
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrapodal iron complexes invoke observable intermediates in nitrate and nitrite reduction†
Jewelianna M.
Moore
 and
Alison R.
Fout
*
and
Alison R.
Fout
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: fout@tamu.edu
First published on 6th December 2024
Abstract
This study investigates the mechanistic pathways of nitrate and nitrite reduction by the tetrapodal iron complex [Py2Py(afamcyp)2Fe]OTf2, revealing key intermediates to elucidate the reaction process. Using UV-Vis, IR, mass and NMR spectroscopies, stable binding of oxyanions to the iron centre was observed, supporting the formation of the iron(III)–hydroxide intermediate [Py2Py(afamcyp)2Fe(OH)]OTf2. This intermediate is less stable than in previous systems, providing insights into the behaviour of metalloenzymes. A bimetallic mechanism is proposed for nitrogen oxyanion reduction where additional iron is required to drive the complete reaction, resulting in the formation of the final nitrosyl complex, Py2Py(pimcyp)2Fe(NO), and water. Our findings enhance the understanding of iron-based reduction processes and contribute to the broader knowledge of oxyanion reduction mechanisms.
Introduction
In cellular respiration, there are two major pathways and several enzymes employed for the reduction of nitrite.1 The dissimilatory nitrite reduction path uses cytochrome c nitrite reductase (ccNir) to reduce nitrite to ammonia without the formation of detectable intermediates. In contrast, the denitrification pathway is considerably more involved. In this sophisticated denitrification route, nitrate or nitrite is reduced to dinitrogen in a stepwise fashion using four separate enzymes. In the initial nitrite reduction step, nitrite is first deoxygenated to form nitric oxide with either copper-containing nitrite reductase (CuNiR) or, more commonly, a d1 heme-containing nitrite reductase (cd1NiR).cd1NiR contains two inequivalent hemes: the electron transfer c-heme and the active site d1-heme (Fig. 1).2 The active site is constituted of an iron centre coordinated to a porphyrin and a histidine in the apical position. The secondary coordination sphere is comprised of two histidine and a tyrosine residue.3 Unsurprisingly, it is these acidic residues that are imperative for nitrite reduction. Mutations of these residues to neutral amino acids result in an inactive enzyme.1 In the resting state, hemes d1 and c are both in the 3+ oxidation state. Upon two successive electron transfers, both hemes are reduced to the ferrous oxidation state. Nitrite is then able to bind to the heme d1 active site. While nitrite has several possible binding modes, the consensus in the field is that nitrite binds to the iron centre through the nitrogen (nitro isomer).1 This nitro isomer has been crystallized in cd1NiR, but only when exposed to an excess of nitrite under single turn over conditions. Additionally, the structure was crystallized at room temperature, drawing into question if it is indeed a reactive intermediate.1,4 An alternative binding mode of nitrite is the O-bound nitrito isomer. While not crystallized in cd1NiR, Richter-Addo and co-workers crystallographically characterized nitrite bound in horse heart myoglobin and human hemoglobin as the nitrito isomer,5,6 making O-bound nitrite a reasonable binding mode in similar heme complexes.
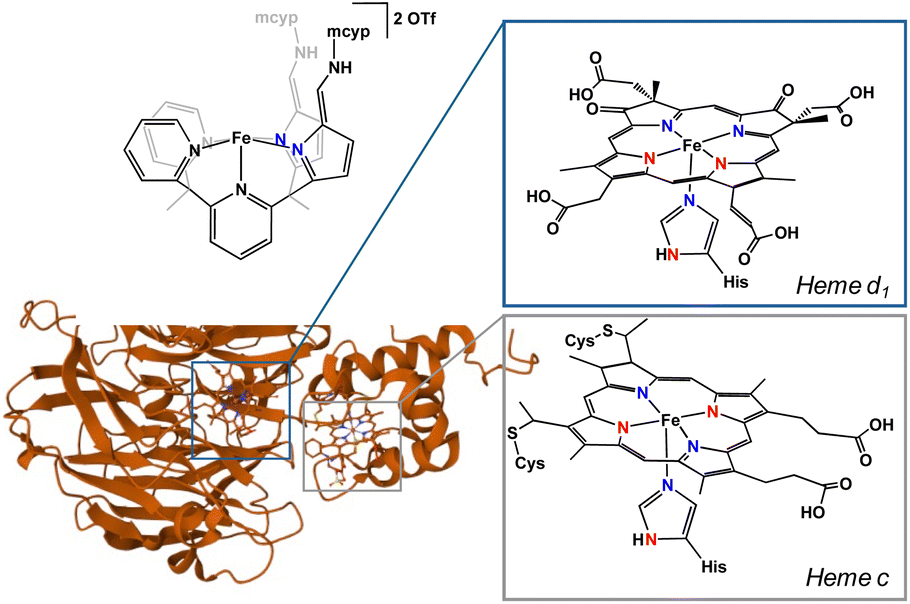 | ||
| Fig. 1 Active (heme c) and electron transfer (heme d1) sites of cd1NiR compared to complex 1. | ||
