
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA recyclable dynamic semiconducting polymer consisting of Pauli-paramagnetic diradicaloids promoted and stabilized by catechol–boron coordination†
Youbing
Mu
*a,
Chenxi
Xiong
a,
Minghui
Cui
b,
Mingxu
Sun
a,
Xinyu
Chen
a,
Biao
Xiao
 a,
Hongqian
Sang
a,
Zhenxing
Wang
c,
Hangxu
Liu
d,
Zhenggang
Lan
d,
You
Song
*b and
Xiaobo
Wan
*a
a,
Hongqian
Sang
a,
Zhenxing
Wang
c,
Hangxu
Liu
d,
Zhenggang
Lan
d,
You
Song
*b and
Xiaobo
Wan
*a
aKey Laboratory of Flexible Optoelectronic Materials and Technology, Ministry of Education, Jianghan University, Wuhan 430056, P. R. China. E-mail: muyb@jhun.edu.cn; wanxb@jhun.edu.cn
bState Key Laboratory of Coordination Chemistry School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: yousong@nju.edu.cn
cWuhan National High Magnetic Field Center, School of Physics, Huazhong University of Science and Technology, Wuhan 430056, P. R. China
dSCNU Environmental Research Institute, Guangdong Provincial Key Laboratory of Chemical Pollution and Environmental Safety, MOE Key Laboratory of Environmental Theoretical Chemistry, School of Environment, South China Normal University, Guangzhou, 510006, P. R. China
First published on 13th December 2024
Abstract
Coordination between 5,5′,6,6′-tetrahydroxyindigo (4OH-ID) and boron tribromide unexpectedly affords a novel dynamic covalent polymer, namely P(ID-O-B), consisting of alternating indigo and indigo diradicaloid units. The catechol–boron dynamic bond plays a vital role in promoting the diradicaloid formation and stabilizing the formed diradicaloid segments. The diradicaloid segment in the polymer has a triplet ground state and a thermally populated doublet state, which has been confirmed by the EPR study. Although not conjugated, the polymer still exhibits an electrical conductivity over 10−6 S cm−1. The SQUID study shows that the polymer is Pauli paramagnetic, indicating that the metallic domain exists in this non-conjugated polymer. This diradicaloid-containing polymer is stable toward long-term storage (over 6 months) and thermal treatment over 200 °C, but can be easily depolymerized when treated with methanol.
Introduction
Radical-containing materials are of great interest due to their potential applications in organic magnetics, redox batteries, and electronic devices as next-generation soft materials.1–4 The most common way is to attach a stable radical pendant, for example, sterically hindered nitroxyl or phenoxyl radicals, either to a non-conjugated polymer backbone (such as polyethers5,6), or to a conjugated backbone (such as polyacetylene7 and polythiophene8,9). The synthesis of a high-spin hydrocarbon polymeric network has been reported; however it suffers from sophisticated synthetic steps and a rather low yield.10,11 Two-dimensional (2D) covalent organic frameworks (COFs) containing stable radical centers have also been reported.12,13 More recently, it was reported that the tris(3,5-dichloro-4-pyridyl) methyl radical can form a 2D inorganic polymeric network with zinc(II) by taking advantage of the coordination capability of the pyridine moiety.14Conjugated polymers offer another possibility, which can generate polarons (radical cations or radical anions) upon doping.15 However, the doping level needs to be well controlled, since overdoping leads to bipolarons (dications or dianions), and the stability of the doped conjugated polymer is highly dopant-dependent, and dedoping may occur after long-time storage or when contacting with reducing (for p-doped polymers) or oxidative (for n-doped polymers) reagents.16
Organic diradicaloids have drawn increased attention in the past decades.17–23 The majority of organic diradicaloids are featured by large extended π-systems containing either quinoidal or non-alternant antiaromatic or pro-aromatic structures, and the driving force to the sextet aromatic ring accounts for their diradical character.19 Most of these stable organic diradicaloids are small molecules, which were seldom incorporated into polymers due to their synthetic complexity. On the other hand, although radicals have been treated as impurities in pristine conjugated polymers, conjugated polymers containing quinoidal structures can be regarded as the diradicaloid-containing materials, and the electron paramagnetic resonance (EPR) signal was clearly observed without doping.24 Some of the pristine donor–acceptor (D–A) type conjugated polymers without quinoidal structures also show clear EPR signals, which is highly dependent on how easy they are prone to form the resonant quinoidal structures and the EPR signal intensity is correlated with the planarity of the polymer: the better the planarity the stronger the ESR signal.25 Nevertheless, all the radical or diradicaloid-containing polymers mentioned above are non-recyclable, which is not eco-friendly.
Here we present an unprecedented discovery of a recyclable dynamic semiconducting polymer that consists of diradicaloids, which are formed and stabilized via catechol–boron coordination. Although the polymer is not conjugated, it still displays an electrical conductivity rivaling pristine polythiophene. Moreover, this polymer is Pauli paramagnetic, indicating its metallic character. The polymer is stable under various conditions such as water treatment, long-time storage, and thermal treatment over 200 °C without appreciable decay of its magnetic properties.
Results and discussion
Polymer synthesis and general characterization
In our initial attempts to demethylate 5,5′,6,6′-tetramethoxyindigo (4MeO-ID) using 4.2 equivalents of boron tribromide (BBr3) in dichloromethane, we found that when aqueous workup conditions were applied to the reaction, an unpurifiable foamy mixture was obtained. Interestingly, a greenish-colored film was obtained simply by evaporating dichloromethane off, which was considered to be a polymer (polymer 1, Fig. 1A). We reasoned that the driving force for the polymer formation is the catechol–boron dynamic bond formed during solvent evaporation. Theoretically, only one equivalent of BBr3 is needed if the polymer has a linear backbone. By treating the film with methanol, we managed to obtain 5,5′,6,6′-tetrahydroxyindigo (4OH-ID), a material insoluble in dichloromethane. A similar metallic greenish-colored film was formed when treating the suspension of 4OH-ID in dichloromethane with one equivalent of BBr3 followed by solvent evaporation. The polymer can be re-dissolved in dichloromethane and extracted with water to remove inorganic impurities, during which the formation of a foamy mixture was not observed. These results indicate that the polymer, named as P(ID-O-B), is not formed immediately after demethylation; instead, it is formed when the solution is concentrated and the dynamic bond between catechol and boron is fully generated. Once the polymer is formed, it exhibits stability toward water treatment. The polymer could be depolymerized simply by treating it with methanol, and the monomeric 4OH-ID is recovered after purification. The recycling cycles can be performed at least five times, depending on how much monomer is lost in the purification step, which varies time to time. The stability of P(ID-O-B) toward water treatment may be attributed to its long hydrophobic alkyl sidechain, which retards the attack of water molecule to the B–O covalent bond.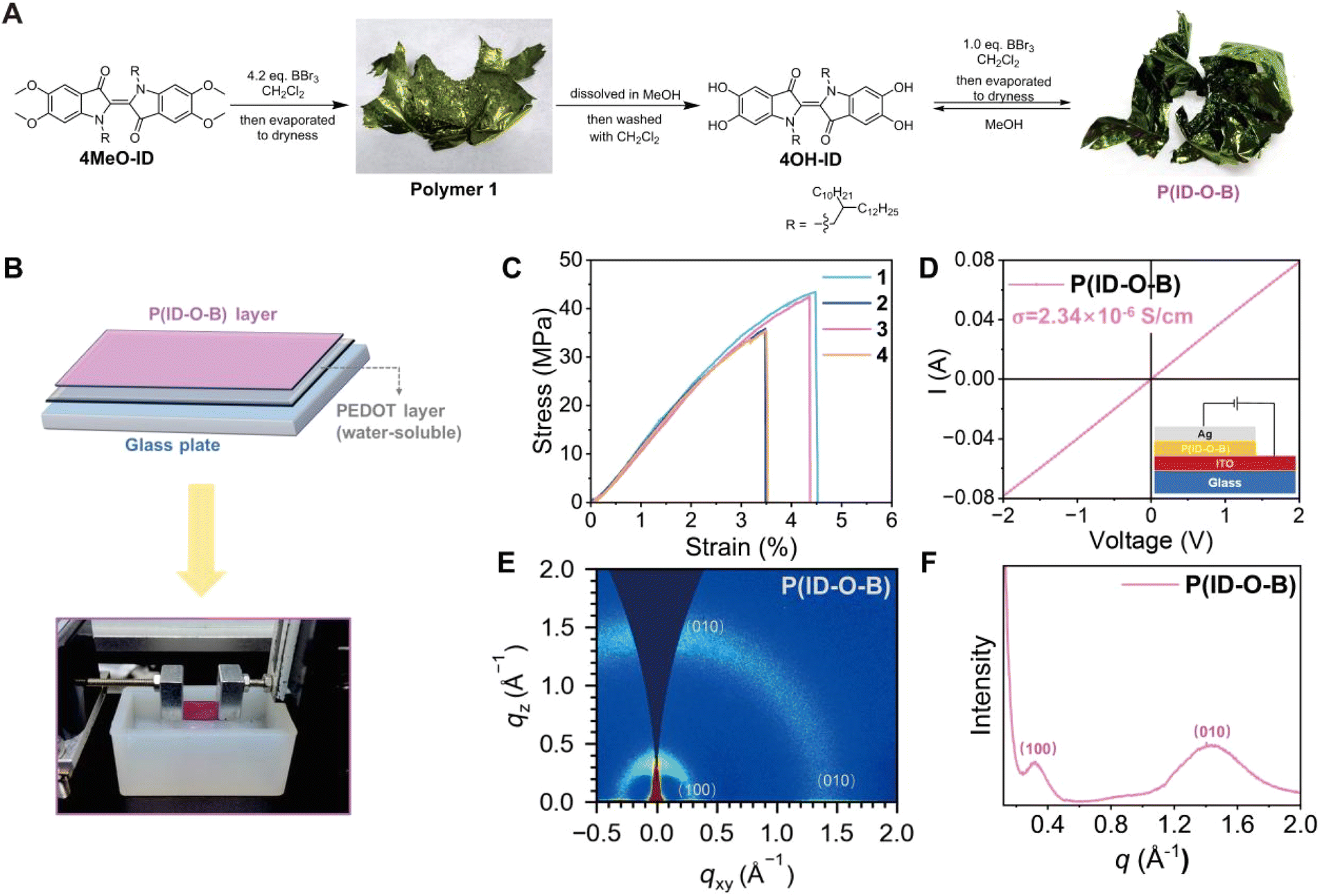 | ||
| Fig. 1 P(ID-O-B) synthesis and general characterization. (A) The accidental finding on the coordination polymerization of 4OH-ID. (B) The illustration of the preparation of a free-standing P(ID-O-B) film and the image of the “film-on-water” mechanical test. (C) Typical stress–strain curves of the polymer. (D) Typical electrical conductivity of the polymer, and the inset shows the architecture of the device for electrical conductivity measurement. (E) The 2D-GIWAXS diffraction pattern, and (F) the 1D diffraction pattern of the polymer. | ||
The molecular weight of the polymer was determined by high temperature gel permeation chromatography (HT-GPC) at 150 °C using polystyrene as the standard. A numeric molecular weight (Mn) of 119–160 kDa was obtained; however, since the polymer is much more rigid than polystyrene, the degree of polymerization could not be simply determined by Mn. Mn may vary batch to batch since controlling the molar ratio between BBr3 and the monomer exact to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is hard; still, a film can easily form if the molar ratio is controlled between 2:1 and 1:1 (BBr3/monomer), indicating that the formation of this dynamic polymer is not so sensitive to the molar ratio. Upon performing a 11B NMR spectrum test on P(ID-O-B) after washing with water, a weak but sharp boron signal observed at 14.83 ppm is assigned to the boron in the polymer, and its low intensity is attributed to its low content of boron in the polymer and the low concentration of P(ID-O-B) in the solution for the 11B NMR test. The broad peak observed in the 11B NMR spectrum is assigned to the impurities existing in the quartz tube. The X-ray diffraction (XRD) result indicates that there is a broad peak around 2θ = 22.4°, corresponding to a periodical distance around 4.0 Å. The polymer has a Tg around 89.8 °C as illustrated by differential scanning calorimetry (DSC) analysis, which may be correlated with the alkyl sidechain movement. Thermogravimetric analysis (TGA) shows that the polymer starts to decompose around 236 °C and the weight loss reaches 5% at 286 °C. It should be pointed out that the BBr3 residue was thoroughly removed by evaporation, and X-ray photoelectron spectroscopy (XPS) analysis reveals that no bromide anions are detectable in the film following exhaustive washing of the polymer solution with water. Conversely, the boron–oxygen bond (ranging from 191 to 194 eV) and the functional groups of elements carbon (ranging from 284 to 290 eV), nitrogen (ranging from 399 to 402 eV), and oxygen (ranging from 529 to 536 eV) are distinctly observable (for these general characterization studies of P(ID-O-B), please see Fig. S1–10, S20 and Tables S1–S5 ESI for details†).
:1 is hard; still, a film can easily form if the molar ratio is controlled between 2:1 and 1:1 (BBr3/monomer), indicating that the formation of this dynamic polymer is not so sensitive to the molar ratio. Upon performing a 11B NMR spectrum test on P(ID-O-B) after washing with water, a weak but sharp boron signal observed at 14.83 ppm is assigned to the boron in the polymer, and its low intensity is attributed to its low content of boron in the polymer and the low concentration of P(ID-O-B) in the solution for the 11B NMR test. The broad peak observed in the 11B NMR spectrum is assigned to the impurities existing in the quartz tube. The X-ray diffraction (XRD) result indicates that there is a broad peak around 2θ = 22.4°, corresponding to a periodical distance around 4.0 Å. The polymer has a Tg around 89.8 °C as illustrated by differential scanning calorimetry (DSC) analysis, which may be correlated with the alkyl sidechain movement. Thermogravimetric analysis (TGA) shows that the polymer starts to decompose around 236 °C and the weight loss reaches 5% at 286 °C. It should be pointed out that the BBr3 residue was thoroughly removed by evaporation, and X-ray photoelectron spectroscopy (XPS) analysis reveals that no bromide anions are detectable in the film following exhaustive washing of the polymer solution with water. Conversely, the boron–oxygen bond (ranging from 191 to 194 eV) and the functional groups of elements carbon (ranging from 284 to 290 eV), nitrogen (ranging from 399 to 402 eV), and oxygen (ranging from 529 to 536 eV) are distinctly observable (for these general characterization studies of P(ID-O-B), please see Fig. S1–10, S20 and Tables S1–S5 ESI for details†).
Since P(ID-O-B) can easily form a free-standing film, its mechanical properties were investigated using a “film-on-water” (FOW) method as shown in Fig. 1B.26 The tensile strength of the film falls into the 35–40 MPa range, with the elongation at break around 3.5–4.5%, and the Young's Modulus of the film surpasses 1 GPa (Fig. 1C and Table S5, ESI†). The mechanical strength of P(ID-O-B) is higher than that of many reported conjugated polymers (10–30 MPa),27,28 while their elongation at break is quite similar. The lack of elasticity is most likely due to the rigidity of both the tetra-coordinated catechol–boron linkage and the indigo core.
P(ID-O-B) was then spin-coated onto an ITO electrode to form a thin film with a thickness around 120 nm and subjected to electrical conductivity measurement, as shown in Fig. 1D. The polymer exhibits an electrical conductivity up to 2.34 × 10−6 S cm−1 (average 2.26 × 10−6 S cm−1, Fig. S11 and Table S6, ESI†), which falls into the upper conductivity range of undoped conjugated polymers (10−10–10−6 S cm−1). It is worthwhile to note that P(ID-O-B) is not conjugated since the adjacent isoindigo units are orthogonal to each other due to the sp3 hybridized catechol–boron linkage. The charge carrier hopping might be the major mechanism for its conductivity, so the adjacent polymeric chains should be closely packed to allow the process to occur. The microstructure of P(ID-O-B) was then investigated by the two-dimensional grazing-incidence wide-angle X-ray scattering (2D-GIWAXS) technique, and the results are shown in Fig. 1E and F. The polymer exhibits two Bragg diffraction arcs, which can be assigned to (100) and (010) peaks, respectively, corresponding to a lamellar packing distance of 19.93 Å and a π–π stacking distance of 4.35 Å (Table S7, ESI†). The results show that structural regularity at the microscopic level exists in P(ID-O-B).
The band gap of polymers is measured by cyclic voltammetry, using a glassy carbon electrode as the working electrode, a platinum sheet electrode as the counter electrode, and Ag/AgCl as the reference electrode in a three-electrode system, with repeated tests conducted under argon protection. The HOMO of the polymer is around −5.09 eV, while the LUMO is around −3.74 eV, and the band gap is 1.35 eV, and results are shown in Fig. S9.†
Elucidation on the polymer structure via various techniques
Then the real structure of this non-conjugated dynamic polymer needs to be clarified and the species accounting for its electrical conductivity needs to be figured out. We initially proposed that the dynamic bond linkage has a structure of a boron tetra-coordinated by two catechol moieties, which means a protonated oxygen is necessary to maintain the overall electroneutrality, as shown in the zwitterion coordination structure in Fig. 2A (structure a). If this is the case, the peak of the acidic proton should be observed in the 1H NMR spectrum of the polymer. However, the results show that while 4OH-ID displays two phenolic peaks at 9.97 ppm and 9.11 ppm (green line, Fig. 2B), the polymer displays no signal in the 12.5–8.5 ppm region (pink line, Fig. 2B). For the full spectra, please see Fig. S12 and S13 in the ESI.† Two broad peaks observed between 4.5 and 3.5 ppm (Fig. S13†) in the polymer were assigned to the hydrogen and carbon attached to the N atom. Similar peaks were also observed in the monomer (Fig. S12†). One possible reason is that one of the protons on this methylene group forms a hydrogen-bond with the nearby carbonyl group which makes it magnetic heterotopic from the other proton on the same methylene group. The FT-IR spectrum of the polymer shows that no vibrational peak exists in the 3600–3100 cm−1 region, indicating that no O–H stretching mode exists in the polymer. The detailed analysis of FT-IR results will be discussed in the later section. These results indicate that the proposed structure a is not correct. | ||
| Fig. 2 Probing the polymer structure via various techniques. (A) Postulated structures of P(ID-O-B), in which structures a and b are excluded, while structure c is the most plausible one. (B) 1H-NMR spectra of 4OH-ID (green line, taken in d6-DMSO), P(ID-O-B) (pink line, taken in CDCl3) and 4O-ID (blue line, taken in CDCl3) in the aromatic and acidic proton region. (C) EPR spectra of P(ID-O-B) in the solid state at different temperatures (purple line 483 K, red line 283 K and blue line 103 K) and the simulated spectrum for the triplet ground state (green line). (D) χMT–T and χM–T curves obtained from the SQUID VSM measurement of P(ID-O-B). | ||
Given that the polymer has a metallic greenish color, which is very similar to that of doped conjugated polymers, we then postulated that radicals may exist in the polymer. Electron paramagnetic resonance spectroscopy (EPR) analysis was then conducted. The polymer in the solid state displays a series of signals in the field range of 3200–3475 gauss at 283 K (red line, Fig. 2C), with two strong peaks in the middle and several weak peaks at lower and higher magnetic fields. Such sophisticated signals, which were not observed for other radical-containing polymers,5–9 indicate that more than one type of spin-species exist in this polymer. Interestingly, increasing temperature simplified the signal, and at 483 K, only one symmetrical peak centered at 3337 gauss is observed (purple line, Fig. 2C), while decreasing temperature (103 K, blue line, Fig. 2C) led to the overall intensity decrease of the two central peaks relative to other peaks. For the full spectra of variable temperature (VT)-EPR, please refer to Fig. S14, ESI.† These results indicate there is an equilibrium between thermally populated spin-species and ground state spin-species. The signal at 483 K corresponds to a g factor of 2.002, which was assigned to unpaired electrons with S = 1/2. We assume that at such a high temperature these unpaired electrons can move nearly freely both along the polymer chain and between the polymer chains, so only one type of EPR signal can be observed. With the decrease of temperature, the interchain movement of electrons may be harnessed, and the difference between intrachain and interchain movements of the unpaired electrons may become more obvious. So two types of S = 1/2 unpaired electrons can be observed at 283 K, which correspond to the two strong EPR peaks (marked with star symbols).
The EPR spectra have confirmed the existence of unpaired electrons in the polymer; however, its concentration is still a question. The spin density in P(ID-O-B) can be calculated relative to a standard sample (2,2,6,6-tetramethylpiperidine 1-oxyl radical, TEMPO), using the signal at 483 K as the integrable one. A spin density of 4.08 × 1018 spin/g was obtained, corresponding to one spin per 146 monomers, if each spin represents one isolated radical. At such a low radical concentration, the polymer should have a structure of mixed zwitterion and radical coordinations with the former as the major linkage, as shown in Fig. 2A (structure b). This hypothesis again contradicts the 1H NMR evidence, which shows that no protonated oxygen exists in the polymer. Furthermore, if structure b represents the actual structure, it is logical to postulate that the radical concentration will increase under harsher oxidation conditions. To test this hypothesis, the polymer was redissolved in dichloromethane and subjected to oxidation in a high pressure autoclave filled with pure oxygen (4.0 MPa) under elevated temperature (60 °C) for one day, after which the polymer was recovered and re-subjected to EPR analysis (Fig. S15, ESI†). Its spectrum shows little change compared to that of the polymer obtained under solvent-evaporating conditions, showing that oxidation under much harsher conditions does not lead to the increase of radical concentration and the postulated structure b can also be ruled out.
We then postulated that the polymer should have structure c as illustrated in Fig. 2A, in which two alternating indigo derivatives coexist: one provides four phenolic anions to coordinate with boron, and the other is converted into a diradicaloid, which provides two phenolic anions and two neutral phenoxy radicals to coordinate with boron, so the electroneutrality of the polymer is maintained. No protonated oxygen is needed to balance the electroneutrality in this structure, which is in accordance with the NMR results. This structure also explains why much harsher oxidation conditions would not cause the increase of spin concentration. Furthermore, the spin density no longer reflects the radical concentration in diradicaloids due to the existence of thermally accessible spin states. The other possible structure, in which each ID can be ruled out, as it would result in a polymer with isolated radicals, and the EPR intensity would to a large extent reflect the radical content. Furthermore, such radicals should be NMR silent in the aromatic region,29 which contradicts with the NMR results.
The other four EPR peaks in Fig. 2C, highlighted with triangle symbols, can be then assigned to the triplet ground state of the diradicaloid units. It is well known that in the solid state, the EPR absorption of the triplet state contains four fine splitting peaks due to the classical dipolar interaction of the magnetic moments of two unpaired electrons: two absorptions in the xy plane (perpendicular to the direction of magnetic field B) and two absorptions in the z direction (parallel to B). The simulated EPR signal of the triplet state is shown as the green line in Fig. 2C, which matches up with the experimental results. The two outermost peaks were assigned to the absorption in the z direction, and the two inner peaks were assigned to the absorption in the xy plane. The gx and gy factors of the triplet state are equal (2.0095), and the gz factor is 1.9949. The zero-field splitting factors D and E are calculated to be 103 gauss and 2.5 gauss, respectively. The fine splitting of the EPR signal of the triplet state disappears at high temperature because such dipolar interaction between two unpaired electrons is averaged out by the enhanced molecular motion. To sum up, the VT-EPR results show that equilibrium exists between the ground triplet state and thermally populated doublet state in this diradicaloid-containing polymer.
To shed more light on the actual structure of P(ID-O-B), superconducting quantum interference device magnetometry (SQUID VSM) measurement was conducted, and the results are shown in Fig. 2D. To rule out the interference of impurities (possible solvent or other small molecules encapsulated in the polymer), the polymer was annealed at 200 °C to constant weight before the measurement. A linear increase of χMT from nearly zero emu K mol−1 at 2 K to 0.87 emu K mol−1 at 300 K was observed; above 300 K, the χMT value continues to increase but at a smaller rate and reaches 0.96 emu K mol−1 at 360 K, close to the value (1.0 emu K mol−1) for a triplet spin system with S = 1. It is well known that strong antiferromagnetic coupling will lead to an open-shell diamagnetic state at low temperatures,30,31 which was not observed in our case. Indeed, the χM–T curve in Fig. 2D (blue line) shows that the molar magnetic susceptibility remains almost unchanged between 300 and 50 K and increases exponentially near 0 K, resembling Pauli paramagnetism of free electrons, which has been observed for highly ordered doped conjugated polymers,32 laddered pernigraniline salt,33 and conjugated polymers with a quinoidal open-shell triplet ground state.34 Based on these lines of evidence, we suggest the magnetic behavior of P(ID-O-B) be ascribed to Pauli paramagnetism due to the existence of a large population of nearly free electrons, which also confirms the presence of doublet states. With the decrease of temperature, the number of electrons thermally populated on the doublet state decreases and the number of electrons located on the triplet ground state increases, accounting for the observed evolution of VT-EPR. After fitting the χMT–T curve according to the equation χTotalT = C + χPauliT, where C is the Curie constant, a Pauli magnetic susceptibility χPauli of 2.90 × 10−3 emu mol−1 and a Curie constant of 1.23 × 10−3 emu K mol−1 were obtained. The density of states (DOS) at the Fermi level of P(ID-O-B) is around 5.27 × 1025 eV−1 mol−1, calculated according to the equation χPauli = μB2N(EF), where μB is the Bohr magneton. The observed Pauli paramagnetism suggests the presence of a metallic diradicaloid lattice33 in P(ID-O-B), which is unexpected for a non-conjugated polymer formed via the catechol–boron tetracoordination linkage; however, it matches up with the metallic greenish color of the polymer and the relatively high electrical conductivity observed for thin films. As a comparison, the Pauli-paramagnetic, fully-conjugated laddered pernigraniline salt oligomer shows an electrical conductivity of 10−5 S cm−1.33
Both EPR and SQUID characterization studies support the presence of diradicaloid units in the polymer. The diradicaloid species are expected to be generated during the evaporating process, where the solution gets exposed to air and a half amount of 4OH-ID units are oxidized in the presence of BBr3. A controlled experiment was then conducted: the suspension of 4OH-ID in dichloromethane was heated to reflux in an oxygen atmosphere to see whether the diradicaloid or similar species could be generated in the absence of BBr3. Although very sluggish (after refluxing for several days), a metallic black-colored species, which is soluble in dichloromethane and could be separated from unreacted 4OH-ID by filtration, was obtained in around 45% yield, as shown in Fig. 3A.
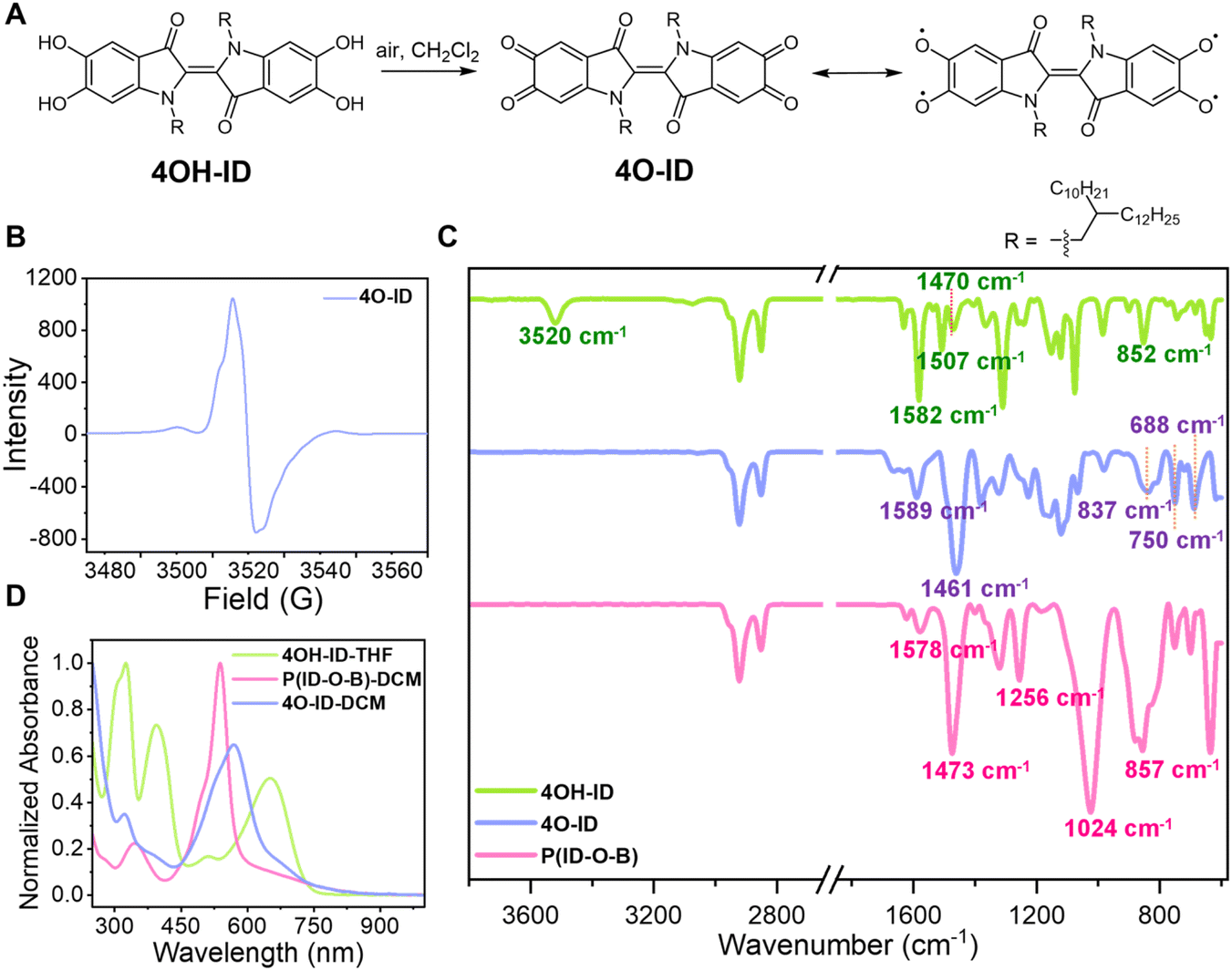 | ||
| Fig. 3 More evidence for the existence of diradicaloids in the polymer. (A) The oxidation of 4OH-ID by air leads to 4O-ID and the illustration of its tetraradicaloid resonance structure. (B) EPR spectrum of 4O-ID. (C) FT-IR spectra of 4OH-ID (green line), 4O-ID (purple line) and P(ID-O-B) (pink line). (D) UV-vis absorption of 4OH-ID (green line) in THF, 4O-ID (purple line) and P(ID-O-B) (pink line) in DCM. | ||
The product was immediately subjected to 1H NMR analysis without further purification, as it would decompose on a silica gel column or on a HPLC column. Only two aromatic protons located at 7.33 and 7.21 ppm were observed without the observation of phenolic protons in the 12.5–6.5 ppm region, showing that all of the four phenolic protons were consumed (Fig. 2B, blue line). We then postulated that the o-quinoidal product 4O-ID was formed. 4O-ID is fairly stable in the solid state for a few days, but can be easily reduced back to 4OH-ID when stored as a solution under ambient conditions. The tetraphenoxy radicaloid was expected as the resonant structure of 4O-ID, so the product was also subjected to EPR analysis. A strong signal centered at g = 2.004 with the spin density around 1.8 × 1018 spins/g was observed, as shown in Fig. 3B, which reflects its resonant tetraradical character.
P(ID-O-B) should resemble 4O-ID to a large extent since they both have a polyradical character. Thus, the FT-IR spectra of 4OH-ID, 4O-ID and P(ID-O-B) were then compared, and the results are shown in Fig. 3C. The FT-IR spectra of 4O-ID and P(ID-O-B) are indeed quite similar to each other: (1) the O–H stretching mode observed for 4OH-ID (3520 cm−1) could not be observed for both 4O-ID and P(ID-O-B); (2) while 4OH-ID displays three C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ring stretching peaks at 1582 cm−1, 1507 and 1470 cm−1, both 4O-ID and P(ID-O-B) display two greatly weakened CC ring stretching modes around 1589–1576 cm−1 and a strongly enhanced absorption around 1461 or 1473 cm−1, showing that the CC stretching modes have been altered due to quinoidal/diradicaloid formation. We noticed that it has been mentioned that the ν(C–O˙) stretching frequency for phenoxy radicals appears around 1500 cm−1,35 so the C–O˙ stretching mode may also contribute to this strongly enhanced peak; (3) in the fingerprint region, both 4O-ID and P(ID-O-B) display very similar out-of-plane aromatic C–H bending absorption, with a strong and broad peak around 850 cm−1 and two sharp peaks located between 750 and 688 cm−1, while 4OH-ID displays one major sharp peak around 852 cm−1, which can also be ascribed to the formation of a quinoidal structure in both 4O-ID and P(ID-O-B); (4) two distinct peaks located at 1256 and 1024 cm−1 were observed for P(ID-O-B), but not for 4OH-ID and 4O-ID, which were assigned to the O–B stretching mode and C–O stretching mode in the C–O–B linkages, respectively.
C ring stretching peaks at 1582 cm−1, 1507 and 1470 cm−1, both 4O-ID and P(ID-O-B) display two greatly weakened CC ring stretching modes around 1589–1576 cm−1 and a strongly enhanced absorption around 1461 or 1473 cm−1, showing that the CC stretching modes have been altered due to quinoidal/diradicaloid formation. We noticed that it has been mentioned that the ν(C–O˙) stretching frequency for phenoxy radicals appears around 1500 cm−1,35 so the C–O˙ stretching mode may also contribute to this strongly enhanced peak; (3) in the fingerprint region, both 4O-ID and P(ID-O-B) display very similar out-of-plane aromatic C–H bending absorption, with a strong and broad peak around 850 cm−1 and two sharp peaks located between 750 and 688 cm−1, while 4OH-ID displays one major sharp peak around 852 cm−1, which can also be ascribed to the formation of a quinoidal structure in both 4O-ID and P(ID-O-B); (4) two distinct peaks located at 1256 and 1024 cm−1 were observed for P(ID-O-B), but not for 4OH-ID and 4O-ID, which were assigned to the O–B stretching mode and C–O stretching mode in the C–O–B linkages, respectively.
The UV-vis spectra of 4OH-ID, 4O-ID and P(ID-O-B) were also compared, and the results are shown in Fig. 3D. Both 4O-ID and P(ID-O-B) display a similar change in the HOMO/LUMO transition compared to 4OH-ID. The major absorption located around 651 nm for 4OH-ID blueshifts to 569 nm for 4O-ID and further to 538 nm for P(ID-O-B), which can be ascribed to quinoidal structure formation. Furthermore, an absorption tail extending to 940 nm was observed for both 4O-ID and P(ID-O-B) but not for 4OH-ID, reflecting their tetraradicaloid/diradicaloid character.36 Overall, FT-IR and UV-vis spectroscopic evidence shows that P(ID-O-B) resembles 4O-ID, which has a strong quinoidal character.
All of the results indicate that the proposed structure c in Fig. 2A is correct. The alternating structure with one oxidized diradicaloid ID unit next to the un-oxidized ID unit is intriguing. As far as we know, P(ID-O-B) is the first diradicaloid-containing polymer reported so far via the formation of the catechol–boron dynamic covalent bond. Although EPR signals have been observed in some metal–organic frameworks (MOFs) formed by catechol-containing ligands with transition metals, however, they were attributed to the structural defects, grain boundaries and edges in these polycrystalline powders or the signals from the transition metal itself.37,38 We believe that boron coordination is necessary for the formation of a diradicaloid in P(ID-O-B) under the mild evaporation conditions, since 4OH-ID is quite stable when stored in air, and a rather long reaction time under elevated temperature is needed to convert it to 4O-ID. It has been reported that the O–B tetracoordinated complex with one quinoidal oxygen can be synthesized, which upon electrochemical reduction can form radicals.39 More recently, a borocyclic radical was synthesized via boron coordination to a quinone compound followed by hydrogen reduction,29 which is stable under 1 atmosphere O2 for several days in the solution state. Similar boron-coordinated di/tri-radicaloids with moderate stability were reported thereafter,40,41 implying that our proposed structure is highly plausible. It needs to be mentioned that in the proposed structure, the assignment of the location of the phenoxy radical is arbitrary, since we could not verify at this stage which phenol moiety (either the one at the para position or the one at the meta position to the carbonyl group) is converted to the phenoxy radical.
The formed diradicaloid-containing polymer could be stored under an ambient environment for more than six months without appreciable decay of its EPR signal (Fig. S16, ESI†), much more stable than the un-coordinated 4O-ID. We also measured the half-life of the polymer in DCM solution exposed to air through UV spectroscopic analysis and found that the half-life of the polymer is around 12 days (Fig. S8†), which is much longer those of many reported phenoxy diradicaloids. This is also intriguing, since shielding radicals or radicaloids with bulky groups has become a common rule to the design of stable phenoxy radicals ever since the discovery of the Galvinoxyl radical,42 even for those phenoxy diradicaloids or polyradicaloids with extended π-conjugation.43–46 It was only reported recently36,47,48 that bulky groups are not necessary for some π-extended aminophenoxy polyradicaloids. We believe that in our case, besides the captodative effect of the ID core, the coordination of boron to the diradicaloid unit plays an important role in improving the stability of the diradicaloid moiety in the polymer. More direct evidence comes from those recently reported stable boron-coordinated radicals/diradicaloids/triradicaloids.29,40,41,49
Theoretical calculation
To better understand the magnetic and electrical properties of P(ID-O-B), density functional simulations were conducted, and the results are shown in Fig. 4. A diradicaloid coordinated with two boron atoms, which are end-capped with two catechol moieties, was used as the model compound to investigate the spin states of the diradicaloid. The calculation results show that the spin density is majorly located on the central phenyl rings (Fig. 4A), and the triplet state is energetically favored compared with the open-shell singlet state by around 9.11 kcal mol−1, which is larger than those of many other diradicaloids with ground triplet states. This can be explained by the resonance structure of the diradicaloids, since no close-shelled Kekulé formula could be deduced from the diradicaloid, so anti-parallel alignment of the two unpaired electrons is strongly energetically disfavored. Instead, the two unpaired electrons would rather stay at two doublet states at high temperatures. The diradicaloid factor y is calculated to be 0.843, a relatively high value for diradicaloids and reflecting its nearly “free radical” character. Moreover, the interaction between two P(ID-O-B) chains is also calculated using tetramers as the model. Two tetramers are capable of forming a unique double helical structure, characterized by a helical pitch of 9.2 nm and an interchain spacing of 3.8 Å, typical for a π–π stacking distance, as shown in Fig. 4B. The calculated π–π stacking distances roughly match up with the GIWAXS result (4.35 Å). The deviation may be derived from the bulky sidechains in the polymer, which increase the steric hindrance and enlarge the distance. Besides the sp3 hybridized catechol–boron linkage, the central double bond of ID units is also not fully planar, which shows a torsion angle of 32.0°, thereby allowing the formation of a helix structure. It should be pointed out that the real interaction scenario in polymeric films is more complicated; however, this simplified simulation shows that the close interaction between the two P(ID-O-B) chains is possible, which allows charge carrier hopping and the formation of metallic domains that accounts for Pauli paramagnetism and electrical conductivity in polymeric films. | ||
| Fig. 4 Theoretical calculations. (A) Simplified model for DFT calculation and the calculation results for spin-density distribution on triplet–singlet states; (B)simulation of the interaction of two P(ID-O-B) chains using two tetramers as the model. Side view and top view are presented. | ||
Conclusion
To summarize, we report here that a π-extended molecule containing the catechol moiety can form an alternating diradicaloid-containing dynamic polymer simply by boron-coordination. Catechol–boron dynamic bond formation plays a vital role in promoting the diradicaloid formation and stabilizing the formed diradicaloid segments. The diradicaloid moiety in the polymer has a triplet ground state and a thermally populated doublet state, which is stable toward thermal treatment over 200 °C. The SQUID study shows that the polymer is Pauli paramagnetic, indicating that the metallic domain exists in this non-conjugated polymer. The polymer exhibits an electrical conductivity over 10−6 S cm−1, although the sp3-hybridized catechol–boron linkage prohibits long-range conjugation along the polymeric backbone. To the best of our knowledge, this is the first non-conjugated dynamic polymer that exhibits both electrical conducting and Pauli paramagnetic properties. We envision that by tuning the electronic structure of the catechol-containing π-conjugated molecule and choosing an appropriate Lewis acid that can coordinate with the catechol moiety, more dynamic polymers with improvable electrical and magnetic properties might be discovered, which will contribute to both organic semiconductors and organic magnetic fields and deserve further investigation.Data availability
The data supporting this article have been included in the main text and the ESI.†Author contributions
Chenxi Xiong did the majority of experimental work, including synthesis, characterization and analysis. Minghui Cui performed the SQUID and part of EPR tests for this experiment. Mingxu Sun and Xinyu Chen participated in the synthesis of both the monomer and polymer. Mingxu Sun also participated in the data analysis and contributed to the revision of the manuscript. Biao Xiao was responsible for mechanical tests. Hongqian Sang calculated the interaction between two tetramers. Hanxu Liu performed the triplet state calculation under the supervision of Zhenggang Lan. Zhenxing Wang was responsible for the analysis of VT-EPR results. You Song was responsible for SQUID analyses. Youbing Mu and Xiaobo Wan conceived the idea, supervised the experimental work, and analyzed the data. Xiaobo Wan wrote and revised the article. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC 22075105 and NSFC 22371093), the Knowledge Innovation Program of Wuhan-Basic Research (2022010801010372), the Hubei Provincial Natural Science Foundation of China (2023AFB1030, 2024AFA031) and the Excellent Discipline Cultivation Project by Jianghan University (2023XKZ035). We also thank the helpful discussions and suggestions from Prof. Yuan Li at South China University of Technology and Prof. Jian Pei at Peiking University.References
- Y. V. Korshak, T. V. Medvedeva, A. A. Ovchinnikov and V. N. Spector, Organic polymer ferromagnet, Nature, 1987, 326, 370–372 CrossRef CAS.
- H. Nishide, Organic redox polymers as electrochemical energy materials, Green Chem., 2022, 24, 4650–4679 RSC.
- X. Hu, W. Wang, D. Wang and Y. Zheng, The electronic applications of stable diradicaloids: present and future, J. Mater. Chem. C, 2018, 6, 11232–11242 RSC.
- A. J. Wingate and B. W. Boudouris, Recent advances in the syntheses of radical-containing macromolecules, J. Polym. Sci., Part A:Polym. Chem., 2016, 54, 1875–1894 CrossRef CAS.
- T. Endo, K. Takuma, T. Takata and C. Hirose, Synthesis and polymerization of 4-(glycidyloxy)-2,2,6,6-tetramethylpiperidine-1-oxyl, Macromolecules, 1993, 26, 3227–3229 CrossRef CAS.
- Y. Joo, V. Agarkar, S. H. Sung, B. M. Savoie and B. W. Boudouris, A nonconjugated radical polymer glass with high electrical conductivity, Science, 2018, 359, 1391–1395 CrossRef CAS PubMed.
- H. Nishide, N. Yoshioka, K. Inagaki and E. Tsuchida, Poly [(3,5-di-tert-butyl-4-hydroxyphenyl)acetylene]: formation of a conjugated stable polyradical, Macromolecules, 1988, 21, 3119–3120 CrossRef CAS.
- M. Miyasaka, T. Yamazaki, E. Tsuchida and H. Nishide, Regioregular polythiophene with pendant phenoxyl radicals: a new high-spin organic polymer, Macromolecules, 2000, 33, 8211–8217 CrossRef CAS.
- M. Otaki and H. Goto, Helical Spin Polymer with Magneto-Electro-Optical Activity, Macromolecules, 2019, 52, 3199–3209 CrossRef CAS.
- A. Rajca, S. Rajca and J. Wongsriratanakul, Very high-spin organic polymer: π-conjugated hydrocarbon network with average spin of S ≥40, J. Am. Chem. Soc., 1999, 121, 6308–6309 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Magnetic Ordering in an Organic Polymer, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- S. Wu, M. Li, H. Phan, D. Wang, T. S. Herng, J. Ding, Z. Lu and J. Wu, Toward Two-Dimensional π-Conjugated Covalent Organic Radical Frameworks, Angew. Chem., Int. Ed., 2018, 57, 8007–8011 CrossRef CAS.
- J. Wang, G. Kim, M. E. Sandoval-Salinas, H. Phan, T. Y. Gopalakrishna, X. Lu, D. Casanova, D. Kim and J. Wu, Stable 2D anti-ferromagnetically coupled fluorenyl radical dendrons, Chem. Sci., 2018, 9, 3395–3400 RSC.
- S. Kimura, M. Uejima, W. Ota, T. Sato, S. Kusaka, R. Matsuda, H. Nishihara and T. Kusamoto, An Open-shell, Luminescent, Two-Dimensional Coordination Polymer with a Honeycomb Lattice and Triangular Organic Radical, J. Am. Chem. Soc., 2021, 143, 4329–4338 CrossRef CAS PubMed.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Polythiophene: from fundamental perspectives to applications, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- H. Koizumi, H. Dougauchi and T. Ichikawa, Mechanism of dedoping processes of conducting poly(3-alkylthiophenes), J. Phys. Chem. B, 2005, 109, 15288–15290 CrossRef CAS PubMed.
- Z. X. Chen, Y. Li and F. Huang, Persistent and stable organic radicals: design, synthesis, and applications, Chem, 2021, 7, 288–332 CAS.
- S. Dong and Z. Li, Recent progress in open-shell organic conjugated materials and their aggregated states, J. Mater. Chem. C, 2022, 10, 2431–2449 RSC.
- S. Moles Quintero, M. M. Haley, M. Kertesz and J. Casado, Polycyclic cydrocarbons from [4n]annulenes: correlation versus hybridization forces in the formation of diradicaloids, Angew. Chem., Int. Ed., 2022, 61, e202209138 CrossRef CAS PubMed.
- J. Guo, Y. Yang, C. Dou and Y. Wang, Boron-containing organic diradicaloids: dynamically modulating singlet diradical character by Lewis acid–base coordination, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS.
- T. Jousselin-Oba, M. Mamada, J. Marrot, A. Maignan, C. Adachi, A. Yassar and M. Frigoli, Excellent semiconductors based on tetracenotetracene and pentacenopentacene: from stable closed-shell to singlet open-shell, J. Am. Chem. Soc., 2019, 141, 9373–9381 CrossRef CAS PubMed.
- H. Hayashi, J. E. Barker, A. Cárdenas Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S.-i. Kato, M. M. Haley and J. Casado, Monoradicals and diradicals of dibenzofluoreno[3,2-b]fluorene isomers: mechanisms of electronic delocalization, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS.
- K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. R. Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, Stable organic diradicals based on fused quinoidal oligothiophene imides with high electrical conductivity, J. Am. Chem. Soc., 2020, 142, 4329–4340 CrossRef CAS.
- Y. Kim, Y.-J. Kim, Y.-A. Kim, E. Jung, Y. Mok, K. Kim, H. Hwang, J.-J. Park, M.-G. Kim, S. Mathur and D.-Y. Kim, Open-Shell and Closed-Shell Quinoid–Aromatic Conjugated Polymers: Unusual Spin Magnetic and High Charge Transport Properties, ACS Appl. Mater. Interfaces, 2021, 13, 2887–2898 CrossRef CAS.
- Y. Li, L. Li, Y. Wu and Y. Li, A Review on the Origin of Synthetic Metal Radical: Singlet Open-Shell Radical Ground State?, J. Phys. Chem. C, 2017, 121, 8579–8588 CrossRef CAS.
- J.-H. Kim, A. Nizami, Y. Hwangbo, B. Jang, H.-J. Lee, C.-S. Woo, S. Hyun and T.-S. Kim, Tensile testing of ultra-thin films on water surface, Nat. Commun., 2013, 4, 2520 CrossRef PubMed.
- T. Zhong, F. Guo, S. Lei, B. Xiao, Q. Li, T. Jia, X. Wang and R. Yang, Multi-scale mechanical properties of bulk-heterojunction films in polymer solar cells, npj Flexible Electron., 2023, 7, 2 CrossRef CAS.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Flexible, highly efficient all-polymer solar cells, Nat. Commun., 2015, 6, 8547 CrossRef CAS.
- L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, Stable borocyclic radicals via frustrated Lewis pair hydrogenations, J. Am. Chem. Soc., 2016, 138, 2500–2503 CrossRef CAS PubMed.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Diindeno-fusion of an anthracene as a design strategy for stable organic biradicals, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed.
- Y. B. Borozdina, E. A. Mostovich, P. T. Cong, L. Postulka, B. Wolf, M. Lang and M. Baumgarten, Spin-dimer networks: engineering tools to adjust the magnetic interactions in biradicals, J. Mater. Chem. C, 2017, 5, 9053–9065 RSC.
- K. Kang, S. Watanabe, K. Broch, A. Sepe, A. Brown, I. Nasrallah, M. Nikolka, Z. Fei, M. Heeney, D. Matsumoto, K. Marumoto, H. Tanaka, S.-i. Kuroda and H. Sirringhaus, 2D coherent charge transport in highly ordered conducting polymers doped by solid state diffusion, Nat. Mater., 2016, 15, 896–902 CrossRef CAS.
- X. Ji, H. Xie, C. Zhu, Y. Zou, A. U. Mu, M. Al-Hashimi, K. R. Dunbar and L. Fang, Pauli paramagnetism of stable analogues of pernigraniline salt featuring ladder-type constitution, J. Am. Chem. Soc., 2020, 142, 641–648 CrossRef CAS PubMed.
- X.-X. Chen, J.-T. Li, Y.-H. Fang, X.-Y. Deng, X.-Q. Wang, G. Liu, Y. Wang, X. Gu, S.-D. Jiang and T. Lei, High-mobility semiconducting polymers with different spin ground states, Nat. Commun., 2022, 13, 2258 CrossRef CAS PubMed.
- A. Sokolowski, J. Müller, T. Weyhermüller, R. Schnepf, P. Hildebrandt, K. Hildenbrand, E. Bothe and K. Wieghardt, Phenoxyl radical complexes of zinc(II), J. Am. Chem. Soc., 1997, 119, 8889–8900 CrossRef CAS.
- Z. Wang, J. Zhou, Y. Zhang, W. Zhu and Y. Li, Accessing highly efficient photothermal conversion with stable open-shell aromatic nitric acid radicals, Angew. Chem., Int. Ed., 2022, 61, e202113653 CrossRef CAS PubMed.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, New porous crystals of extended metal-catecholates, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- Q. Chen, O. Adeniran, Z.-F. Liu, Z. Zhang and K. Awaga, Graphite-like Charge Storage Mechanism in a 2D π–d Conjugated Metal–Organic Framework Revealed by Stepwise Magnetic Monitoring, J. Am. Chem. Soc., 2023, 145, 1062–1071 CrossRef CAS PubMed.
- R. C. Haddon, S. V. Chichester and J. H. Marshall, Electron delocalization in 9-oxidophenalenone complexes of boron and beryllium, Tetrahedron, 1986, 42, 6293–6300 CrossRef CAS.
- J. Wang, H. Cui, H. Ruan, Y. Zhao, Y. Zhao, L. Zhang and X. Wang, The Lewis acid induced formation of a stable diradical with an intramolecular ion pairing state, J. Am. Chem. Soc., 2022, 144, 7978–7982 CrossRef CAS.
- X. Dong, Q.-C. Luo, Y. Zhao, T. Wang, Q. Sun, R. Pei, Y. Zhao, Y.-Z. Zheng and X. Wang, A dynamic triradical: synthesis, crystal structure, and spin frustration, J. Am. Chem. Soc., 2023, 145, 17292–17298 CrossRef CAS PubMed.
- G. M. Coppinger, A stable phenoxy radical inert to oxygen, J. Am. Chem. Soc., 1957, 79, 501–502 CrossRef CAS.
- E. V. Canesi, D. Fazzi, L. Colella, C. Bertarelli and C. Castiglioni, Tuning the quinoid versus biradicaloid character of thiophene-based heteroquaterphenoquinones by means of functional groups, J. Am. Chem. Soc., 2012, 134, 19070–19083 CrossRef CAS PubMed.
- G. Li, H. Phan, T. S. Herng, T. Y. Gopalakrishna, C. Liu, W. Zeng, J. Ding and J. Wu, Toward stable superbenzoquinone diradicaloids, Angew. Chem., Int. Ed., 2017, 56, 5012–5016 CrossRef CAS PubMed.
- W. Wang, L. Ge, G. Xue, F. Miao, P. Chen, H. Chen, Y. Lin, Y. Ni, J. Xiong, Y. Hu, J. Wu and Y. Zheng, Fine-tuning the diradical character of molecular systems via the heteroatom effect, Chem. Commun., 2020, 56, 1405–1408 RSC.
- G. Li, Y. Han, Y. Zou, J. J. C. Lee, Y. Ni and J. Wu, Dearomatization approach toward a superbenzoquinone-based diradicaloid, tetraradicaloid, and hexaradicaloid, Angew. Chem., Int. Ed., 2019, 58, 14319–14326 CrossRef CAS PubMed.
- Y. Xue, P. Guo, H.-L. Yip, Y. Li and Y. Cao, General design of self-doped small molecules as efficient hole extraction materials for polymer solar cells, J. Mater. Chem. A, 2017, 5, 3780–3785 RSC.
- C. Chen, Q. Liang, Z. Chen, W. Zhu, Z. Wang, Y. Li, X. Wu and X. Xiong, Phenoxy radical-induced formation of dual-layered protection film for high-rate and dendrite-free lithium-metal anodes, Angew. Chem., Int. Ed., 2021, 60, 26718–26724 CrossRef CAS PubMed.
- H. Wei, L. Zhang, H. Phan, X. Huang, T. S. Herng, J. Zhou, W. Zeng, J. Ding, S. Luo, J. Wu and Z. Zeng, A stable N-annulated perylene-bridged bisphenoxyl diradicaloid and the corresponding boron trifluoride complex, Chem. Eur. J., 2017, 23, 9419–9424 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06910b |
| This journal is © The Royal Society of Chemistry 2025 |