
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalyzed three-component tandem remote C–H functionalization of naphthalenes: modular and concise synthesis of multifunctional naphthalenes†
Mao-Gui
Huang‡
a,
Yue-Liu-Ting
Fu‡
a,
Jia-Wei
Li
b and
Yue-Jin
Liu
 *a
*a
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Ministry of Education Key Laboratory for the Synthesis and Application of Organic Functional Molecules, College of Chemistry and Chemical Engineering, Hubei University, Wuhan 430062, P. R. China. E-mail: liuyuejin@hubu.edu.cn
bInstitute of Medicinal Development and Application for Aquatic Disease Control, Zhoukou Key Laboratory of Small Molecule Drug Development and Application, School of Chemistry and Chemical Engineering, Zhoukou Normal University, Zhoukou 466001, P. R. China
First published on 12th December 2024
Abstract
The prevalence of naphthalene compounds in biologically active natural products, organic ligands and approved drugs has motivated investigators to develop efficient strategies for their selective synthesis. C–H functionalization of naphthalene has been frequently deployed, but mainly involves two-component reactions, while multiple-component C–H functionalization for the synthesis of naphthalene compounds has thus far proven elusive. Herein, we disclose a versatile three-component protocol for the modular synthesis of multifunctional naphthalenes from readily available simple naphthalenes, olefins and alkyl bromides via P(III)-assisted ruthenium-catalyzed remote C–H functionalization. This protocol not only tolerates various functional groups, but can be applied to many natural product and drug derivatives, and can involve a three-component reaction with two different bioactive molecules. Mechanism studies indicated that the utilization of tertiary phosphines as auxiliary groups is the key to achieving the three-component free-radical reaction.
Naphthalene and its derivatives frequently occur in natural products and functional materials.1 They also play prominent roles in ligands (BINAP) as well as pharmaceuticals such as the commercial drug Naproxen (Fig. 1A).2 Traditional preparation of substituted naphthalene derivatives is achieved through intermolecular or intramolecular annulation reaction from complex multiple-step synthesized substrates.3 In contrast, the C–H activation technique has in recent years emerged as an increasingly viable alternative for late-stage functionalization, avoiding the use of complicated starting materials.4 In this context, C–H functionalization of naphthalenes has seen remarkable development (Fig. 1B).5 Chemists have in recent years successfully achieved selective C–H functionalization of naphthalene rings from adjacent C2 and C8 positions, as well as remote C4, C5, C6 and C7 positions with different strategies.6 Despite these advances, the present methods are limited to two-component coupling reactions that yield structurally uncomplicated naphthalenes. Conceptually, the direct functionalization of naphthalene molecules through multi-component reactions (MCRs) would be more attractive because it could efficiently synthesize valuable complicated naphthalene derivatives from multiple inexpensive and abundant starting materials, in particular those difficult to prepare using traditional methods (Fig. 1C).
