
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical upcycling of polybutadiene into size controlled α,ω-dienes and diesters via sequential hydrogenation and cross-metathesis†
Christophe
Vos
 ,
Igor
Beckers
,
Galahad
O'Rourke
and
Dirk
De Vos
*
,
Igor
Beckers
,
Galahad
O'Rourke
and
Dirk
De Vos
*
Centre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Celestijnenlaan 200F, Post Box 2454, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
First published on 23rd January 2025
Abstract
Plastic waste conversion into valuable chemicals is a promising alternative to landfill or incineration. In particular, the chemical upcycling of polybutadiene rubber (PBR) could provide a renewable route towards highly desirable α,ω-dienes with varying chain lengths, which can find ample industrial application. While previous research has shown that the treatment of polybutadiene with a consecutive hydrogenation and ethenolysis reaction can afford long-chain α,ω-dienes, achieving precise control over the product chain length remains an important bottleneck. In this work, it was discovered that undesired isomerization during the initial hydrogenation step compromises the product selectivity after ethenolysis, leading to a distribution of α,ω-dienes that covers the full range of chain lengths from C6 to C22. Based on this insight, we show that the suppression of isomerization affords a well-defined product distribution predominantly consisting of C4n+2-dienes (with n = 1–5). With tight control over both the hydrogenation degree and isomerization in the studied PBD samples, we demonstrate that rational modifications to the reaction conditions can steer the selectivity towards the desired chain lengths of the α,ω-diene products. In addition, these insights were expanded to cross-metathesis (CM), giving access to a diverse range of high-value bifunctional products.
1 Introduction
Since plastic is an indispensable material in our modern society, its production has drastically increased over the past decades to a scale of 380 million tons per year.1 However, only a small fraction of these materials is effectively being recycled, which leads to an ever growing waste pile. Environmental concerns emerge from traditional landfilling of plastics that show virtually no degradation and eventually lead to microplastic pollution in terrestrial and aquatic environments. While greenhouse gas emissions are a problem accompanying incineration of plastics, more sustainable routes such as mechanical recycling generally yield lower value products (i.e. downcycling). A promising alternative is the development of chemical recycling routes that can ideally convert waste polymers into value-added products (i.e. upcycling).2 This large asset of chemical recycling technology could drive future efforts in the treatment of plastic waste and could provide a more sustainable solution to the current plastic waste problem.Yearly, 2.8 million metric tons of PBR are produced.3 A major fraction is used in the tire industry, whereas the remaining part is largely used in the production of high-impact polystyrene (HIPS) and acrylonitrile-butadiene-styrene (ABS) polymers. Other applications include the manufacture of rubber goods, conveyer belts and the cores of golf balls.3,4 While end-of-life rubber materials in the US are still primarily handled by landfill and combustion for energy recovery, recycling rates are only gradually increasing due to the complexity of the material.5,6 In terms of chemical recycling of plastics, it was recently found that polyene materials can be obtained by bromination–dehydrobromination of polyethylene (PE).7 An interesting technique to chemically break down these polyenes is via cross-metathesis (CM). Because of the similarity of the chemical structure of PBR with a polyene-type polymer, double bonds of rubbers may provide an opportunity for well controlled chemical recycling. By cross-metathesis of polyenes with shorter alkenes like ethylene (i.e. ethenolysis) or other cross-metathesis partners such as methyl acrylate (MA), the double bonds in the polyene can be cleaved.8–10
Polybutadiene (PBD) is a synthetic rubber, of which the chemical structure contains one double bond for every four bonds. Therefore, upon ethenolysis of several types of PBD, the formed products predominantly consist of 1,5-hexadiene, vinylcyclohexene and allylcyclopentene.9 α,ω-Dienes such as 1,5-hexadiene can be used as monomers in copolymerization reactions11–13 or can be employed in the production of polyalfaolefins (PAO's) to produce lubricants.14 Upon reaction with functional thiols, α,ω-dienes can be transformed into telechelic polyethylenes.15 α,ω-Dienes also serve as the source for numerous bifunctional compounds that can eventually end up in fragrances, agrochemicals, and pharmaceuticals.16 Examples of these bifunctional compounds are di-esters which are important intermediates for the production of polyesters and polyamides.17 After reaction of the ester with LiAlH4, also α,ω-diols can be obtained18 which are used in the production of polyurethane.
1,5-Hexadiene, 1,9-decadiene and 1,13-tetradecadiene can be produced via ethenolysis of cyclic alkenes derived from oil derived butadiene.16 The SHOP process only produces α-olefins with even carbon numbers; odd ones are consequently much harder to synthesize and are more expensive.19 Using (waste) PBD as an alternative feedstock towards these α,ω-difunctionalized molecules of different lengths and functionalities would be a more green and circular approach.
Hydrogenation-metathesis reaction sequences have been explored in the context of plastic recycling as a route to non-terminal alkenes.20–23 In these works, metathesis using 4-octene as metathesis partner was performed on homogeneously and heterogeneously hydrogenated PBD with WCl6/Sn(Me)4 or WCl6/EtAlCl2 as catalyst system. To the best of our knowledge, the hydrogenation-ethenolysis reaction sequence for the upcycling of PBD has only been explored once by Shiono et al.;24 however no detailed information was obtained about the product distribution.
In this work, we explore a consecutive hydrogenation-metathesis route to upcycle high-molecular weight (HMW) PBD into α,ω-dienes, of which the length can be controlled. Isomerization decreases the yield of α,ω-functionalized molecules with well-defined chain lengths and drastically broadens the product distribution. Therefore, we herein seek for ways to control and minimize the isomerization to provide substantially higher yields of C4n+2-dienes (n = 1–5). Additionally, strict control over the degree of hydrogenation of PBD is used to guide the product distribution towards shorter or longer α,ω-dienes after the subsequent metathesis. Both hydrogenation and metathesis are performed with homogeneous Ru-catalysts, with a readily available Ru(PPh3)3Cl2 complex for the initial partial hydrogenation of HMW PBD. Beside α,ω-diene formation after cross-metathesis with ethylene using second generation Grubbs catalysts, the product scope is expanded by cross-metathesis with dimethyl maleate (DMM) to form diesters with controlled chain lengths from the hydrogenated PBD (Fig. 1).
 | ||
| Fig. 1 Concept of consecutive hydrogenation-metathesis of polybutadiene (PBD) rubber. Tuning the hydrogenation degree for the intermediate hydrogenated PBD polymer and controlling isomerization by the Ru-catalysts in both reaction steps allows excellent control over the upcycled α,ω-dienes after ethenolysis. | ||
2 Results and discussion
2.1 Enhancing ethenolysis activity to avoid isomerization
In view of performing controlled ethenolysis and cross-metathesis (CM) with hydrogenated PBD (hPBD), optimal ethenolysis conditions were identified by studying the conversion of trans-5-decene as a model compound. Since isomerization is a known side reaction during ethenolysis,25,26 optimizing the selectivity of the reaction to the 1-hexene product over products formed by isomerization (C10 or C6 isomers) and ensuing ethenolysis and isomerization reactions towards C3–C5 alkenes (via C7–C9 intermediates) is desirable (Fig. 2, scheme). | ||
| Fig. 2 Ethenolysis (E) reaction of trans-5-decene by Grubbs M202 or M220 catalyst with possible routes of isomerization (I) side reactions (scheme). Conversion of trans-5-decene without BQ (A) and with BQ (B). Mol% 1-hexene vs. total products after 1 h (C) and 2 h (D). Conditions: 100 μmol trans-5-decene (1 eq.), 1 mL cyclopentyl methyl ether (CPME) as solvent, 1 mol% Grubbs Ru-catalyst, 0.185 eq. BQ, 8 bar C2H4. | ||
Initially, the conversion of trans-5-decene by two second generation Grubbs catalysts (M202 and M220) was monitored at various temperatures without additives (Fig. 2A). Temperatures of 60 °C and 80 °C were chosen when using the M202 catalyst, while the M220 catalyst, with a phosphite ligand instead of a phosphine ligand, required higher temperatures of 80–100 °C (conversion was only 25% after 4 h reaction at 60 °C). A previous report suggests that M202 has a very high activity during the early phase of the reaction at 70 °C.27 The same behaviour was seen here for both M202 and M220. Although increasing the temperature from 60 °C to 80 °C for M202 and from 80 °C to 100 °C for M220 had a positive impact on conversion for both catalysts, the observed selectivity for 1-hexene decreased (Fig. 2C). Isomerization by 2nd generation Grubbs catalysts is known to occur at temperatures as low as 50 °C and could be caused by the activity of formed ruthenium hydride species. Earlier research pointed out that addition of 1,4-benzoquinone (BQ) suppresses isomerization via reduction of BQ to hydroquinone, which consumes any hydrides that would be formed on the Ru.25,26 Therefore, the ethenolysis reactions using both catalysts were repeated with 0.185 eq. of BQ vs. trans-5-decene as an additive. While the conversion of 1-hexene remained similar during the first 2 h of reaction (Fig. 2B), the addition of BQ completely suppressed isomerization during the course of the reaction (Fig. 2C) and (Fig. 2D) and led to 100 mol% 1-hexene. When using either the M202 or M220 catalyst, the mol% of obtained 1-hexene was lower at higher temperature without BQ. This indicates that isomerization is more likely to occur at higher temperatures, but this undesirable side reaction can be completely suppressed by applying BQ as an auxiliary. These insights are important in the ethenolysis of actual hPBD, since avoiding isomerization can lead to well-defined splitting sites on the polymer backbone.
2.2 Influence of partial hydrogenation of PBD on formed α,ω-dienes after ethenolysis
With the objective to convert PBD into α,ω-dienes with well-controlled chain lengths via sequential hydrogenation-ethenolysis, we investigated the effect of hydrogen pressure on the partial hydrogenation of PBD (see ESI Table S3 and Fig. S3† for characterization). Two hydrogenation conditions were compared. Firstly, the PBD was partially hydrogenated under 8 bar H2 pressure at 80 °C with a Ru loading of 0.25 mol% (low H2-pressure, high temperature); secondly, a combination of higher H2-pressure (30 bar) at lower temperature (40 °C) with a Ru loading of 0.125 mol% was investigated. Both sets of conditions were applied to hydrogenate PBD to yield products with a varying amount of residual double bonds as determined by 1H NMR analysis of the partially hydrogenated PBD (hPBD) after precipitation from the product mixture. Table 1 gives the residual fraction of unsaturated double bonds per 100 carbon atoms (%C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in the polymer product and the corresponding mmol H2 required in this partial hydrogenation.
C bonds) at 80 °C under 8 bar H2 using 0.25 mol% Ru(PPh3)3Cl2vs. CC. Conditions high P, low T: hydrogenation of 8 mmol 25% PBD (which is equal to a total of 2 mmol CC bonds) at 40 °C under 30 bar H2 using 0.125 mol% Ru(PPh3)3Cl2vs. CC. Ethenolysis of 100 μmol CC (1 eq.) at 100 °C under 8 bar C2H4 using 1 mol% Ru M220 vs. CC and 0.185 eq. BQ for 1 h
C) in the polymer product and the corresponding mmol H2 required in this partial hydrogenation.
C bonds) at 80 °C under 8 bar H2 using 0.25 mol% Ru(PPh3)3Cl2vs. CC. Conditions high P, low T: hydrogenation of 8 mmol 25% PBD (which is equal to a total of 2 mmol CC bonds) at 40 °C under 30 bar H2 using 0.125 mol% Ru(PPh3)3Cl2vs. CC. Ethenolysis of 100 μmol CC (1 eq.) at 100 °C under 8 bar C2H4 using 1 mol% Ru M220 vs. CC and 0.185 eq. BQ for 1 h
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditions hydrogenation | %CC in hPBDa (%) calculated by 1H NMR |
Corresponding H2 (mmol) required | Conversion CC after ethenolysis hPBDa (%) calculated by 1H NMR |
Vinyl groups after ethenolysis hPBDa (%) calculated by 1H NMR | Mass fraction non-detected products in GCb (%) |
|
a As determined by 1H NMR. % Values denote number of CC or vinyl groups per 100 carbon atoms. See ESI for calculation details.
b Determined via GC mass balance with 100 μmol n-nonane internal standard.
|
||||||
| 0 | — | 25.0 | 0.00 | 80 | 28 | 10 |
| 1 | Low P, high T | 20.8 | 0.34 | 92 | 26 | 18 |
| 2 | Low P, high T | 17.1 | 0.63 | 92 | 25 | 16 |
| 3 | Low P, high T | 14.9 | 0.81 | 91 | 19 | 40 |
| 4 | Low P, high T | 13.1 | 0.95 | 90 | 18 | 39 |
| 5 | Low P, high T | 9.8 | 1.22 | 88 | 13 | 55 |
| 6 | Low P, high T | 7.0 | 1.44 | 88 | 10 | 78 |
| 7 | High P, low T | 22.8 | 0.18 | 85 | 26 | 0 |
| 8 | High P, low T | 19.1 | 0.47 | 90 | 24 | 13 |
| 9 | High P, low T | 16.5 | 0.68 | 90 | 21 | 22 |
| 10 | High P, low T | 14.9 | 0.81 | 86 | 20 | 22 |
| 11 | High P, low T | 12.8 | 0.98 | 89 | 17 | 26 |
| 12 | High P, low T | 10.8 | 1.14 | 77 | 13 | 45 |
| 13 | High P, low T | 9.3 | 1.26 | 84 | 12 | 58 |

Subsequently, the hPBD samples were subjected to the optimal, non-isomerizing ethenolysis conditions (100 °C, 1 mol% M220, 0.185 eq. BQ vs. CC) as determined for trans-5-decene. Table 1 shows that the conversion of internal double bonds in the ethenolysis is very high, up to 92%, which is comparable to the 95% conversion of trans-5-decene. The table also shows the number of vinyl groups per 100C atoms, after incorporation of extra CH2 groups from ethylene, as determined based on integration of the methine protons vs. the total signal in the 1H NMR spectra (see ESI Fig. S4, S5, Tables S4 and S5† for calculation). The number of vinyl groups decreases monotonously as the hydrogenation of PBD proceeds in both of the investigated hydrogenation conditions. Clearly, more deeply hydrogenated PBD contains fewer internal double bonds that can be converted to vinyl groups. While the NMR analysis detects all dissolved products containing H-atoms, Table 1 also shows the estimated mass fraction of products that are too high-boiling to be detectable by GC. In the employed GC-analysis, products with maximally 22 carbon atoms can be detected. This fraction is higher in ethenolysis reactions that were performed on hPBD with higher degrees of hydrogenation, which is caused by the fact that fewer double bonds lead to less ethenolysis splitting sites. Hence, high-boiling fractions of α,ω-dienes with longer chains (>22 carbons) result from this. Similar conversions of the CC bonds were obtained for hPBD obtained either under low or high H2-pressure in the hydrogenation; however, the observed product selectivity after metathesis was different. Additional tests were performed to further evaluate the utility of the method for PBD upcycling. The reactions were scaled up to a multigram scale using CPME as the solvent for the hydrogenation step (see ESI Fig. S8 and S9†). It was shown that both reactions are also feasible in one pot, using chloroform as the reaction solvent (see ESI Fig. S10 and S11†). In addition, the sequential partial hydrogenation and ethenolysis reactions were also successfully demonstrated on the PBD fraction originating from “high impact polystyrene” as a real-life polymer material (see ESI Fig. S12†).
The α,ω-diene product distribution that was obtained after ethenolysis of partially hydrogenated PBD under low H2 pressure is illustrated in Fig. 3A. As the hydrogenation degree in the first step increases, the fraction of C6 dienes that is obtained after ethenolysis decreases in favour of the heavier dienes such as C10, C14 and C18. A second observation is that the share of trienes and tetraenes similarly decreases as the hydrogenation degree increases. When PBD is subjected to ethenolysis without any hydrogenation, only decatriene is formed within the C10 fraction, but as the hydrogenation degree increases, more and more decadiene is formed. This also holds true for the C14 fraction. Especially when the polymer is subjected to a deeper hydrogenation, products with other carbon numbers than (4n + 2) are formed. Due to the significant isomerization under reaction conditions with low H2 pressure, the product spectrum is broad and contains all lengths of α,ω-dienes. On the one hand, this decreases the yields of the useful C10, C14, C18 but on the other hand also produces the expensive and hard-to-synthesize α,ω-dienes with uneven C-numbers. Since the isomerization during the ethenolysis is suppressed by addition of BQ, the observed isomerization is hypothesized to be happening during the hydrogenation reaction. However, when producing hPBD under high H2-pressure and at lower T, the product distribution (Fig. 3B) shows ostensibly less isomerization products. While under low hydrogenation pressure, up to 60 mol% isomerization products can be obtained, the observed isomerization side reactions yielded a limited amount of maximally 15 mol% when applying high pressure. These observations indicate that an isomerization side reaction is competing with the hydrogenation reaction at low pressure and high temperature.
 | ||
| Fig. 3 Consecutive conversion of PBD to α,ω-dienes (scheme) with the distribution of α,ω-dienes formed in the ethenolysis, starting from hPBD produced (A) under low H2-pressure and at high temperature (8 bar, 80 °C); (B) under high H2-pressure and at low temperature (30 bar, 40 °C). | ||
In Fig. 4 the distribution of hPBD ethenolysis products over the different carbon numbers is plotted vs. the %CC in the hPBD before ethenolysis. The product share is displayed in Fig. 4A for the isomerization products, and in Fig. 4B–D for the C6, C10 and C14 fractions, respectively. The effect of high vs. low pressure hydrogenation becomes more apparent at higher hydrogenation degrees (hPBD with lower %CC). Fig. 4A shows that ethenolysis of hPBD with about 9% CC produces about 3× more isomerization products (see ESI Fig. S7† for product distribution of isomerization products) when hydrogenated at low pressure as opposed to high pressure (60% vs. 20%). Variations within the C6, C10 and C14 fractions are also most pronounced at high hydrogenation degrees. 80 mol% of C6 can be formed without hydrogenation, while in case of C10 and C14, partial hydrogenation can increase the share in the product mixture from 15 mol% to 30 mol% for C10 and from 2.5 mol% to about 12.5 mol% for C14.
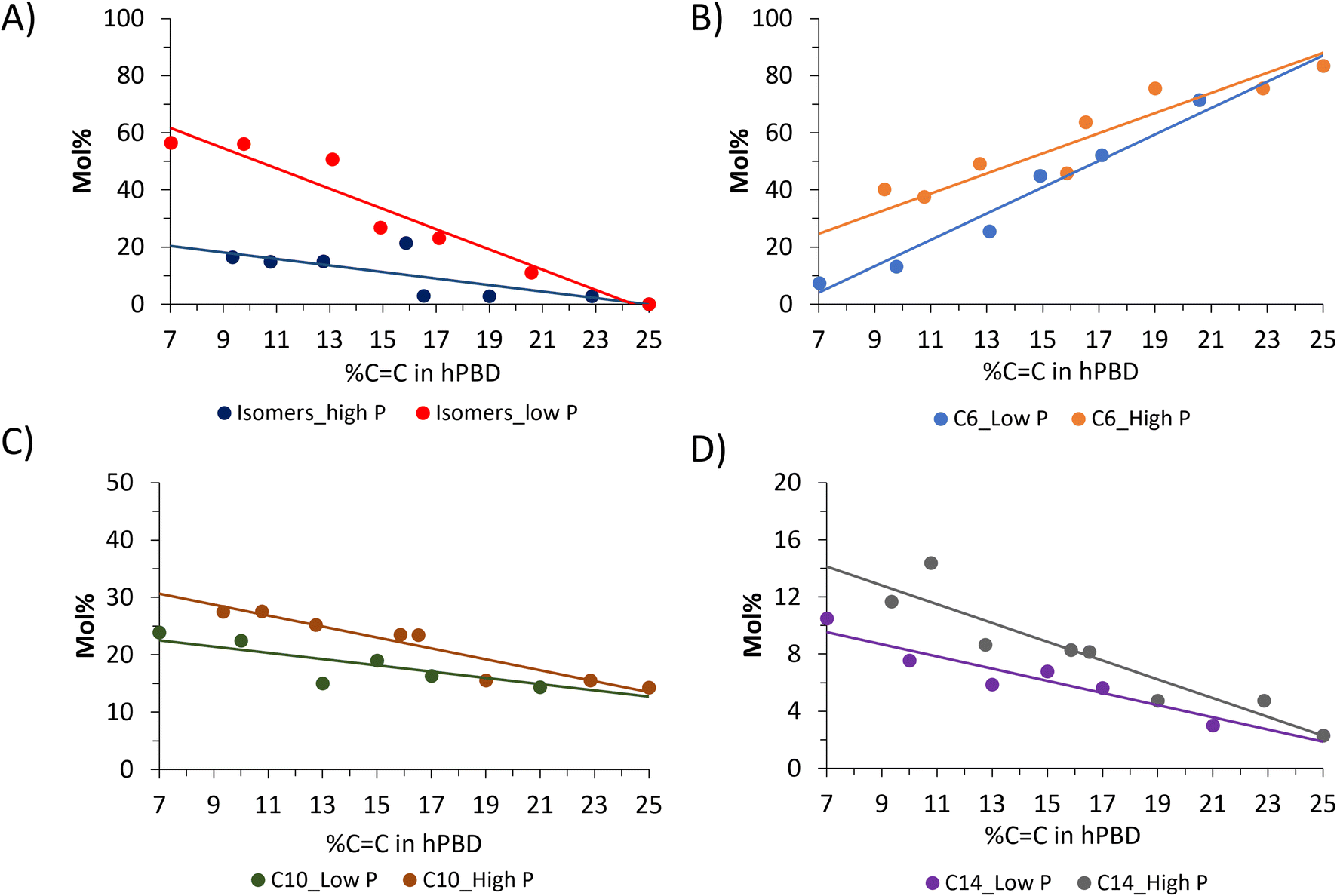 | ||
| Fig. 4 Product distribution over various carbon numbers after ethenolysis of hPBD under low P hydrogenation and high P hydrogenation. Fraction of isomerized products (A), fraction C6 (B), fraction C10 (C), fraction C14 (D). | ||
A plausible reaction mechanism28 for hydrogenation and simultaneous isomerization is displayed in Fig. 5A. Starting from a Ru-monohydride, coordination of the CC bond followed by migratory insertion of the olefin results in a Ru-alkyl species. The Ru-alkyl can undergo oxidative addition of H2 producing a Ru-dihydride from which a saturated fragment is reductively eliminated, regenerating the Ru-monohydride.28 However, if insufficient H2 is available, the Ru-alkyl intermediate can also undergo a β-H elimination that can form an olefinic product in which the double bond has migrated. Subsequent ethenolysis then yields the products with carbon numbers different from 4n + 2. By increasing the hydrogen pressure, it can be expected that the hydrogen activation step is favoured over the β-H elimination, thereby avoiding isomerization. This is confirmed by the observations that less isomerization occurs when applying high H2-pressures, which has a significant effect on the α,ω-diene product distribution after the ensuing ethenolysis reaction. While hPBD material with high or low %CC can be obtained by monitoring the H2 pressure decrease in the hydrogenation step, the H2 pressure determines whether isomerization occurs (Fig. 5B). Even while limiting isomerization during the ethenolysis when using BQ as an auxiliary, an entire range of Cn-containing product fractions is obtained at low H2 pressure. Primarily C4n+2-carbon fractions, thus fractions with an even number of carbons, are formed when using high H2 pressure. Hence an optimal route to control isomerization in both reaction steps is found, which opens up a pathway to α,ω-dienes with tuneable chain length.
 | ||
| Fig. 5 Possible reaction mechanism of Ru(PPh3)3Cl2 for hydrogenation of PBD (A). Low H2-pressure leads to isomerization by a β-hydride elimination pathway. High H2-pressure leads to primarily the oxidative addition and reductive elimination route, thereby giving less isomerization. The hydrogen pressure consequently has an influence on α,ω-diene product outcome (B). | ||
2.3 Diester formation by cross-metathesis of hPBD with dimethyl maleate
After demonstrating that the hydrogenation-ethenolysis sequence with suppression of the isomerization leads to the selective formation of dienes with specific chain lengths of C4n+2 (n = 1,2,3, …), we sought to further extend this promising strategy towards cross-metathesis (CM). A hPBD sample (17 %CC, Table 1 entry 9) obtained by partial hydrogenation with 30 bar H2 pressure was subjected to cross-metathesis with dimethyl maleate (DMM) to produce unsaturated diesters of chain lengths C4n+6 (n = 1,2,3, …) (Fig. 6A). When comparing the produced chain lengths of products under ethenolysis conditions (C4n+2-fragments) with the chain lengths obtained with DMM for cross-metathesis (Fig. 6B), there are only slight differences in the distribution. Similar chain lengths of diesters can be produced by the hydrogenation-cross metathesis route of PBD rubber as in ethenolysis.
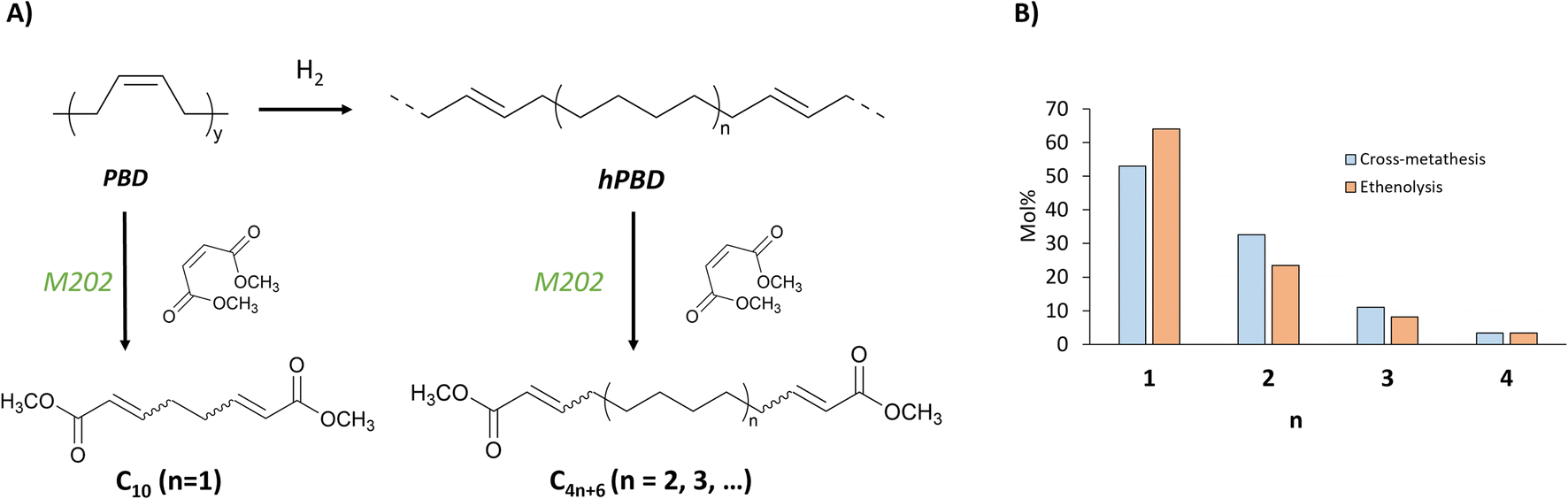 | ||
| Fig. 6 Conversion of PBD and hPBD by cross-metathesis with dimethyl maleate to diesters comprising C4n+6 carbon fractions (A). Chain length distribution with n ranging from 1–4 of formed dienes (C4n+2) and diesters (C4n+6), by ethenolysis and cross-metathesis of a hPBD with 17% CC by high P, low T hydrogenation (entry 9, Table 1) (B). | ||
Finally, the 1H NMR spectra of hPBD (Fig. 7, top) and the product after cross-metathesis (Fig. 7, bottom) demonstrate the incorporation of ester functional moieties in the product. The conversion of the internal double bonds towards diester bonds was calculated via1H NMR to be 86%, which is comparable to the 90% after ethenolysis of this sample. Both GC and NMR analyses were supported by reference reactions (see ESI Fig. S13 and S14†).
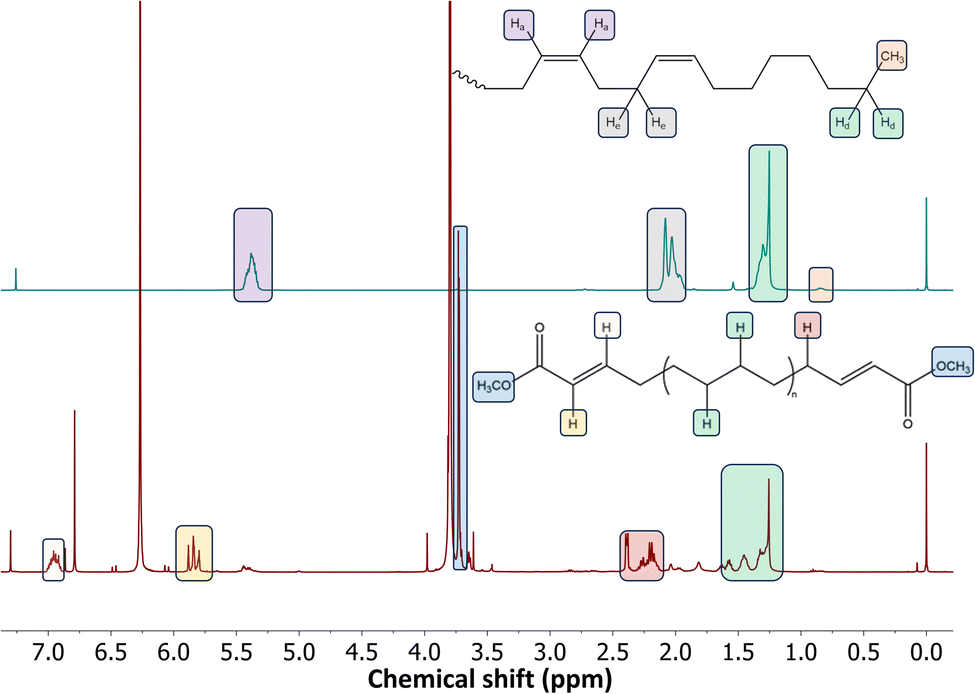 | ||
| Fig. 7
1H NMR spectra (400 MHz, CDCl3) of (top) high P, low T hPBD with 17% CC; and (bottom) cross-metathesis product after 4 h reaction of hPBD (100 μmol CC) with 500 μmol dimethyl maleate in 1 mL CDCl3 using 1 mol% Grubbs M202 at 80 °C. | ||
3 Conclusion
In this work, a hydrogenation protocol was developed that allows partial hydrogenation of high molecular weight polybutadiene (PBD). During hydrogenation under high H2 pressure (30 bar) and at low temperature (40 °C), isomerization can be limited, in contrast to hydrogenation under low pressure (8 bar) and at high temperature (80 °C). Ethenolysis with Grubbs M220 using BQ suppressed the isomerization during ethenolysis completely while still ensuring a high conversion of the internal double bonds into useful α,ω-dienes. By combining partial hydrogenation under high pressure and ethenolysis with BQ, the yield of the C10 and C14 fraction in the resulting product mix can be improved vs. ethenolysis of virgin PBD. Low pressure hydrogenation in combination with ethenolysis can on the other hand give access to expensive and hard to synthesize α,ω-dienes with an uneven number of C-atoms. Cross-metathesis with DMM on a partially hydrogenated PBD obtained by high pressure hydrogenation also selectively produced valuable unsaturated diesters of specific chain lengths.Data availability
Original data are available within KULeuven's OneDrive system. The ESI† contains additional characterization data and conditions of experiments.Author contributions
C. V. has conceived the research concept and performed the experiments. G. O. designed the figures for the manuscript. D. D. V. has supervised the project and contributed to the research concept. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work in this research was funded by the KU Leuven IBOF project IBOF/21/016 “Catalysts and Polymer Chemistry for Carbon Circularity: Innovative chemistry for upgrading plastic waste (C3)”. I. B. is grateful to FWO (grant 1291724N) and the KU Leuven Research Council Award for Science & Technology 2024 for funding. D. D. V. is grateful to FWO for project funding G0D3721N. The authors thank Dr Ir. Robin Coeck for his help with the operation of the Parr reactor.References
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Expanding Plastics Recycling Technologies: Chemical Aspects, Technology Status and Challenges, Green Chem., 2022, 24(23), 8899–9002, 10.1039/D2GC02588D.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Critical Advances and Future Opportunities in Upcycling Commodity Polymers, Nature, 2022, 603(7903), 803–814, DOI:10.1038/s41586-021-04350-0.
- L. Friebe, O. Nuyken and W. Obrecht, Neodymium-Based Ziegler/Natta Catalysts and Their Application in Diene Polymerization, in Neodymium Based Ziegler Catalysts – Fundamental Chemistry, Springer, Berlin Heidelberg, 2006, vol. 204, pp. 1–154, DOI:10.1007/12_094.
- D. F. Graves, Rubber, in Kent and Riegel's Handbook of Industrial Chemistry and Biotechnology, Springer US, Boston, MA, 2007, pp. 689–718, DOI:10.1007/978-0-387-27843-8_16.
- Rubber and Leather: Material-Specific Data, US EPA, https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/rubber-and-leather-material-specific-data, accessed 2024-05-31.
- Recycling Tires is Difficult, Interco, https://intercotradingco.com/recycling-tires-difficult/, accessed 2024-05-31.
- M. Zeng, Y. H. Lee, G. Strong, A. M. Lapointe, A. L. Kocen, Z. Qu, G. W. Coates, S. L. Scott and M. M. Abu-Omar, Chemical Upcycling of Polyethylene to Value-Added α,ω-Divinyl-Functionalized Oligomers, ACS Sustain. Chem. Eng., 2021, 9(41), 13926–13936, DOI:10.1021/acssuschemeng.1c05272.
- R. F. Smith, S. C. Boothroyd, R. L. Thompson and E. Khosravi, A Facile Route for Rubber Breakdown via Cross Metathesis Reactions, Green Chem., 2016, 18(11), 3448–3455, 10.1039/C5GC03075G.
- M. D. Watson and K. B. Wagener, Acyclic Diene Metathesis (ADMET) Depolymerization: Ethenolysis of 1,4-Polybutadiene Using a Ruthenium Complex, J. Polym. Sci., Part A: Polym. Chem., 1999, 37(12), 1857–1861, DOI:10.1002/(SICI)1099-0518(19990615)37:12<1857::AID-POLA15>3.0.CO;2-C.
- M. D. Schulz, R. R. Ford and K. B. Wagener, Insertion Metathesis Depolymerization, Polym. Chem., 2013, 4(13), 3656, 10.1039/c3py00531c.
- B. Palucci, G. Zanchin, G. Ricci, L. Vendier, C. Lorber and G. Leone, Vanadium-Catalyzed Terpolymerization of α,ω-Dienes with Ethylene and Cyclic Olefins: Ready Access to Polar-Functionalized Polyolefins, Macromolecules, 2021, 54(23), 10700–10711, DOI:10.1021/acs.macromol.1c02142.
- X. Shi, Y. Wang, J. Liu, D. Cui, Y. Men and Y. Li, Stereospecific Cyclopolymerization of α,ω-Diolefins by Pyridylamidohafnium Catalyst with the Highest Activity, Macromolecules, 2011, 44(4), 1062–1065, DOI:10.1021/ma1023268.
- W. Hou, D. Zhang, M. A. Camacho-Fernandez, Y. Zhang, G. Liu, Y. Tang, Z. Guan and Z. Huang, Double-Linear Insertion Mode of α,ω-Dienes Enabled by Thio-Imino-Quinoline Iron Catalyst, ACS Catal., 2020, 10(24), 15092–15103, DOI:10.1021/acscatal.0c04567.
- S. Ray, P. V. C. Rao and N. V. Choudary, Poly-α-olefin-based Synthetic Lubricants: A Short Review on Various Synthetic Routes, Lubr. Sci., 2012, 24(1), 23–44, DOI:10.1002/ls.166.
- S. Norsic, C. Thomas, F. D'Agosto and C. Boisson, Divinyl-End-Functionalized Polyethylenes: Ready Access to a Range of Telechelic Polyethylenes through Thiol–Ene Reactions, Angew. Chem., 2015, 127(15), 4714–4718, DOI:10.1002/ange.201411223.
- L. Delaude and A. F. Noels, Metathesis, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2005, DOI:10.1002/0471238961.metanoel.a01.
- A. Behr, S. Toepell and S. Harmuth, Cross-Metathesis of Methyl 10-Undecenoate with Dimethyl Maleate: An Efficient Protocol with Nearly Quantitative Yields, RSC Adv., 2014, 4(31), 16320, 10.1039/C3RA47671E.
- T. Witt, M. Häußler, S. Kulpa and S. Mecking, Chain Multiplication of Fatty Acids to Precise Telechelic Polyethylene, Angew. Chem., Int. Ed., 2017, 56(26), 7589–7594, DOI:10.1002/anie.201702796.
- M. A. Bruening, S. Xiong and T. Agapie, α,ω-Diene Generation from Ethylene and Butadiene by Copolymer Upcycling, ACS Sustain. Chem. Eng., 2023, 11(27), 9918–9923, DOI:10.1021/acssuschemeng.3c02569.
- W. Ast, C. Zott, H. Bosch and R. Kerber, Aspects Of Polymer Characterization By Olefine-Metathesis, Recl. Trav. Chim. Pays-Bas, 1977, 96(11), M81–M85 Search PubMed.
- W. Ast, C. Zott and R. Kerber, Bestimmung von Sequenzlängenverteilungen Hydrierter Polyalkenylene Durch Metathese-Abbau, 1 Homogen Hydriertes Polybutadien, Makromol. Chem., 1979, 315–323 CrossRef CAS.
- D. Wewerka and K. Hummel, Abbau von Teilweise Hydriertem Polybutadien Durch Olefin-Metathese, Colloid Polym. Sci., 1976, 254(1), 116–117, DOI:10.1007/BF01526749.
- K. Hummel and W. Ast, Metathese-Abbau von Polybutadien Mit 4-Octen, Die Makromolekulare Chem., 1972, 166, 39–44 CrossRef.
- T. Shiono, N. Naga and K. Soga, Synthesis of α,Ω-divinylpolyethylene-like Polymers from Cis-1,4-polybutadiene Using Partial Hydrogenation and Metathesis Degradation with Ethylene, Makromol. Chem. Rapid Commun., 1993, 14(6), 323–327, DOI:10.1002/marc.1993.030140601.
- S. H. Hong, D. P. Sanders, C. W. Lee and R. H. Grubbs, Prevention of Undesirable Isomerization during Olefin Metathesis, J. Am. Chem. Soc., 2005, 127(49), 17160–17161, DOI:10.1021/ja052939w.
- P. A. Fokou and M. A. R. Meier, Studying and Suppressing Olefin Isomerization Side Reactions During ADMET Polymerizations, Macromol. Rapid Commun., 2010, 31(4), 368–373, DOI:10.1002/marc.200900678.
- V. B. Patil, K. O. Saliu, R. M. Jenkins, E. M. Carnahan, E. J. Kramer, G. H. Fredrickson, G. C. Bazan, V. B. Patil, K. O. Saliu, E. J. Kramer, G. H. Fredrickson, G. C. Bazan, R. M. Jenkins and E. M. Carnahan, Efficient Synthesis of α,ω-Divinyl-Functionalized Polyolefins, Macromol. Chem. Phys., 2014, 215(11), 1140–1145, DOI:10.1002/MACP.201400139.
- S. J. Lippard and F. H. Jardine, The Chemical and Catalytic Reactions of Dichlorotris(Triphenylphosphine)Ruthenium(II) and Its Major Derivatives, in Progress in inorganic chemistry, Interscience, 1984, vol. 31, pp. 265–370 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06826b |
| This journal is © The Royal Society of Chemistry 2025 |