
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P+ addition and transfer involving a tetraphosphenium ion†
Roman
Franz
a,
Máté
Bartek
 b,
Clemens
Bruhn
a,
Zsolt
Kelemen
*b and
Rudolf
Pietschnig
*a
b,
Clemens
Bruhn
a,
Zsolt
Kelemen
*b and
Rudolf
Pietschnig
*a
aInstitute for Chemistry, CINSaT, University of Kassel, Heinrich-Plett-Straße 40, 34132 Kassel, Germany. E-mail: pietschnig@uni-kassel.de; Web: https://www.uni-kassel.de/go/hybrid
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Műegyetem Rkp 3, 1111 Budapest, Hungary. E-mail: kelemen.zsolt@vbk.bme.hu
First published on 25th November 2024
Abstract
Triphospha[3]ferrocenophane Fe(C5H4-PTip)2PCl (Tip = 2,4,6-tri(isopropyl)phenyl) has been prepared and its suitability to generate the corresponding bisphosphanylphosphenium ion has been explored. By formal addition of P+ to the latter, an unprecedented tetraphosphenium ion forms which likewise is capable of P+ transfer and qualifies as Lewis superacid based on its computed fluoride ion affinity. As a solid, this species is stable and conveniently storable, featuring a remarkably long P–P bond (2.335(5) Å). From this tetraphosphenium ion, known and unprecedented triphosphenium ions have been generated via P+ transfer in solution, including a triphosphenium ion with P–H functionalities. Moreover, the latter has been obtained by tautomeric rearrangement from the corresponding hydrophosphane precursor. The bonding situation and details of the P+ transfer have been investigated by DFT calculations and experimental methods like multinuclear NMR spectroscopy and SC-XRD.
Introduction
The transfer of single phosphorus atoms has emerged as a hot topic in recent years with a special focus on P(0) or P− fragments.1–14 By contrast, P+ ion transfer is far less explored,15–23 and in addition needs to be distinguished from the transfer of substituted P(I) fragments with more than one atom, such as phosphinidenes. So called triphosphenium ions are established reagents for the transfer of formally unsubstituted P+ and have been carefully studied by Macdonald and co-workers in recent years.15–24 The first triphosphenium ions have been reported by Schmidpeter et al. in the 1980s as the first isolable P(I) compound in which the formal charge of P+ is also preserved as a net charge.25,26 It needs to be pointed out that – despite their similar name – the above mentioned triphosphenium ions are only distantly related to actual phosphenium ions [PR2]+ with the latter featuring an electron sextet, in contrast to triphosphenium ions with an electron octet at the central phosphorus atom (Fig. 1).27 Phosphenium ions are valence isoelectronic and isolobal to neutral carbene analogues such as [ER2] (E = C, Si, Ge, Sn, Pb).28,29 In addition, the positive charge imparts pronounced electrophilic character to phosphenium ions, comparable to Lewis acidic carbenium or silicenium ions [R3E]+ (E = C, Si) which in turn lack an electron lone pair, however.30 Consequently, phosphenium ions are highly reactive species which, for a long time, could only be detected in the gas phase until the development of thermodynamically stabilized examples,31–42 with the term NHP being in use for N-heterocyclic phosphenium ions of the type I+ (Fig. 1) in analogy to isolobal NHCs.40,43–46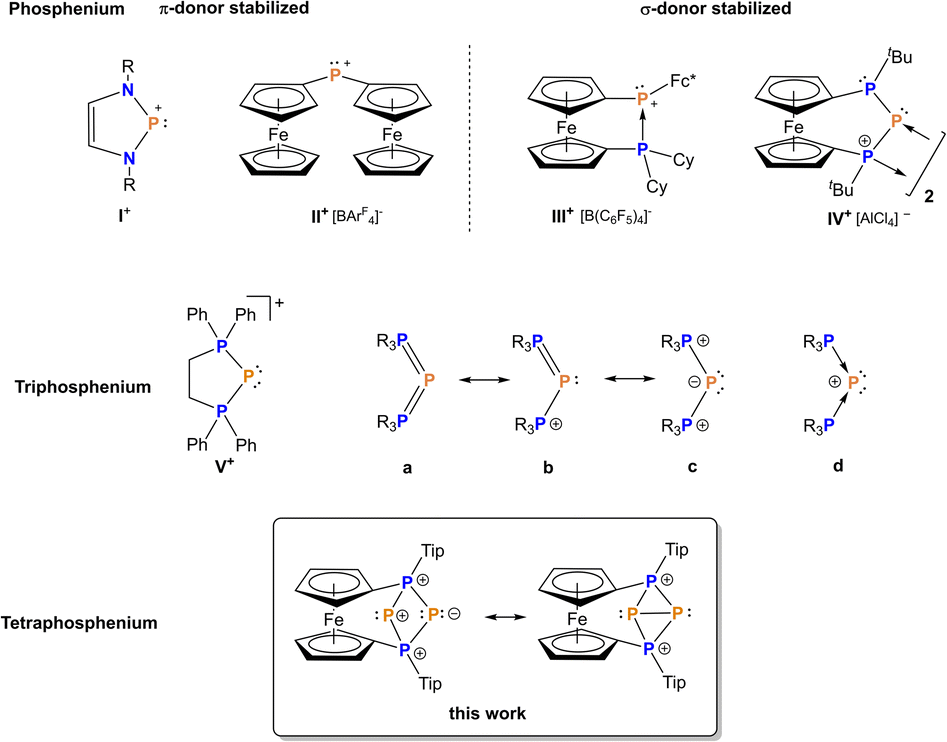 | ||
| Fig. 1 Comparison of phosphenium ions I+, II+,50III+,42 and IV+,54 (top) to triphosphenium ions V+ (middle) and a tetraphosphenium ion (bottom). | ||
Thermodynamic stabilization has been achieved for carbon substituted (nitrogen-free) phosphenium ions as well, using ylidic47,48 or electron-rich olefinic carbon atoms,49 or ferrocenyl substituted phosphenium ions such as II+ and III+ (Fig. 1).42,50–52 Moreover, Beckmann et al. succeeded in isolating a kinetically stabilized phosphenium ion recently.53 For increased reactivity, in situ-formed, non-stabilized phosphenium ions have been employed as versatile reaction partners to construct polyphosphorus compounds, activate small molecules and alkenes.34,54–61 In line with this, bisphosphanylphosphenium ions IV+, which could be referred to as P-heterocyclic phosphenium ions (PHPs) in analogy to the above mentioned NHPs, feature insufficient stabilization by adjacent phosphanyl units and consequently undergo either dimerization or fragmentation54,62 involving P–C bond activation, with the P3-unit remaining intact (Fig. 1).54,62
Using an in situ generated bisphosphanylphosphenium ion equipped with a suitable substitution pattern, we report here the first transfer of formally monoatomic P+ to a phosphenium cation, resulting in the formation of a characteristic tetraphosphorus dication which we refer to as tetraphosphenium ion. This contrasts other known examples of P+ transfer proceeding to nucleophilic substrates such amines, phosphanes, NHCs or carbonylmetallates to name just a few.15–17,63
Results and discussion
As a starting point, we introduced 2,4,6-tri(isopropyl)phenyl (Tip) as a so far unexplored substituent at the phosphorus atoms of the [3]ferrocenophane scaffold following a previous established substitution and condensation sequence (Scheme 1).64 We anticipated that the increased steric congestion would supply sufficient steric protection to prevent dimerization observed for the tBu substituted heterocarbenes.54,65 | ||
| Scheme 1 Synthetic pathway to phosphenium ion 3[AlCl4], stabilized phosphenium ion 6[AlCl4] and tetraphosphenium ion 4[AlCl4]2 with rearrangement to triphosphenium ion 5[Al2Cl7]. | ||
To this end 1,1-bis(dichlorophosphanyl)ferrocene66 is reacted with two equivalents of 2,4,6-tri(isopropyl)phenyl lithium (Tip–Li) then the resulting chlorophosphane is transformed into the secondary hydrophosphane 2 which subsequently condenses with PCl3 in the presence of base to form [3]ferrocenophane 3 (Scheme 1). Given the stereogenic nature of the phosphorus atoms in the terminal positions, it should be mentioned that prior to the ring closure the ferrocenophane precursor is a mixture of the corresponding rac and meso diastereomers of 2 for which rac2 epimerizes upon reaction to 3. The ferrocene bridged triphosphanyl scaffold limits the number of diastereomers originating from the presence of the P-stereogenic centers.64 Interestingly, for Tip-substituted 3 exclusive formation of the meso-trans-isomer is observed, where trans refers to the orientation of the chlorine with respect to the Tip substituents. The computed Gibbs free energy difference between the cis and its corresponding trans isomer is small (ΔG = 2.9 kcal mol−1 see Computational details in ESI†), comparable to the value obtained in case of the tBu-substituted counterpart, where both isomers were observed.64 Assuming that interconversion of the isomers is unlikely due to the high inversion barrier of the phosphorus centers, the selective formation of the trans isomer of 3 can be attributed to kinetic factors rather than to its thermodynamic stability.
The 31P NMR spectrum of [3]ferrocenophane 3 features an AX2 spin system, with the signal of the central phosphorus atom (δ(31P) = 91.2 ppm) split into a triplet, whereas the two outer phosphorus nuclei resonating at −28.1 ppm feature a doublet splitting. The low 1JPP coupling constant of 177 Hz in 3 is in agreement with the formation of the meso-trans diastereomer since significantly higher values (ca. 350 Hz) are generally observed for meso-cis-triphospha[3]ferrocenophanes.64 Although we were able to grow small orange crystals of 3, these were not suitable for X-ray crystallography owing to extremely poor scattering. The identity and purity of 3 were further confirmed by 1H, 13C-NMR spectroscopy and elemental analysis. The 31P resonances of both diastereomers of 2 are located at −97.9 ppm (as two overlapping resonances) featuring 1JPH-coupling of 226 Hz. Identity and purity of 2 were further confirmed by 1H, 13C-NMR spectroscopy, mass spectrometry, elemental analysis and X-ray crystallography (Fig. S1†). The latter findings support significant steric congestion, with the angular sum at the phosphorus atoms (∑P1 = 332(4)° and ∑P2 = 325(3)°) in 2 being remarkably high and comparable to extremely bulky triarylphosphanes such as P(Mes)3 (330°)67 or P(Tip)3 (337°).67
With chlorophosphane 3 in hand, we set out to investigate chloride abstraction to the respective phosphonium ion. While with GaCl3 or Li[Al(OC(CF3)3)4] no selective reactivity was found, for AlCl3 complete and selective P+ transfer to dicationic 42+ and diphosphane Fc'(TipP)2 was observed (Scheme 1). As intended the bulky Tip substituents successfully prevent dimerization of 3+, which had been observed for its tBu substituted analog IV+ (Fig. 1).54 However, stabilization of the flanking phosphanyl units is insufficient to isolate the free phosphenium ion 3+,68 resulting in formal elimination of “P+” which is transferred to a second unit of transient phosphenium cation 3+ forming the thermodynamically more stable dication 42+. This hypothesis is thermodynamically plausible, as confirmed by the exergonic computed reaction Gibbs free energy for the “P+” transfer to cation 3+ (ΔG = −5.1 kcal mol−1, Computational details in ESI†). At this point, it is important to highlight that the formation of the corresponding bisphosphanylphosphenium dimer (analogue of (IV+)2) is thermodynamically favored (ΔG = −12.5 kcal mol−1) over the formation of 42+ (ΔG = −5.1 kcal mol−1). The exergonic dimerization process, even in the presence of bulky groups, indicates the outstanding stability of this dimeric structural motif54,69,70 and implies that the formation of 42+ is under kinetic control (proposed mechanism in Scheme S1 in the ESI†). Again, 31P NMR spectra are helpful to establish the connectivity in 42+ featuring two triplet resonances centered at δ = −213.9 ppm (central phosphanyl units) and δ = 24.0 ppm (tetracoordinated phosphorus atoms) with a 1JPP coupling constant of 304 Hz, which is consistent with a rigid tetraphosphabicyclobutane. The identity and purity of 4[AlCl4]2 was confirmed by 1H, 13C, 27Al NMR spectroscopy and elemental analysis. The constitution of 4[AlCl4]2 has been further corroborated by single crystal X-ray diffraction (Fig. 2).
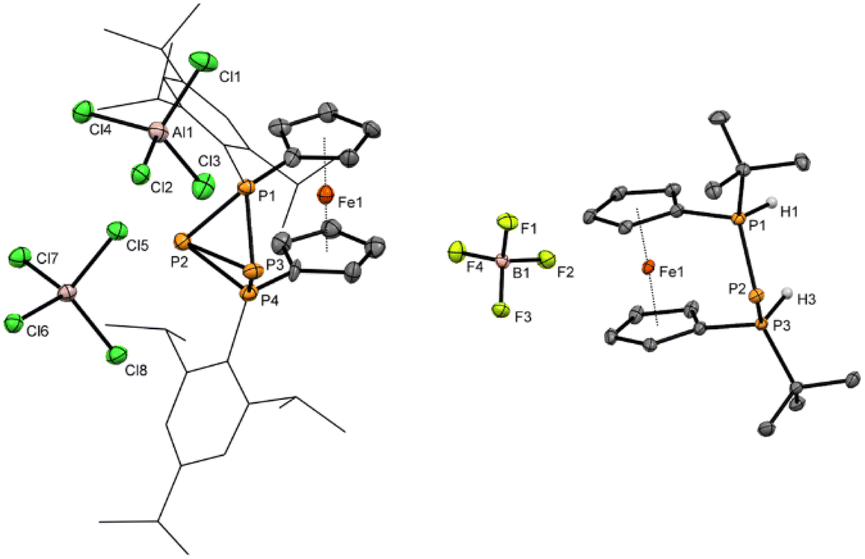 | ||
| Fig. 2 Molecular structures of 4[AlCl4]2 and 8c[BF4] in the solid state. Ellipsoids are shown at 30% probability. All hydrogen atoms except H1 and H3, which are bonded to phosphorus atoms are omitted for clarity. The Tip substituents at P1 and P4 in 4[AlCl4]2 are shown as wireframe. Selected bond lengths and angles in 4[AlCl4]2: P1–P2 2.184(5) Å, P1–P3 2.171(5) Å, P2–P3 2.335(5) Å, P2–P4 2.194(5) Å, P3–P4 2.173(5) Å; 8c[BF4]: P1–P2 2.1284(7) Å, P2–P3 2.1265(7) Å, P1–H1 1.28(2) Å, P2–H2 1.26(2) Å, PPP-angle 89.50(2)°. | ||
The bisphosphonium ion 42+ crystallizes without direct contact to its [AlCl4]− counter anions and with almost plane-parallel arrangement of the Cp ligands (α = 2.7(2)°) indicating an unstrained ferrocenophane structure. The molecular structure in the solid state of 4[AlCl4]2 shows a P4 fold angle of 72.1(2)°, which is usually found for strongly puckered P4 rings in diaryltetraphosphabicyclobutanes.71–73 In line with this a strongly distorted trigonal pyramidal geometry is found at P2 and P3 with an angular sum of ΣP2 = 200.2(5)° and ΣP3 = 202.5(5)°. The P–P bond lengths [2.184(5) Å (P1–P2) and 2.171(5) Å (P1–P3)] between the tetrasubstituted outer and trisubstituted central phosphorus atoms are in the range of common P–P single bonds. Interestingly the bond between the two central phosphorus atoms is significantly elongated [2.335(5) Å P2–P3]. To the best of our knowledge, the tetraphosphenium salt 4[AlCl4]2 contains the longest documented P–P single bond between two tricoordinated phosphorus atoms in a functionalized “P4”. Closely related to 42+ are P4-adducts with cations such as Ag+, NO+, or PPh2+ reported by Krossing, Weigand and Riedel or the edge protonated P4H+ reported by Müller and Riedel.57,74–77
The bonding situation of 42+ was explored by DFT calculations. Topological analysis was performed by the means of Bader's atoms in molecules (AIM) theory, and a bond critical point with electron density of 0.092 au was found between P2 and P3. In agreement with the shorter bond length, the electron density is higher (0.122 a.u.) in the bond critical points between atoms P1 and P2 (or P1 and P3). By examining the dimensionless ratios defined by Bader78 and Espinosa79 and other computed parameters, the interaction can be classified as a covalent bond (Tables S2–S5†). The Laplacian of the electron density along the P2–P3 bond path (∇2(r) function) shows a high degree of symmetry, with a local maximum of −0.0314 au at the bond critical point (see Fig. S2†). The rather weak character of the P2–P3 bond is reflected by the computed quantum theory of atoms in molecules (QTAIM) data, Mayer bond, and delocalization indices (see Tables S2–S4 in the ESI†) and the gradient plot of the Laplacian plot (see Fig. S3 and S4 in the ESI†). Natural resonance theory analysis proposes only one leading resonance structure for tetraphosphenium ion 42+ with 70% weight, which is represented by the Lewis structure depicted in Scheme 1 and in Fig. 4 (structure D) (more details in the ESI†).
The analysis of Kohn–Sham orbitals shows that all four phosphorus atoms make significant contributions to the unoccupied orbitals (Fig. 3). The LUMO is basically the combination of the empty pz orbitals at the central phosphorus atoms and the antibonding σ* of the P2–P3 bond, which foreshadows the reactivity of the central phosphorus atoms towards nucleophiles. The orbital-weighed Fukui function (which is a useful tool to identify the reactive site of a system)80 verified the most electrophilic character of the central phosphorus atoms as well (Fig. S2 and S3 in the ESI†). The computed natural charge at each of the central phosphorus atoms is 0.21 which is in close agreement with the charges computed within QTAIM atomic basins. In view of these data, a possible alternative description of 42+ could be a phosphenium ion intramolecularly stabilized by a triphosphenium ion (cf.C in Fig. 4), where the triphosphenium part emerges from twofold coordination of P+ by the phosphanyl groups of former 3+ (cf.A and B in Fig. 4). However, the natural bond order in D, and QTAIM results indicate a purely covalent central P–P bond in line with the above-mentioned NRT results for 42+. Nonetheless, polar structures B or C, whilst not representative of the bonding situation, are useful to understand the reactivity of 42+ (Schemes 1–3).
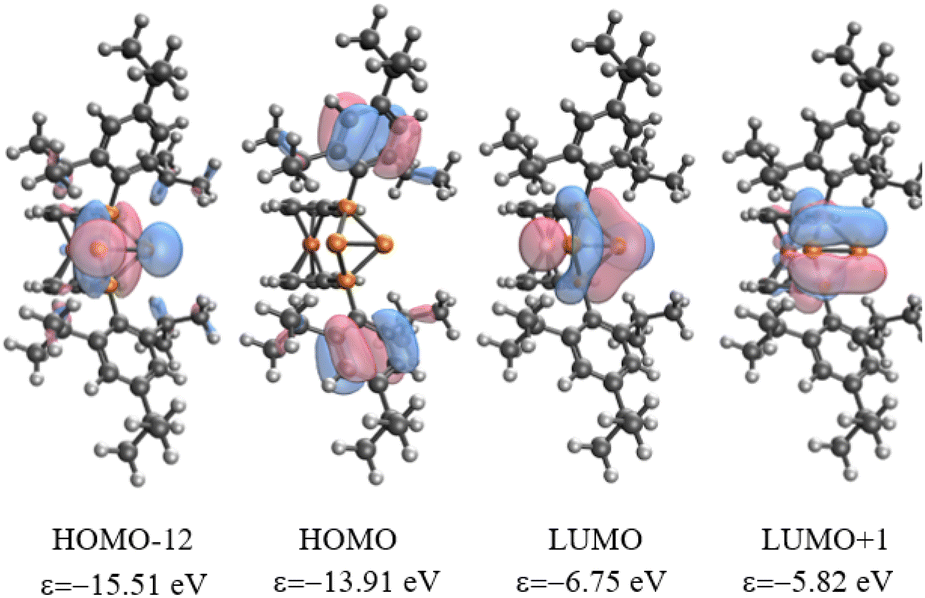 | ||
| Fig. 3 Selected Kohn–Sham molecular orbitals of 42+ at ωB97X-D/def2-TZVP. | ||
 | ||
| Fig. 4 Alternative Lewis-type descriptions of 42+. | ||
In order to evaluate the Lewis acid strength of 42+, its fluoride ion affinity (FIA) was computed (for single point energy calculations PCM = THF solvent model was applied), which is 87.2 kcal mol−1. This value is significantly higher than the average of 27 dicationic compounds (40.8 kcal mol−1) recently investigated by Greb et al.81 and suggests super Lewis acidic character. (The corresponding vacuum phase FIA is 239.0 kcal mol−1 for 42+ and the average reported by Greb is 215 kcal mol−1). In order to get a more precise picture (applying the same computational protocol) we have calculated phosphorus containing dicationic systems, where the positive charge is not localized at one center similar to 42+. Among the investigated cases (Table S6 in the ESI†) Dielmann's tricoordinate phosphorus dication [P(Nim)3]2+ (im = 1,3-diisopropyl-4,5-dimethylimidazolin-2-yliden) exhibits a very similar FIA value (87.9 kcal mol−1), which was considered as Lewis superacid as well.82
Tetraphosphenium salt 4[AlCl4]2 is poorly soluble in halogenated solvents like chloroform, dichloromethane or 1,2-difluorobenzene. Isolated 4[AlCl4]2 can be stored as solid under inert atmosphere for several weeks without decomposition but has a limited lifetime in solution. In solution, 4[AlCl4]2 transforms quantitatively within several hours to a new product, which could be assigned to its chloride adduct 5[Al2Cl7] representing the first bicyclic triphosphenium ion (Scheme 1). The calculated reaction Gibbs free energy of the process is only −0.1 kcal mol−1. Since the small negative ΔG value does not exclude the reversibility of the process, the quantitative back-reaction to tetraphosphenium ion 42+ was tested and after the addition of further Lewis acid (AlCl3 or Li[Al(OC(CF3)3)4]) 42+ was detected. The mono cation 5[Al2Cl7] shows characteristic signals in the 31P NMR spectrum (Table 1).
| Compound | δ(31P) [ppm] | |JPP| [Hz] | Spin system |
|---|---|---|---|
| 3 | 91.2(t), −28.1(d) | 177 (1JAX) | AX2 |
| 4 | 24.0(t), −213.9(t) | 304 (1JAX) | A2X2 |
| 5 | 19.5(dd), −39.8(dt), −130.7(td) | 372 (1JAX), 337 (1JMX), 108 (2JAM) | AMX2 |
| 6 | 43.2(dd), 22.3(dt), 5.2(dt) | 174 (1JAX), 72 (2JMX), 379 (1JAM) | AMX2 |
Thus, the resonance of the two chemically equivalent terminal phosphorus atoms at 19.5 ppm splits into a doublet of doublets owing to coupling with the central phosphanide center (1JPP = 372 Hz) and the central P–Cl unit (1JPP = 337 Hz). The same couplings are found for the signals of the central disubstituted phosphorus atom resonating at −130.7 ppm (1JPP = 372 Hz, 2JPP = 108 Hz) and the chlorine substituted phosphorus atom at −39.8 ppm (1JPP = 337 Hz, 2JPP = 108 Hz) which in turn split into doublets of triplets. The identity and purity of 5[Al2Cl7] was further confirmed by 1H, 13C NMR spectroscopy and elemental analysis. Since initially formed 3+ was not directly observed spectroscopically, the formation of 3+ as an intermediate was verified by trapping the phosphenium ion with the established Lewis base PPh3, leading to the formation of the expected triphenylphosphane-stabilized phosphenium ion 6[AlCl4] (Scheme 1). Interestingly, 6+ exhibits higher stability by 15.0 kcal mol−1 (Scheme S2 in ESI†), than its tBu substituted counterpart.54 The PPh3-adduct 6[AlCl4] shows three different resonances in the 31P NMR spectra with expected splitting patterns similar to a triphenylphosphane-stabilized phosphenium ion reported only recently.54 The two chemically equivalent outer phosphorus nuclei show a resonance at 43.2 ppm, split into a doublet of doublets due to the coupling to the central phosphorus atom (1JPP = 174 Hz) and the PPh3 group (2JPP = 72 Hz). The signal of the central phosphorus atom resonates at 5.2 ppm and splits into a doublet of triplets owing to coupling with the two chemically equivalent outer phosphanyl groups and the PPh3 fragment (1JPP = 379 Hz). The signal of the PPh3 group in 6[AlCl4] is found at 22.3 ppm with the expected doublet of triplet pattern resulting from the P–P couplings described above. Identity and purity of 6[AlCl4] was further confirmed by 1H, 13C, 27Al NMR spectroscopy and elemental analysis.
The bonding situation along with the exceptionally long P–P bond in 4[AlCl4]2 sparked us to explore the reactivity of this unprecedented species. Since 42+ originates from P+ transfer itself and its formation was not highly exergonic (ΔG = −5.1 kcal mol−1), we were wondering whether this P+ fragment may be further transferred to other substrates. Thus, we reacted 4[AlCl4]2 with different bisphosphanes such as dppe (1,2-bis(diphenylphosphino)ethane), dppf (1,1′-bis(diphenylphosphino)ferrocene) and prochiral 1,1′-bis(tert-butylphosphino)ferrocene 7. In all cases a formal P+ transfer was obtained leading to selective formation of the corresponding triphosphenium ions 8a–c (Scheme 2). As a by-product, the corresponding phosphenium cations stabilized by a second equivalent of bisphosphane were observed.
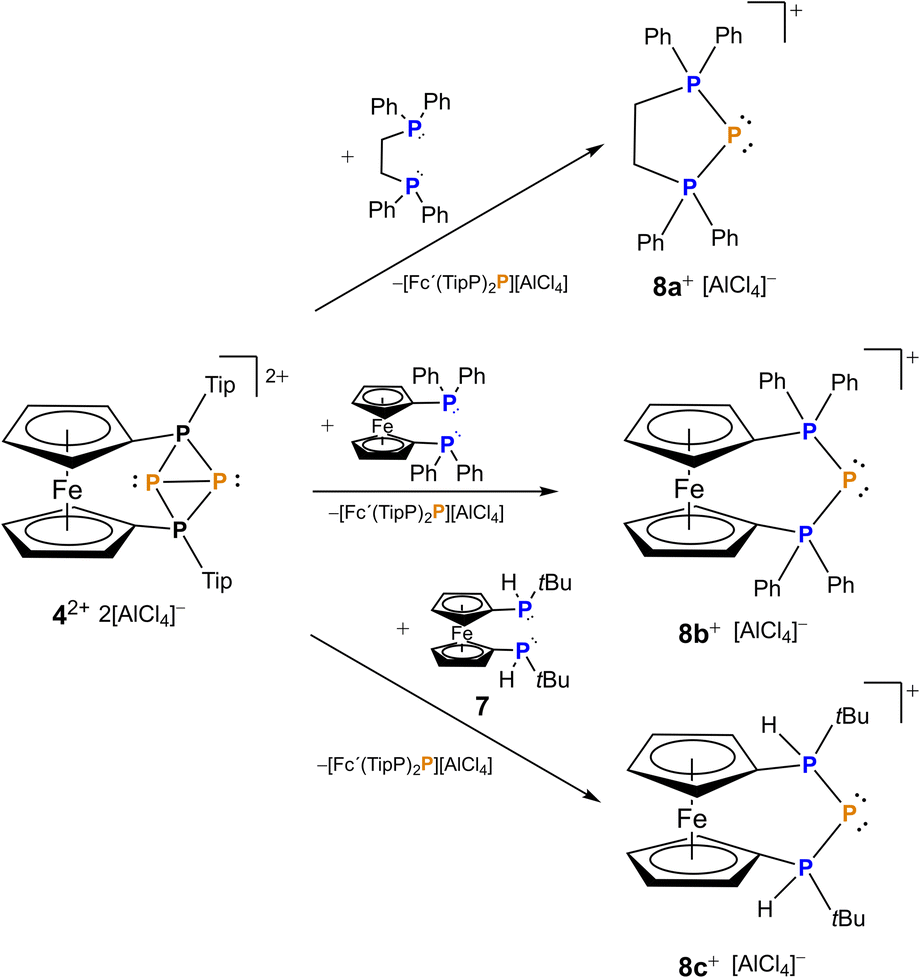 | ||
| Scheme 2 Synthesis of triphosphenium ions 8a–cvia formal P+ transfer using 4[AlCl4]2 starting from the corresponding bisphosphane. | ||
Our DFT calculations (Scheme 3, a model system with Me substituents at the phosphorus atoms was used, ωB97X-D/def2-SVP) reveal that the nucleophilic attack of the bisphosphane to one of the central phosphorus atoms (P2) is a barrierless process, resulting in the cleavage of the central P2–P3 bond. The addition of a second bisphosphane to the other central phosphorus atom (P3) leads to the breakage of the bond between the central and the outer phosphorus atoms (P1–P3) and similar to the first addition it proceeds without any significant barrier. In the next step, the P3–P4 bond cleaves, and at the same time, the second phosphane unit of the bisphosphane closes the ring with the originally P3 phosphorus atom. The overall process has a significant thermodynamic driving force (−53.8 kcal mol−1).
 | ||
| Scheme 3 Calculated reaction mechanism of the P+ transfer reaction. | ||
While triphosphenium ions 8a,b are already known in the literature and their successful formation was verified by 31P-NMR spectroscopy,83,848c represents the first triphosphenium ion carrying hydrogen substituents at both phosphonium centers, in addition to one tert-butyl group. Given the unique possibilities of attaching or abstracting protic hydrogen atoms we explored an alternative access to 8c starting from literature known 9. The calculated proton affinity of 9 is 270.7 kcal mol−1, and we found that HBF4 is able to protonate secondary triphosphane 9 inducing a [1,2-H] shift of the proton already present at the central phosphorus atom resulting in selective formation of 8c (Scheme 4), which is more stable by 1.9 kcal mol−1, than the originally formed intermediate (8c′). Despite its simplicity this straightforward approach to triphosphenium ions via protonation of a secondary triphosphane is unprecedented in the current literature. A clear advantage over previously published protocols emerges from the selectivity of the reaction, as no byproducts are formed and 8c can be isolated in good yields (65%) after first recrystallization. Regardless of whether 8c is formed via a formal “P+” transfer to bisphosphane 7 or protonation of triphospha[3]ferrocenophane 9 the formation proceeds stereoselectively and 8c is obtained in the meso form only, while there is no evidence for the appearance of its rac diastereomer neither in solution nor in solid state. In the 31P-NMR spectra, protonation of the outer phosphorus atoms entails a significant downfield shift of these nuclei by more than 50 ppm resulting in a chemical shift at 42.5 ppm in 8c. In turn, the central phosphorus atom encounters a drastic high field shift and resonates at −195.2 ppm in 8c and the AX2 spin system features a 1JPP-coupling constant of 456 Hz. These values are fully consistent with those of literature known triphosphenium ions reported by Schmidpeter or Macdonald.16,26,83,85 In addition the signal of the two outer phosphorus atoms in 8c splits into a doublet with a 1JPH coupling constant of 468 Hz, which is typical for proton substituted phosphonium ions, as is the δ(1H) = 7.07 ppm. The identity and purity of isolated 8c was further corroborated by 1H-, 11B-, 13C, 19F-NMR, mass spectrometry and elemental analysis.
 | ||
| Scheme 4 Synthesis of triphosphenium ion 8cvia protonation of triphosphane 9 with HBF4. | ||
In the solid state the meso arrangement in 8c was confirmed by single crystal X-ray diffraction (Fig. 2). The ferrocenophane based triphosphenium ion 8c crystallizes in an almost ecliptic conformation of the ferrocene unit (τ = 4.58(8)°) and plane-parallel arrangement of the Cp rings (α = 1.24(8)°). For the central phosphorus atom P2 of the P3-bridge, symmetric contacts to the adjacent phosphonium centers P1 (2.1284(7) Å (P1–P2)) and P3 (2.1265(7) Å (P2–P3)) are found. The P–P bond lengths in 8c are somewhat shorter compared with other phosphorus-rich ferrocenophanes,54,64,65 indicative for delocalization of the negative charge across the ansa-bridge comparable to other cyclic triphosphenium ions.24,26,278c was investigated computationally to get further insight into its bonding situation. As expected, the HOMO is localized mainly (72%) at the central phosphorus and polarized towards the two outer phosphorus atoms, similar to other triphosphenium systems (Fig. S7 in the ESI†). In agreement with the shorter P–P bond lengths second order perturbation analysis on NBO basis revealed backdonation (∼60 kcal mol−1) from the central phosphorus atom (Table S7 in the ESI†).
Conclusion
To conclude, we demonstrated P+ transfer from and to a transient phosphenium ion. The dication 42+ resulting from P+ addition to this phosphenium ion is stable and its salt 4[AlCl4]2 is storable in the solid state for prolonged time. The latter serves as P+ source itself and transforms various bisphosphanes to the corresponding triphosphenium ions, which was demonstrated for known as well as for unprecedented examples. In this context the first triphosphenium ion bearing hydrogen substituents at both phosphonium centers was obtained and a new synthetic pathway was demonstrated via simple protonation of a secondary triphosphane. In analogy to the existing class of triphosphenium ions we suggest the name tetraphosphenium ion for species 42+. The latter features the longest structurally characterized P–P bond involving tricoordinate phosphorus atoms reported so far. Moreover, its FIA value suggests Lewis superacidic character. Based on these new insights we aim at transferring P+ ions to other cationic or electron deficient molecules and element fragments in future investigations. In terms of bonding situation, the here presented tetraphosphenium dication 42+ is featuring a non-polar central P–P bond, whereas its reactivity (Schemes 1 and 3) involves heterolytic cleavage of the same bond with an alternative description assuming a polar nature of the electron configurations (sextet and octet) at the central phosphorus atoms in 42+ (cf.C, Fig. 4). It is well established, that closed-shell divalent phosphorus compounds (i.e. PR2+ and PR2−) may be regarded as a continuum of species, where depending on the electronic properties of the substituents R the reactivity ranges from nucleophilic to electrophilic.86,87 In this picture, the substituents R would be the phosphonio groups of 42+, concurrently allowing configurations with opposite polarity at the adjacent phosphorus atoms.Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Crystallographic data for 2, 4[AlCl4]2 and 8c[BF4] have been deposited at the CCDC under 2328571–2328573 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 328961117 – SFB 1319 ELCH. ZK is grateful for the general support of János Bolyai Research Scholarship and OTKA (FK-145841) provided by the Hungarian National Research Development and Innovation Office.References
- D. Heift, Z. Benkő, H. Grützmacher, A. R. Jupp and J. M. Goicoechea, Chem. Sci., 2015, 6, 4017–4024 RSC.
- T. P. Robinson, M. J. Cowley, D. Scheschkewitz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 683–686 CrossRef CAS PubMed.
- L. N. Grant, B. Pinter, B. C. Manor, R. Suter, H. Grützmacher and D. J. Mindiola, Chem.–Eur. J., 2017, 23, 6272–6276 CrossRef CAS PubMed.
- M. Joost, W. J. Transue and C. C. Cummins, Chem. Commun., 2017, 53, 10731–10733 RSC.
- G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431–436 CrossRef CAS PubMed.
- A. Hinz and J. M. Goicoechea, Dalton Trans., 2018, 47, 8879–8883 RSC.
- A. Hinz and J. M. Goicoechea, Chem.–Eur. J., 2018, 24, 7358–7363 CrossRef CAS.
- J. Abbenseth, M. Diefenbach, A. Hinz, L. Alig, C. Würtele, J. M. Goicoechea, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 10966–10970 CrossRef CAS.
- Y. Wang, T. Szilvási, S. Yao and M. Driess, Nat. Chem., 2020, 801–807 CrossRef CAS.
- S. Bestgen, M. Mehta, T. C. Johnstone, P. W. Roesky and J. M. Goicoechea, Chem.–Eur. J., 2020, 26, 9024–9031 CrossRef CAS PubMed.
- A. Reinholdt, M. G. Jafari, C. Sandoval-Pauker, E. Ballestero-Martínez, M. R. Gau, M. Driess, B. Pinter and D. J. Mindiola, Angew. Chem., Int. Ed., 2021, 60, 17595–17600 CrossRef CAS PubMed.
- M. G. Jafari, Y. Park, B. Pudasaini, T. Kurogi, P. J. Carroll, D. M. Kaphan, J. Kropf, M. Delferro, M.-H. Baik and D. J. Mindiola, Angew. Chem., Int. Ed., 2021, 60, 24411–24417 CrossRef CAS.
- B. L. Frenette, J. Trach, M. J. Ferguson and E. Rivard, Angew. Chem., Int. Ed., 2023, 62, e202218587 CrossRef CAS PubMed.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- J. H. W. LaFortune, A. Swidan, J. S. Ovens, Z.-W. Qu, S. Grimme, M. A. Land and C. L. B. Macdonald, Chem.–Eur. J., 2023, 29, e202302558 CrossRef CAS PubMed.
- E. M. Dionisi, J. F. Binder, J. H. W. LaFortune and C. L. B. Macdonald, Dalton Trans., 2020, 49, 12115–12127 RSC.
- J. F. Binder, A. Swidan and C. L. B. Macdonald, Inorg. Chem., 2018, 57, 11717–11725 CrossRef CAS.
- S. C. Kosnik, M. C. Nascimento, J. F. Binder and C. L. B. Macdonald, Dalton Trans., 2017, 46, 17080–17092 RSC.
- C. L. B. Macdonald, J. F. Binder, A. Swidan, J. H. Nguyen, S. C. Kosnik and B. D. Ellis, Inorg. Chem., 2016, 55, 7152–7166 CrossRef CAS PubMed.
- S. C. Kosnik and C. L. B. Macdonald, Dalton Trans., 2016, 45, 6251–6258 RSC.
- F. O. Elnajjar, J. F. Binder, S. C. Kosnik and C. L. B. Macdonald, Z. Anorg. Allg. Chem., 2016, 642, 1251–1258 CrossRef CAS.
- J. F. Binder, A. Swidan, M. Tang, J. H. Nguyen and C. L. B. Macdonald, Chem. Commun., 2015, 51, 7741–7744 RSC.
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, Chem. Commun., 2005, 15, 1965–1967 RSC.
- J. W. Dube, C. L. B. Macdonald and P. J. Ragogna, Angew. Chem., Int. Ed., 2012, 51, 13026–13030 CrossRef CAS PubMed.
- A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., Int. Ed., 1982, 21, 63–64 CrossRef.
- A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., Int. Ed., 1985, 24, 226–227 CrossRef.
- E. L. Norton, K. L. S. Szekely, J. W. Dube, P. G. Bomben and C. L. B. Macdonald, Inorg. Chem., 2008, 47, 1196–1203 CrossRef CAS PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS.
- M. He, C. Hu, R. Wei, X.-F. Wang and L. L. Liu, Chem. Soc. Rev., 2024, 53, 3896–3951 RSC.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS.
- S. Fleming, M. K. Lupton and K. Jekot, Inorg. Chem., 1972, 11, 2534–2540 CrossRef CAS.
- B. E. Maryanoff and R. O. Hutchins, J. Org. Chem., 1972, 37, 3475–3480 CrossRef CAS.
- D. Gudat, in Comprehensive Inorganic Chemistry II, eds. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 587–621 Search PubMed.
- A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367–382 CrossRef CAS.
- N. Burford, B. W. Royan, A. Linden and T. S. Cameron, J. Chem. Soc., Chem. Commun., 1988, 842–844 RSC.
- N. Burford, T. S. Cameron, P. J. Ragogna, E. Ocando-Mavarez, M. Gee, R. McDonald and R. E. Wasylishen, J. Am. Chem. Soc., 2001, 123, 7947–7948 CrossRef CAS PubMed.
- N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS PubMed.
- R. Pietschnig, J. Organomet. Chem., 2007, 692, 3363–3369 CrossRef CAS.
- Y.-y. Carpenter, C. A. Dyker, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2008, 130, 15732–15741 CrossRef CAS PubMed.
- S. Weller, S. H. Schlindwein, C. M. Feil, Z. Kelemen, D. Buzsáki, L. Nyulászi, S. Isenberg, R. Pietschnig, M. Nieger and D. Gudat, Organometallics, 2019, 38, 4717–4725 CrossRef CAS.
- J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808–1815 RSC.
- T. Zhang, V. Y. Lee, S. Morisako, S. Aoyagi and T. Sasamori, Eur. J. Inorg. Chem., 2021, 2021, 3988–3991 CrossRef CAS.
- A. L. Brazeau, C. A. Caputo, C. D. Martin, N. D. Jones and P. J. Ragogna, Dalton Trans., 2010, 39, 11069–11073 RSC.
- B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed.
- C. M. Feil, T. D. Hettich, K. Beyer, C. Sondermann, S. H. Schlindwein, M. Nieger and D. Gudat, Inorg. Chem., 2019, 58, 6517–6528 CrossRef CAS PubMed.
- M. Papendick and D. Gudat, Chem.–Eur. J., 2023, 29, e202302525 CrossRef CAS PubMed.
- A. Schmidpeter and M. Thiele, Angew. Chem., Int. Ed., 1991, 30, 308–310 CrossRef.
- D. Gudat, M. Schrott and M. Nieger, Chem. Commun., 1995, 1541–1542 RSC.
- C. C. Chong, B. Rao, R. Ganguly, Y. Li and R. Kinjo, Inorg. Chem., 2017, 56, 8608–8614 CrossRef CAS PubMed.
- M. Olaru, A. Mischin, L. A. Malaspina, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2020, 59, 1581–1584 CrossRef CAS.
- S. G. Baxter, R. L. Collins, A. H. Cowley and S. F. Sena, J. Am. Chem. Soc., 1981, 103, 714–715 CrossRef CAS.
- A. Kraft, J. Beck and I. Krossing, Chem.–Eur. J., 2011, 17, 12975–12980 CrossRef CAS PubMed.
- M. Olaru, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2021, 60, 19133–19138 CrossRef CAS PubMed.
- R. Franz, B. Szathmári, C. Bruhn, Z. Kelemen and R. Pietschnig, Inorg. Chem., 2023, 62, 4341–4350 CrossRef CAS PubMed.
- C. Riesinger, A. Erhard and M. Scheer, Chem. Commun., 2023, 59, 10117–10120 RSC.
- S. Reichl, E. Mädl, F. Riedlberger, M. Piesch, G. Balázs, M. Seidl and M. Scheer, Nat. Commun., 2021, 12, 5774 CrossRef CAS.
- J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295–298 CrossRef CAS PubMed.
- D. Gudat, Coord. Chem. Rev., 1997, 163, 71–106 CrossRef CAS.
- M. Weber, G. Balázs, A. V. Virovets, E. Peresypkina and M. Scheer, Molecules, 2021, 26, 3920 CrossRef CAS PubMed.
- G. Balázs, A. Seitz and M. Scheer, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 1105–1132 Search PubMed.
- A. H. Cowley, R. A. Kemp, J. G. Lasch, N. C. Norman and C. A. Stewart, J. Am. Chem. Soc., 1983, 105, 7444–7445 CrossRef CAS.
- K. Izod, P. Evans and P. G. Waddell, Angew Chem., Int. Ed., 2019, 58, 11007–11012 CrossRef CAS.
- R. Franz, D. Gál, C. Bruhn, Z. Kelemen and R. Pietschnig, Adv. Sci., 2024, 11, 2306805 CrossRef CAS.
- S. Borucki, Z. Kelemen, M. Maurer, C. Bruhn, L. Nyulaszi and R. Pietschnig, Chem.–Eur. J., 2017, 23, 10438–10450 CrossRef CAS PubMed.
- S. Isenberg, S. Weller, D. Kargin, S. Valić, B. Schwederski, Z. Kelemen, C. Bruhn, K. Krekić, M. Maurer, C. M. Feil, M. Nieger, D. Gudat, L. Nyulászi and R. Pietschnig, ChemistryOpen, 2019, 8, 1235–1243 CrossRef CAS.
- C. Moser, A. Orthaber, M. Nieger, F. Belaj and R. Pietschnig, Dalton Trans., 2006, 3879–3885 RSC.
- R. T. Boeré and Y. Zhang, J. Organomet. Chem., 2005, 690, 2651–2657 CrossRef.
- D. Buzsáki, L. Nyulászi, R. Pietschnig, D. Gudat and Z. Kelemen, Organometallics, 2022, 41, 2551–2561 CrossRef.
- S. Nasemann, R. Franz, D. Kargin, C. Bruhn, Z. Kelemen, T. Gutmann and R. Pietschnig, Chem.–Asian J., 2024, 19, e202300950 CrossRef CAS.
- D. Kargin, Z. Kelemen, K. Krekić, L. Nyulászi and R. Pietschnig, Chem.–Eur. J., 2018, 24, 16774–16778 CrossRef CAS.
- A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2005, 44, 7729–7733 CrossRef CAS PubMed.
- R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2016, 55, 14798–14801 CrossRef CAS.
- J. Bresien, K. Faust, C. Hering-Junghans, J. Rothe, A. Schulz and A. Villinger, Dalton Trans., 2016, 45, 1998–2007 RSC.
- A. Wiesner, S. Steinhauer, H. Beckers, C. Müller and S. Riedel, Chem. Sci., 2018, 9, 7169–7173 RSC.
- K. F. Hoffmann, A. Wiesner, N. Subat, S. Steinhauer and S. Riedel, Z. Anorg. Allg. Chem., 2018, 644, 1344–1348 CrossRef CAS.
- T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139–8143 CrossRef.
- I. Krossing and L. van Wüllen, Chem.–Eur. J., 2002, 8, 700–711 CrossRef CAS.
- R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- R. Pino-Rios, O. Yañez, D. Inostroza, L. Ruiz, C. Cardenas, P. Fuentealba and W. Tiznado, J. Comput. Chem., 2017, 38, 481–488 CrossRef CAS PubMed.
- P. Erdmann, M. Schmitt, L. M. Sigmund, F. Krämer, F. Breher and L. Greb, Angew. Chem., Int. Ed., 2024, 63, e202403356 CrossRef CAS PubMed.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS PubMed.
- S. Lochschmidt and A. Schmidpeter, Z. Naturforsch. B, 1985, 40, 765–773 CrossRef.
- J. D. Burton, R. M. K. Deng, K. B. Dillon, P. K. Monks and R. J. Olivey, Heteroat. Chem., 2005, 16, 447–452 CrossRef CAS.
- S. Kosnik, J. F. Binder, M. C. Nascimento and C. L. B. Macdonald, J. Visualized Exp., 2016, e55021 Search PubMed.
- D. Gudat, Coord. Chem. Rev., 1997, 163, 71–106 CrossRef CAS.
- L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606–626 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis and characterization of the compounds, experimental procedures, SCXRD details, molecular structure of 2 in the solid state, computational details, NMR spectra. CCDC 2328571–2328573. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06823h |
| This journal is © The Royal Society of Chemistry 2025 |