Although less studied, a proposed mechanism for nitrite reduction via the nitrito isomer is as follows: nitrite binds through oxygen to the ferric d1 centre. Then, the acidic histidine residue or water protonates the bound oxygen, causing release of nitric oxide and formation of an iron(III)–hydroxide.7 To further support the possible existence of the importance of a nitrito isomer, in 2004, Silaghi-Dumitrescu published computational studies finding that while the nitro isomer is thermodynamically favoured by 6 kcal mol−1, the nitro and nitrito isomers are nearly identical in energy when strong hydrogen bonds are accounted for in the computations.8 Further, the presence of hydrogen bonds render N–O bond breakage from the O-bound nitrito isomer to be more facile than from the N-bound nitro isomer, suggesting the nitrito species to be more catalytically active. To shine light on the complexities of nitrite bound iron species, Dissanayake et al. synthesized a bis-nitrite iron pentadentate “N5” complex featuring NH bonds capable of hydrogen bonding. The ligand scaffold in this “N5” complex allows only for one nitrite to engage in hydrogen bonding to the ligand framework. This hydrogen bound nitrite is O-bound to the iron centre, while the second nitrite does not benefit from hydrogen bonding and is N-bound, showcasing the significant role the small nuances of hydrogen bonding play in orienting nitrite ligands.9
Results and discussion
Nitrate and nitrite reduction have been extensively studied in iron,10–21 copper,22–35 and electrochemical systems.36–40 Additionally, other transition metals41–49 and lanthanides50,51 have garnered interest due to their oxyanion reduction capabilities. Previous studies from our group using a tripodal iron system, [N(afaCy)3Fe]OTf2 (N(afaCy)3 = tris(5-cyclohexyl-amineazafulvene-2-methyl)amine)52 further demonstrated the role hydrogen bonding plays in nitrate and nitrite reduction.10–12 The resultant iron species from deoxygenation of the nitrogen containing oxyanion is an iron(III)–oxo when cyclohexyls are in the periphery of the ligand, however, when the cyclohexyl moieties are changed to mesityl, the corresponding iron(III)–OH is formed with ligand tautomerization. DFT calculations showed substantial variations in H-bond strengths between the cyclohexyl and mesityl iron complexes. This difference in H-bond strength and the increase in steric bulk of the mesityl moieties resulted in isolation of an iron(III)–OH.12 This system is rather unique for iron-based nitrate and nitrite reductions, where the final products are typically water and an iron nitrosyl complex.13–21,53Interrogation of the mechanism for these oxyanion reductions in the tripodal system has been relatively difficult due to the lack of observable intermediates, despite these being rather sluggish reactions. We previously hypothesized that the nitrite binds as the nitrito isomer due to crystallographic characterization studies of a Zn–ONO complex, [N(afaCy)3Zn(ONO)]OTf.10 In order to better probe intermediates, we sought to destabilize the resultant iron–oxo containing product through perturbing the geometry at the metal centre.
The geometry of cd1NiR is geometrically more similar to a tetrapodal ligand like 2,2′,2′-methyl-bis-pyridyl-6-(2,2′,2′-methylbis-5-cyclohexy-liminopyrrol)-pyridine (Py2Py(piCy)2),54 or the more soluble 2,2′,2′-methyl-bis-pyridyl-6-(2,2′,2′-methylbis-5-methyl-cyclopropyl-liminopyrrol)-pyridine (Py2Py(pimcyp)2) system, in comparison to the aforementioned tripodal system. Modification of the previously reported (Py2Py(piCy)2)54 ligand with methylcyclopropane was easily achieved in the last step of the reported ligand synthesis via use of methylcyclopropyl amine. In order to investigate nitrate and nitrite reduction, the Py2Py(pimcyp)2 ligand was metalated with Fe(OTf)2(MeCN)2 following a modified literature procedure54 to form [Py2Py(afamcyp)2Fe]OTf2 (1). Upon exposure of 1 to one equiv. of [TBA]NO2 (TBA = tetrabutylammonium) in acetonitrile, an immediate colour change from yellow to brown occurred along with precipitation of an orange powder. Allowing the reaction to continue stirring for several hours, the precipitate resolubilized and the solution changed to green (Fig. 2).
