
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferent p-block elements induce C3[111] octahedral distortion in titanium to generate an intense nonlinear effect†
Zhenhua
Li‡
a,
Zhengli
Liang‡
 b,
Jiahao
Wan
a,
Lehui
Liu
c,
Chunxiang
Wu
a,
Ping
Wang
a,
Xingxing
Jiang
*b,
Zheshuai
Lin
b and
Hongming
Liu
*a
b,
Jiahao
Wan
a,
Lehui
Liu
c,
Chunxiang
Wu
a,
Ping
Wang
a,
Xingxing
Jiang
*b,
Zheshuai
Lin
b and
Hongming
Liu
*a
aGuangxi Key Laboratory of Electrochemical Energy Materials, School of Chemistry and Chemical Engineering, Guangxi University, Nanning, Guangxi 530004, China. E-mail: hongming9224@126.com
bTechnical Institute of Physics and Chemistry, Chinese Academy of Science, Beijing 100190, China. E-mail: xxjiang@mail.ipc.ac.cn
cKey Laboratory of Optoelectronic Materials Chemistry, Physics Fujian Institute of Research on the Fujian, Fuzhou, 350002, China
First published on 15th January 2025
Abstract
Acentric crystalline materials are the cornerstone of numerous cutting-edge technologies and have been highly sought-after, but they are difficult to construct controllably. Herein, by introducing a new p-block element to break the symmetrical environment of the d0 transition metal in the centric matrix TiTe3O8, a novel acentric tellurite sulfate, namely Ti(TeO3)(SO4), was successfully constructed. In its structure, two types of p-block element-centered oxo-anionic groups, i.e. [TeO3] and [SO4], endow [TiO6] with an out-of-center distortion along the local C3[111] direction, which is rare in titanium oxides containing a lone-pair cation. The synergy of the distorted [TiO6] octahedron, lone-pair [TeO3] pyramid and rigid [SO4] tetrahedron within its structure induces a strong second harmonic generation (SHG) response of 11.6 × KDP (KH2PO4), the largest value among mercury-free sulfates. Additionally, Ti(TeO3)(SO4) also shows the largest birefringence (0.145) among sulfates possessing an SHG response that is more than ten times that of KDP, showing huge potential as a nonlinear optical material. The successful implementation of the strategy of inducing intra-octahedral distortion in a d0 transition metal by different p-block elements provides new opportunities for constructing acentric structures and exploiting outstanding nonlinear optically active sulfates.
Introduction
Crystalline materials featuring a non-centrosymmetric (NCS) structure are the cornerstone of some crucial properties such as piezoelectricity, ferroelectricity, second-order nonlinear optical (NLO) phenomena, and dielectric behavior.1–3 Among these properties, the second-order NLO effect is the key to the frequency conversion of solid-state laser devices.4–11 Though acentric crystals are highly sought after for their fascinating versatility, the vast majority of inorganic compounds have a tendency to crystallize in the centric space groups. Over the past few decades, numerous strategies have been proposed to facilitate the construction of NCS structures, e.g., the combination of multiple anionic groups,12–14 cation compensation,15,16 multi-component modification,17 heterovalent substitution,18–21 the condensation of anionic units,22–24etc.25–31 Their implementation gave rise to the discovery of plentiful NLO crystals that demonstrate outstanding performance. However, the application of these strategies does not always ensure the formation of an NCS structure. Generally, the symmetric arrangement of anionic groups within the structure still leads to a centric space group. The crucial step in constructing NCS structures lies in designing more practical and feasible strategies to directly disrupt the spatially symmetric alignment.According to the anionic group theory,32,33 the macroscopic second harmonic generation (SHG) effect of NLO crystals stems from the geometric addition of the microscopic second-order susceptibility tensor of the NLO-active basic building units (BBUs) within the structure. Distorted octahedra consisting of a d0 transition metal (Md0) bonded to six O atoms (Md0O6) represent a category of NLO-active BBUs with spontaneous polarization; the introduction of these units facilitates the formation of NCS structures and may potentially lead to a strong SHG response.34–37 Therefore, the combination of distorted [Md0O6] with other NLO-active BBUs has been widely applied to construct NLO crystals, including iodate, selenite, tellurite, and germanate.38–44 For [Md0O6] octahedra, most of their out-of-center distortion can be ascribed to the second-order Jahn–Teller (SOJT) electronic effect,45,46 which is categorized as a primary distortive effect and occurs when the empty d-orbitals of the metal mix with the filled p-orbitals of the ligands.35 Further, the intra-octahedral distortion of [Md0O6] can also be influenced by the bond networks and lattice stresses from other directly connected oxo-anionic groups, which is called secondary distortion.35 Normally, the secondary distortion is weaker and serves to enhance the primary distortion, as reflected by the Md0-oxides containing a lone-pair cation. When an [Md0O6] octahedron bridges both the lone-pair cation and other [Md0O6] octahedra simultaneously, an intra-distortion is highly likely to occur (Fig. 1a). Nevertheless, the secondary distortion can sometimes override the primary distortion, as exemplified by the [TiO6] octahedron in the winstanleyite TiTe3O8,47 and its out-of-center distortion is inhibited due to the coordination with six identical [TeO4] polyhedra. This symmetric configuration is unfavorable to the formation of acentric structures.
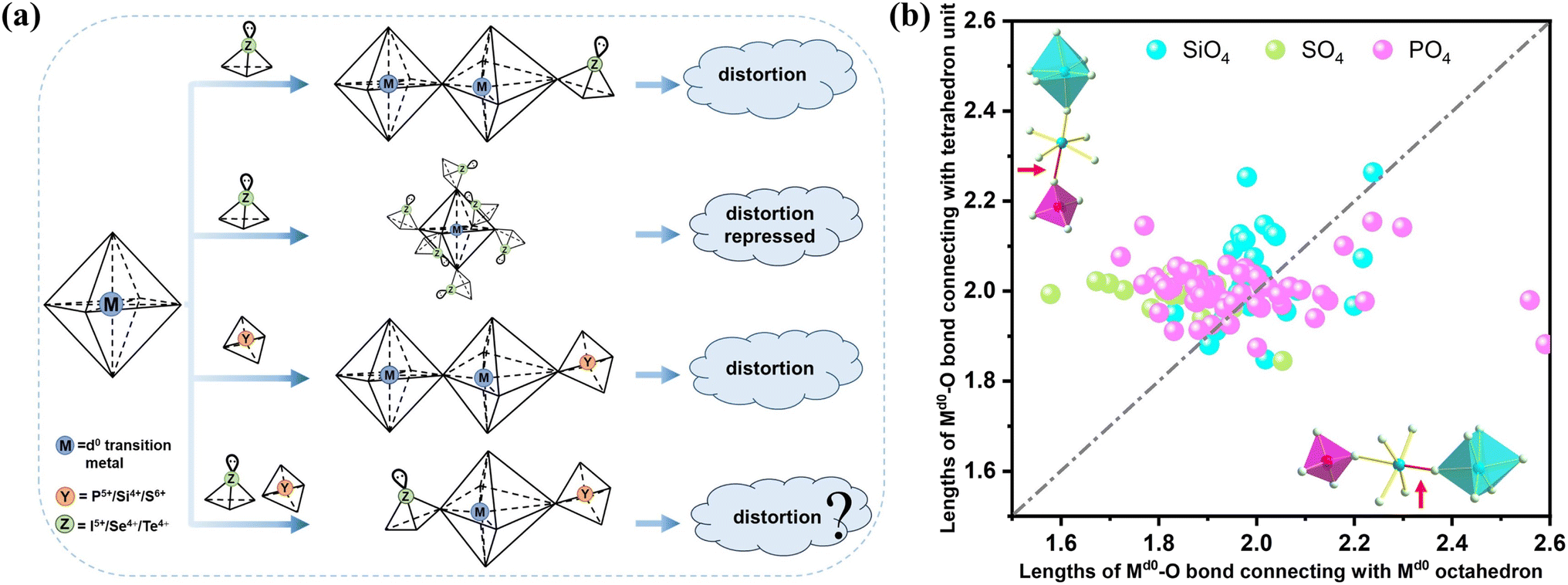 | ||
| Fig. 1 (a) Schematic diagram of [Md0O6] octahedral distortion induced by different connections. (b) Length distribution of Md0–O bonds in the [Md0O6] octahedron connected to other [Md0O6] octahedra and tetrahedral units simultaneously in phosphates, sulfates, and silicates containing Md0. | ||
For those [Md0O6] octahedra with repressed distortion, introducing an additional BBU to break their symmetric coordination may be a viable route to activate their secondary distortion. In order to explore the feasibility of this assumption, the aforementioned centric TiTe3O8 was selected as the matrix. In terms of the selection of appropriate BBUs, the tetrahedral [PO4], [SO4], and [SiO4] came into our sight for the following reasons: (1) tetrahedral BBUs can also exert a profound influence on the intra-octahedral distortion of [Md0O6], as exemplified by KTiOPO4.48 Additionally, we systematically examined the distortion characteristics of the [Md0O6] octahedra in phosphates, sulfates, and silicates containing Md0, and the result showed that inside the [Md0O6] octahedra, the length of the Md0–O bonds connecting to the tetrahedral groups differed greatly from that connecting to other [Md0O6] octahedra, which induced universal distortion inside the [Md0O6] octahedra (Fig. 1b and S1†), just as the situation of the intra-octahedral [Md0O6] distortion in Md0-oxides containing a lone-pair cation.35 (2) The introduced tetrahedral groups are expected to induce a symmetry-breaking of the [TiO6] coordination (Fig. 1a), and the subsequently distorted [TiO6] unit can act as an NLO-active BBU to participate in the SHG response contribution of the target crystals. (3) The introduction of tetrahedral groups may also break the symmetric arrangement of the [TeO4] polyhedra in the TiTe3O8 structure and give rise to a surprisingly strong SHG in the resulting phosphates, sulfates, or silicates, as most of them are beset with low SHG intensities owing to the small polarizability of [PO4], [SO4], and [SiO4] tetrahedra.
Following the strategy of introducing a tetrahedral unit into the structure of TiTe3O8, a novel tellurite–sulfate, namely Ti(TeO3)(SO4), was synthesized. It represents a rare Md0-containing acentric crystal where the [Md0O6] octahedron directly connects to both the tetrahedral anionic group and lone-pair cation, and it is also a rare titanium oxide in which the [TiO6] octahedron is linked to lone-pair cations but features C3[111] (face) distortion, which results from its simultaneous connection with two types of p-block elements, Te and S. Furthermore, experimental and calculational investigations suggest that the unique distorted [TiO6] octahedron collaborates with the [TeO3] and [SO4] groups, giving rise to an intense SHG response of 11.6 × KDP for Ti(TeO3)(SO4), which is the maximum among mercury-free sulfates.
Results and discussion
Pure Ti(TeO3)(SO4) crystals were prepared via a hydrothermal process at 230 °C with raw materials of TiO2, TeO2, and sulfuric acid solution. The purity of the synthetic samples was confirmed by powder X-ray diffraction (XRD) analysis (Fig. S2†). Energy-dispersive X-ray spectroscopy (EDS) performed on Ti(TeO3)(SO4) crystals suggests an average molar ratio of 1.08![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.16:1:7.15 for Ti, Te, S, and O elements (Fig. S3†), matching well with the chemical formula determined by single-crystal XRD. The IR spectrum of Ti(TeO3)(SO4) powder shows no obvious absorption in the range of 4000–1370 cm−1 (Fig. S4†), and the absorption peaks between 1200 and 950 cm−1 can be attributed to the stretching vibrations of S–O bonds, accurately proving the existence of the [SO4] tetrahedron, while these peaks between 900 and 620 cm−1 are assigned to the stretching vibration of Te–O and Ti–O bonds. These assignments are consistent with the reported articles.
:1.16:1:7.15 for Ti, Te, S, and O elements (Fig. S3†), matching well with the chemical formula determined by single-crystal XRD. The IR spectrum of Ti(TeO3)(SO4) powder shows no obvious absorption in the range of 4000–1370 cm−1 (Fig. S4†), and the absorption peaks between 1200 and 950 cm−1 can be attributed to the stretching vibrations of S–O bonds, accurately proving the existence of the [SO4] tetrahedron, while these peaks between 900 and 620 cm−1 are assigned to the stretching vibration of Te–O and Ti–O bonds. These assignments are consistent with the reported articles.
