
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating built-in electric field via Br induced partial phase transition for robust alkaline freshwater and seawater electrolysis†
Lei
Jin
,
Hui
Xu
 *,
Kun
Wang
,
Yang
Liu
,
Xingyue
Qian
,
Haiqun
Chen
* and
Guangyu
He
*
*,
Kun
Wang
,
Yang
Liu
,
Xingyue
Qian
,
Haiqun
Chen
* and
Guangyu
He
*
Key Laboratory of Advanced Catalytic Materials and Technology, Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University, Changzhou, Jiangsu Province 213164, China. E-mail: xuhui006@cczu.edu.cn; hegy@cczu.edu.cn; chenhq@cczu.edu.cn
First published on 27th November 2024
Abstract
Repulsing Cl− to reduce its negative effects during seawater electrolysis is a promising strategy to guard against the corrosion of high-valence metal sites. Herein, we synthesized Fe2P/Ni2P by a facile Br-induced partial in situ phase transition strategy. This Fe2P/Ni2P possessed intensified built-in electric field (BEF) due to large work function difference (ΔΦ), demonstrating outstanding OER and HER activity in alkaline freshwater/seawater solution and exhibiting a low cell voltage for an anion exchange membrane water electrolyzer (AEMWE) system. Both experiments and theoretical results verify that the interfacial charge redistribution induced by the enhanced BEF optimizes the adsorption strength for the intermediates. Moreover, the appropriate phosphorus–oxygen anion self-transformation can protect the NiOOH active species from corrosion by repulsing Cl− in alkaline seawater. This work not only proposes a fresh perception of the water/seawater splitting mechanism but also provides new design principles to defend active sites in seawater-to-H2 conversion systems.
1. Introduction
Electrolysis of water powered by renewable energy constitutes a sustainable strategy for the production of transportable and green hydrogen (H2).1,2 Apart from considering the expense of electrolyzers and the intermittence of renewable electricity, a supply of freshwater may be a practical problem for deploying large-scale electrocatalytic H2 production.3,4 If saline water, particularly seawater—which constitutes nearly 96.5% of Earth's total water resources—could serve as an ideal feedstock for electrocatalytic H2 production, it would help alleviate the shortage of freshwater resources. While the transformation of seawater to H2 is an intriguing prospect, chloride ions (Cl−) and their derivatives (such as Cl2 or HClO/ClO−) can significantly accelerate the corrosion or deactivation of electrocatalysts. This occurs through surface adsorption or coordination mechanisms, especially during the anodic oxygen evolution reaction (OER) associated with seawater splitting.5,6 Moreover, the standard thermodynamic voltage of OER is 0.48 V lower than that of the chlorine oxidation reaction (ClOR) at pH > 7.5. This means that superior water splitting performance can significantly suppress Cl− transformation into Cl2 or HClO/ClO−. Even if the electrode shows excellent activity in inhibiting Cl− oxidation, Cl− ions themselves can etch electron-poor transition metals, leading to metal leaching and a deterioration in performance.7,8 Therefore, the exploration and construction of catalysts that suppress the loss of reaction sites to maintain high seawater splitting activity and optimize adsorption behaviors for intermediates to safeguard the electrode against Cl− attack are of paramount importance.The exquisite construction of a built-in electric field (BEF) between two hetero substances with a difference in Fermi level, would be a promising way to manipulate the electronic state of active sites and stabilize the surficial dynamical adsorption balance.9,10 For example, a Co LDH/Cu3P composite can accelerate charge transport and improve active sites due to an enhanced built-in potential (EBI).11 Zhang et al. have constructed N–Ni5P4/CoP nanowires with a strong BEF for hydrazine-assisted H2 production.12 Very recently, Xu et al. demonstrated that the enhanced BEF at the Fe2P/NiCoP interface facilitates the surface transformation of NiCoP into NiCoOOH active species, exhibiting high freshwater and seawater oxidation activity.13 Essentially, the constructed BEF can facilitate the division of positive and negative charges, inducing their separation in opposite directions at the heterointerface to accelerate regional charge polarization.14 Consequently, manipulating the features of the BEF can achieve an asymmetric distribution of charge that is promising for realizing the stable adsorption of intermediates on the heterointerface or active sites.15 However, while coupling transition metal phosphides (TMPs) with other components (nitrides or sulfides), charge carriers would experience severe localization due to the overlap of the electron cloud across the heterointerface, where the electrons are completely delocalized via the metal whereas the protons are still confined and greatly weaken the BEF.16,17 Fortunately, by manipulating the difference in the work function (ΔΦ) between TMP and other substances, the charge transfer direction across the heterointerface can be controlled.18 Based on the above discussion, it is rational and promising to simultaneously regulate the adsorption behaviors for intermediates by exquisitely designing the appropriate components with suitable work functions to control and modulate the BEF. Moreover, revealing the relationship between BEF and ΔΦ and its effect on catalytic activity and selectivity is important.
Herein, for the first time, we have exquisitely designed and manipulated the Br-induced partial in situ phase transition from Fe2P/Ni5P4 to Fe2P/Ni2P under the phosphorization process, which strongly influences the ΔΦ. Specifically, Fe2P/Ni2P, with a large ΔΦ (0.5 eV) compared with Fe2P/Ni5P4 (0.3 eV), demonstrates an enhanced BEF, as confirmed by various electrochemical tests. Furthermore, in situ/ex situ spectroscopic investigations confirm that the enhanced BEF plays an important role in subtly tailoring the intermediates and phosphate absorption strength, which is essential for water and seawater splitting. As a result, the Fe2P/Ni2P catalyst reveals remarkable OER and HER activity with low overpotentials of 196 mV and 108 mV for freshwater and 229, 203 mV for seawater, respectively, and can be used in an anion exchange membrane water electrolyzer (AEMWE) with low cell voltage. This work paves the way for synthesizing bifunctional water-splitting electrocatalysts by exquisitely designing and modulating the interfacial BEF.
2. Experimental section
2.1 Synthesis of NiFe LDH
First, 0.582 g of Ni(NO3)2·6H2O, 0.404 g of Fe(NO3)3·9H2O, and 0.841 g of C6H12N4 were weighed and fully dissolved in a Teflon autoclave with 45 mL of ethanol solution, and stirred for 2 h. After reaction for 12 h at 120 °C, a brown product (NiFe LDH) was obtained by centrifugation with ethanol and dried at 60 °C all night.2.2 Synthesis of Fe2P/Ni2P
The as-prepared NiFe LDH precursors, a small amount of hexabromobenzene (HBB) and 300 mg of NaH2PO2·H2O were placed in three separate positions in a ceramic boat inside a tube furnace, where NaH2PO2·H2O was at the midstream of the gas flow, NiFe LDH was placed on the downstream side, and one piece of HBB flake (pressurized at 14 MPa at room temperature) was located upstream. Then the temperature was raised to 350 °C for 2 h. After cooling down to room temperature, a black product consisting of thick Fe2P/Ni2P nanosheets was obtained. For the synthesis of Fe2P/Ni5P4, the same synthetic method was adopted as for the Fe2P/Ni2P sample without the addition of HBB flake.2.3 Material characterizations and electrochemical methods
The material characterizations and electrochemical methods are documented in detail in the ESI.†3. Results and discussion
3.1 Material synthesis and characterization
In general, thermodynamic stability can be altered when materials are doped with heteroatoms. Bromine, being more electronegative than phosphorus, was chosen as a candidate to induce the formation of a P-poor phase. The relatively large size of Br favors the creation of phosphorus vacancies due to the strain field it generates.19,20 As schematically illustrated in Fig. 1, an Fe2P/Ni2P catalyst could be successfully synthesized by a two-step route. Firstly, NiFe LDH nanosheets with a large surface area were obtained via an appropriate solvothermal pathway. Next, the NiFe LDH was completely transformed into Fe2P/Ni5P4 (the ratio of P/Ni = 0.8) via a thermal phosphorization process at 350 °C. However, Fe2P/Ni5P4 adopts the bulk nanosheet structure, which may be attributed to the introduction of molten NaH2PO2 salt gas with high viscosity, causing the NiFe LDH to agglomerate easily. In addition, during the process of synthesizing Fe2P/Ni2P, a small amount of Br atoms and more P atoms first form an Fe2P/Ni5P4Br4−x intermediate. Subsequently, the remaining P atoms struggle with Br and consume each other. Eventually Br atoms are completely replaced by P atoms, causing a partial phase transition, thus forming a P-poor-phase Fe2P/Ni2P (P/Ni = 0.5) catalyst.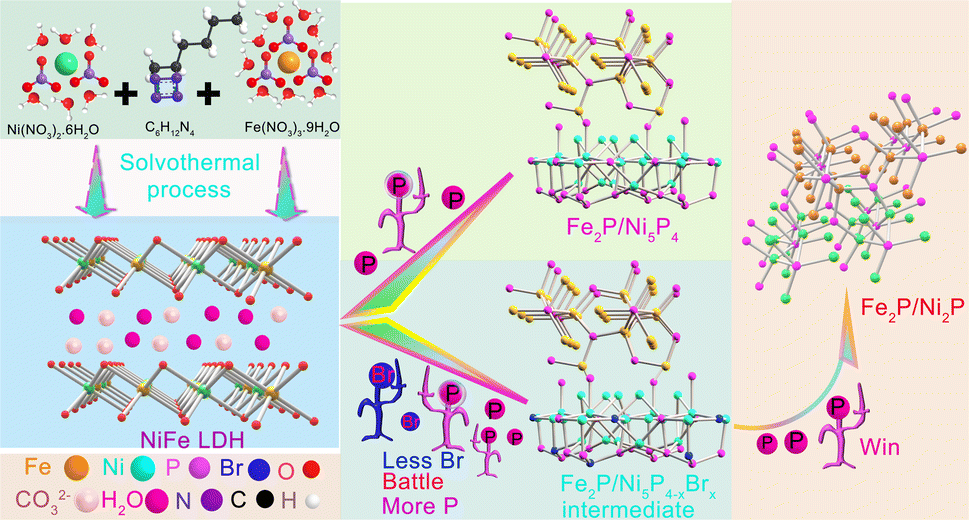 | ||
| Fig. 1 Schematic illustration of the formation of Fe2P/Ni5P4 and Fe2P/Ni2P. | ||
The morphology of the as-prepared catalysts was characterized via scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As illustrated in Fig. 2a and S1,† the NiFe LDH exhibits a nanosheet morphology. After being subjected to a phosphorylation process, both with and without Br atoms, the synthesized Fe2P/Ni2P and Fe2P/Ni5P4 catalysts display a bulk nanosheet structure, attributed to the introduction of molten NaH2PO2 salt gas with high viscosity, causing the NiFe LDH to agglomerate easily (Fig. 2b and c). Moreover, the high-resolution TEM (HRTEM) images in Fig. 2d–f and S2a–c† show that both Fe2P/Ni2P and Fe2P/Ni5P4 contain common clear lattice fringes with an approximate spacing of 0.17 nm (Fig. 2e, e-1, S2c and c-1†) that are indexed to the (300) crystal plane of Fe2P. While, two clear lattice fringes with approximate spacings of 0.2 (Fig. 2f and f-1) and 0.22 nm (Fig. S2b and b-1†) are indexed to the (201) and (210) crystal planes of Ni2P and Ni5P4 in the Fe2P/Ni2P and Fe2P/Ni5P4 catalysts, respectively, confirming the successful fabrication of the Fe2P/Ni2P and Fe2P/Ni5P4 structures.21–24 These results can be further confirmed via selected area electron diffraction (SAED) patterns (Fig. 2g and S2d†). Moreover, the EDS analysis of the as-synthesized Fe2P/Ni2P proved that Ni, Fe, and P were the main elemental components with Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:P:Br mass ratios of 1:1.04:2.73:0 (Fig. S3†), which may be ascribed to a small amount of Br atoms and more P atoms first forming an Fe2P/Ni5P4Br4−x intermediate, and the remaining P atoms struggle with Br atoms and consume each other. Eventually, the Br is replaced by P, causing a phase transition. This result strongly proves that the Br atoms play a brief intervention role in synthesizing Fe2P/Ni2P. The element mapping images of the Fe2P/Ni2P and Fe2P/Ni5P4 catalysts also confirmed the uniform dispersion of the elements of Fe, Ni and P (Fig. 2h and S2f†).
:Fe:P:Br mass ratios of 1:1.04:2.73:0 (Fig. S3†), which may be ascribed to a small amount of Br atoms and more P atoms first forming an Fe2P/Ni5P4Br4−x intermediate, and the remaining P atoms struggle with Br atoms and consume each other. Eventually, the Br is replaced by P, causing a phase transition. This result strongly proves that the Br atoms play a brief intervention role in synthesizing Fe2P/Ni2P. The element mapping images of the Fe2P/Ni2P and Fe2P/Ni5P4 catalysts also confirmed the uniform dispersion of the elements of Fe, Ni and P (Fig. 2h and S2f†).
 | ||
| Fig. 2 TEM images of (a) NiFe LDH, (b) Fe2P/Ni2P and (c) Fe2P/Ni2P. (d–f) High-resolution TEM images of Fe2P/Ni2P, integrated pixel intensities (e-1 and f-1) of Fe2P and Ni2P (taken from the green dotted rectangles in (e) and (f)). (g) SAED pattern of Fe2P/Ni2P. (h) Elemental mapping images of Fe2P/Ni2P. | ||
To verify this portion-limited phase-transition mechanism, X-ray diffraction (XRD) patterns of NiFe LDH, Fe2P/Ni5P4 and Fe2P/Ni2P were recorded first. As shown in Fig. S4,† four prominent peaks can be observed at approximately 33.5°, 34.4°, 59.9° 61.3°, attributed to the (110), (012), (101) and (113) crystal planes of NiFe LDH (JCPDS no. 40-0215), respectively.25 Notably, the peaks located at 40.3°, 44.2°, 47.3°, 54.1° and 54.6° belonging to the (111), (201), (210), (300) and (211) crystal planesof Fe2P (JCPDS no. 51-0943), respecitvely, which can be observed in both Fe2P/Ni5P4 and Fe2P/Ni2P samples,26 while peaks located at approximately 31.5°, 36.1°, 40.6°, 41.4°, 43.9°, 45.1°, 47.8°, 53.9° and 56.4° belong to the (201), (104), (210), (211), (212), (204), (213), (220) and (310) crystalline planes of Ni5P4 (JCPDS no. 18-0883), respectively.23 However, after a brief intervention by the Br atoms, the Ni5P4 phase disappears and new peaks located at 40.8°, 44.6°, 47.3°, 54.2°, 54.9° and 74.7° correspondly ascribed to the (111) (201), (210), (300), (211) and (400) crystal planes of Ni2P (JCPDS no. 03-0953) can be found (Fig. 3a).27 The above results confirm the successful synthesis of Fe2P/Ni5P4 and Fe2P/Ni2P, and also strongly verify the phase transition from Ni5P4 to Ni2P. Moreover, to reveal the structural characteristics and bonding properties of Fe2P/Ni5P4 and Fe2P/Ni2P, the Fourier transform infrared (FT-IR) spectra were measured. As illustrated in Fig. 3b, two dominant absorption bands of M–P at 920 and 1024 cm−1 can be observed, and one broad band at 551 cm−1 is ascribed to bending vibration of the ν4 (O–P–O) bond.28–30 Furthermore, to reveal the role of the portion-limited phase transition in tuning the electronic structure, electron paramagnetic resonance (EPR) spectra of Fe2P/Ni5P4 and Fe2P/Ni2P were documented (Fig. 3c). The relatively large size of Br favors the formation of abundant P vacancies due to the strain field. Fe2P/Ni2P displays a pair of prominent signals, suggesting the existence of abundant unpaired electrons derived from dangling bonds in the portion-limited phase-transition structure compared with Fe2P/Ni5P4, also confirming that it may form an Fe2P/Ni5P4Br4−x intermediate first and then phase change into Fe2P/Ni2P, or directly form Fe2P/Ni2PBr1−x and Fe2P/Ni5P4Br4−x. However, the XRD proves the phase transition from Ni5P4 to Ni2P, and EDS demonstrates that there are no Br atoms. Thus, it is directly proved that Fe2P/Ni5P4Br4−x is formed first and then converted into Fe2P/Ni2P, instead of directly forming Fe2P/Ni2PBr1−x and Fe2P/Ni5P4Br4−x.19,20,31 In fact, numerous dangling bonds offer more accessible active sites for electrochemical water splitting.32 Accordingly, X-ray photoelectron spectroscopy (XPS) was employed to reveal the chemical composition and bonding configuration of Fe2P/Ni5P4 and Fe2P/Ni2P. The XPS full spectra (Fig. S5a†) reveal the coexistence of Fe, Ni and P elements, which correspond to the above elemental mapping.33 As shown in the high-resolution Fe 2p spectrum (Fig. 3d), for Fe2P/Ni2P, the prominent peak at 705.9 eV belongs to Fe–P, while three peak doublets located at (710.6 and 723.5 eV), (713.2 and 726.7 eV), and (717.2 and 730.6 eV), consistent with Fe2+, Fe3+, and satellite signals, respectively. The peaks of Fe 2p display a slight positive shift compared with Fe2P/Ni5P4, demonstrating greater electron transfer from Fe to the P atom.34 Likewise, three pairs of peaks corresponding to satellite peaks (861.1, 879.9 eV), Ni–O (855.9, 874.3 eV), and Ni–P (852.7, 870.1 eV) are also observed in the high-resolution Ni 2p spectra (Fig. 3e), which exhibit a remarkable positive shift (about 1.0 eV), implying a reduction in charge density around the metal atom.18 Meanwhile, for P 2p orbitals, the peaks located at 129.1, 130.0 and 133.6 eV in Fig. 3f are ascribed to P 2p3/2, P 2p1/2 and the P–O bonding state, respectively.35 Compared with Fe2P/Ni5P4, the peaks of Fe2P/Ni2P exhibit a slight negative shift (about 0.42 eV), indicating more accumulated electrons on P due to the Br and P consuming each other and thus introducing abundant P vacancies.36 These shifts confirm the abundant electron transfer from the Ni atom to the P atom compared with the Fe atom. In order to balance the charge, the Fe atom spontaneously transfers partial electrons to the Ni atom, which further confirms the electron transfers from Fe2P to Ni2P, thus signifying the formation of a BEF with electron-rich Ni atoms and electron-poor Fe atoms, optimizing the adsorption energy of the intermediate in the water-splitting process.
 | ||
| Fig. 3 (a) XRD patterns of Fe2P/Ni5P4 and Fe2P/Ni2P. (b) FT-IR spectra of Fe2P/Ni5P and Fe2P/Ni2P. (c) EPR spectra of Fe2P/Ni5P and Fe2P/Ni2P. High-resolution XPS spectra of (d) Fe 2p, (e) Ni 2p and (f) P 2p in Fe2P/Ni5P4 and Fe2P/Ni2P. (g) UPS spectra of Ni5P4, Fe2P and Ni2P. (h) Energy-band alignment diagram of Ni5P4 and Ni2P with respect to Fe2P. (i) Schematic diagram of the BEF based on the interfaces between Fe2P and Ni2P. | ||
Generally, the charge transfer direction is closely correlated with the difference ΔΦ in the semiconductor heterostructure.37,38 Ultraviolet photoelectron spectroscopy (UPS) was undertaken to analyze the values for Fe2P, Ni5P4 and Ni2P. As illustrated in Fig. 3g and Table S1,† the Φ value of Ni2P is increased by 0.8 eV compared with Ni5P4, suggesting that a brief intervention by the Br atom can decrease the Fermi level. Accordingly, the measured Φ values of Ni5P4, Fe2P and Ni2P are 5.92, 6.22, and 6.72 eV, respectively (Fig. 3h). Compared with a relatively small ΔΦ of 0.3 eV at the Fe2P/Ni5P4 heterogeneous interface, the Fe2P/Ni2P interface possesses a relatively large ΔΦ of 0.5 eV. Moreover, the Mott–Schottky (M–S) plots of Ni5P4, Fe2P and Ni2P were calculated to construct energy diagrams. As shown in Fig. S5b–d,† Ni5P4, Fe2P and Ni2P possess a positive slope, indicating that these catalysts are n-type semiconductors, and the flat band potentials (EFB) of Ni5P4, Fe2P and Ni2P can be tested to be 0.64, 0.59 and 0.66 (vs. Hg/HgO), respectively; thus, the conduction band potential (ECB) values of Ni5P4, Fe2P and Ni2P can be calculated as 0.64, 0.59 and 0.66 V vs. NHE (ENHE = EHg/HgO + 0.098 V).39,40 Furthermore, the valence band (EVB) maximum values were measured as 3.96, 2.47 and 3.67 eV for Ni5P4, Fe2P and Ni2P, respectively. Therefore, the band gap (Eg) values of Ni5P4, Fe2P and Ni2P were calculated as 3.32 eV, 1.88 eV and 1.5 eV by using the formula: EVB = ECB + Eg; the smaller Eg of Ni2P (1.5 eV) confirms the faster charger transfer across the Fermi level.41 Based on the above results, the interfacial electronic structure in the Fe2P/Ni2P heterojunction can be modified, and the large discrepancy in Φ forms an n–n heterojunction, where ΔΦ would drive the charge migration from high level to low until the heterojunction interface recovers balance.42 Therefore, the charge in Fe2P will flow into Ni2P, forming a positively charged donor. Concurrently, a BEF is formed with the orientation pointing from Fe2P to Ni2P (Fig. 3i). Generally, the largest ΔΦ indicates the presence of the strongest BEF between the two semiconductors. A BEF with electron flow in a single direction can effectively adjust the charge redistribution and can concurrently induce an electron-rich Ni2P zone and an electron-poor Fe2P zone, thus promoting the adsorption process for intermediates and PO43−.43,44
3.2 OER and HER in freshwater
The electrochemical performance of the as-prepared catalysts was documented by a standard three-electrode system in 1 M KOH solution. For comparison, RuO2 and Pt/C catalysts were employed as reference HER and OER catalysts, respectively. Linear sweep voltammetry (LSV) curves in Fig. 4a show that the Fe2P/Ni2P catalyst displays superior electrocatalytic performance for OER and HER compared with Fe2P/Ni5P4 and NiFe LDH. Concretely, it requires ultra-small overpotentials of 196 and 224 mV to achieve η10 and η20 for OER (Fig. 4b), which are superior to those of Fe2P/Ni5P4 (237 and 258 mV), NiFe LDH (272 and 307 mV), and RuO2 (327 and 389 mV). Additionally, Fe2P/Ni2P shows the highest HER activity, requiring overpotentials of only 108 and 151 mV to attain η10 and η20, also being superior to Fe2P/Ni5P4 (204 and 250 mV) and NiFe LDH (422 and 475 mV). These results confirm that the introduction of Br induces a phase change and constructs a stronger BEF due to the larger ΔΦ. This, in turn, substantially enhances the OER and HER activities,45 also outperforming the majority of reported electrocatalysts (Tables S2 and S3†). The Tafel slope is usually used to reveal OER and HER kinetics, and a low Tafel slope means favorable reaction kinetics. As shown in Fig. 4c and d, the Tafel slopes of the Fe2P/Ni2P catalyst are determined to be 89.1 and 121.5 mV dec−1 for OER and HER, respectively, which are smaller than those of Fe2P/Ni5P4 (91.2 and 134.4 mV dec−1) and NiFe LDH (96.3 and 148.1 mV dec−1), indicating a Volmer–Heyrovsky water-splitting procedure. Moreover, the electrochemically active surface area (ECSA) was probed by using double-layer capacitance (Cdl) to understand the surface properties of the samples. Fig. S6 and S7† show that the Cdl for Fe2P/Ni5P4–Ov are calculated to be 1.0 and 13.9 mF cm−2 for OER and HER, respectively, suggesting a greater electrochemically active surface area. Furthermore, the electrochemical stability of Fe2P/Ni2P for OER and HER was assessed via a cyclic voltammetry (CV) test. The LSV curve of Fe2P/Ni2P after 1000 CV cycles coincides well with the initial curve, indicating its outstanding cyclability (Fig. 4f). To estimate the intrinsic OER and HER activity, Fig. S8† documents the turnover frequency (TOF) values of Fe2P/Ni2P and referenced samples, where Fe2P/Ni2P demonstrates higher TOF values for OER (0.029 s−1) and HER (0.195 s−1).46,47 The superb long-term stability was evaluated using chronoamperometry measurements, in which the electrochemical OER and HER activities of Fe2P/Ni2P were conducted smoothly for more than 135 h without a remarkable change (Fig. 4e and g), demonstrating its high stability and good corrosion resistance properties in anodic and cathodic conditions. Multi-current curves of Fe2P/Ni2P were tested by changing the current density from 10 to 50 mA cm−2 for OER and from −10 to −50 mA cm−2 for HER per 1000 s. The high mass transport capability and stability can be further confirmed by the instantaneous potential response of each step when different currents are applied, suggesting the superb durability and fast charge/mass transport in the Fe2P/Ni2P sample. | ||
| Fig. 4 (a) Electrocatalytic OER and HER performance of Fe2P/Ni2P and the references in 1.0 M KOH solution. (b) Overpotentials of as-prepared samples at 10 and 20 mA cm−2 for OER and HER. (c and d) Corresponding Tafel plots of Fe2P/Ni2P for OER and HER. (f) Polarization curves for OER and HER after 1000 cycles. (e and g) Chronopotentiometric (CP) curves and multi-step chronopotentiometry tests results of Fe2P/Ni2P for OER and HER. | ||
3.3 OER and HER in alkaline seawater
With the shortage of fresh water resources in the world, seawater electrolysis and sustainable hydrogen energy have attracted increasing attention. One of the dominant challenges in seawater splitting is the chlorine evolution reaction (CER) on the anode, which is driven by the existence of Cl− ions. This reaction competes with the OER and leads to the formation of insoluble precipitates, such as calcium hydroxide, on the catalyst surface.48 To verify the selective inhibition of CER and shielding from impurities in seawater, the OER and HER performance of the samples were also measured in alkaline seawater solution. As shown in Fig. 5a–d, the Fe2P/Ni2P catalyst requires a smaller OER η10 of only 229 mV with faster kinetics of 55.1 mV dec−1 compared to Fe2P/Ni5P4 (260 mV, 60.0 mV dec−1), NiFe LDH (277 mV, 62.0 mV dec−1) and RuO2 (345 mV, 134.6 mV dec−1), outperforming some reported electrocatalysts (Table S6†). The activity of Fe2P/Ni2P is almost unaffected compared with freshwater due to the construction of a stronger BEF and the appearance of the surface of PO43− that effectively suppresses Cl− oxidation, indicating its highly selective inhibition and shielding from impurities.49 In addition, Fe2P/Ni2P exhibits better HER activity, which requires overpotentials of only 203 and 247 mV to attain η10 and η20, respectively, smaller than those of Fe2P/Ni5P4 (293 and 339 mV), and NiFe LDH (448 and 496 mV). Fig. 5d demonstrates that the Tafel slop of Fe2P/Ni2P is 127 mV dec−1, which is lower than that of other as-prepared samples, indicating faster kinetics for HER. Fe2P/Ni2P displays apparent Cdl values of 0.71 and 13.4 mF cm−2 for OER and HER beyond those of Fe2P/Ni5P4 (0.29 and 6.2 mF cm−2) and NiFe LDH (0.48 and 1.5 mF cm−2), which can be ascribed to the abundant exposed active sites of the nanosheets (Fig. S9 and S10†). The TOF values of Fe2P/Ni2P, Fe2P/Ni5P4 and NiFe LDH follow the same trend as the apparent electrochemical activities in alkaline seawater solution (Fig. S11†). Furthermore, the cycling stability of the Fe2P/Ni2P sample was further evaluated through continuous CV test, in which the LSV curves after 1000 cycles almost coincide with the initial curve, indicating its remarkable cyclability. Additionally, as shown in Fig. 5e and g, the CP test demonstrates that Fe2P/Ni2P maintains stable operation without remarkable current degradation for 70 and 140 h for OER and HER. The figures also display consecutive multi-step CP tests for Fe2P/Ni2P, where the instant response in potential almost remains steady at each step. These results confirm that Fe2P/Ni2P exhibits exceptional OER/HER performance and enduring stability in harsh conditions. This is ascribed to the stronger BEF, and the existence of negatively charged anionic (PO43−) layers that can repel Cl− and thus protect the electrode from corrosion. | ||
| Fig. 5 (a) Electrocatalytic OER and HER performance of Fe2P/Ni2P and references in alkaline seawater solution. (b) Overpotential of the as-prepared samples at 10 and 20 mA cm−2 for OER and HER. (c and d) Corresponding Tafel plots of Fe2P/Ni2P for OER and HER. (f) Polarization curves for OER and HER after 1000 cycles. (e and g) CP curves and multi-step chronopotentiometry tests results of Fe2P/Ni2P for OER and HER. | ||
3.4 Mechanism discussion
Operando electrochemical impedance spectroscopy (EIS) was documented to gain a better understanding of the OER kinetics in the different catalyst surfaces.50 As illustrated in Fig. 6a and d, the Nyquist plots of Fe2P/Ni2P and Fe2P/Ni5P4 under different applied potentials were obtained, indicating various electrochemical reaction properties. The equivalent circuits of two continuous electrochemical processes were employed for data fitting (Fig. 6c), and the best fitting parameters are shown in Tables S4 and S5.† The high-frequency (HF) region is considered to be the oxidation processes occurring within the electrode, whereas the low-frequency (LF) region is concerned with the asymmetric distribution of interface charges caused by the oxidized materials (Fig. 6b and e).10R1 and R2 embody the oxidation resistance of the catalysts under electrochemical operation in the HF and LF zones, respectively. When the potential shifts from 1.2 to 1.6 V vs. RHE, after 1.4 V (Fe2P/Ni2P) and 1.45 V (Fe2P/Ni5P4) in the HF region, the catalyst reconstruction concludes, R1 becomes remarkably smaller and the OER occurs in the LF interface, indicating that Fe2P/Ni5P4 undergoes a severe surface electrooxidation process. Moreover, the phase angle of the LF region symbolizing the OER decreases earlier for Fe2P/Ni2P (at 1.35 V) than for Fe2P/Ni5P4 (at 1.4 V), suggesting that Fe2P/Ni2P is more susceptible to polarization. The changes in R1 and R2 with potentials in Fig. 6f indicate that Fe2P/Ni2P undergoes an accelerated rate of electrooxidation compared with Fe2P/Ni5P4, and the OER process is faster.51–53 | ||
| Fig. 6 (a and d) Operando Nyquist plots, (b and e) Bode-phase plots of Fe2P/Ni2P and Fe2P/Ni5P4. (c) Electrical equivalent circuit model used for analyzing the interfacial charge transfer. (f) Correlation of the equivalent resistances (R1 and R2) and potentials for Fe2P/Ni2P and Fe2P/Ni5P4. | ||
In situ Raman techniques were conducted to explore the origin of the activity enhancement of the as-prepared black products of Fe2P/Ni5P4 and Fe2P/Ni2P catalysts (Fig. 7a and b). As shown in Fig. 7c–f, the spectra of Fe2P/Ni5P4 and Fe2P/Ni2P exhibit a prominent pair of peaks at 476 cm−1 (eg bending vibration) and 554 cm−1 (A1g NiIII–O stretching vibration) corresponding to characteristic peaks in the OER-active phase of γ-NiOOH that appear at potentials of 1.5 and 1.4 V vs. RHE, respectively. No characteristic peaks assigned to FeOOH appear, suggesting Ni2P undergoes a surface reconstruction process and is transformed into the Ni–OOH active species during the OER test, while Fe2P is still robust (white/black dotted oval frame in Fig. 7c).54 Moreover, a weak band located in the range of 1000–1100 cm−1 can be observed in Fe2P/Ni2P, which can be ascribed to PO43−.49 The fitted band intensities of Fe2P/Ni2P are stronger than those of Fe2P/Ni5P4 (Fig. 7d), implying an accelerated partial phase transformation to disordered NiOOH. Furthermore, FT-IR spectra of Fe2P/Ni2P were recorded to further confirm the phase transition before and after the OER test. Fig. 7g shows that the dominant absorption bands belonging to M–P and ν4 (O–P–O) become weaker after OER operation. Three new absorption peaks appear at 630 cm−1 (Ni–O–H), 1150 cm−1 (PO43−) and 1230 cm−1 (NiOOH) after the reaction.55,56 These above results suggest that Fe2P/Ni2P will interact with OH− to form Fe2P/Ni(OH)2 first, and will then be further oxidized and evolve to Fe2P/NiOOH during the OER process (Fe2P/Ni(OH)2 + OH− → Fe2P/NiOOH + H2O + e−). The in situ Raman and FT-IR confirm the existence of NiOOH and the formation and adsorption of PO43− on the electrode surface, where the negatively charged PO43− layer can protect the active species NiOOH from corrosion by repulsing Cl− (Fig. 7a).7 After the OER test, Fe2P/Ni2P shows negligible change in the XRD pattern, except that the peak intensity becomes weaker than that before the test, indicating that the electrode undergoes surface reconstruction and partial transformation into the corresponding amorphous NiOOH (Fig. S12†). It is particularly important to note that Fe2P/Ni5P4 displays a potential loss of 42 mV, whereas the loss of activity of Fe2P/Ni2P can be ignored under 50 mA cm−2 for 30 h, showing that the PO43− of the electrode surface plays a crucial role in maintaining OER active sites (Fig. S13†). In addition, partial Fe elements were dissolved from Fe2P/Ni2P in the initial period of the stability test (from 0 to 20 h), but the loss of Fe element had not remarkably increased after 20 h, showing that Fe leaching had increasingly stopped, attaining a dynamic balance process. However, the Ni element could not be detected in seawater electrolyte after 30 h of continuous operation, confirming that the formed negatively charged PO43− layer at the electrode surface can protect the NiOOH active species from corrosion and dissolution by repulsing Cl− ions (Fig. S14†). Fe2P/Ni5P4 shows a negative zeta potential (−17.86 eV), while Fe2P/Ni2P exhibits higher surface electronegativity (−18.93 eV), indicating the adsorption of a PO43− protective layer on the surface of catalyst (Fig. S15†). Furthermore, the active chlorine concentration in alkaline seawater solution was investigated and detected using the colorimetric method and UV/Vis spectroscopy (Fig. S16–S18†). The hypochlorite content did not increase after 30 h of CP testing, suggesting that the Cl− evolution was significantly inhibited.40 Corrosion polarization plots (Fig. S19†) demonstrate a higher potential and a smaller j for Fe2P/Ni2P than for Fe2P/Ni5P4, indicating that Fe2P/Ni2P with the strongest BEF can facilitate the adsorption of intermediates, thus showing stronger resistance against chloride corrosion.7 As a result, the Fe2P/Ni5P4 catalysts with weaker BEF dissolve successively, while Fe2P/Ni2P is very robust (Fig. S20†).
 | ||
| Fig. 7 (a) Schematic diagram of the possible evolution of catalyst. (b) The in situ Raman measurement device. (c) The image of Fe2P/Ni2P in the in situ Raman test process. (d and e) In situ Raman spectra of Fe2P/Ni5P4 and Fe2P/Ni2P with different operating potentials (vs. RHE). (f) Comparison of the ratios of band intensity (I476/I554). (g) FT-IR spectra of Fe2P/Ni2P before and after test. | ||
To emphasize the tremendous potential of Fe2P/Ni2P for practical application, a lab-scale AEMWE device was built (Fig. 8a). The overall water-splitting (OWS) performance of the Fe2P/Ni2P catalyst in the AEMWE system was documented using LSV in 1 M KOH electrolyte. As shown in Fig. 8b, the Fe2P/Ni2P catalyst requires only a voltage of 1.63 V to achieve η10, which is superior to other recently reported bifunctional electrocatalysts (Fig. 8c and Table S7†). Moreover, in Fig. 8d, the Fe2P/Ni2P catalyst demonstrates good stability over 80 h in the AEMWE system, indicating highly promising potential for practical application. Previous tests have demonstrated that the Fe2P/Ni2P catalyst exhibits excellent activity in alkaline HER, OER, and OWS, suggesting the superior all-around catalytic activity of the target catalyst. The mechanisms of improved OER and HER performance for Fe2P/Ni2P can be summarized, as schematically illustrated in Fig. 8e. The large ΔΦ will drive spontaneous electron transfer from Fe2P to Ni2P in heterogeneous Fe2P/Ni2P, and concurrently a strong interfacial BEF with an asymmetric charge distribution is constructed.57,58 During the water-splitting process, water molecules are first adsorbed on the catalyst surface and dissociated into intermediates (Volmer step). The negative-charge-rich Ni2P region prefers to absorb hydrogen intermediates, and subsequently generates hydrogen molecules by combining with two electrons. Meanwhile, the oxygen species are attracted by the positive-charge-enriched Ni2P side for catalyzing OER, which facilitates the shedding of hydrogen protons by decreasing the free energy in Fe2P/Ni2P. Therefore, significant bifunctionality for OWS activity is developed. For comparison, the adsorption of hydrogen protons by Ni2P and Fe2P is either too strong or too weak for HER, while the high energy barrier for the shedding of hydrogen protons for OER is attributed to the conventional charge distribution.
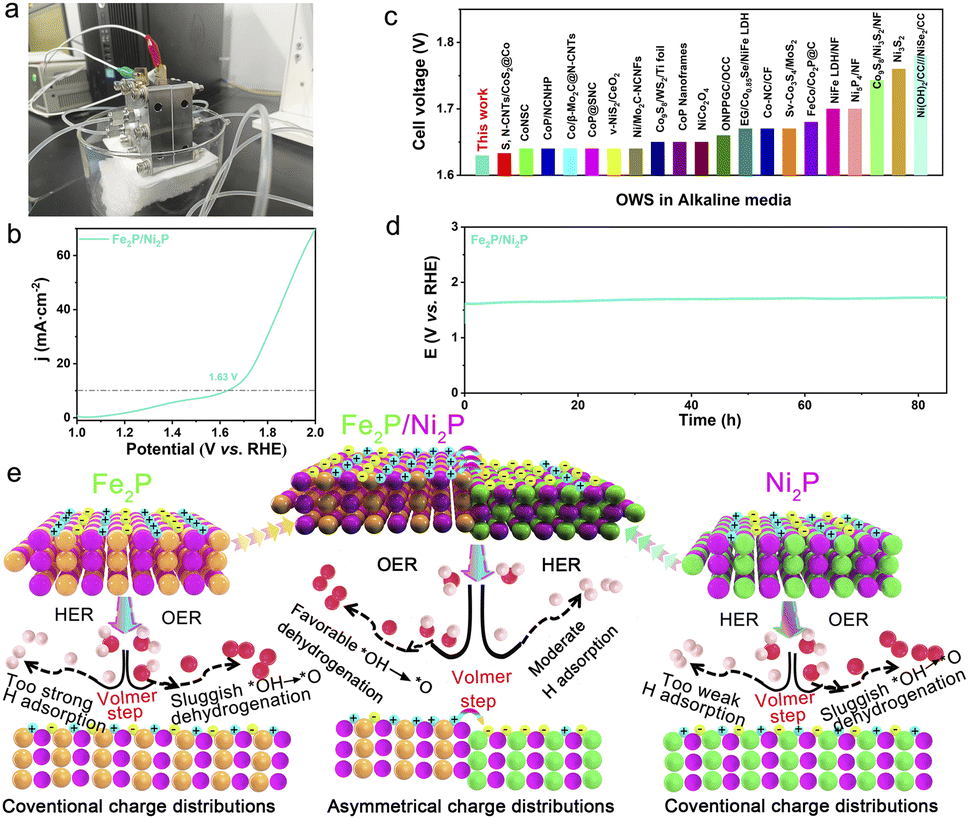 | ||
| Fig. 8 (a) The AEMWE device. (b) Polarization curves of AEM water electrolyzer with Fe2P/Ni2P. (c) Comparison of Fe2P/Ni2P with reported bifunctional electrocatalysts at 10 mA cm−2. (d) Stability of Fe2P/Ni2P//Fe2P/Ni2P at 10 mA cm−2 in AEMWE. (e) Schematic illustration of OER and HER mechanisms for Fe2P/Ni2P bifunctional electrocatalyst. | ||
4. Conclusions
We have exquisitely designed and manipulated a Br-induced partial in situ phase transition from Fe2P/Ni5P4 to Fe2P/Ni2P under the phosphorization process, which leads to a strong BEF due to the large ΔΦ difference. As a result, the Fe2P/Ni2P catalyst demonstrates remarkable OER and HER activity with low overpotentials of 196, 108 mV for freshwater and 229, 203 mV for seawater, respectively. Experiments and in situ/ex situ spectroscopic investigations confirm that the enhanced BEF plays an important role in subtly engineering absorption strength for reaction intermediates and phosphate intermediates due to an asymmetric charge distribution at the Fe2P/Ni2P interface. In particular, the existence of high-valence NiOOH and the co-existence of adsorbed PO43− at the electrode surface confirm that the negatively charged PO43− layer can protect the NiOOH active species from corrosion by repulsing Cl−. Moreover, Fe2P/Ni2P also exhibits a low cell voltage for an AEMWE system. This work paves the way for synthesizing bifunctional seawater/water-splitting electrocatalysts by exquisitely designing and modulating the interfacial BEF.Data availability
All data that support the findings of this study are included within the article.Author contributions
Lei Jin: conceptualization, methodology, writing – original draft, data curation, visualization. Hui Xu: methodology, formal analysis, writing – review & editing. Kun Wang: data curation. Yang Liu: investigation. Xingyue Qian: formal analysis, supervision, writing – review & editing. Haiqun Chen: project administration. Guangyu He: validation, funding acquisition, resources.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 22305025, 21978026, 22078028) and the Natural Science Foundation of Jiangsu Province (BK20230640). We also thank the Analysis and Testing Center of Changzhou University for assistance in characterizations.Notes and references
- S. Zhang, C. Tan, R. Yan, X. Zou, F. L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed., 2023, 135, e202302795 CrossRef.
- B. Huang, J. Yan, Z. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2024, 63, e202409419 CAS.
- H. Xu, Y. Zhao, Q. Wang, G. He and H. Chen, Coord. Chem. Rev., 2022, 451, 214261 CrossRef CAS.
- S. Zhao, H. Li, J. Dai, Y. Jiang, G. Zhan, M. Liao, H. Sun, Y. Shi, C. Ling and Y. Yao, Nat. Sustain., 2024, 7, 148–157 CrossRef.
- Y. Yang, J. Lou, Y. Zhao, J. Wei, Y. Zhou, C. Zhang, M. Wu, Y. Zhang, Q. Wang, L. Wang, T. Yang and X. Song, Chem. Eng. J., 2023, 477, 146900 CrossRef CAS.
- H. Xu, K. Wang, G. He and H. Chen, J. Mater. Chem. A, 2023, 11, 17609–17615 Search PubMed.
- J. Wei, J. Lou, W. Hu, X. Song, H. Wang, Y. Yang, Y. Zhang, Z. Jiang, B. Mei, L. Wang, T. Yang, Q. Wang and X. Li, Small, 2024, 20, e202308956 Search PubMed.
- H. Xu, Y. Liu, K. Wang, L. Jin, J. Chen, G. He and H. Chen, Inorg. Chem., 2024, 63, 16037–16046 CrossRef CAS PubMed.
- H. Xu, L. Yang, Y. Liu, L. Jin, K. Wang, G. He and H. Chen, Fuel, 2024, 377, 132796 CrossRef CAS.
- S. Zhao, Y. Wang, Y. Hao, L. Yin, C. H. Kuo, H. Y. Chen, L. Li and S. Peng, Adv. Mater., 2024, 36, 2308925 CrossRef CAS.
- X. Xu, A. Cao, W. You, Z. Tao, L. Kang and J. Liu, Small, 2021, 17, 2101725 CrossRef CAS PubMed.
- S. Zhang, C. Zhang, X. Zheng, G. Su, H. Wang and M. Huang, Appl. Catal., B, 2023, 324, 122207 CrossRef CAS.
- H. Xu, L. Jin, K. Wang, L. Yang, Y. Liu, G. He and H. Chen, Fuel, 2024, 369, 131716 CrossRef CAS.
- X. Zhao, M. Liu, Y. Wang, Y. Xiong, P. Yang, J. Qin, X. Xiong and Y. Lei, ACS Nano, 2022, 16, 19959–19979 CrossRef CAS PubMed.
- J. Yao, W. Huang, W. Fang, M. Kuang, N. Jia, H. Ren, D. Liu, C. Lv, C. Liu and J. Xu, Small Methods, 2020, 4, 2000494 CrossRef CAS.
- H. Xu, L. Yang, L. Jin, Y. Liu, K. Wang, J. Chen, G. He and H. Chen, J. Colloid Interface Sci., 2025, 677, 158–166 CrossRef CAS.
- C. Guo, H. Xue, J. Sun, N. Guo, T. Song, J. Sun, Y.-R. Hao and Q. Wang, Chem. Eng. J., 2023, 470, 144242 CrossRef CAS.
- H. Xu, J. Li and X. Chu, Nanoscale Horiz., 2023, 8, 441–452 RSC.
- T. Xu, D. Jiao, L. Zhang, H. Zhang, L. Zheng, D. J. Singh, J. Zhao, W. Zheng and X. Cui, Appl. Catal., B, 2022, 316, 121686 CrossRef CAS.
- K. Ren, W. J. Xu, K. Li, J. M. Cao, Z. Y. Gu, D. H. Liu, D. M. Dai, W. L. Li and X. L. Wu, Adv. Funct. Mater., 2024, 2415585 CrossRef.
- L. Jin, H. Xu, K. Wang, L. Yang, Y. Liu, X. Qian, G. He and H. Chen, Appl. Surf. Sci., 2024, 159777 CrossRef CAS.
- H.-M. Yang, H.-Y. Wang, S. Zhai, J.-T. Ren and Z.-Y. Yuan, Chem. Eng. J., 2024, 489, 151236 Search PubMed.
- Y. Li, Y. Wu, H. Hao, M. Yuan, Z. Lv, L. Xu and B. Wei, Appl. Catal., B, 2022, 305, 121033 Search PubMed.
- D. Li, C. Zhou, R. Yang, Y. Xing, S. Xu, D. Jiang, D. Tian and W. Shi, ACS Sustain. Chem. Eng., 2021, 9, 7737–7748 CrossRef CAS.
- H. T. Dao, S. Sidra, M. Mai, M. Zharnikov and D. H. Kim, Chem. Eng. J., 2024, 150054 CrossRef CAS.
- H. Liu, L. Jiang, Y. Sun, J. Khan, B. Feng, J. Xiao, H. Zhang, H. Xie, L. Li and S. Wang, Adv. Energy Mater., 2023, 13, 2301223 CrossRef CAS.
- D. Yue, T. Feng, Z. Zhu, S. Lu and B. Yang, ACS Catal., 2024, 14, 3006–3017 CrossRef CAS.
- J. Li, G. Wei, Y. Zhu, Y. Xi, X. Pan, Y. Ji, I. V. Zatovsky and W. Han, J. Mater. Chem. A, 2017, 5, 14828–14837 RSC.
- S. Mondal, D. Bagchi, M. Riyaz, S. Sarkar, A. K. Singh, C. Vinod and S. C. Peter, J. Mater. Chem. A, 2022, 144, 11859–11869 CAS.
- M. Asim, B. Maryam, S. Zhang, M. Sajid, A. Kurbanov, L. Pan and J.-J. Zou, J. Colloid Interface Sci., 2023, 638, 14–25 CrossRef CAS.
- J. Zhu, J. Chi, X. Wang, T. Cui, L. Guo, B. Dong, X. Liu and L. Wang, Nano Energy, 2024, 121, 109249 CrossRef CAS.
- K. Wang, S. He, B. Li, H. Du, T. Wang, Z. Du, L. Xie and W. Ai, Appl. Catal., B, 2023, 339, 123136 CrossRef CAS.
- Y. Li, X. Yu, J. Gao and Y. Ma, Chem. Eng. J., 2023, 470, 144373 CrossRef CAS.
- K. Chang, D. T. Tran, J. Wang, K. Dong, S. Prabhakaran, D. H. Kim, N. H. Kim and J. H. Lee, Appl. Catal., B, 2023, 338, 123016 CrossRef CAS.
- Z. Li, C. Xu, Z. Zhang, S. Xia, D. Li, L. Liu, P. Chen and X. Dong, Adv. Sci., 2024, 2308477 CrossRef CAS.
- X. Pei, J. Bian, W. Zhang, Z. Hu, Y. H. Ng, Y. Dong, X. Zhai, Z. Wei, Y. Liu and J. Deng, Adv. Funct. Mater., 2024, 2400542 CrossRef CAS.
- H. Y. Chen, L. Yang, R. X. Wang, W. J. Zhang, R. Liu, Y. Z. Yun, N. Wang, S. Ramakrishna, L. Jiao and Y. Z. Long, Small, 2023, 19, 2304086 CrossRef CAS.
- L. Yang, X. Cao, X. Wang, Q. Wang and L. Jiao, Appl. Catal., B, 2023, 329, 122551 CrossRef CAS.
- M. Gu, L. Jiang, S. Zhao, H. Wang, M. Lin, X. Deng, X. Huang, A. Gao, X. Liu and P. Sun, ACS Nano, 2022, 16, 15425–15439 CrossRef CAS PubMed.
- X. Yan and Z. Jin, Chem. Eng. J., 2021, 420, 127682 CrossRef CAS.
- Y. Song, M. Sun, S. Zhang, X. Zhang, P. Yi, J. Liu, B. Huang, M. Huang and L. Zhang, Adv. Funct. Mater., 2023, 33, 2214081 CrossRef CAS.
- P. Zhao, S. Fu, Y. Luo, C. Peng, L. Cheng and Z. Jiao, Small, 2023, 19, 2305241 CrossRef CAS.
- W. Chen, W. Wei, F. Li, Y. Wang, M. Liu, S. Dong, J. Cui, Y. Zhang, R. Wang and K. Ostrikov, Adv. Funct. Mater., 2024, 34, 2310690 CrossRef CAS.
- S. Zhang, X. Wei, S. Dai, H. Wang and M. Huang, Adv. Funct. Mater., 2024, 34, 2311370 CrossRef CAS.
- S. Zhang, Y. Wang, X. Wei, L. Chu, W. Tian, H. Wang and M. Huang, Appl. Catal., B, 2023, 336, 122926 CrossRef CAS.
- S. M. El-Refaei, P. A. Russo, T. Schultz, Z. N. Chen, P. Amsalem, N. Koch and N. Pinna, Carbon Energy, 2023, e556 Search PubMed.
- Y. Shi, S. Zhou, J. Liu, X. Zhang, J. Yin, T. Zhan, Y. Yang, G. Li, J. Lai and L. Wang, Appl. Catal., B, 2024, 341, 123326 CrossRef CAS.
- J. Liu, S. Duan, H. Shi, T. Wang, X. Yang, Y. Huang, G. Wu and Q. Li, Angew. Chem., Int. Ed., 2022, 134, e202210753 CrossRef.
- W. Liu, J. Yu, M. G. Sendeku, T. Li, W. Gao, G. Yang, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 135, e202309882 CrossRef.
- S. Fu, Y. Ma, X. Yang, X. Yao, Z. Jiao, L. Cheng and P. Zhao, Appl. Catal., B, 2023, 333, 122813 CrossRef CAS.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS.
- Y. Li, Y. Wu, M. Yuan, H. Hao, Z. Lv, L. Xu and B. Wei, Appl. Catal., B, 2022, 318, 121825 CrossRef CAS.
- N. Zhang, Y. Hu, L. An, Q. Li, J. Yin, J. Li, R. Yang, M. Lu, S. Zhang and P. Xi, Angew. Chem., Int. Ed., 2022, 61, e202207217 CrossRef CAS PubMed.
- B. Wu, S. Gong, Y. Lin, T. Li, A. Chen, M. Zhao, Q. Zhang and L. Chen, Adv. Mater., 2022, 34, 2108619 CrossRef CAS.
- S. Wang, P. Yang, X. Sun, H. Xing, J. Hu, P. Chen, Z. Cui, W. Zhu and Z. Ma, Appl. Catal., B, 2021, 297, 120386 CrossRef CAS.
- Z.-Y. Yu, Y. Duan, J.-D. Liu, Y. Chen, X.-K. Liu, W. Liu, T. Ma, Y. Li, X.-S. Zheng and T. Yao, Nat. Commun., 2019, 10, 2799 CrossRef.
- D. Chen, R. Lu, R. Yu, Y. Dai, H. Zhao, D. Wu, P. Wang, J. Zhu, Z. Pu and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202208642 CrossRef CAS PubMed.
- W. Zhang, L. Yang, Z. Li, G. Nie, X. Cao, Z. Fang, X. Wang, S. Ramakrishna, Y. Long and L. Jiao, Angew. Chem., Int. Ed., 2024, 63, e202400888 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06673a |
| This journal is © The Royal Society of Chemistry 2025 |