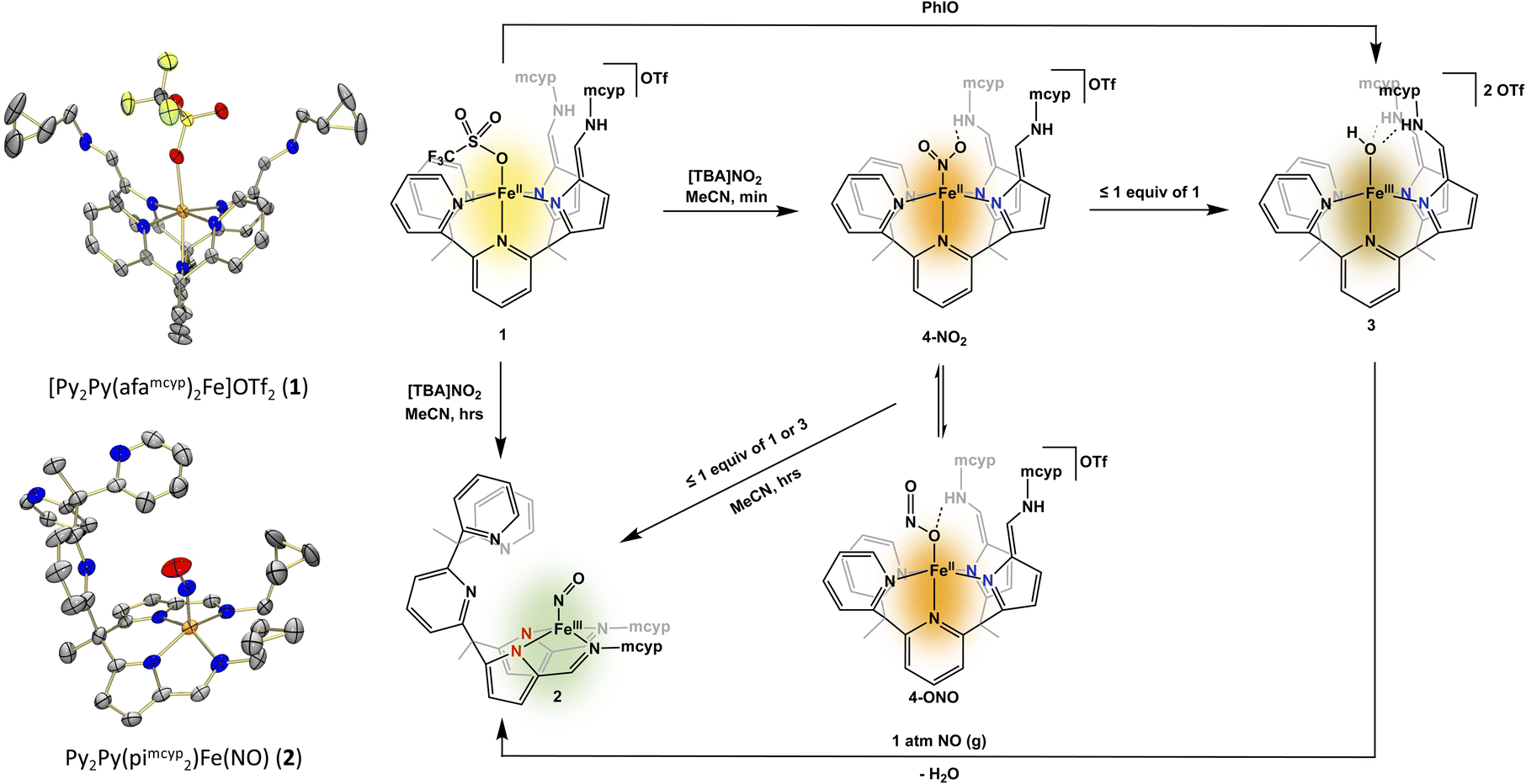 | ||
| Fig. 2 Crystal structures of complexes 1 and 2 at the 50% probability level. Hydrogen atoms and a triflate in 1 have been removed for clarity (left). Synthesis of complexes 2–4 from 1 with the actual colours of the compounds depicted within the scheme (right). | ||
As the reaction of nitrite with 1 features several intense colour changes, we sought to analyse the reaction by UV-visible (UV-Vis) spectroscopy. Under an inert atmosphere, a solution of [TBA]NO2 was added to 1 and a new absorbance at 475 nm immediately grew in followed by disappearance of this new absorbance and appearance of a broad absorbance at 750 nm over several hours (Fig. 3). The identity of the various products observed by UV-Vis spectroscopy was initially unclear, and we sought further characterization via other spectroscopic techniques and independent syntheses.
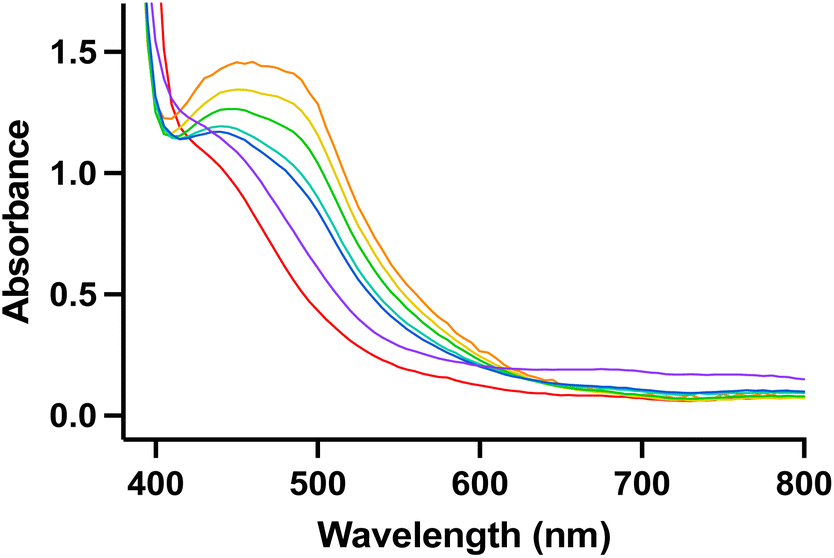 | ||
| Fig. 3 UV-visible spectrum of nitrite reduction (1.0 mM in DMA). 1 (red line) reacts to form a mixture of 3 and 4 (orange line) immediately upon addition of nitrite. 3 and 4 decompose to 2 (purple line) over several hours. Yellow, green, and blue lines show disappearance of 3 and 4 before final formation of 2. | ||
Analysis of the green solution via1H NMR spectroscopy showed complete consumption of 1 and formation of a new complex with no distinct resonances. Interestingly, the reaction head space did not show production of nitric oxide or other nitrogen containing gases, suggesting nitric oxide is either not formed or is consumed to form the final product. While previously in our tripodal system the reduction of nitrite by [N(afaR)3Fe]OTf2 (R = Cy, Mes) resulted in an iron–O(H), most synthetic iron based nitrite reduction systems produce water and iron–nitrosyl complexes.20,21 To interrogate the formation of an Fe–NO complex we sought first IR spectroscopy, where we observe a new stretch at 1666 cm−1, in line with either a linear or non-linear nitrosyl. Further, the formation of an iron–nitrosyl, Py2Py(pimcyp)2Fe(NO) (2), was confirmed via X-ray crystallography. Complex 2 showcases a novel binding mode of the ligand to iron, where iron is bound out of plane to the four pyrrole-imine nitrogens with no ligation of iron to the pyridines in the ligand system. The geometry at the iron centre can best be described as distorted square pyramidal. The Fe–N–O angle of 159.6(6)° is consistent with a bent “NO−” ligand.55–60
We next sought to characterize the colour changes, or intermediates, observed on the way to isolation of 2. Stopping the reaction after the initial colour change featuring a brown solution with an orange precipitate allowed for analysis of both species. The analysis of both species by UV-Vis spectroscopy showed no distinct features for either product, making it difficult to assess the reaction. However, when the brown filtrate was characterized by 1H NMR spectroscopy, broad resonances nearly identical to the previously published54 [Py2Py(afaCy)2Fe(OH)]OTf2 were observed along with formation of a new paramagnetic iron(II) complex. Due to similarities in the 1H NMR spectra, we hypothesized that the complex corresponding to the broad 1H NMR resonances was an iron(III)–hydroxide product. To confirm formation of an iron(III)–oxygen containing product, complex 1 was stirred with an equivalent of iodosobenzene for 16 hours, resulting in an immediate colour change from yellow to brown with no noticeable precipitate. Characterization by 1H NMR spectroscopy confirmed that the brown product obtained was indeed the same product as observed in our nitrite reduction studies. While this complex has not been crystallographically characterized, the IR spectrum contained one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch (1669 cm−1) consistent with two azafulvene-amine (afa) ligand arms. Additionally, a mass spectrum consistent with the formation of [Py2Py(afamcyp)2Fe(OH)]2+ allows for the assignment of [Py2Py(afamcyp)2Fe(OH)]OTf2 (3) as an initial product in nitrite reduction (Fig. 2).
N stretch (1669 cm−1) consistent with two azafulvene-amine (afa) ligand arms. Additionally, a mass spectrum consistent with the formation of [Py2Py(afamcyp)2Fe(OH)]2+ allows for the assignment of [Py2Py(afamcyp)2Fe(OH)]OTf2 (3) as an initial product in nitrite reduction (Fig. 2).
While the identity of the orange precipitate remained unclear, we were able to isolate it in low yield (10%) after filtration and several washings with tetrahydrofuran and acetonitrile to remove any 1 or 3. As the complex is sparingly soluble in acetonitrile, analysis of the complex by 1H NMR spectroscopy was possible, showing formation of the same sharp iron(II) complex seen in conjunction with 3 in the reaction filtrate. The IR spectrum of the orange precipitate contain new stretches at 1212 cm−1, 1419 cm−1, and 1646 cm−1, the latter corresponding to the afa tautomer of the ligand framework. The former two IR stretches are in line with reported iron–nitrite complexes, giving insight that this may be a bound nitrite species.61
To investigate the existence of a bound nitrite, the corresponding 15N labelled iron nitrosyl complex was synthesized. Upon addition of [TBA]15NO2 to complex 1, we observed immediate formation of a brown solution with orange precipitate, as expected. The precipitate was isolated by filtration and washed well with tetrahydrofuran and acetonitrile. Analysis of the complex by 1H NMR spectroscopy showed an identical spectrum to the proposed nitrite species. The IR spectrum was quite telling, showing the expected shifts in the proposed nitrite stretches to 1192 cm−1 and 1391 cm−1 due to the 15N labelling.
While we do not have direct evidence of the nitrite binding mode, we propose an N-bound nitrite complex. This assignment was initially surprising, as an O-bound isomer is more consistent with the binding mode in the related zinc complex. Additionally, a nitrito isomer is consistent with initial formation of a hydroxide followed by loss of water and ultimately binding of NO to form a nitrosyl complex. Despite this, the IR stretches are far more similar to reported iron nitro species61 and are quite dissimilar from the nitrito N–O stretch observed in the related zinc compound, [N(afaCy)3Zn(ONO)]OTf,10 (1455 cm−1 (νNO = 1431 cm−1 with 15N labelling)). Based on these results, and mass spectrometry, we hypothesize that the orange precipitate is [Py2Py(afamcyp)2Fe(NO2)]OTf (4-NO2).
Since complexes 2 and 3 could be independently synthesized, we took UV-Vis spectra of these complexes, and they matched the various species observed while monitoring the reaction with UV-Vis (Fig. 2), giving greater insight into what was observed via1H NMR spectroscopy. Similar to nitrite reduction studies with [N(afaCy)3Fe]OTf2, the initial product is 4 followed by deoxygenation of nitrite, producing 3 and nitric oxide. However, 3 is not the final product in nitrite reduction, going on to react with nitric oxide to form 2 and release water. The production of 2 from 3 and nitric oxide was confirmed through the independent addition of pure nitric oxide to isolated 3 (Fig. 2), showing clean formation of 2 by IR and UV-Vis spectroscopies.
To further study the oxyanion reduction abilities of complex 1, we tested reactivity with nitrate, which had similar reactivity to nitrite. When complex 1 is stirred with one equivalent of [TBA]NO3, an orange powder immediately precipitates out of solution. Like with the proposed nitrite bound complex 4, the precipitate is stable when isolated and has a 1H NMR spectrum with sharp paramagnetic peaks characteristic of an iron(II) complex in our system. Due to the spectroscopic similarities to the nitrite analogue 4, we propose an iron bound nitrate complex, [Py2Py(afamcyp)2Fe(NO3)]OTf (5).
To probe the complete list of products from nitrate reduction by 1, three equivalents of 1 and one equivalent of [TBA]NO3 were stirred in acetonitrile overnight (Scheme 1). The analogous colour changes are seen with nitrate as with nitrite. Immediately, an orange powder precipitates out of solution. However, if the reaction is stopped after only a few minutes, compound 3 is not visible by 1H NMR spectroscopy. After stirring overnight, a homogeneous dark green solution is observed. Upon extraction with THF and acetonitrile, brown and green residues, respectively, were obtained. Analysis of the brown product by 1H NMR spectroscopy showed formation of compound 3 in 70% yield. The IR spectrum of the green product cleanly overlayed with the IR spectrum of 2, illustrating that in nitrate reduction, 2 is also the final product. Complex 1 can not only successfully reduce nitrite to an iron bound NO complex, but can also reduce the more difficult oxyanion nitrate.
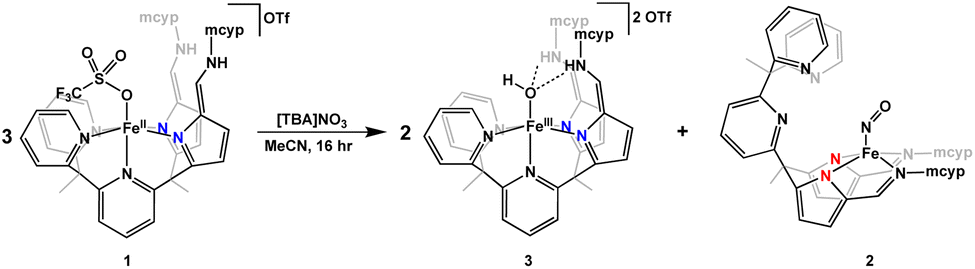 | ||
| Scheme 1 Reduction of nitrate by 1 to form 3 and 2. | ||
Interestingly, stirring the iron-bound nitrite or nitrate complexes 4 or 5, respectively, in acetonitrile for several hours did not result in solubilization or any further reactivity. Instead, upon addition of any amount of 1 (more or less than one equiv.) to the orange precipitate (4 or 5), an immediate solubilization and colour change to brown was noted. This colour change to brown was consistent with the formation of 3 by 1H NMR spectroscopy. Over the course of several hours, the colour changed to green and complex 2 was observed by IR and UV-Vis spectroscopies. The need for an additional iron centre not bound to nitrate or nitrite to reduce 4 and 5 is consistent with a bimolecular mechanism, where nitrite or nitrate binds to one iron centre and is not reduced until a second equivalent of 1 reacts with 4 or 5. The need for two iron centres for the reduction of one nitrogen oxyanion is reminiscent of bimetallic cd1NiR.
Conclusions
In conclusion, we have elucidated possible reduction pathways of nitrate and nitrite to form iron–bound nitrosyl complexes, highlighting key intermediates in these reactions. Our findings are consistent with a bimetallic mechanism for nitrite reduction, where an additional iron equivalent is necessary for the complete reaction, leading to the formation of an iron(III)–hydroxide intermediate (3). This intermediate is less stable in our system compared to previous models and further reacts with nitric oxide to yield a final nitrosyl complex (2) and water. These results enhance our understanding of iron-based reduction processes and provide valuable insights into metalloenzyme behaviour. The work contributes to the broader knowledge of oxyanion reduction mechanisms and the development of more efficient reduction complexes.Data availability
The data that support the findings of this study are available in the ESI† of this article. Deposition numbers 2387394–2387395 contain the supplementary crystallographic data for this paper.Author contributions
J. M. M. carried out the synthetic work, performed and interpreted the analytical characterizations. A. R. F. provided funding and supervised the studies. All authors contributed to writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Sciences, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under award number DOE DE-SC0025026. J. M. M. is thankful for a Hagler Fellowship from Texas A&M University. We thank Dr N. Bhuvanesh (TAMU X-ray Diffraction Laboratory) for support in solving crystal structures. We also thank Shrivignesh S. for artwork.Notes and references
- L. B. Maia and J. J. G. Moura, Chem. Rev., 2014, 114, 5273–5357 CrossRef CAS PubMed.
- S. Rinaldo and F. Cutruzzolà, Biology of the Nitrogen Cycle, 2006, pp. 37–55 Search PubMed.
- I. Moura, J. J. G. Moura, S. R. Pauleta, L. B. Maia, S. Rinaldo, G. Giardina and F. Cutruzzolà, Metalloenzymes in Denitrification: Applications and Environmental Impacts, 2016 Search PubMed.
- P. A. Williams, V. Fülöp, E. F. Carman, N. F. W. Saunders, S. J. Ferguson and J. Hajdu, Nature, 1997, 389, 406–412 Search PubMed.
- J. Yi, M. K. Safo and G. B. Richter-Addo, Biochemistry, 2008, 47, 8247–8249 CrossRef CAS PubMed.
- D. M. Copeland, A. S. Soares, A. H. West and G. B. Richter-Addo, J. Inorg. Biochem., 2006, 100, 1413–1425 CrossRef CAS PubMed.
- T. T. Zhang, Y. D. Liu and R. G. Zhong, J. Inorg. Biochem., 2015, 150, 126–132 CrossRef CAS.
- R. Silaghi-Dumitrescu, Inorg. Chem., 2004, 43, 3715–3718 CrossRef CAS.
- D. M. M. M. Dissanayake, B. E. Petel, W. W. Brennessel, K. L. Bren and E. M. Matson, J. Coord. Chem., 2020, 73, 2664–2676 CrossRef CAS.
- E. M. Matson, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2014, 136, 17398–17401 CrossRef CAS.
- C. L. Ford, Y. J. Park, E. M. Matson, Z. Gordon and A. R. Fout, Science, 2016, 354, 741–743 CrossRef CAS.
- Y. J. Park, M. N. Peñas-Defrutos, M. J. Drummond, Z. Gordon, O. R. Kelly, I. J. Garvey, K. L. Gullett, M. García-Melchor and A. R. Fout, Inorg. Chem., 2022, 61, 8182–8192 CrossRef CAS.
- M. Delgado and J. D. Gilbertson, Chem. Commun., 2017, 53, 11249–11252 RSC.
- Y. M. Kwon, M. Delgado, L. N. Zakharov, T. Seda and J. D. Gilbertson, Chem. Commun., 2016, 52, 11016–11019 RSC.
- K. T. Burns, W. R. Marks, P. M. Cheung, T. Seda, L. N. Zakharov and J. D. Gilbertson, Inorg. Chem., 2018, 57, 9601–9610 CrossRef CAS PubMed.
- P. M. Cheung, K. T. Burns, Y. M. Kwon, M. Y. Deshaye, K. J. Aguayo, V. F. Oswald, T. Seda, L. N. Zakharov, T. Kowalczyk and J. D. Gilbertson, J. Am. Chem. Soc., 2018, 140, 17040–17050 CrossRef CAS PubMed.
- C. C. Tsou, W. L. Yang and W. F. Liaw, J. Am. Chem. Soc., 2013, 135, 18758–18761 CrossRef CAS PubMed.
- F. Te Tsai, P. L. Chen and W. F. Liaw, J. Am. Chem. Soc., 2010, 132, 5290–5299 CrossRef PubMed.
- F. Te Tsai, Y. C. Lee, M. H. Chiang and W. F. Liaw, Inorg. Chem., 2013, 52, 464–473 CrossRef PubMed.
- N. Kulbir, S. Das, T. Devi, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2023, 14, 2935–2942 RSC.
- W. M. Ching, P. P. Y. Chen and C. H. Hung, Dalton Trans., 2017, 46, 15087–15094 RSC.
- C. M. Moore and N. K. Szymczak, Chem. Sci., 2015, 6, 3373–3377 RSC.
- H. Yokoyama, K. Yamaguchi, M. Sugimoto and S. Suzuki, Eur. J. Inorg. Chem., 2005, 1435–1441 CrossRef CAS.
- E. Monzaní, G. J. Anthony, A. Koolhaas, A. Spandré, E. Leggierí, L. Casella, M. Gullottí, G. N. Nardin, L. Randaccio, M. Fontaní, P. Zanelló and J. Reedijk, J. Biol. Inorg. Chem., 2000, 251–261 CrossRef.
- Z. Sakhaei, S. Kundu, J. M. Donnelly, J. A. Bertke, W. Y. Kim and T. H. Warren, Chem. Commun., 2017, 53, 549–552 RSC.
- P. Ghosh, M. Stauffer, M. E. Ahmed, J. A. Bertke, R. J. Staples and T. H. Warren, J. Am. Chem. Soc., 2023, 12007–12012 CrossRef CAS.
- S. Kundu, W. Y. Kim, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2017, 139, 1045–1048 CrossRef CAS.
- M. Kujime, C. Izumi, M. Tomura, M. Hada and H. Fujii, J. Am. Chem. Soc., 2008, 130, 6088–6098 CAS.
- J. A. Halfen, S. Mahapatra, E. C. Wilkinson, A. J. Gengenbach, V. G. Young, L. Que and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 763–776 CAS.
- K. Shi, L. Mathivathanan, A. K. Boudalis, P. Turek, I. Chakraborty and R. G. Raptis, Inorg. Chem., 2019, 58, 7537–7544 CrossRef CAS PubMed.
- A. C. Merkle and N. Lehnert, Dalton Trans., 2012, 41, 3355–3368 CAS.
- S. Gupta, S. Arora, A. Mondal, S. C. E. Stieber, P. Gupta and S. Kundu, Eur. J. Inorg. Chem., 2022, e202200105 CAS.
- W. Tao, J. K. Bower, C. E. Moore and S. Zhang, J. Am. Chem. Soc., 2019, 141, 10159–10164 CrossRef CAS PubMed.
- S. C. N. Hsu, Y. L. Chang, W. J. Chuang, H. Y. Chen, I. J. Lin, M. Y. Chiang, C. L. Kao and H. Y. Chen, Inorg. Chem., 2012, 51, 9297–9308 CrossRef CAS.
- H. Kunkely and A. Vogler, J. Am. Chem. Soc., 1995, 117, 540–541 CrossRef CAS.
- J. Zhou, S. Gao and G. Hu, Energy Fuels, 2024, 38, 6701–6722 CrossRef CAS.
- Z. Zhang, N. Zhang, J. Zhang, B. Deng, Z. Cao, Z. Wang, G. Wei, Q. Zhang, R. Jia, P. Xiang and S. Xia, Chem. Eng. J., 2024, 483, 148952 Search PubMed.
- X. Zhang, Y. Wang, Y. Wang, Y. Guo, X. Xie, Y. Yu and B. Zhang, Chem. Commun., 2022, 58, 2777–2787 RSC.
- M. Qiao, D. Zhu and C. Guo, Chem. Commun., 2024, 60, 11642–11654 RSC.
- Y. Wu, K. K. Lu and L. H. Xu, J. Mater. Chem. A, 2023, 11, 17392–17417 RSC.
- S. D. Kulbir, T. Devi, M. Goswami, M. Yenuganti, P. Bhardwaj, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2021, 12, 10605–10612 RSC.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, J. Am. Chem. Soc., 2018, 140, 842–847 CrossRef CAS.
- S. Gupta, S. Vijayan, J. A. Bertke and S. Kundu, Inorg. Chem., 2024, 61, 8477–8483 CrossRef PubMed.
- J. Gwak, S. Ahn, M. H. Baik and Y. Lee, Chem. Sci., 2019, 10, 4767–4774 RSC.
- S. Padmanaban, J. Choi, H. Vazquez-Lima, D. Ko, D. Yoo, J. Gwak, K. Bin Cho and Y. Lee, J. Am. Chem. Soc., 2022, 144, 4585–4593 CrossRef CAS PubMed.
- K. S. Suslick and R. A. Watson, Inorg. Chem., 1991, 30, 912–919 CrossRef CAS.
- A. R. Corcos, J. S. Pap, T. Yang and J. F. Berry, J. Am. Chem. Soc., 2016, 138, 10032–10040 CrossRef CAS PubMed.
- D. M. Beagan, A. C. Cabelof, R. Pepin, M. Pink, V. Carta and K. G. Caulton, Inorg. Chem., 2021, 60, 17241–17248 CrossRef CAS PubMed.
- A. C. Cabelof, V. Carta and K. G. Caulton, Chem. Commun., 2021, 57, 2780–2783 RSC.
- W. R. Marks, D. F. Baumgardner, E. W. Reinheimer and J. D. Gilbertson, Chem. Commun., 2020, 56, 11441–11444 RSC.
- P. L. Damon, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 12743–12746 CrossRef CAS.
- E. M. Matson, J. A. Bertke and A. R. Fout, Inorg. Chem., 2014, 53, 4450–4458 CrossRef CAS.
- C. H. Hsieh, S. Ding, Ö. F. Erdem, D. J. Crouthers, T. Liu, C. C. L. McCrory, W. Lubitz, C. V. Popescu, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Nat. Commun., 2014, 5, 3684 CrossRef CAS PubMed.
- M. J. Drummond, C. L. Ford, D. L. Gray, C. V. Popescu and A. R. Fout, J. Am. Chem. Soc., 2019, 141, 6639–6650 CrossRef CAS.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- M. Ray and A. P. Golombek, Inorg. Chem., 1999, 38, 3110–3115 CrossRef CAS.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121, 14682–14905 CrossRef CAS PubMed.
- D. M. P. Mingos, Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine I, 2014 Search PubMed.
- D. J. Thomas and N. Lehnert, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2017 Search PubMed.
- J. B. Harland, E. C. Manickas, A. P. Hunt and N. Lehnert, Comprehensive Coordination Chemistry III, 2021, pp. 806–874 Search PubMed.
- Z. N. Nilsson, B. L. Mandella, K. Sen, D. Kekilli, M. A. Hough, P. Moënne-Loccoz, R. W. Strange and C. R. Andrew, Inorg. Chem., 2017, 56, 13205 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06570k |
| This journal is © The Royal Society of Chemistry 2025 |