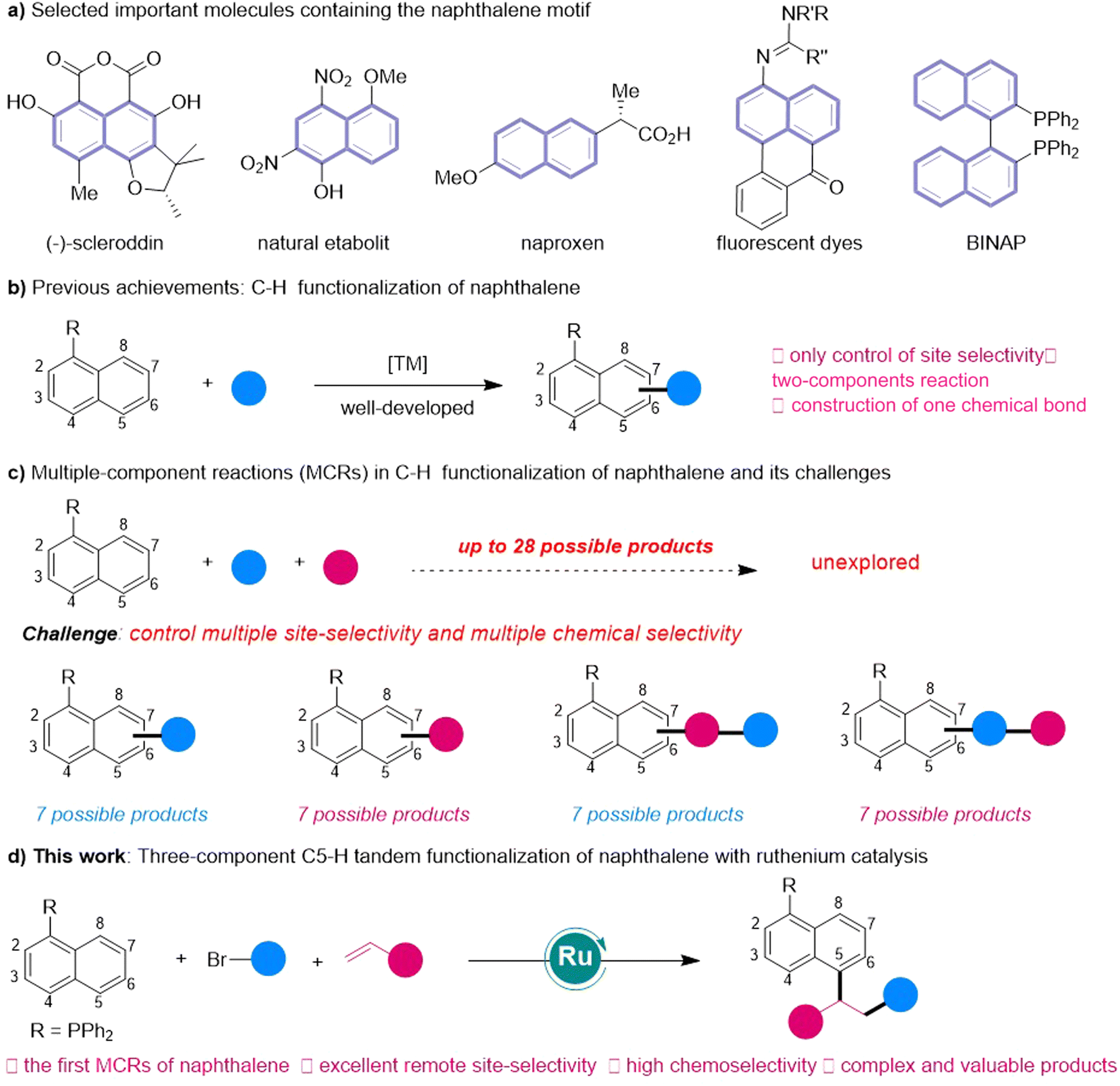 | ||
| Fig. 1 Important naphthalene-containing molecules and their synthesis via C–H functionalization of naphthalene. | ||
MCRs of olefins constitute one of the most important organic reactions for constructing complex molecules from simple raw materials.7 Remarkable advances have been made in metal-, light- and electricity-initiated MCRs of alkenes with different coupling reagents.8 In these reactions, the other two components usually need to be prefunctionalized to enable the success of reactions and control the selectivity. Deployment of MCRs involving the coupling of olefins with nonprefunctionalized substrates via C–H activation is a more attractive strategy for efficiently synthesizing complicated molecules. Although a few elegant works have been reported using nonprefunctionalized aromatic compounds,9 it remains a significant challenge to develop MCRs of substituted naphthalenes with more subtle C–H bonds (up to 7), especially for those C–H bonds at remote positions.
Very recently, we reported the first remote C5-functionalization of naphthalenes6ovia ruthenium-catalyzed δ-activation.10 We found, while carrying out mechanism studies, that owing to the strong electron-donating effect of tertiary phosphine, the P(III)-coordinated cyclicoruthenium intermediate could react with bromoalkanes to trigger formation of alkyl radicals under mild conditions and finally produce two-component C5-functionalized products. Inspired by this study, we assumed that the three-component radical reaction of alkenes with naphthalenes and alkyl bromides may be achieved using a strategy involving phosphine-assisted11 ruthenium-catalyzed δ-activation. Yet, achieving an MCR C–H functionalization of naphthalene is a great challenge, as it does not only require controlling the chemical selectivity of two-component coupling reactions, but also controlling the sequence selectivity and site selectivity of multi-component reactions, such as by (1) inhibiting C8-hydrogenarylation of alkenes with naphthalenes as previously reported,6q (2) overcoming two-component C5-functionalization of naphthalenes with alkyl bromides,6o and (3) avoiding radical polymerization of olefins. Thus, whether the three-component remote C–H tandem reaction of naphthalenes with simple olefins and alkyl bromides could be realized is still not determined.
In the current work, we tested the above speculation by developing a three-component reaction of naphthalenes with olefins and alkyl bromides, with this reaction involving P(III)-assisted Ru-catalyzed remote C5–H activation (Fig. 1D). The reaction features excellent chemoselectivity and high remote C5-site selectivity. This achievement provides a concise and modular protocol for the synthesis of complicated and valuable 1,5-disubstituted naphthalenes from simple and inexpensive materials and corresponding synthesis of a series of natural products and drugs. Moreover, this three-component strategy can be expanded to involve polycyclic aromatic hydrocarbons (PHAs) for synthesizing challenging and complex PHAs.
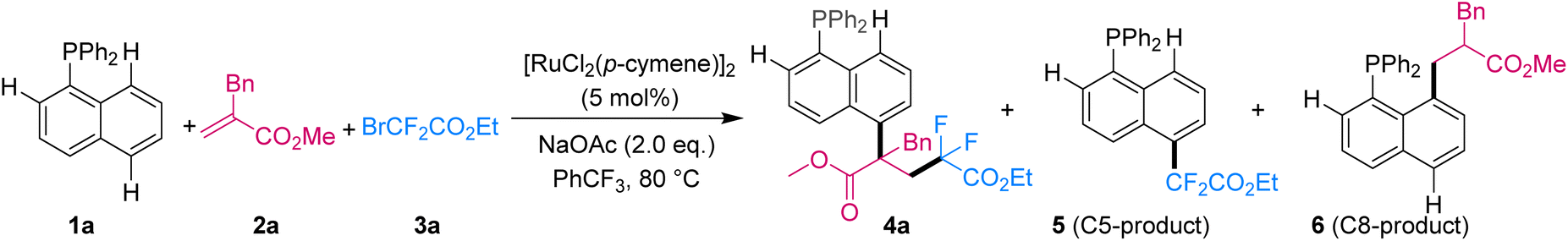
We initiated our studies of three-component reactions with commercially available naphthalene 1a, olefin 2a and fluoroalkyl bromide 3a. After extensive investigation of various reaction parameters (Table 1), the desired three-component C5-tandem product (4a) was obtained in 85% yield with excellent site-selectivity (C5/C8 > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) and chemoselectivity (C5/C5′ > 95:5) when the reaction was assisted by the phosphine group and used [RuCl2(p-cymene)]2 (5 mol%) and NaOAc (2.0 equiv.) at 80 °C in (trifluoromethyl)benzene (see ESI† for detailed optimization) (entry 1). Other assisting groups such as Cy2P–, Ph2N–, PhO– and pyridine, as well as bidentate quinoline amide were also tested, but they were all ineffective for this reaction (entries 2–6), demonstrating the key role of the phosphine group in this transformation. Notably, only C8-hydroarylation product 6 was obtained when methyl acrylate was used (entry 7), proving the key role of alkene for the three-component tandem reaction of naphthalenes. When 2-bromoisobutyrate was selected as the coupling reagent (entry 8), the two-component C5-functionalized product 5 was obtained. This result may have been due to the tertiary alkyl radical having attacked the naphthalene faster than its having attacked the olefin. No product was detected without addition of [RuCl2(p-cymene)]2 or with palladium and rhodium catalysts instead of [RuCl2(p-cymene)]2 (entries 9–11), indicating the significant and unique ruthenium catalytic activity for this three-component remote C5-tandem reaction.
:5) and chemoselectivity (C5/C5′ > 95:5) when the reaction was assisted by the phosphine group and used [RuCl2(p-cymene)]2 (5 mol%) and NaOAc (2.0 equiv.) at 80 °C in (trifluoromethyl)benzene (see ESI† for detailed optimization) (entry 1). Other assisting groups such as Cy2P–, Ph2N–, PhO– and pyridine, as well as bidentate quinoline amide were also tested, but they were all ineffective for this reaction (entries 2–6), demonstrating the key role of the phosphine group in this transformation. Notably, only C8-hydroarylation product 6 was obtained when methyl acrylate was used (entry 7), proving the key role of alkene for the three-component tandem reaction of naphthalenes. When 2-bromoisobutyrate was selected as the coupling reagent (entry 8), the two-component C5-functionalized product 5 was obtained. This result may have been due to the tertiary alkyl radical having attacked the naphthalene faster than its having attacked the olefin. No product was detected without addition of [RuCl2(p-cymene)]2 or with palladium and rhodium catalysts instead of [RuCl2(p-cymene)]2 (entries 9–11), indicating the significant and unique ruthenium catalytic activity for this three-component remote C5-tandem reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Variation from the optimal conditions | Yield (%) of 4a | Yield (%) of 5 | Yield (%) of 6 |
a Reaction conditions: 1a (0.10 mmol), 2a (0.30 mmol), 3a (0.30 mmol) 5 mol% [RuCl2(p-cymene)]2, NaOAc (2 equiv.), 1 mL PhCF3, 80 °C in sealed Schlenk tubes, 12 h, under argon. Yields were determined using 1H NMR, R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H or Bn.
b Isolated yield. H or Bn.
b Isolated yield.