Single-crystal XRD determination shows that Ti(TeO3)(SO4) crystallizes into the NCS space group P21 (Table S1†). There are crystallographically independent one Te atom, one S atom, one Ti atom, and seven O atoms in its asymmetric unit (Table S2†). Bond valence sum (BVS) calculation49 yields values of 4.34, 6.07 and 3.96 for Ti, S and Te atoms, respectively (Table S2†), matching well with their oxidation states. All these atoms are located in ordinary positions (Fig. S5†). As shown in Fig. 2a, the Te atom is bonded with three O atoms to form a [TeO3] pyramid containing lone-pair electrons, with the Te–O bond lengths ranging from 1.858 Å to 1.897 Å (Table S3†). The S atom coordinates with four O atoms to form a typical [SO4] tetrahedron with the S–O bond lengths in the range of 1.459–1.476 Å. The Ti atom is coordinated by six O atoms to compose a typical [TiO6] octahedron with three long (2.018–2.072 Å) and three short (1.813–1.876 Å) Ti–O bonds. According to the classification proposed by Halasyamani,35 the out-of-center distortion of the [TiO6] octahedron in this structure belongs to the C3[111] direction. To our knowledge, this C3[111] distortion is rare in Ti4+ oxides containing a lone-pair cation. Further structural analysis indicates that the three long Ti–O bonds are linked by [SO4] groups, while the three short ones are connected by [TeO3] groups (Fig. S6†). This implies that the C3[111] distortion in the [TiO6] octahedron in this structure results from the connection of Ti–O bonds to different types of p-block element-centered oxo-anionic groups. It breaks the existing view that, for the Md0 oxides containing a lone-pair cation, the intra-octahedral distortion of [Md0O6] only occurs when it directly connects with at least one other Md0 cation or contains at least one terminal O atom.35 The [TiO6] octahedron links with three [SO4] groups and three [TeO3] groups (Fig. 2b), and both the [TeO3] pyramid and [SO4] tetrahedron are three-connected by [TiO6] octahedra, with the remaining O atom in the [SO4] group being pensile alone (Fig. S6†). The [TiO6] octahedra and [TeO3] pyramids are interlaced to form a [(TiO3O3/2)(TeO3/2)] layer parallel to the ab-plane (Fig. 2d). The [(TiO3O3/2)(TeO3/2)] layers are stacked along the c-axis through the connection of [SO4] tetrahedra to build the final three-dimensional structure of Ti(TeO3)(SO4) (Fig. 2c). In the connection mode, it can be characterized to [(TiO6/2)(TeO3/2)(SO3/2O)].
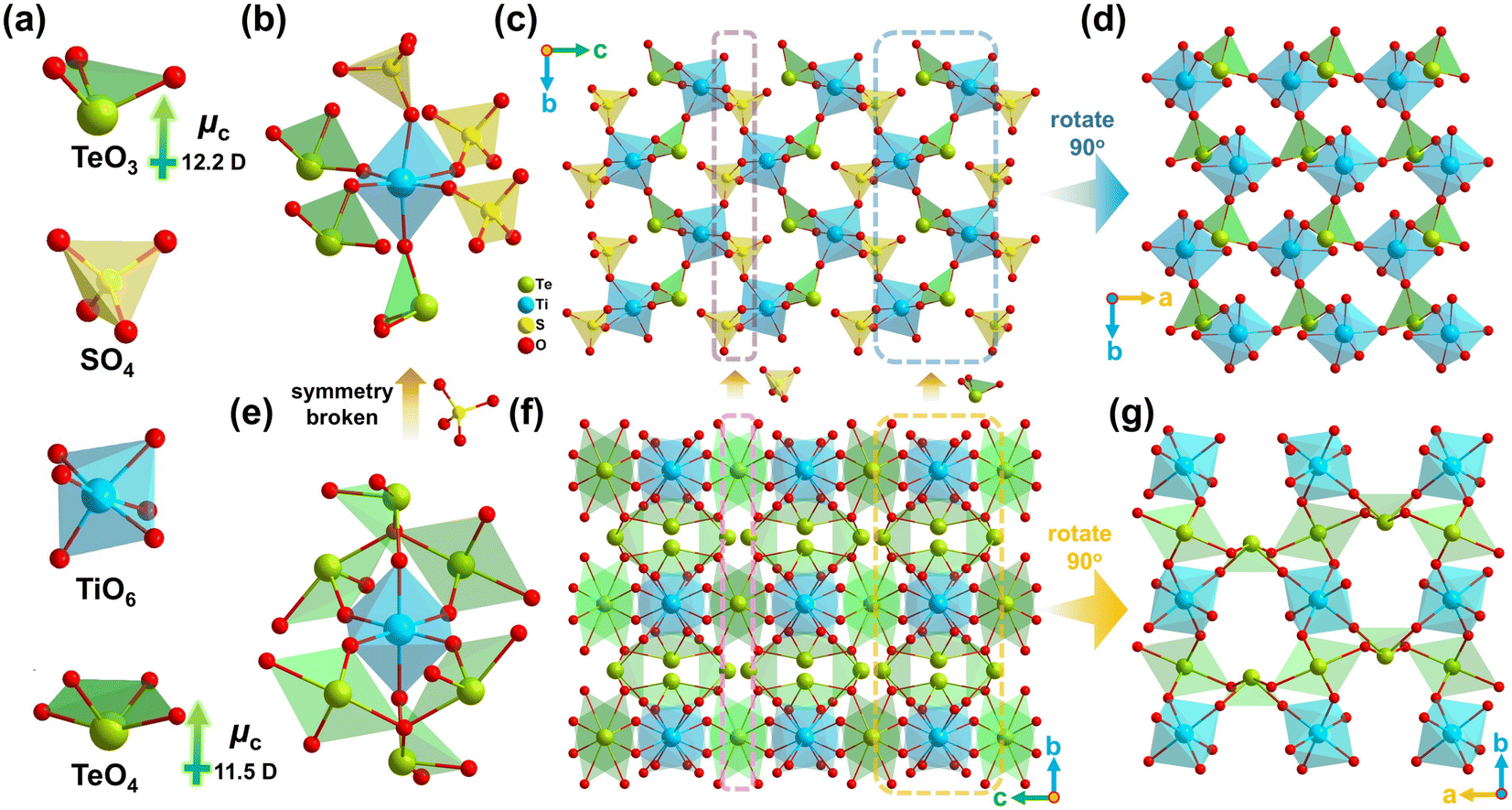 | ||
| Fig. 2 Structural diagram of Ti(TeO3)(SO4) and TiTe3O8. (a) The anionic groups; Ti(TeO3)(SO4) structure: (b) the Ti(TeO3)3(SO4)3 unit, (c) the structure viewed along the [100] direction, (d) the [(TiO3O3/2)(TeO3/2)] layer viewed along the [001] direction; TiTe3O8 structure: (e) the coordination environment of [TiO6], (f) the structure viewed along the [100] direction, (g) the [(TiO2O4/2)(TeO4/2)2] layer viewed along the [001] direction. | ||
A detailed structural comparison suggests that the introduction of the [SO4] group leads to obvious structural changes in contrast to the matrix TiTe3O8. Firstly, the introduced [SO4] groups substitute three of the six [TeO4] groups surrounding the [TiO6] octahedron (Fig. 2e, b), and the remaining three [TeO4] groups with relatively small local dipole moment (LDM, μc = 11.5 D) are also converted to [TeO3] groups with larger LDM (μc = 12.2 D) (Fig. 2a). Secondly, the [SO4] groups combined with the [TeO3] groups induce the C3[111] distortion of the [TiO6] octahedron in Ti(TeO3)(SO4) (Fig. 2b), which is different from the undistorted [TiO6] octahedron in TiTe3O8 (Fig. 2e). The magnitude of out-of-center distortion (Δd) calculated by the method proposed by Halasyamani35 gives a value of Δd = 0.62 for the [TiO6] octahedron in Ti(TeO3)(SO4), which can be quantified as moderate distortion. This value is much larger than the average magnitude of Ti4+ (Δd = 0.34), which is often considered to be a weak distorter and is the weakest distorted Md0 cation among Mo6+, V5+, W6+, Nb5+, Ta5+ and Ti4+.35 A reasonable spatial arrangement of such distorted [TiO6] octahedra within the structure is expected to enhance the NLO effect of the resultant Ti(TeO3)(SO4). Thirdly, the introduction of [SO4] groups also changes the arrangement of the lone-pair anionic groups. As shown in Fig. 2f and g, the structure of TiTe3O8 can be seen as the stacking of [(TiO2O4/2)(TeO4/2)2] layers along the c-axis through the interlayer connection of [TeO4] groups, while the structure of Ti(TeO3)(SO4) can be approximately regarded as a derivation of the TiTe3O8 structure, obtained by replacing the interlayer [TeO4] groups with the [SO4] groups, and the [TeO4] groups within the layer are replaced by the [TeO3] groups (Fig. 2c). All the [TeO4] groups are anti-parallelly oriented in the TiTe3O8 structure (Fig. 2f and g). Combined with the symmetric [TiO6] octahedron, it crystallizes in the cubic Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group.47 However, the introduced [SO4] groups break the highly symmetric arrangement of the [TeO4] groups, leading to a non-antiparallel orientation of the [TeO3] groups (Fig. 2c and d), and ultimately result in symmetry-breaking in the structure of Ti(TeO3)(SO4). The [TeO3] groups in its structure have a distinct net polarization vector on the b-axis. This will undoubtedly lead to a drastic change in the optical properties of Ti(TeO3)(SO4) compared to TiTe3O8. In addition, the composition of Ti(TeO3)(SO4) implies its possible similarities to TiO2 polymorphs. Comparative analysis of structures suggests that Ti(TeO3)(SO4) can be regarded as a derived product of rutile TiO2, which is obtained by replacing half of the O atoms of the [TiO6] octahedron with [TeO3] units and the other half of the O atoms with [SO4] units in its structure (Fig. S7†).
space group.47 However, the introduced [SO4] groups break the highly symmetric arrangement of the [TeO4] groups, leading to a non-antiparallel orientation of the [TeO3] groups (Fig. 2c and d), and ultimately result in symmetry-breaking in the structure of Ti(TeO3)(SO4). The [TeO3] groups in its structure have a distinct net polarization vector on the b-axis. This will undoubtedly lead to a drastic change in the optical properties of Ti(TeO3)(SO4) compared to TiTe3O8. In addition, the composition of Ti(TeO3)(SO4) implies its possible similarities to TiO2 polymorphs. Comparative analysis of structures suggests that Ti(TeO3)(SO4) can be regarded as a derived product of rutile TiO2, which is obtained by replacing half of the O atoms of the [TiO6] octahedron with [TeO3] units and the other half of the O atoms with [SO4] units in its structure (Fig. S7†).
Powder SHG measurement shows that the SHG intensity of Ti(TeO3)(SO4) enhances gradually as the particle size increases under 1064 nm laser irradiation, implying that it is type-I phase-matching at 1064 nm (Fig. 3a).50 Furthermore, it exhibits a strong SHG intensity that is about 11.6 times that of the reference KDP (Fig. 3b). The calculated SHG tensors (Table S4†) taking into account the constraints of Kleinman's symmetry and the space group (P21) also afford a large value (d222 = 3.16 pm V−1), in rough agreement with the measured value. As is known, due to the small microscopic second-order susceptibility tensor of the tetrahedral [SO4] unit, the majority of sulfates suffer from the issue of weak SHG intensity, despite several tactics having been implemented recently to enhance the SHG response of sulfate, such as introducing lone-pair and lanthanide-polyhedron anionic groups, which leads to the finding of improved SHG response of CeF2(SO4) (8 × KDP),51 Te2O3(SO4) (6 × KDP),52 K2Bi2(SO4)2Cl4 (5.5 × KDP),53 Rb2Bi2(SO4)2Cl4 (5.3 × KDP),53 and Nb2O3(IO3)2(SO4) (6 × KDP).42 Only two age-old mercury-based acentric crystals were lately found to possess SHG responses stronger than 10 × KDP, including HgSO4 (11 × KDP)54 and Hg3O2SO4 (14 × KDP).55 However, the presence of recognized highly toxic mercury will greatly limit their application expansion as NLO materials. By contrast, the application expansion of Ti(TeO3)(SO4) only suffers from the concern about the suspected hazards of tellurium compounds such as teratogenicity.56,57 Partial density of states (DOS) calculation reveals that the top of the valence band is dominated by the non-bonding O-2p and Te-5p orbitals from −7 eV to 0 eV, and the bottom of the conduction band is composed of the unoccupied Ti-3d and Te-5p orbitals located from +2 eV to +8 eV (Fig. S8†), suggesting that the Ti–O and Te–O interactions determine the strong SHG response of Ti(TeO3)(SO4). The SHG-weighted electron density maps also show that the occupied and unoccupied states are mostly concentrated on the [TiO6] and [TeO3] units in the virtual electron process, but are nearly absent in the [SO4] groups (Fig. 3c and d), implying the predominant contribution of [TiO6] and [TeO3] groups to the strong SHG response of Ti(TeO3)(SO4). This result is also supported by the local dipole moment (LDM) calculation.
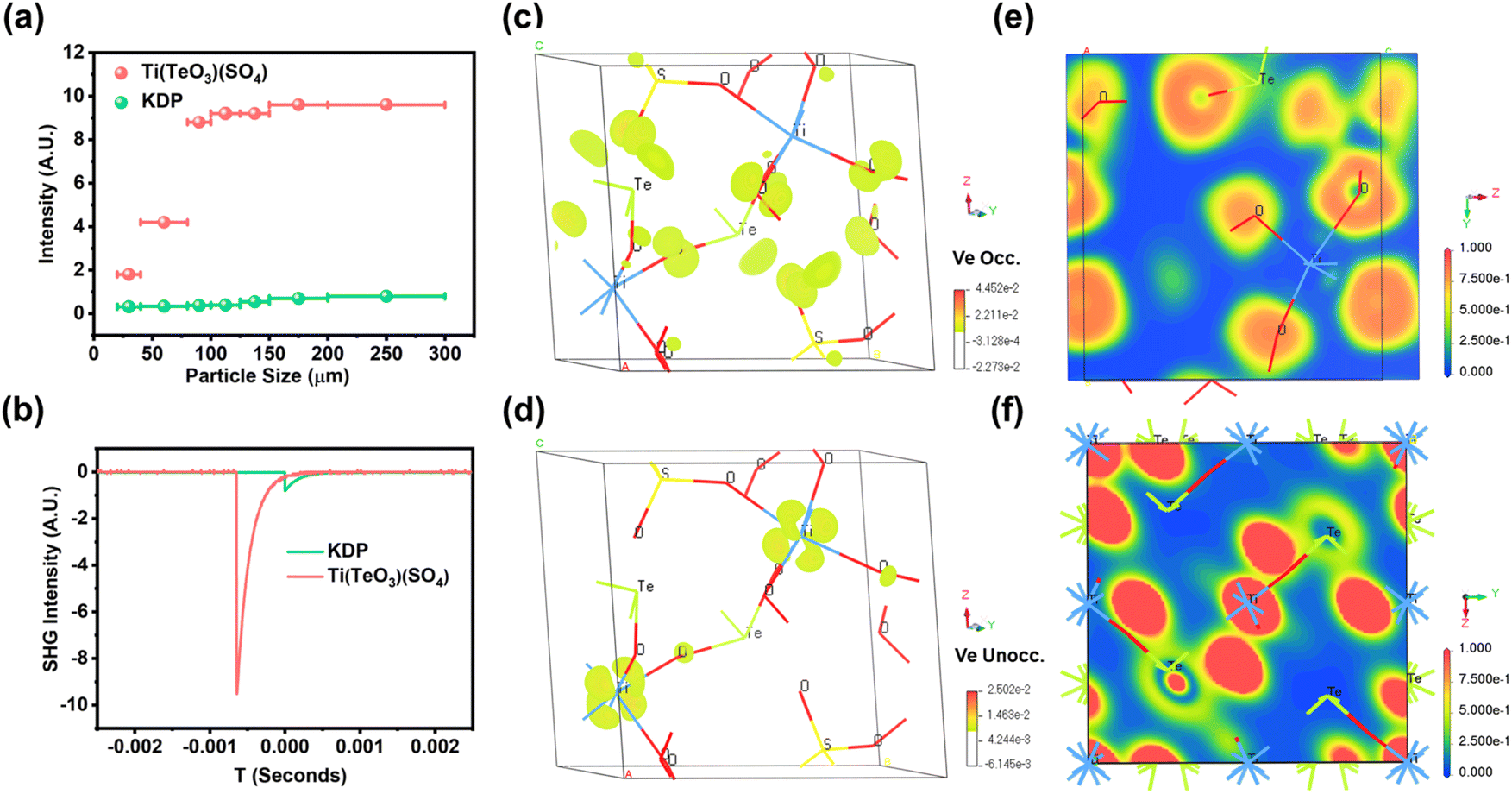 | ||
| Fig. 3 (a) SHG intensity versus particle size under laser irradiation at 1064 nm with KDP as the reference; (b) oscilloscope traces of the SHG signals (at the sample size range of 200–300 μm); SHG-weighted density map of (c) occupied and (d) unoccupied states in the virtual electron process; calculated electron-density difference of (e) Ti(TeO3)(SO4) (100) and (f) TiTe3O8 (100). | ||
As listed in Table S5,† the polarizations of the [TiO6], [TeO3], and [SO4] units entirely neutralize each other on the x- and z-components, while they are superimposed on the y-component, giving a total dipole moment of 16.864 D within the unit cell (Fig. S9†). This indicates that the strong SHG response of Ti(TeO3)(SO4) can be ascribed to the synergy of the [TiO6] octahedron, [TeO3] pyramid, and [SO4] tetrahedron. It is a compound with an excellent SHG response obtained by the combination of three types of NLO-active BBUs. Taking into account the unit cell volume, the calculated density of dipole moment is 64.5 × 10−21 esu cm Å−3 for Ti(TeO3)(SO4). Contrastively, the same calculation approach gives the renowned NLO crystal KTiOPO4 a dipole moment density of 32.9 × 10−21 esu cm Å−3 (Table S6†). Additionally, the net dipole moments of the isolated [TiO6] and [TeO3] units were calculated to be 2.224 D and 15.842 D, respectively, contributing predominantly to the total dipole moment of Ti(TeO3)(SO4). Besides, the electron density calculation results also reveal that the electron densities around Ti and O in TiTe3O8 are close (Fig. 3f). In stark contrast, an apparent difference in electron density is found around Ti and O in Ti(TeO3)(SO4) (Fig. 3e). This implies that the introduction of an [SO4] tetrahedron enhances the polarization of the Ti–O covalent bonds, consistent with the high distortion and enhanced dipole moment of the [TiO6] octahedron in Ti(TeO3)(SO4) compared to that in TiTe3O8. All the above analyses imply that [TiO6] and [TeO3] units could be enormously conducive to sulfates to generate a strong SHG response. This provides a novel avenue to achieve large NLO coefficients in sulfates.
For a potential NLO crystal, a large-enough birefringence (usually >0.02) is a requisite to guarantee its phase-matching character. Under a polarizing microscope, the observed interference color for a crystal plate with a thickness of 7.6 μm is II-order blue, corresponding to an optical path difference of about 1.1 μm (Fig. 4a),58 so the measured birefringence is 0.145@visible light for Ti(TeO3)(SO4). This value is competitive to those of most reported asymmetric sulfates (Table S7†). Among acentric sulfates possessing an SHG response stronger than 10 × KDP, Ti(TeO3)(SO4) exhibits the largest birefringence (Fig. 4b), ensuring phase matchability of its crystal over a wide wavelength range. The DFT calculation gave a refractive index relationship of nX1 > nX2 > nX3 between the three optical main axes, a birefringence of 0.137@1064 nm and the shortest phase-matchable wavelength of 413 nm (Fig. 4c), well consistent with the experimental results. As is known, most sulfates suffer from small birefringences owing to the weak optical anisotropy resulting from the nearly isotropic configuration of the tetrahedral [SO4] group, hence hindering their phase-matchability in the short wavelength region.59,60 Compared with the non-birefringence characteristics of the matrix TiTe3O8 (isotropic cubic space group), the birefringence of Ti(TeO3)(SO4) has gained a great enhancement. This confirms the superiority of the strategy of introducing the [SO4] unit to induce symmetry-breaking in developing large birefringent sulfates. In the structure of Ti(TeO3)(SO4), the [TiO6] features an obvious out-of-center distortion (Δd = 0.62), while the [TeO3] possesses lone-pair and a large local dipole moment (μc = 12.2 D), both of which exhibit larger optical anisotropy than the [SO4] group, hence the [TiO6] and [TeO3] groups contribute predominantly to its large birefringence, consistent with the results of some other sulfates.42,51,61 The large birefrigence of some tellurites and TiO2 crystal can support this speculation as well.62,63
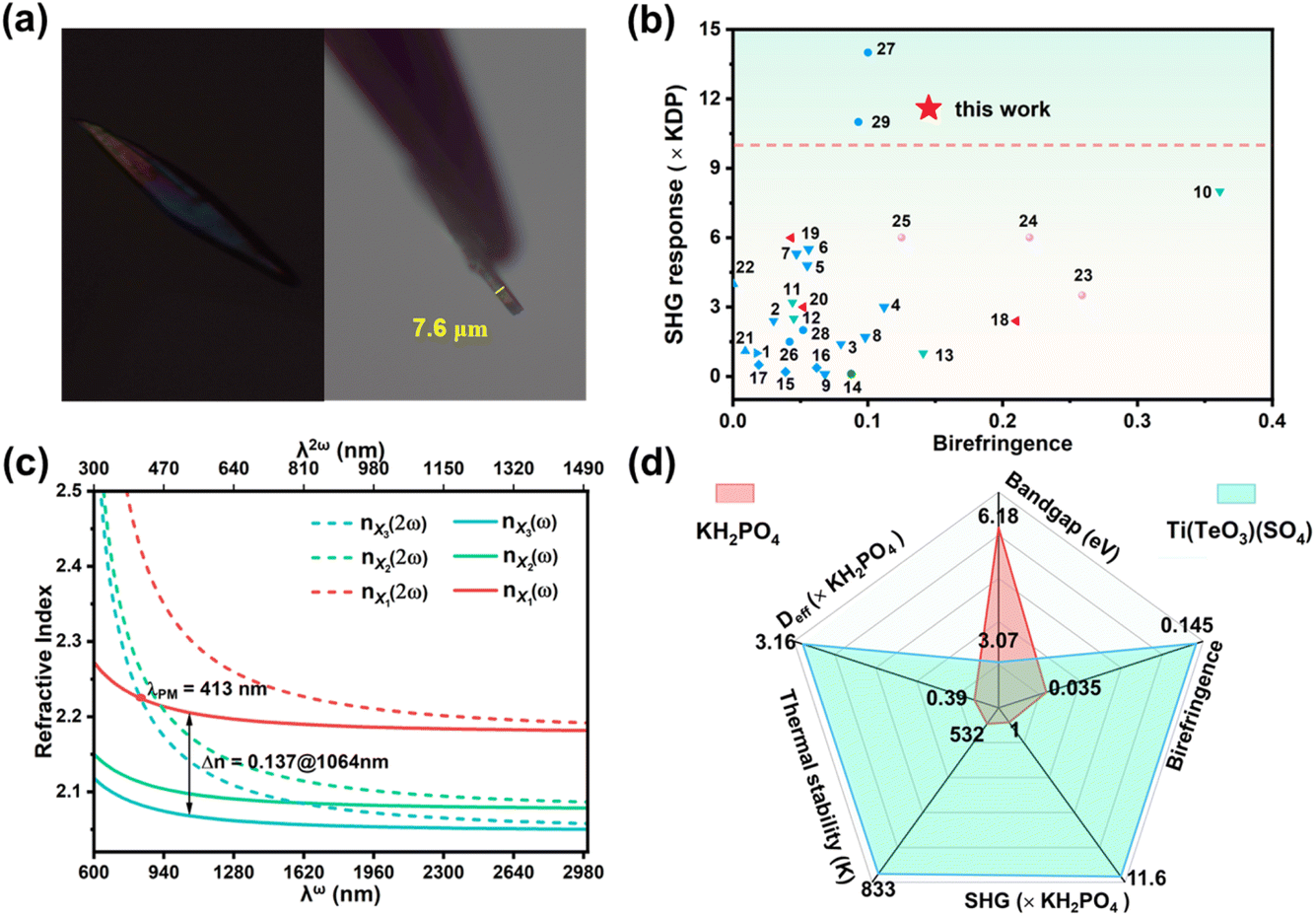 | ||
| Fig. 4 (a) The original interference color under orthogonal polarized light and thickness of the selected crystal plate of Ti(TeO3)(SO4); (b) SHG responses and birefringence of some reported sulfates (the numbers insides correspond to the serial numbers of the sulfates listed in Table S7†); (c) the calculated refractive index dispersion curves; (d) properties comparison of Ti(TeO3)(SO4) with the commercial KH2PO4. | ||
Thermogravimetric analysis reveals that Ti(TeO3)(SO4) can be stable up to 560 °C (Fig. S10†). The differential scanning calorimetry curve also illustrates that there is no phase transition prior to its decomposition, as verified by the powder XRD pattern of the samples after thermal annealing at 500 °C (Fig. S11†). Its stability is superior to that of most tellurite sulfates, as exemplified by Te2O3SO4 (540 °C),52 Te(OH)3(SO4)·H3O (110 °C),52 and Y3(TeO3)2(SO4)2(OH)(H2O) (434 °C).64 The powder XRD pattern of the calcined product at 700 °C shows that it decomposes into TiTe3O8 and TiO2 (Fig. S12†). To find out the water, acid, and alkali resistance of Ti(TeO3)(SO4), the morphological evolution of its crystals in pure water, 0.1 M nitric acid and 0.1 M sodium hydroxide solutions was recorded, respectively. The crystals preserve their full morphology and high transparency after being immersed either in water for 15 days or in an acid/alkali solution for 24 h (Fig. S13–S15†), and the PXRD patterns also remain unchanged (Fig. S16–S18†). This preliminarily result indicates that it has outstanding resistance to water-solubility as well as stability in acidic and alkaline environments. In addition, the SHG intensity of the powder samples also confirms that the water/acid/alkali environment will not weaken its NLO intensity (Fig. S19 and S20†), ensuring optical stability in multiple usage scenarios. The UV-vis-NIR spectrum indicates a UV cut-off edge of 312 nm for Ti(TeO3)(SO4) (Fig. S21†), and the fitted Tauc plot gives a direct band gap of 3.61 eV and an indirect band gap of 3.07 eV (Fig. S22†). The calculated band structure diagram (Fig. S23†) shows that it is an indirect band gap (D → Y) crystal, and the calculated value is 2.75 eV, a little lower than the experimental indirect band gap value (3.07 eV) due to the limitation of the DFT method. This band gap is clearly wider than that of the commercial NLO crystals AgGaSe2 (1.83 eV) and AgGaS2 (2.6 eV), and is comparable to some remarkable sulfates, such as CeF2(SO4) (2.71 eV)51 and Nb2O3(IO3)2(SO4) (3.25 eV).42 A test on powder samples gives a laser damage threshold of up to 50 MW cm−2 for Ti(TeO3)(SO4), which is 12.5 times higher than that of AgGaS2 (4 MW cm−2), in line with its wide band gap.65
Since Ti(TeO3)(SO4) represents one of the NLO crystals with potential application value, its NLO-related properties were systematically compared with those of KH2PO4, one of the broadly applied SHG materials. The comparison focused on several key aspects: SHG intensity, birefringence, band gap, thermal stability, and NLO coefficient (deff). The results show that Ti(TeO3)(SO4) presents an outstanding comprehensive performance as a potential NLO crystal (Fig. 4d).
Conclusions
By introducing an [SO4] group into a centric titanium tellurate, an acentric titanium-containing tellurate–sulfate, Ti(TeO3)(SO4), was gained through a hydrothermal process. Within its three-dimensional structure, the [TiO6] octahedron is connected to three lone-pair [TeO3] pyramids and three [SO4] tetrahedra. The two types of p-block element-centered oxo-anionic groups induce an out-of-center distortion in the [TiO6] along the local C3[111] direction, which is rare in titanium oxides containing lone-pair cation. This also proves that the octahedral distortion of [TiO6] in titanium oxides containing a lone-pair cation can also be induced by the direct attachment of two types of p-block elements, rather than by the requisite involvement of a [TiO6] octahedron or terminal O atom as previously thought. Ti(TeO3)(SO4) exhibits the strongest second harmonic generation response (11.6 × KDP) among mercury-free sulfates and a large birefringence (0.145) owing to the favorable arrangement of the [TiO6] octahedron, [TeO3] pyramid, and [SO4] tetrahedron within the structure, suggesting its high application potential as an NLO material. This work also paves the way for the design of novel NLO materials by introducing various p-block elements to induce distortion in [Md0O6] units, thereby potentially achieving a strong SHG response.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2364994 have been deposited at the CCDC.Author contributions
Zhenhua Li: conceptualization, investigation, data curation, and writing – original draft; Zhengli Liang: data curation and writing – original draft; Jiahao Wan: data curation and visualization, Lehui Liu: data curation; Chunxiang Wu: data curation; Ping Wang: data curation; Xingxing Jiang: formal analysis, writing – review & editing and supervision; Zheshuai Lin: software; Hongming Liu: funding acquisition, writing – review & editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for financial support from the National Natural Science Foundation of China (grant no. 22105046) and the Natural Science Foundation of Guangxi Province (no. 2022GXNSFBA035484).References
- W. Q. Liao, D. Zhao, Y. Y. Tang, Y. Zhang, P. F. Li, P. P. Shi, X. G. Chen, Y. M. You and R. G. Xiong, Science, 2019, 363, 1206–1210 CrossRef CAS PubMed.
- H. Y. Ye, Y. Y. Tang, P. F. Li, W. Q. Liao, J. X. Gao, X. N. Hua, H. Cai, P. P. Shi, Y. M. You and R. G. Xiong, Science, 2018, 361, 151–155 CrossRef CAS.
- D. F. Eaton, Science, 1991, 253, 281–287 CrossRef CAS PubMed.
- X. Dong, H. Huang, L. Huang, Y. Zhou, B. Zhang, H. Zeng, Z. Lin and G. Zou, Angew. Chem., Int. Ed., 2024, 63, e202318976 CrossRef CAS PubMed.
- X. Wang, X. Leng, Y. Kuk, J. Lee, Q. Jing and K. M. Ok, Angew. Chem., Int. Ed., 2024, 63, e202315434 CrossRef CAS PubMed.
- L. Qi, X. Jiang, K. Duanmu, C. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202309365 CrossRef CAS PubMed.
- L. Kang and Z. Lin, Light:Sci. Appl., 2022, 11, 201 CrossRef CAS.
- M. Mutailipu, F. Li, C. Jin, Z. Yang, K. R. Poeppelmeier and S. Pan, Angew. Chem., Int. Ed., 2022, 61, e202202096 CrossRef CAS PubMed.
- H. Tian, N. Ye and M. Luo, Angew. Chem., Int. Ed., 2022, 61, e202200395 CrossRef CAS PubMed.
- M. Bugajski and M. Malinowski, Open Phys., 2010, 8, 157–158 CrossRef.
- D. Cyranoski, Nature, 2009, 457, 953–955 CrossRef CAS PubMed.
- S. Zhao, Y. Yang, Y. Shen, B. Zhao, L. Li, C. Ji, Z. Wu, D. Yuan, Z. Lin, M. Hong and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 540–544 CrossRef CAS.
- M. Xia, X. Jiang, Z. Lin and R. Li, J. Am. Chem. Soc., 2016, 138, 14190–14193 CrossRef CAS.
- C. Wu, X. Jiang, Z. Wang, L. Lin, Z. Lin, Z. Huang, X. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3464–3468 CrossRef CAS PubMed.
- C. Wu, C. Jiang, G. Wei, X. Jiang, Z. Wang, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2023, 145, 3040–3046 CrossRef CAS PubMed.
- C. Wu, X. Jiang, Y. Hu, C. Jiang, T. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202115855 CrossRef CAS.
- G. Peng, C. Lin, H. Fan, K. Chen, B. Li, G. Zhang and N. Ye, Angew. Chem., Int. Ed., 2021, 60, 17415–17418 CrossRef CAS.
- H. Liu, Q. Wu, L. Liu, Z. Lin, P. S. Halasyamani, X. Chen and J. Qin, Chem. Commun., 2021, 57, 3712–3715 RSC.
- F. You, F. Liang, Q. Huang, Z. Hu, Y. Wu and Z. Lin, J. Am. Chem. Soc., 2019, 141, 748–752 CrossRef CAS PubMed.
- H. Liu, Q. Wu, X. Jiang, Z. Lin, X. Meng, X. Chen and J. Qin, Angew. Chem., Int. Ed., 2017, 56, 9492–9496 CrossRef CAS PubMed.
- M. Liang, C. Hu, F. Kong and J. Mao, J. Am. Chem. Soc., 2016, 138, 9433–9436 CrossRef CAS.
- N. Ma, J. Chen, B. Li, C. Hu and J. Mao, Small, 2023, 19, 2304388 CrossRef CAS.
- Q. Chen, C. Hu, J. Chen, Y. Li, B. Li and J. Mao, Angew. Chem., Int. Ed., 2021, 60, 17426–17429 CrossRef CAS.
- D. Phanon and I. Gautier-Luneau, Angew. Chem., Int. Ed., 2007, 46, 8488–8491 CrossRef CAS.
- E. L. Belokoneva, S. Y. Stefanovich, O. V. Dimitrova, A. S. Karamysheva and A. S. Volkov, Inorg. Chem., 2017, 56, 1186–1192 CrossRef CAS.
- D. Phanon, Y. Suffren, M. B. Taouti, D. Benbertal, A. Brenier and I. Gautier-Luneau, J. Mater. Chem. C, 2014, 2, 2715–2723 RSC.
- K. Bouchouit, H. Bougharraf, B. Derkowska-Zielinska, N. Benali-cherif and B. Sahraoui, Opt. Mater., 2015, 48, 215–221 CrossRef CAS.
- D. O. Charkin, V. Y. Grishaev, T. A. Omelchenko, E. V. Nazarchuk, S. Y. Stefanovich and O. I. Siidra, Solid State Sci., 2023, 137, 107116 CrossRef CAS.
- R. Robert, V. Balisetty, K. Mohanrao, M. Mannamala Pappan, S. Mangalassery, D. Rao and K. Vidyasagar, Inorg. Chem., 2023, 62, 7890–7897 CrossRef CAS.
- S. Pal, R. Nagarajan and S. Uma, Chem. Mater., 2023, 35, 1249–1258 CrossRef CAS.
- S. Katrych, A. Arakcheeva, A. Sienkiewicz, L. Forró and T. Ivšić, Inorg. Chem., 2021, 60, 286–291 CrossRef CAS.
- C. Chen, Y. Wu and R. Li, Int. Rev. Phys. Chem., 1989, 8, 65–91 Search PubMed.
- N. Ye, Q. Chen, B. Wu and C. Chen, J. Appl. Phys., 1998, 84, 555 CrossRef CAS.
- K. M. Ok, P. S. Halasyamani, D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. Mater., 2006, 18, 3176–3183 CrossRef CAS.
- P. S. Halasyamani, Chem. Mater., 2004, 16, 3586–3592 CrossRef CAS.
- P. S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS.
- M. Kunz and I. D. Brown, J. Solid State Chem., 1995, 115, 395–406 CrossRef CAS.
- T. Wu, X. Jiang, K. Duanmu, C. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202318107 CrossRef CAS PubMed.
- Y. X. Ma, P. F. Li, C. L. Hu, J. G. Mao and F. Kong, Adv. Sci., 2023, 10, 2304463 CrossRef CAS.
- Q. Wang, X. H. Dong, L. Huang, K. M. Ok, Z. E. Lin and G. H. Zou, Small, 2023, 19, 2302797 CrossRef CAS PubMed.
- K. Chen, C. Lin, J. Chen, G. Yang, H. Tian, M. Luo, T. Yan, Z. Hu, J. Wang, Y. Wu, N. Ye and G. Peng, Angew. Chem., Int. Ed., 2023, 62, e202217039 CrossRef CAS PubMed.
- H. X. Tang, Y. X. Zhang, C. Zhuo, R. B. Fu, H. Lin, Z. J. Ma and X. T. Wu, Angew. Chem., Int. Ed., 2019, 58, 3824–3828 CrossRef CAS PubMed.
- H. Yu, M. L. Nisbet and K. R. Poeppelmeier, J. Am. Chem. Soc., 2018, 140, 8868–8876 CrossRef CAS.
- H. Ra, K. M. Ok and P. S. Halasyamani, J. Am. Chem. Soc., 2003, 125, 7764–7765 CrossRef CAS PubMed.
- M. E. Welk, A. J. Norquist, F. P. Arnold, C. L. Stern and K. R. Poeppelmeier, Inorg. Chem., 2002, 41, 5119–5125 CrossRef CAS PubMed.
- J. B. Goodenough, Annu. Rev. Mater. Sci., 1998, 28, 1–27 CrossRef CAS.
- L. Bindi and C. Cipriani, Can. Mineral., 2003, 41, 1469–1473 CrossRef CAS.
- F. C. Zumsteg, J. D. Bierlein and T. E. Gier, J. Appl. Phys., 1976, 47, 4980–4985 CrossRef CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B:Struct. Sci., 1991, 47, 192–197 CrossRef.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 8, 3798–3813 CrossRef.
- C. Wu, T. Wu, X. Jiang, Z. Wang, H. Sha, L. Lin, Z. Lin, Z. Huang, X. Long, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2021, 143, 4138–4142 CrossRef CAS.
- Y. Song, X. Hao, C. Lin, D. Lin, M. Luo and N. Ye, Inorg. Chem., 2021, 60, 11412–11418 CrossRef CAS PubMed.
- K. Chen, Y. Yang, G. Peng, S. Yang, T. Yan, H. Fan, Z. Lin and N. Ye, J. Mater. Chem. C, 2019, 7, 9900–9907 RSC.
- Y. Han, X. Zhao, F. Xu, B. Li, N. Ye and M. Luo, J. Alloys Compd., 2022, 902, 163727 CrossRef CAS.
- X. Dong, L. Huang, H. Zeng, Z. Lin, K. M. Ok and G. Zou, Angew. Chem., Int. Ed., 2022, 61, e202116790 CrossRef CAS PubMed.
- R. E. Perez-D'Gregorio and R. K. Miller, Teratology, 1988, 37, 307–316 CrossRef PubMed.
- N. Kanematsu, M. Hara and T. Kada, Mutat. Res., 1980, 77, 109–116 CAS.
- B. E. Sørensen, Eur. J. Mineral., 2013, 25, 5–10 CrossRef.
- Y. Shang, J. Xu, H. Sha, Z. Wang, C. He, R. Su, X. Yang and X. Long, Coord. Chem. Rev., 2023, 494, 215345 CrossRef CAS.
- W. Huang, S. Zhao and J. Luo, Chem. Mater., 2022, 34, 5–28 CrossRef CAS.
- P. Li, C. Hu, Y. Li, J. Mao and F. Kong, J. Am. Chem. Soc., 2024, 146, 7868–7874 CrossRef CAS PubMed.
- T. Wu, X. Jiang, K. DuanMu, C. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, Adv. Sci., 2024, 11, 2306670 CrossRef CAS.
- W. M. Sinton, J. Opt. Soc. Am., 1961, 51, 1301–1309 CrossRef.
- P. Li, C. Hu, F. Kong, S. Ying and J. Mao, Inorg. Chem. Front., 2021, 8, 164–172 RSC.
- L. Kang, D. M. Ramo, Z. S. Lin, P. D. Bristowe, J. G. Qin and C. T. Chen, J. Mater. Chem. C, 2013, 1, 7363–7370 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic data, information on some sulfates, XRD patterns, EDS data, IR spectrum, additional crystal structure, TG-DSC curves, crystal photos, UV-vis-NIR spectra, dipole moment calculation, and computational results. CCDC 2364994 contains the supplementary crystallographic data for Ti(TeO3)(SO4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06620k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |