Y. X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13267–13272 CrossRef CAS.
- T. Visnes, M. Grube, B. M. F. Hanna, C. Benitez-Buelga, A. Cázares-Körner and T. Helleday, DNA Repair, 2018, 71, 118–126 CrossRef CAS PubMed.
- W. Zhong and J. T. Sczepanski, J. Am. Chem. Soc., 2023, 145, 17066–17074 CrossRef CAS.
- J. C. Park, Y. J. Kim, G. H. Hwang, C. Y. Kang, S. Bae and H. J. Cha, Nat. Commun., 2024, 15, 4002 CrossRef CAS.
- K. M. Schermerhorn and S. Delaney, Acc. Chem. Res., 2014, 47, 1238–1246 CrossRef CAS PubMed.
- A. C. Drohat and C. T. Coey, Chem. Rev., 2016, 116, 12711–12729 CrossRef CAS PubMed.
- C. C. Li, W. X. Liu, J. Hu and C. Y. Zhang, Chem. Sci., 2019, 10, 8675 RSC.
- M. R. Szymanski, A. Karlowicz, G. K. Herrmann, Y. Cen and Y. W. Yin, J. Am. Chem. Soc., 2022, 144, 23543–23550 CrossRef CAS PubMed.
- Z. P. Chen, W. J. Zeng, Y. M. Lei, W. B. Liang, X. Yang, R. Yuan, C. Y. Yang and Y. Zhuo, Angew. Chem., Int. Ed., 2024, e202414064 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and related experimental data. See DOI: https://doi.org/10.1039/d4sc06493c |
| This journal is © The Royal Society of Chemistry 2025 |