|
||||
| 1 | No | 85b | <5 | <5 |
| 2 | Cy2P– instead of Ph2P– | 0 | 0 | 0 |
| 3 | Ph2N– instead of Ph2P– | 0 | 0 | 0 |
| 4 | PhO– instead of Ph2P– | 0 | 0 | 0 |
| 5 | Py instead of Ph2P– | 0 | 0 | 0 |
| 6 | PA instead of Ph2P– | 0 | 0 | 0 |
| 7 | CH2CHCO2Me instead of 2a |
<5 | <5 | 56 |
| 8 | BrC(CH3)2CO2Et instead of 3a | <5 | 67 | <5 |
| 9 | Without [RuCl2(p-cymene)]2 | 0 | 0 | 0 |
| 10 | With Pd(OAc)2 instead of [RuCl2(p-cymene)]2 | 0 | 0 | 0 |
| 11 | With Rh(Cp*)Cl2 instead of [RuC12(p-cymene)]2 | 0 | 0 | 0 |
With the optimized conditions in hand, we probed the versatility of the three-component C5-tandem reaction of naphthalenes, olefins and alkyl bromides (Scheme 1). First, the olefin scope was investigated. Modified 2-benzylacrylate esters were evaluated and it turned out that electron-donating alkoxyl (3b) and electron-withdrawing ester (3c), acetyl (3d), and formyl (3e) groups were tolerated, affording the desired products 4b–e in 82–84% yields, respectively. Furthermore, an array of 2-alkylacrylate esters could be converted smoothly into the corresponding products 4f–i, 4m in moderate to good yields. Moreover, acrylamide and styrenes (3j–l) were also accepted and delivered the products in 51–65% yields with excellent regio- and chemoselectivity levels (>20:1). However, mono-substituted and cyclic olefins were not suitable for the three-component reaction of naphthalenes. Next, we turned our attention to the scope of alkyl bromides. A variety of bromodifluoroacetates were applicable to the reaction and generated the desired products 4o–u in 50–89% yields, respectively. Additionally, a series of bromodifluoro acetamides underwent the reaction in the standard conditions, delivering products 4v–4ab, respectively, in 48–63% yields. Pleasingly, we also found that monofluoroalkyl and secondary alkyl bromides could be employed as the alkylating reagents, affording products 4ac and 4ad in good yields without diastereocontrol of the newly formed consecutive chiral centers, which proved these reaction to involve a radical addition process. Substituted naphthalenes and even anthracene were also investigated, giving the corresponding products 4ae–4ai in 38–76% yields, respectively. Given the excellent applicability of the three-component tandem transformation, diversification of bioactive molecules via this strategy was investigated (Scheme 2).
 | ||
| Scheme 1 Scope of three-component C5-tandem functionalization of naphthalenes with olefins and alkyl bromides. aReaction conditions: 1 (0.10 mmol), 2 (0.30 mmol), 3 (0.30 mmol), 5 mol% [RuCl2(p-cymene)]2, NaOAc (2 equiv.), 1 mL PhCF3, in sealed Schlenk tubes, 12 h, 80 °C, under argon; selectivity was determined using 31P NMR. bOxidized by H2O2. | ||
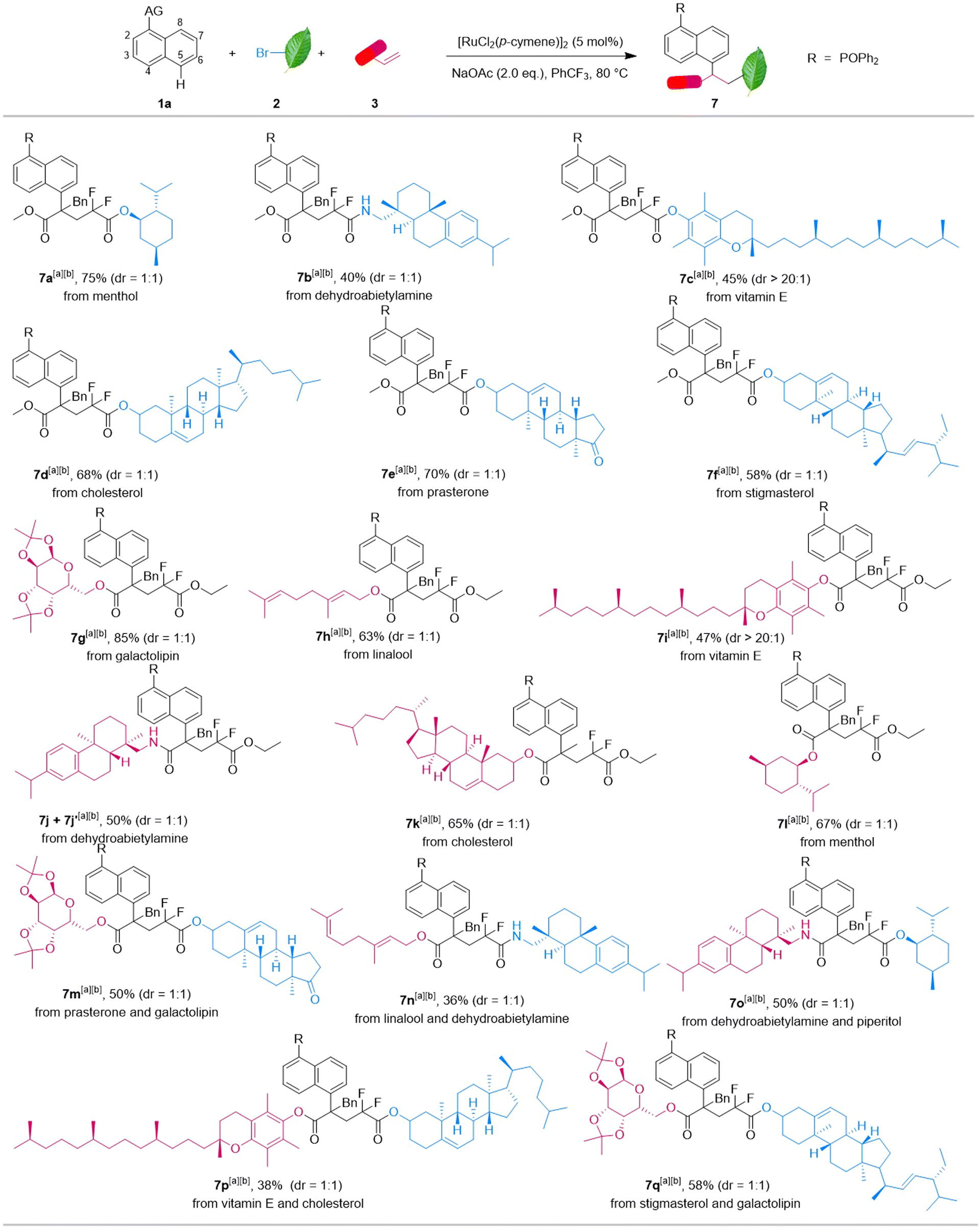 | ||
| Scheme 2 Reaction with natural products and drugs. aReaction conditions: 1 (0.10 mmol), 2 (0.30 mmol), 3 (0.30 mmol), 5 mol% [RuCl2(p-cymene)]2, NaOAc (2 equiv.), 1 mL PhCF3, in sealed Schlenk tubes, 12 h, 80 °C, under argon, selectivity was determined by 31P NMR. bOxidized by H2O2. | ||
Reactions with modified bromodifluoroacetate and bromodifluoro acetamide derived from piperitol, dehydroabietylamine, vitamin E, etc. could proceed under the standard conditions, and afforded the corresponding products 7a–f in 40–75% yields. Moreover, diverse acrylate ester and acrylamide species were well tolerated, affording the coupling products 7g–l in moderate to good yields. Notably, modified alkyl bromides and diverse acrylate esters turned out to be suitable and reacted with naphthalene 1a in one pot smoothly to give 7m–q in 36–58% yields, respectively. Therefore, this strategy may provide a serviceable way for achieving post-functionalization and modification of bioactive molecules in drug development and innovation.
To prove the practicability of this protocol, a scaled-up reaction, specifically on a 3 mmol scale, was conducted; here, a mass of 1.37 g (75% yield) of product 4a was obtained (Fig. 2a). The product 4a could be further converted into valuable lactone product 8via selective reduction of the ester group and a subsequent intramolecular esterification reaction (Fig. 2a).
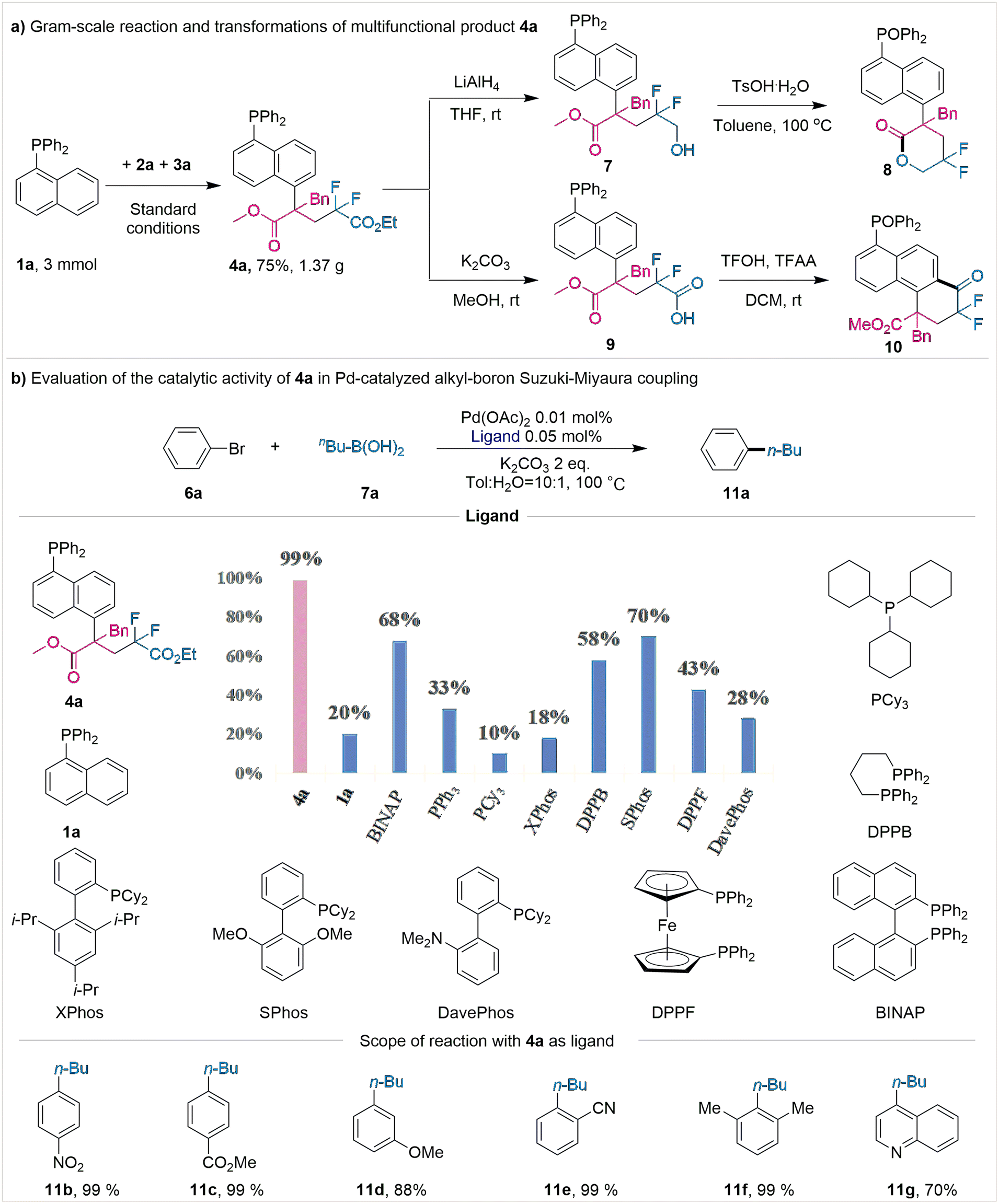 | ||
| Fig. 2 Synthetic applications. | ||
Additionally, the product could be transformed into complicated and otherwise synthetically challenging fluorine-containing ketone 10 through hydrolysis and intramolecular Friedel–Crafts acylation steps (Fig. 2a) (see ESI† for detailed conditions). In consideration of the unique activity of phosphine ligands in Pd-catalyzed Suzuki–Miyaura cross-couplings (SMCs), control experiments were conducted (Fig. 2b). In comparison with the formation of C(sp2)–C(sp2), the C(sp3)–C(sp2) SMCs involving alkyl borons proved to be more challenging and less developed, as a result of the C(sp3)–Pd bond being more unstable thermodynamically.
We assumed that the modification of 1-naphthphosphine at the C5 position may enhance the stability of palladium intermediates, thereby improving the catalytic efficiency. Therefore, we tested the effect of product 4a on palladium-catalyzed Suzuki–Miyaura coupling of alkylboronic acids and aryl bromides with low catalyst loading. As shown in Fig. 2b, using 4a as the ligand, we obtained up to 99% yield of product (11a) in the coupling of bromobenzene (6a) with n-butylboronic acid (7a) at a concentration of 100 ppm of palladium catalyst. The good reactivity of product 4a was shown in the Suzuki cross-coupling reaction, probably owing to that the bulky and strong electron-donating alkyl substituent was introduced at the C-5 position of naphthalene phosphine, increasing the electron cloud density at the center of the P atom and promoting the catalytic activity of the palladium catalyst. However, only a low yield (20%) was observed using unmodified 1-naphthphosphine (1a), which indicated the importance of C5 substitution of 1-naphthphosphine. Other widely used commercial phosphine ligands, including dppb, PCy3, and XPhos, were also tested. However, these ligands gave only low to moderate yields (10–70%) with 0.01 mol% of Pd(OAc)2. Then we investigated the scope of aryl bromides with n-butylboronic acid (7a) using 4a as ligand. Surprisingly, not only substituted bromobenzenes (11b–f) but the heterocycle bromide quinoline (11g) also could undergo the Suzuki–Miyaura coupling, generating the product in 70–99% yields, respectively. These results demonstrated the high performance of our C5-alkylated phosphines, which may have a wide application in the field of metal catalysis.
To shed light on the mechanism of this reaction, free radical verification experiments were first carried out (Fig. 3). When including 1.0 equivalents of radical trapping agent (TEMPO), the reaction was completely inhibited and a radical adduct product of 2a and TEMPO was observed (Fig. 3a) (see ESI† for detailed information), indicating that the reaction involved a radical process. Next, we conducted controlled experiments of auxiliary groups. The results showed that the diphenylamine (Ph2N–) group and phenoxy (PhO–) group did not promote the three-component reaction, proving the key role of the tertiary phosphine auxiliary group in this design (Fig. 3b). Excitingly, Int A prepared using our previous method could react with 2a and 3a stoichiometrically under standard conditions and furnish product 4a in 20% yield. Moreover, examination of the catalytic reaction of Int A also suggested that it might be a viable intermediate for this transformation (Fig. 3c). Then, the kinetic isotope effect (KIE) of this transformation was investigated. High (1.9) and low (1.1) values of KIE were observed for the C5 and C8 positions, respectively (Fig. 3d), suggesting C5–H bond activation to be the rate-determining step of this ruthenium-catalyzed three-component C5-functionalization of naphthalene. Next, an H/D exchange experiment of 1a was conducted with CD3OD. A high level of deuterium was found at the C8 position of the product (55%) and recovered raw materials 1a (56%), but the C5–H bond was not deuterated (Fig. 3e). This result showed that C8–H activation was reversible while activation of the C5–H bond was an irreversible process. At last, a competition experiment was carried out (Fig. 3f). The results showed the electron-donating group to be more conducive than to the reaction.
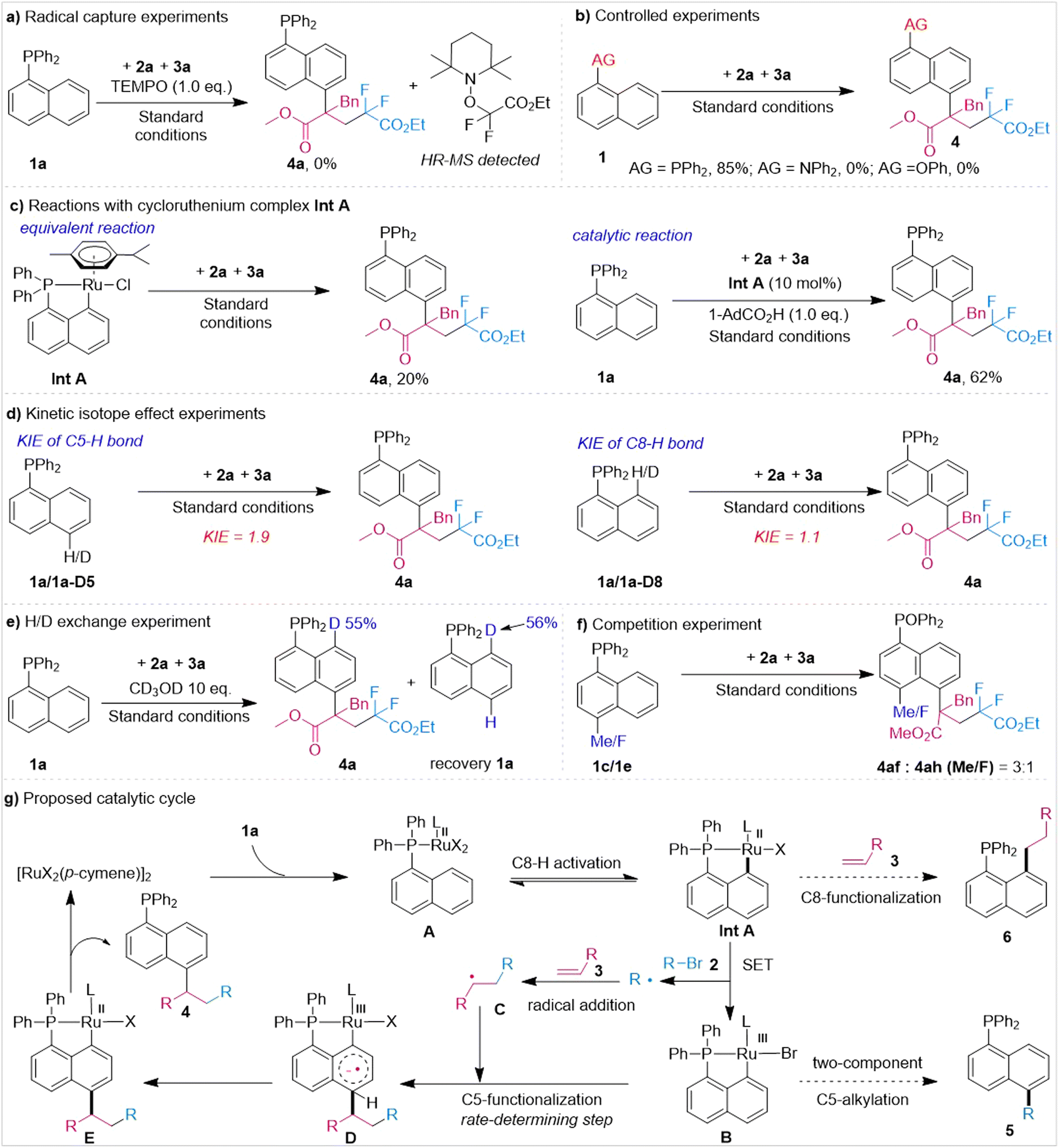 | ||
| Fig. 3 Mechanistic investigations. | ||
Based on the above results and previous literature,12 a plausible process was derived, and is shown in Fig. 3g. According to this process, first substrate 1a coordinates with [RuCl2(p-cymene)]2 to generate ruthenium complex A, which then undergoes C8–H activation to form the five-membered cyclometalated complex Int A. Next, Int A is oxidized by alkyl bromides 2via single-electron transfer to form trivalent ruthenium complex B and releases the first alkyl radical R, which is trapped by olefin 3 to form secondary alkyl radical species C. Subsequently, radical C attacks the para-position of the Ru–C8 bond of intermediate B to generate intermediate D. Then, intermediate D generates the C5-functionalized intermediate E via a dehydroaromatization process. Finally, intermediate E undergoes protonation reactions to afford product 4 and regenerates the catalyst [RuX2(p-cymene)]2. During this process, insertion of alkene into the Ru–C bond in intermediate Int A followed by a protonation process could deliver the C8-alkylated by-product 6. Moreover, the alkyl radical may attack the para-position of the Ru–C bond in intermediate B, producing C5-alkylated by-product 5.
In summary, we have developed the first three-component tandem reaction of naphthalenes with olefins and alkyl bromides by using a phosphine-coordinated ruthenium catalytic system. This system overcomes the otherwise favorable two-component reaction, offering a modular and concise strategy for accessing complex and valuable multifunctional naphthalenes with high chemoselectivity and site-selectivity. Notably, this protocol features a broad substrate scope, wide functional group tolerance, mild reaction conditions and high universality including disubstituted olefins and trisubstituted olefins. The multifunctional products not only can serve as highly efficient ligands in Suzuki–Miyaura coupling reactions, but can also be further converted into fluorinated naphthohexacyclic ketones and fluorinated hexacyclic intramolecular lipids. Moreover, the synthetic value of this methodology was demonstrated by the synthesis of a series of complicated products including many natural products and drugs, which may contribute to research and discovery of new drugs containing naphthalene. Mechanistic studies disclosed that the C5–H bond activation is the rate-determining step in this three-component radical tandem naphthyl C5-functionalization, which is different from the previous two-component C5-functionalization. Further research on developing new types of multi-component C–H functionalization reactions is underway in our laboratory.
Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
Yue-Jin Liu conceived and directed the project and wrote the manuscript. Mao-Gui Huang conducted most of the experiments. Yue-Liu-Ting Fu performed preliminary experiments. Jia-Wei Li conducted nuclear magnetic resonance test. All the authors discussed the results, commented on and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Scientific and Technological Project of Henan Province (232102310376) and the Specialized Research Fund for the Doctoral Program of Zhoukou Normal University (ZKNUC2021021).Notes and references
-
(a) W. A. Ayer, M. Kamada and Y.-T. Ma, Can. J. Chem., 1989, 67, 2089–2094 CrossRef CAS
; (b) M. A. Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef CAS
-
(a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS
- M. Maheswari and N. Hussain, Synthesis, 2024, 56, 2145–2182 CrossRef
-
(a) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC
-
(a) S. Prévost, ChemPlusChem, 2020, 85, 476–486 CrossRef PubMed
-
(a) A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752–14755 CrossRef CAS
-
(a) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083–4088 RSC
-
(a) N. Huang, J. Luo, L. Liao and X. Zhao, J. Am. Chem. Soc., 2024, 146, 7029–7038 CrossRef CAS PubMed
-
(a) X.-L. Lai and H.-C. Xu, J. Am. Chem. Soc., 2023, 145, 18753–18759 CrossRef CAS PubMed
-
(a) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed
-
(a) X. Lv, M. Wang, Y. Zhao and Z. Shi, J. Am. Chem. Soc., 2024, 146, 3483–3491 CrossRef CAS PubMed
-
(a) R. Sun, X. Chu, S. Zhang, T. Li, Z. Wang and B. Zhu, Eur. J. Inorg. Chem., 2017, 2017, 3174–3183 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2368667. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06846g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |