
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolving the relationship between capacity/voltage decay and the phase transition by accelerating the layered to spinel transition†
Qi
Pang
a,
Mengke
Zhang
a,
Yang
Song
 a,
Yueying
Liu
a,
Manqi
Tang
a,
Sunqi
Su
b,
Lang
Qiu
*a,
Yao
Xiao
*bc and
Xiaodong
Guo
*a
a,
Yueying
Liu
a,
Manqi
Tang
a,
Sunqi
Su
b,
Lang
Qiu
*a,
Yao
Xiao
*bc and
Xiaodong
Guo
*a
aSchool of Chemical Engineering, Sichuan University, Chengdu 610065, China. E-mail: qiulang2023@scu.edu.cn; xiaodong2009@163.com
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, China. E-mail: xiaoyao@wzu.edu.cn
cKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
First published on 25th January 2025
Abstract
Lithium-rich cathode materials are some of the most promising choices for lithium-ion batteries due to their excellent energy density (>900 W h kg−1). However, severe voltage/capacity degradation during cycling has seriously hindered the further commercialization of lithium-rich cathode materials. Current research efforts are focused on enhancing their voltage and capacity retention. Here, the coating of FeF3 on specific crystal planes is utilized to achieve a degradation trend that is very different from that of the as-received material. Using this as an entry point, the relationship between voltage and capacity degradation was studied in depth. The oriented coated material undergoes a more drastic phase transition during cycling, yet its voltage decay remains basically the same as that of the original sample (769.6 mV after 200 cycles, compared to 723.5 mV for the original sample). Notably, the capacity retention rate is significantly improved (97% after 200 cycles vs. 75% for the pristine material). These findings suggest that the capacity degradation and the voltage decay do not interact with each other and that the phase transition during cycling does not seem to negatively affect the voltage. This conclusion can also be extrapolated to other oxygen-reducing oxide systems to help understand the relationship between capacity and voltage decay. The modification is generalized and applicable to other cathode materials.
Introduction
Nowadays, in the face of problems such as environmental pollution and climate warming caused by the overexploitation of fossil energy sources around the world, there is an urgent need to replace non-renewable fossil energy sources with renewable energy sources. Therefore, researchers are looking for ways to improve the efficiency of renewable energy sources. In the current field of cathode materials, Li-rich manganese-based cathode materials (LRMs) with the chemical formula Li1+xTM1−xO2 (LLOs, TM = Ni, Co, Mn, etc.) have emerged as the most promising cathode materials due to their high specific capacity over 250 mA h g−1.1–5 Compared with traditional lithium-ion battery cathode materials (e.g. LiCoO2, LiFePO4, etc.), which realize chemical energy storage through the redox of metal cations, LRMs can take advantage of the unique Li–O–Li structural features to achieve the oxidation–reduction of oxygen atoms and thus express an additional capacity contribution on the basis of metal ion redox. However, its inherent low first-time coulombic efficiency and voltage/capacity decay during cycling not only lead to the continuous decrease of battery energy density, but also seriously hinder the further commercialization and application of LRMs.6Many studies have shown that capacity decay is related to various structural degradations caused by oxygen loss, including layered-to-spinel/rock phase transitions, cracks, and surface side reactions.7–9 Voltage decay is related to the occurrence of low-potential redox pairs during cycling, layered-to-spinel structural transition, and increased interfacial resistance. Hu et al.10 found that the voltage plateau of LRMs continued to decline during cycling despite little change in capacity. This suggests that the capacity compensation effect between elements can maintain the overall capacity to a certain extent, but the voltage decay is not effectively controlled, implying that the relationship between voltage decay and capacity decay is not simply linear. In contrast, Sun et al.11 pointed out that irreversible lattice oxygen loss exacerbates structural deformation and phase transition, leading to simultaneous voltage and capacity decay. This observation emphasizes the coupled relationship between voltage decay and capacity decay, i.e., the dual effect of structural changes on both. Thus, these two perspectives demonstrate a significantly different relationship between voltage and capacity decay: Hu et al.10 focused on the compensating effect of capacity on voltage, while Sun et al.11 emphasized the synchronous decay triggered by structural deformation. However, the existing literature provides no clear explanation for the correlation between capacity and voltage decay. Therefore, further research is required to analyze the relationship between voltage and capacity decay, in order to reveal their intrinsic connection and interaction mechanism.
Herein, we prepared two Li-rich manganese-based cathode materials with different capacity/voltage decays to reveal the relationship between capacity decay and voltage decay. Modified materials (referred to as LFF3) were obtained by coating the pristine materials (referred to as LRMs) with FeF3. First, a premise is considered that the introduced substance does not affect the original internal structure of the material, and due to the low content, the chemical reaction involved does not have an effect on the overall material. Characterization by XRD, XPS, Raman spectroscopy etc., combined with electrochemical performance analysis, revealed that the presence of the spinel phase in LFF3 contributes to stable capacity retention (97% after 200 cycles), representing a significant improvement over the 75% capacity retention observed in the LRM. After cycling, the spinel phase content in the LRM and LFF3 was 1.03% and 9.34%, respectively. However, the voltage retention was close, 78.36% for the LRM and 79.62% for LFF3. The results confirm that there is no clear linear relationship between voltage decay and capacity decay for LRMs and break the traditional concept that the transition from a layered to a spinel structure does affect the voltage decay. This work could help understand the relationship between capacity and voltage decay in other oxide systems containing oxygen redox and facilitated the development of high-performance cathode designs.
Results and discussion
Structures of the LRM and LFF3
Fig. 1a shows primary particles of LFF3 with FeF3 coated on its specific crystal surface. Fig. 1b and c show the XRD patterns of the LRM and LFF3. The samples show good crystallinity. A clear peak of the hexagonal a-NaFeO2 structure belonging to the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m can be observed. Several weak peaks located at ≈20–25° belong to the monoclinic Li2MnO3 phase with the space group C2/m, which has a unique superlattice honeycomb-ordering structure.12,13 No peaks for FeF3 are found in the X-ray Powder Diffraction (XRD) patterns of LFF3 due to the relatively low amount of the FeF3 phase (3%).14 In addition, the (006)/(102) and (108)/(110) diffraction peaks for all samples are well split, signifying no disruption of the higher ordered layered structure after the surface modification treatment.15 The I(003)/I(104) values of all samples are greater than 1.2.16 Detailed refined lattice parameters are listed in Table S1.† The results show that both samples have similar cell parameters and phase occupancy, which suggests that the crystal structure of LFF3 has not been changed.17 In order to further explore the surface chemical states and elemental valence states of the materials, X-ray photoelectron spectroscopy (XPS) analyses of Ni 2p, Mn 2p, O 1s, F 1s and Fe 2p were performed. The specific information is in Fig. S1† and the results show that the Mn valence states of the LRM and LFF3 remain basically the same, indicating that the modification does not lead to a change in the chemical state of Mn3+.18 And the binding energy peak belonging to Fe–F in FeF3 was found in Fe 2p (Fig. S1e†).14 Raman spectroscopy is sensitive to short-range ordering, which is favorable for phase identification, so this test was also carried out to detect the local structure. The results of the Raman spectra of the LRM and LFF3 are shown in Fig. 1d and e. The peaks near 477 cm−1 and 595 cm−1 belong to the Eg and A1g vibrations of the Rm space group; in addition, no peaks appeared near 650 cm−1, suggesting that the spinel phase hardly exists before and after coating.19
m can be observed. Several weak peaks located at ≈20–25° belong to the monoclinic Li2MnO3 phase with the space group C2/m, which has a unique superlattice honeycomb-ordering structure.12,13 No peaks for FeF3 are found in the X-ray Powder Diffraction (XRD) patterns of LFF3 due to the relatively low amount of the FeF3 phase (3%).14 In addition, the (006)/(102) and (108)/(110) diffraction peaks for all samples are well split, signifying no disruption of the higher ordered layered structure after the surface modification treatment.15 The I(003)/I(104) values of all samples are greater than 1.2.16 Detailed refined lattice parameters are listed in Table S1.† The results show that both samples have similar cell parameters and phase occupancy, which suggests that the crystal structure of LFF3 has not been changed.17 In order to further explore the surface chemical states and elemental valence states of the materials, X-ray photoelectron spectroscopy (XPS) analyses of Ni 2p, Mn 2p, O 1s, F 1s and Fe 2p were performed. The specific information is in Fig. S1† and the results show that the Mn valence states of the LRM and LFF3 remain basically the same, indicating that the modification does not lead to a change in the chemical state of Mn3+.18 And the binding energy peak belonging to Fe–F in FeF3 was found in Fe 2p (Fig. S1e†).14 Raman spectroscopy is sensitive to short-range ordering, which is favorable for phase identification, so this test was also carried out to detect the local structure. The results of the Raman spectra of the LRM and LFF3 are shown in Fig. 1d and e. The peaks near 477 cm−1 and 595 cm−1 belong to the Eg and A1g vibrations of the Rm space group; in addition, no peaks appeared near 650 cm−1, suggesting that the spinel phase hardly exists before and after coating.19
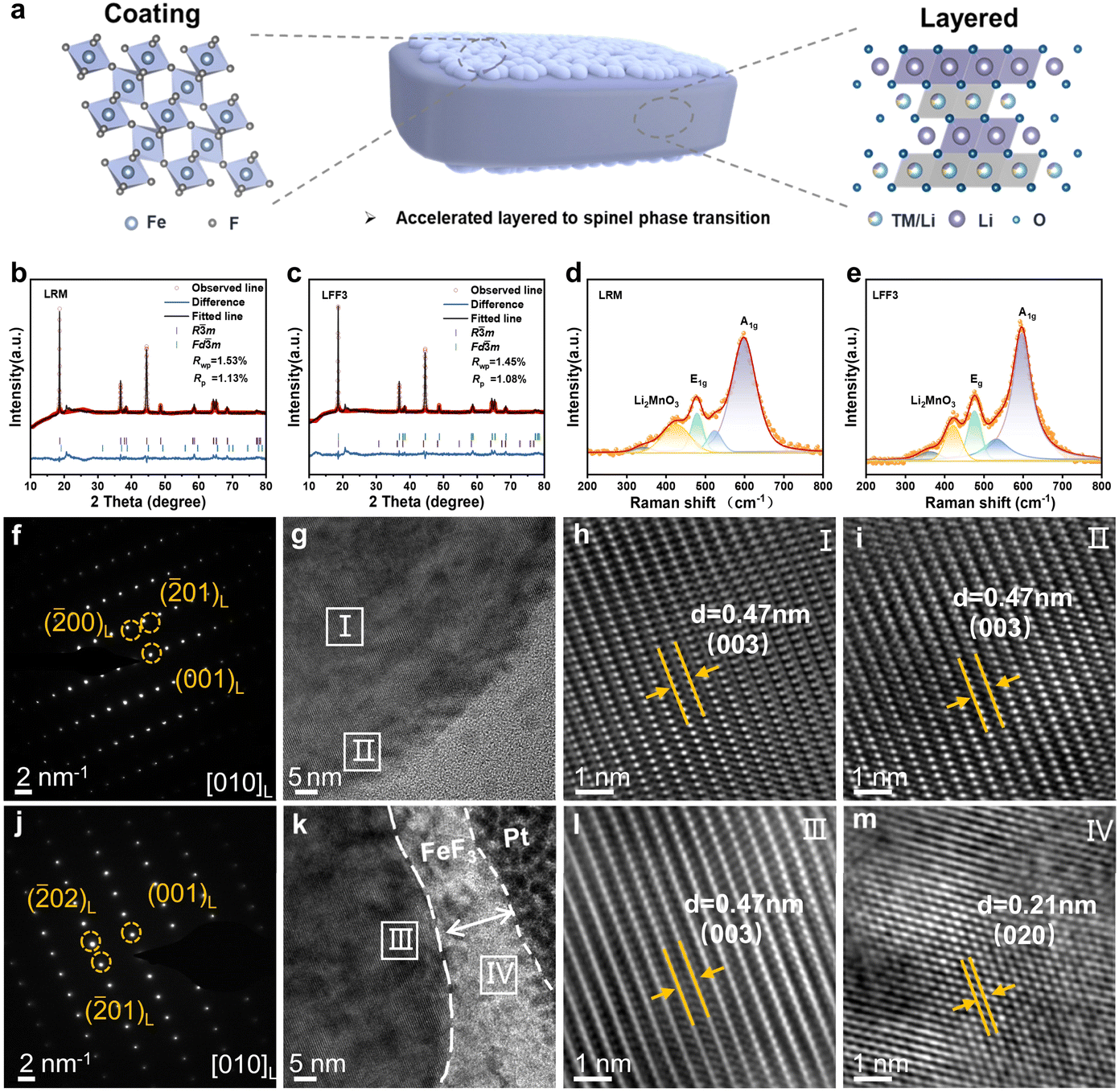 | ||
| Fig. 1 Structural characterization of the LRM and LFF3. (a) Schematic of primary particles of LFF3. The XRD pattern of (b) LRM and (c) LFF3 with Rietveld refinement. Raman spectra of (d) LRM and (e) LFF3. (f) SAED, (g) TEM, and (h and i) HRTEM images of the LRM. (j) SAED, (k) TEM, and (l and m) HRTEM images of LFF3. | ||
The surface morphology of the LRM and LFF3 samples was investigated by scanning electron microscopy (SEM). As shown in Fig. S2,† all samples exhibit spherical secondary particles, which are composed of aggregated primary particles, with an approximate size of 5 μm. The LFF3 surface (001) crystal plane is rough with the presence of a coating layer (Fig. S2b†) compared to the LRM (Fig. S2a†). The SEM mapping plots of LRM and LFF3 particles are shown in Fig. S3 and S4.† The changes in the surface crystal structure after surface modification were explored by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) characterization along the [010] band axis direction as shown in Fig. 1f–j. Fig. 1g–k show the HRTEM images of the LRM and LFF3 extracted from Fig. S5†, respectively. By fast Fourier transform (FFT) analysis (local FFT of the rectangular region in Fig. 1g), it can be found that both inside and outside of the LRM (Fig. 1h and i) exhibit a planar spacing of 0.47 nm, which corresponds to the layer structure with the Rm space group (003) surface. In contrast, crystals with an average thickness of ≈12 nm were found attached to the surface of FeF3 particles in LFF3 (Fig. 1k).The interior of the LFF3 particles (Fig. 1l) remains a layer structure with a spacing of 0.47 nm, corresponding to the (003) crystal plane of Rm, and the crystals on the surface (Fig. 1m) correspond to the (020) crystal plane of FeF3 (plane spacing of 0.21 nm).14,20
This orientated coating may impose additional stresses on the material surface, thus affecting the phase transition pathways of the material. In addition to this, microstructural variations can have a significant impact on the electrochemical behavior of the materials. In order to fully understand the practical impact on performance, the focus is shifted to the evaluation of electrochemical performance, exploring how structural differences affect the capacity and voltage stability of the materials during cycling.
Electrochemical performance
Subtle differences in microstructures result in the LRM and LFF3 exhibiting different electrochemical performances. Coin cells are assembled and measured in the voltage range of 2.0–4.8 V (relative to Li/Li+) at 30 °C to evaluate the electrochemical performance. Fig. 2a shows the initial charging/discharging curves of the LRM and LFF3. The long platform period observed at 4.5 V corresponds to the electrochemical activation process of Li2MnO3, while the sloped region below 4.5 V represents the oxidation process of transition metals.21 It is noteworthy that during the first charging of LFF3, the potential increases rapidly to 4.5 V, followed by a distinct long platform corresponding to an oxygen redox providing a capacity of 197.96 mA h g−1, and this fraction of the oxygen in the LRM provides a capacity of 190.68 mA h g−1. Since the redox behavior of the lattice oxygen is not fully reversible at high pressures (>4.5 V), the release of oxygen from the lattice results in the formation of oxygen vacancies, which greatly reduces the diffusion energy barrier of TM, accelerates the migration of TM, and ultimately accelerates the phase transition process.22 LFF3 exhibits a higher discharge capacity and a higher initial coulombic efficiency of 76.88% compared to 64.51% for the LRM.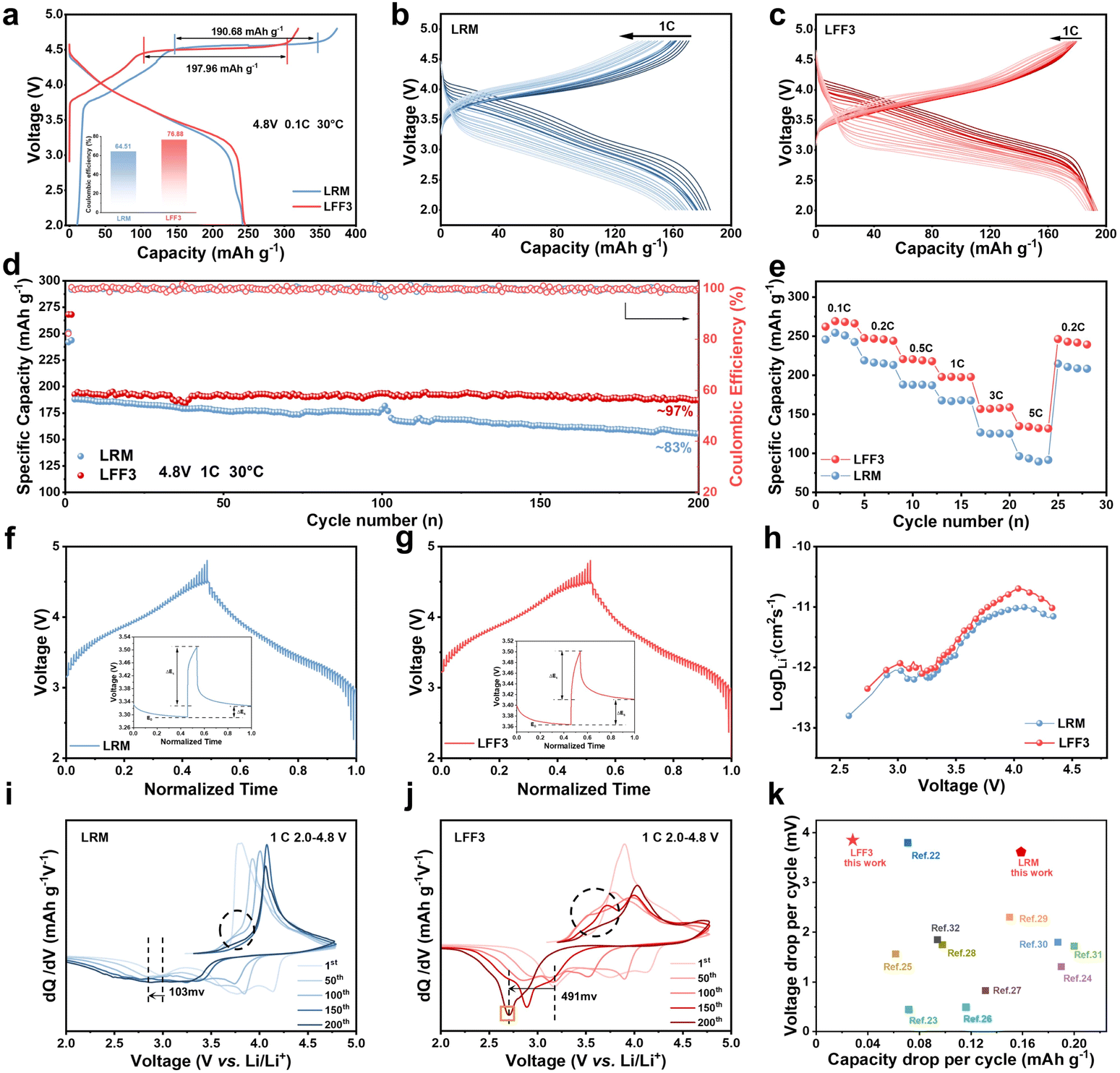 | ||
| Fig. 2 Electrochemical performance of the LRM and LFF3. (a) The initial charging and discharging curves for the LRM and LFF3. Capacity–voltage curve for 1–200 cycles for (b) LRM and (c) LFF3 at 1C after two turns of activation at 0.1C. (d) Cycling performance at 1C for the LRM and LFF3. (e) Rate capability. (f and g) The galvanostatic intermittent titration technique (GITT) curves of the LRM and LFF3 samples measured during the charge–discharge process. (h) The calculated lithium diffusion coefficient. The corresponding dQ/dV curves of (i) LRM and (j) LFF3 cathode at different stages of cycling at a 1C rate. (k) Comparison of the LRM, LFF3 and the literature. | ||
As shown in Fig. 2b and c, there is no significant difference in voltage degradation of LFF3 after coating compared to the LRM, but the capacity degradation is more significantly improved. The cycling performance of the LRM and LFF3 was further compared in the voltage interval of 2.0–4.8 V at 1C. As shown in Fig. 2d, the difference in the discharge capacity between the LRM and LFF3 was not large in the first cycle at 1C. After 200 cycles, the discharge capacity of the LRM decreased from 187.83 mA h g−1 to 155.01 mA h g−1, with a capacity retention of 75%, while the discharge capacity of LFF3 decreased from 192.48 mA h g−1 to 187.04 mA h g−1, with a capacity retention of 97%. It is noteworthy that although the LRM and LFF3 show a great difference in capacity retention, they exhibit nearly the same decay in voltage. Fig. S6† shows the discharge curves of the LRM and LFF3 in the 1st, 50th, 100th, 150th, and 200th cycles at 1C, respectively. The horizontal coordinates are normalized so that the voltage–capacity curves of the two materials are compared on a unified scale, thus showing more clearly the difference in the performance of the materials in the voltage range. After 200 cycles, the voltage drop is 769.6 mV for the LRM and 723.5 mV for LFF3. For a comprehensive comparison of cycling stability and voltage decay, the energy density of the LRM and LFF3 over 200 cycles was plotted (Fig. S7†). The data show that the energy retention of LFF3 (80%) is higher than that of the LRM (66%), and accordingly, the energy density loss decreases from 224 W h kg−1 to 127 W h kg−1. And the energy efficiency of LFF3 is significantly higher than that of the LRM throughout the cycle (Fig. S8†).
The rate capability of the two materials is shown in Fig. 2e and S9,† which confirms the better rate capability of LFF3 compared to the pristine material. The discharge specific capacities of LFF3 at 0.1C, 0.2C, 0.5C, 1C, 3C, and 5C are 266.25 mA h g−1, 245.25 mA h g−1, 219.36 mA h g−1, 197.67 mA h g−1, 157.51 mA h g−1, and 133.07 mA h g−1. Additionally, even after experiencing a high discharge rate of 5C, the specific discharge capacity of LFF3 was able to recover 98.85% of its initial capacity at 1C. To explore the kinetic properties of the LRM and LFF3 samples, the corona electrostatic intermittent titration technique (GITT) was employed. The results show that LFF3 has a higher Li+ diffusion rate (Fig. 2f–h). This result is consistent with the results analyzed above; the introduction of FeF3 accelerates the phase transition from the layered structure to the spinel phase during cycling, contributing to improved Li+ diffusion and electrochemical performance.23Fig. 2i and j show the dQ/dV plots of the LRM and LFF3 at 1C, respectively, further explaining the role of phase transition on stability. The anodic peaks between 2.5 V and 3.5 V correspond to the reduction of Mn4+ to Mn3+.24 The anodic peak between 3.5 V and 4.5 V corresponds to Ni4+ reduction to Ni2+. In addition, LFF3 shows a new peak at ∼2.7 V (orange arrow) during cycling, which is a distinctive feature of low-voltage spinel phase formation. As cycling proceeds, the reduction peak of Mn4+/Mn3+ is gradually shifted to lower voltages, which is widely believed to be related to the structural transition from layered to spinel phases.25,26Fig. 2i shows that the reduction peak of the LRM located at 2.983 V shifted to 2.880 V (Δ = 103 mV) after 200 cycles, whereas a much larger potential shift clearly occurred for LFF3 (Fig. 2j), Δ = 491 mV (from 3.172 V to 2.681 V). As the cycle proceeds, LFF3 shows an oxidation peak near 3.25–3.75 V, which is related to the phase transition, while the peak here is significantly lower in the LRM, suggesting that LFF3 undergoes a more severe phase transition process during cycling.20 As shown in Fig. 2k, the modified material did not suppress the voltage degradation, but its capacity per cycle degradation is excellent in the literature.27–36 The phase transition resulted in the formation of a larger amount of spinel phase. Due to the highly reversible nature of the manganese redox process within the spinel structure, the formation of the spinel phase in the modified sample provided an additional contribution to the overall capacity.
The surface chemical environment
Following the acquisition of electrical performance data, further research will be conducted on structural degradation with the objective of decoupling the relationship between structure and performance. In order to explore the changes in the phases of the material after cycling, X-ray diffraction (XRD) was used to probe the material post-cycling. Fig. 3a and b show the Rietveld refinement results of the XRD patterns of the LRM and LFF3 after 200 cycles (the detailed cell parameters are given in Tables S2 and S3†). It is clear that the XRD profiles are well-fitted based on the three-phase model (layered, spinel and rock salt phase structures). The results show that after 200 cycles, the main phases in both the LRM and LFF3 are still layered (Fig. 3c) and the proportions of spinel and rock salt phases in the LRM are 1.03% and 3.04%, respectively, while the proportions of spinel and rock salt phases in LFF3 are 9.34% and 2.33%, which suggests that LFF3 undergoes a more severe phase transformation during cycling, with more layered phases transforming into spinel phases.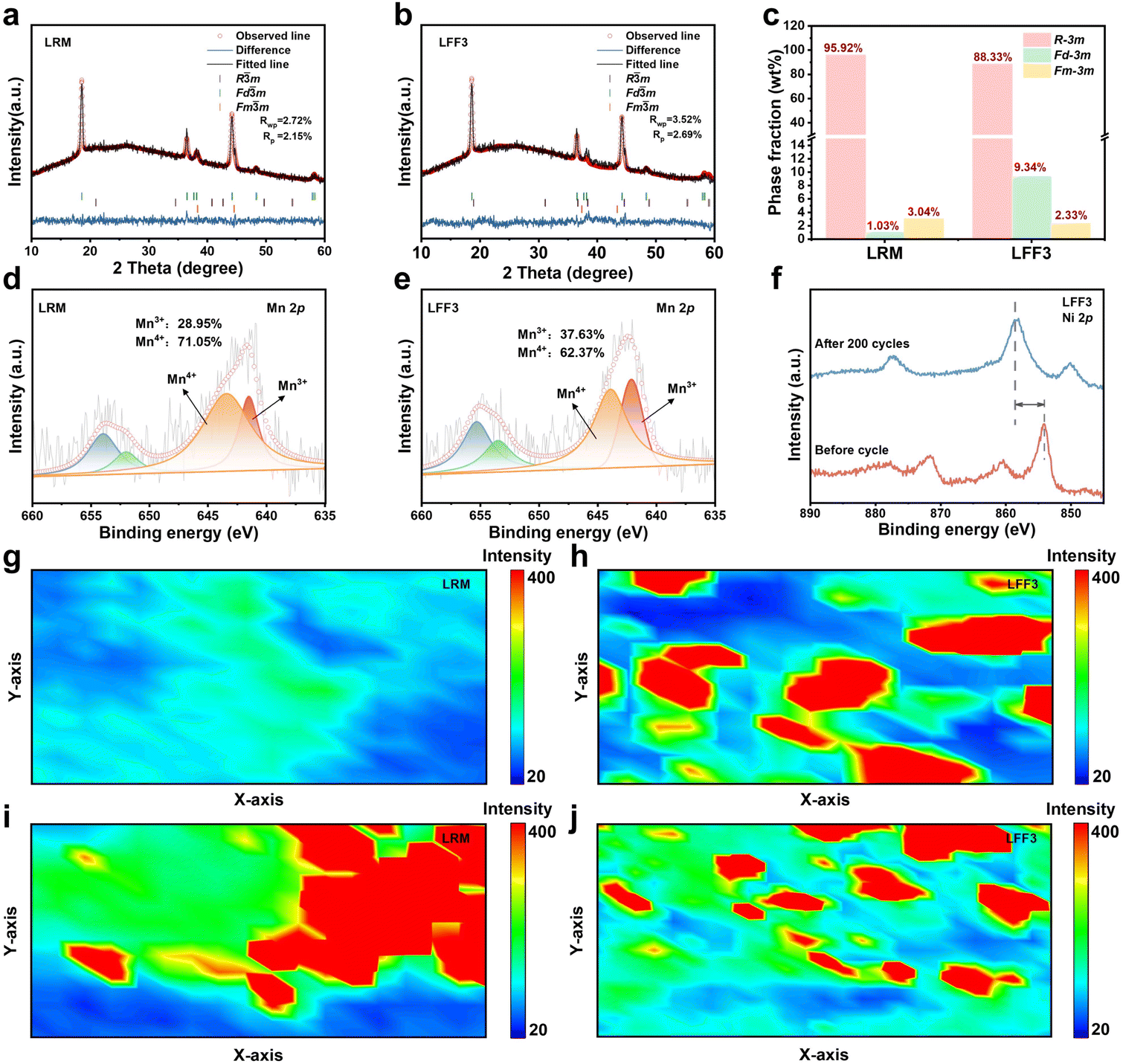 | ||
| Fig. 3 The surface chemical environment of the LRM and LFF3. XRD patterns of (a) LRM and (b) LFF3 with Rietveld refinement after 200 cycles. (c) The phase fraction (wt%) of the LRM and LFF3. Mn 2p XPS spectra of (d) LRM and (e) LFF3. (f) Ni 2p XPS spectra of LFF3. Raman mapping at 630.91 cm−1 of (g) LRM and (h) LFF3 after 200 cycles. Raman mapping at 595.98 cm−1 of (i) LRM and (j) LFF3 after 200 cycles. | ||
The change in elemental valence can reflect the stability of transition metal ions. The valence and phase composition changes of Mn after 200 cycles were investigated by XPS and Raman spectroscopy, respectively. Fig. 3d and e show the Mn 2p XPS spectra of the LRM and LFF3 after cycling, respectively. By comparing the ratio of Mn4+ and Mn3+ peak areas in the Mn 2p spectra, the percentage of Mn3+ increased from 28.48% to 28.95% for LRM and from 27.78% to 37.63% for LFF3. For Ni 2p, both the LRM (Fig. S10†) and LFF3 (Fig. 3f) samples moved to higher binding energies after 200 cycles. However, LFF3 changed more, indicating an increase in the oxidation state after cycling. This change in the Ni oxidation state may be a combination of the Mn oxidation state and oxygen loss.37,38 Raman mapping (Fig. 3g and h) can clearly show that the spinel content in LFF3 is significantly higher than that in the LRM after cycling, and the distribution of the spinel phase is not uniform due to the directionality of the coating. Accordingly, the proportion of layered phases in the LRM after cycling is slightly higher than that in LFF3 and the distribution is relatively concentrated, whereas in LFF3, the distribution of layered phases is not as regular as that in the LRM due to oriented coating as shown in Fig. 3i and j (detailed Raman mapping is shown in Fig. S11†).39 Combined with the capacity retention analysis of the two materials, specifically, while the intensification of the phase transformation of the material may lead to changes in the internal structure of the material, this does not necessarily lead to a reduction in capacity. Instead, this intensified phase transformation may have introduced new, stable phases in the modified material or enhanced the structural stability of the material, thereby improving the capacity retention of the battery.
Structural evolution
After analyzing the material bulk phase, we delved into the evolution of the material bulk phase at the microscopic level. SEM and TEM images of the LRM and LFF3 after cycling provide more visual evidence of the structural degradation of the materials after cycling. The low magnification SEM images are shown in Fig. S12.† Apparently, the spherical particles of LFF3 remain more intact, but more cracks are present on the surface of the particles. This is because FeF3 is coated around specific crystal planes, which exacerbates the inhomogeneous expansion and contraction of the material during discharging, thus leading to the evolution of the phase structure.40This inhomogeneous expansion and contraction also lead to lattice distortion during cycling, resulting in severe nano-defects, as can be observed in the FIB-SEM cross-section images in, which obvious nanoscale pore structures are present inside the primary particles of both the LRM (Fig. 4a) and LFF3 (Fig. 4e), and such nano-defects limit the diffusion of Li+ within the grains and exacerbate the transformation of the layered into the spinel phase.41,42Fig. 4b shows a high-resolution transmission electron microscopy (HRTEM) image of LRM corresponding to the yellow-circled portion in Fig. 4a. The bulk phase exhibits a lattice stripe of 0.47 nm, indicating that the material still maintains a lamellar structure inside.23Fig. 4d shows the spinel phase on the surface of the particles corresponding to the [121] band axis, with a thickness of ∼6 nm. In contrast, the content of the irregular spinel phase is higher in LLF3. And Fig. 4f shows the TEM image of LFF3. It can be clearly observed that the atomic arrangement in the surface region is different from that of the inner region. The inner region (II) and surface region (IV) of the STEM image were zoomed in, and the corresponding fast Fourier transform (FFT) patterns are provided. Fig. 4g shows a hierarchical arrangement of TM atoms (bright spots) with a spacing of 4.7 Å, close to d003 in the LiTMO2 structure. The corresponding FFT patterns can be matched along the direction onto the layered structure. In contrast, the STEM image in Fig. 4h shows distinct TM atoms in the interplanar arrangement, and the FFT pattern matches the spinel [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] band axis along the direction. And the irregular spinel structure on its surface is as thick as 10 nm in some regions, and the presence of spinel phases (high-energy boundaries) at the grain boundaries promotes the dissolution of Mn and the migration of the TM to the octahedral sites of Li during cycling, accelerating the phase transition.43 The TEM results confirm that both the LRM and LFF3 have undergone some degree of structural evolution during cycling and that the degree of phase transformation of LFF3 is more drastic. Fig. 4i is able to show well the results of the phase transition of the internal particles after cycling.
10] band axis along the direction. And the irregular spinel structure on its surface is as thick as 10 nm in some regions, and the presence of spinel phases (high-energy boundaries) at the grain boundaries promotes the dissolution of Mn and the migration of the TM to the octahedral sites of Li during cycling, accelerating the phase transition.43 The TEM results confirm that both the LRM and LFF3 have undergone some degree of structural evolution during cycling and that the degree of phase transformation of LFF3 is more drastic. Fig. 4i is able to show well the results of the phase transition of the internal particles after cycling.
 | ||
| Fig. 4 Structural evolution of the LRM and LFF3. (a) The FIB-SEM image, (b) TEM images and (c and d) HRTEM images of the LRM. (e) The FIB-SEM images, (f) TEM images and (g and h) HRTEM images of LFF3. (i) Schematic diagram of the phase transition after cycling. | ||
Conclusions
In summary, a material LFF3 with a different voltage/capacity decay was constructed from the LRM by using FeF3 coated on specific crystal planes. The relationship between capacity decay, voltage decay, and the phase transition is revealed by analyzing the electrical properties and structure of both materials. This work emphasizes that there is no specific link between voltage and capacity decay for LRMs, and furthermore, the phase transition from layered to spinel phases during cycling does not seem to have an effect on voltage decay. Although LFF3 undergoes a more drastic phase transformation during cycling, it instead shows higher discharge capacity (268.06 mA g−1 at 0.1C) and superior capacity retention (97% after 200 cycles), but its voltage retention remains essentially unchanged (79.62% compared to 78.36% for LRM), and the relationship between the improved capacity retention, similarity in voltage retention, and intensified phase transformation suggests that there is no specific relationship between capacity retention, voltage retention and phase transformation. The relationship between the increase in capacity retention, similarity in voltage retention, and increase in the phase transition indicates that there is no positive correlation between voltage and capacity decay. This implies that the capacity retention can be maintained at a relatively stable level even if the voltage of the material decays during the cycling process. This finding suggests that voltage decay and capacity decay are two relatively independent phenomena and that phase transformation does not negatively affect the stability of voltage retention. Although the intensification of the phase transition changes the structure of the material, this change does not appear to have an impact on voltage stability while improving capacity retention. This discovery also provides a new modification idea for cathode materials such as lithium cobalt oxide and nickel–cobalt–aluminum oxide, which have a stable voltage retention rate but a fast capacity degradation, to improve their capacity retention rate while maintaining their excellent voltage retention rate. Further research should focus on these potential mechanisms to fully understand the relationship between voltage and capacity decay in order to optimize the long-term stability and performance of the materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Qi Pang: conceptualization, data curation, writing-original draft. Yang Song: supervision, funding acquisition. Yueying Liu and Sunqi Su: formal analysis. Mengke Zhang: investigation. Manqi Tang: validation. Lang Qiu and Yao Xiao: project administration, visualization, resources. Xiaodong Guo: supervision, writing-review & editing, funding acquisition.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by projects from the National Natural Science Foundation of China (22425804, U20A20145, 22108183, and 52402301), the Sichuan Science and Technology Program (2022YFG0124, 2022ZHCG0121, 2023NSFSC1098, 2024NSFTD0001, and 2024NSFSC1158), the Natural Science Foundation of Zhejiang Province (LQ23E020002), the Wenzhou Key Scientific and Technological Innovation Research Project (ZG2023053), the Wenzhou Natural Science Foundation (G20220019), the Sichuan Province Innovative Talent Funding Project for Postdoctoral Fellows (BX202306), and the Postdoctoral Fellowship Program of CPSF under Grant Number GZB20240476.Notes and references
- D. Luo, H. Zhu, Y. Xia, Z. Yin, Y. Qin, T. Li, Q. Zhang, L. Gu, Y. Peng, J. Zhang, K. M. Wiaderek, Y. Huang, T. Yang, Y. Tang, S. Lan, Y. Ren, W. Lu, C. M. Wolverton and Q. Liu, Nat. Energy, 2023, 8, 1078–1087 CrossRef CAS.
- W. Zuo, M. Luo, X. Liu, J. Wu, H. Liu, J. Li, M. Winter, R. Fu, W. Yang and Y. Yang, Energy Environ. Sci., 2020, 13, 4450–4497 RSC.
- M. Zhang, L. Qiu, W. Hua, Y. Song, Y. Deng, Z. Wu, Y. Zhu, B. Zhong, S. Chou, S. Dou, Y. Xiao and X. Guo, Adv. Mater., 2024, 36, 2311814 CrossRef CAS PubMed.
- G. Yang, X. Liang, S. Zheng, H. Chen, W. Zhang, S. Li and F. Pan, eScience, 2022, 2, 79–86 CrossRef.
- W. Hua, X. Yang, N. P. M. Casati, L. Liu, S. Wang, V. Baran, M. Knapp, H. Ehrenberg and S. Indris, eScience, 2022, 2, 183–191 CrossRef.
- W. He, W. Guo, H. Wu, L. Lin, Q. Liu, X. Han, Q. Xie, P. Liu, H. Zheng, L. Wang, X. Yu and D. L. Peng, Adv. Mater., 2021, 33, 2005937 CrossRef CAS PubMed.
- Q. Huang, X. Zhang, X. Lv, X. Wang, W. Wen, F. Wu, R. Chen and L. Li, Small, 2023, 19, 2302086 CrossRef CAS PubMed.
- L. Qiu, M. Zhang, Y. Song, Z. Wu, Y. F. Zhu, J. Zhang, D. Wang, H. Y. Hu, H. W. Li, H. R. Liu, X. B. Jia, J. Peng, S. Chen, Z. Yang, Y. Xiao and X. Guo, Carbon Energy, 2022, 5, e298 CrossRef.
- M. Saubanère, E. McCalla, J.-M. Tarascon and M.-L. Doublet, Energy Environ. Sci., 2016, 9, 984–991 RSC.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- J. Sun, C. Sheng, X. Cao, P. Wang, P. He, H. Yang, Z. Chang, X. Yue and H. Zhou, Adv. Funct. Mater., 2021, 32, 2110295 CrossRef.
- J. Zhang, F. Cheng, S. Chou, J. Wang, L. Gu, H. Wang, H. Yoshikawa, Y. Lu and J. Chen, Adv. Mater., 2019, 31, 1901808 CrossRef CAS PubMed.
- S. Huang, Y. Sun, T. Yuan, H. Che, Q. Zheng, Y. Zhang, P. Li, J. Qiu, Y. Pang, J. Yang, Z. F. Ma and S. Zheng, Carbon Neutralization, 2024, 3, 584–596 CrossRef CAS.
- Y. Chu, Y. Mu, L. Zou, Y. Hu, J. Cheng, B. Wu, M. Han, S. Xi, Q. Zhang and L. Zeng, Adv. Mater., 2023, 35, 2212308 CrossRef CAS PubMed.
- X. Gao, L. Wang, J. Guo, S. Li, H. Zhang, L. Chen, Y. Zhang, Y. Lai and Z. Zhang, Adv. Funct. Mater., 2024, 34, 2407692 CrossRef CAS.
- C. Shen, Y. Liu, L. Hu, W. Li, X. Liu, Y. Shi, Y. Jiang, B. Zhao and J. Zhang, Nano Energy, 2022, 101, 107555 CrossRef CAS.
- B. Cao, T. Li, W. Zhao, L. Yin, H. Cao, D. Chen, L. Li, F. Pan and M. Zhang, Small, 2023, 19, 2301834 CrossRef CAS PubMed.
- Z. Xu, X. Guo, W. Song, J. Wang, T. Qin, Y. Yuan and J. Lu, Adv. Mater., 2024, 36, 2303612 CrossRef CAS PubMed.
- Y. Zhang, X. Shi, S. Zheng, Y. Ouyang, M. Li, C. Meng, Y. Yu and Z.-S. Wu, Energy Environ. Sci., 2023, 16, 5043–5051 RSC.
- Y. Yang, Q. Zhu, J. Yang, H. Liu, Y. Ren, X. Sui, P. Wang, G. Sun and Z. Wang, Adv. Funct. Mater., 2023, 33, 2304979 CrossRef CAS.
- T. Zeng, M. Yang, F. Sun, Z. Huang, W. Zhao, Z. Chen, D. Zou, J. Qiu, L. Wang, R. Wang, C. Zhang, T. Yang, W. Ji, J. Xu, W. Yin, R. Li, H. Meng and Y. Xiao, Adv. Funct. Mater., 2024, 34, 2314528 CrossRef CAS.
- L. Wang, L. Xu, W. Xue, Q. Fang, H. Liu, Y. Liu, K. Zhou, Y. Li, X. Wang, X. Wang, X. Yang, X. Yu and X. Wang, Nano Energy, 2024, 121, 109241 CrossRef CAS.
- Z. Hao, H. Sun, Y. Ni, G. Yang, Z. Yang, Z. Hao, R. Wang, P. Yang, Y. Lu, Q. Zhao, W. Xie, Z. Yan, W. Zhang and J. Chen, Adv. Mater., 2023, 36, 2307617 CrossRef PubMed.
- Z. Ye, B. Zhang, T. Chen, Z. Wu, D. Wang, W. Xiang, Y. Sun, Y. Liu, Y. Liu, J. Zhang, Y. Song and X. Guo, Angew. Chem., Int. Ed., 2021, 60, 23248–23255 CrossRef CAS PubMed.
- D. Wang, Z. Wu, W. Xiang, Y. Liu, G. Wang, K. Hu, Q. Xu, Y. Song and X. Guo, J. Energy Chem., 2022, 64, 344–353 CrossRef CAS.
- S. Chen, P. Zhang, X. Zhou, W. Wu, X. Liu, Y. Liu, G. Feng, B. Zhang, W. Xing, M. Zuo, P. Zhang, G. Lv, Y. Xiao, S. Dou and W. Xiang, Chem. Sci., 2024, 15, 14415–14424 RSC.
- Z. B. Li, Y. W. Li, M. J. Zhang, Z. W. Yin, L. Yin, S. Y. Xu, C. J. Zuo, R. Qi, H. Y. Xue, J. T. Hu, B. Cao, M. H. Chu, W. G. Zhao, Y. Ren, L. Xie, G. X. Ren and F. Pan, Adv. Energy Mater., 2021, 11, 2101962 CrossRef CAS.
- Y. Fan, E. Olsson, G. Liang, Z. Wang, A. M. D'Angelo, B. Johannessen, L. Thomsen, B. Cowie, J. Li, F. Zhang, Y. Zhao, W. K. Pang, Q. Cai and Z. Guo, Angew. Chem., Int. Ed., 2022, 62, e202213806 CrossRef PubMed.
- R. Q. Zhao, M. M. Wu, P. X. Jiao, X. T. Wang, J. Zhu, Y. Zhao, H. T. Zhang, K. Zhan, C. X. Li, Y. F. Ma and Y. S. Chen, Nano Res., 2023, 16, 6805–6814 CrossRef CAS.
- Y. Chen, Q. Li, Z. Chen, W. Zeng, Z. Liu, M. Wang, F. Xia, G. Wang and J. Wu, Adv. Funct. Mater., 2024, 2411542 CrossRef CAS.
- W. Zeng, F. Liu, J. Yang, B. Zhang, F. Cao, W. Tian, J. Wang, R. Yu, F. Xia, H. Peng, J. Ma, Z. Wang, S. Mu and J. Wu, Energy Storage Mater., 2023, 54, 651–660 CrossRef.
- K. Chai, J. Zhang, Q. Li, D. Wong, L. Zheng, C. Schulz, M. Bartkowiak, D. Smirnov and X. Liu, Small, 2022, 18, 2201014 CrossRef CAS PubMed.
- Y. Pei, Q. Chen, M. Wang, B. Li, P. Wang, G. Henkelman, L. Zhen, G. Cao and C.-Y. Xu, Nano Energy, 2020, 71, 104644 CrossRef CAS.
- W. Jiang, C. Zhang, Y. Feng, B. Wei, L. Chen, R. Zhang, D. G. Ivey, P. Wang and W. Wei, Energy Storage Mater., 2020, 32, 37–45 CrossRef.
- C. Zhang, Y. Feng, B. Wei, C. Liang, L. Zhou, D. G. Ivey, P. Wang and W. Wei, Nano Energy, 2020, 75, 104995 CrossRef CAS.
- Y. Liu, H. Zhu, H. Zhu, Y. Ren, Y. Zhu, Y. Huang, L. Dai, S. Dou, J. Xu, C.-J. Sun, X.-L. Wang, Y. Deng, Q. Yuan, X. Liu, J. Wu, Y. Chen and Q. Liu, Adv. Energy Mater., 2021, 11, 2003479 CrossRef CAS.
- X. Sun, C. Qin, B. Zhao, S. Jia, Z. Wang, T. Yang, X. Liu, L. Pan, L. Zheng, D. Luo and Y. Zhang, Energy Storage Mater., 2024, 70, 103559 CrossRef.
- Y. Ouyang, Y. Zhang, G. Wang, X. Wei, A. Zhang, J. Sun, S. Wei, L. Song, F. Dai and Z. S. Wu, Adv. Funct. Mater., 2024, 34, 2401249 CrossRef CAS.
- M. Tang, S. Dong, J. Wang, L. Cheng, Q. Zhu, Y. Li, X. Yang, L. Guo and H. Wang, Nat. Commun., 2023, 14, 6006 CrossRef CAS PubMed.
- T. Liu, J. Liu, L. Li, L. Yu, J. Diao, T. Zhou, S. Li, A. Dai, W. Zhao, S. Xu, Y. Ren, L. Wang, T. Wu, R. Qi, Y. Xiao, J. Zheng, W. Cha, R. Harder, I. Robinson, J. Wen, J. Lu, F. Pan and K. Amine, Nature, 2022, 606, 305–312 CrossRef CAS PubMed.
- J.-J. Marie, R. A. House, G. J. Rees, A. W. Robertson, M. Jenkins, J. Chen, S. Agrestini, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Mater., 2024, 23, 818–825 CrossRef CAS PubMed.
- J.-Y. Li, H.-Y. Hu, H.-W. Li, Y.-F. Liu, Y. Su, X.-B. Jia, L.-F. Zhao, Y.-M. Fan, Q.-F. Gu, H. Zhang, W. K. Pang, Y.-F. Zhu, J.-Z. Wang, S.-X. Dou, S.-L. Chou and Y. Xiao, ACS Nano, 2024, 18, 12945–12956 CrossRef CAS PubMed.
- S. Sharifi-Asl, V. Yurkiv, A. Gutierrez, M. Cheng, M. Balasubramanian, F. Mashayek, J. Croy and R. Shahbazian-Yassar, Nano Lett., 2019, 20, 1208–1217 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental section and additional figures. See DOI: https://doi.org/10.1039/d4sc07037b |
| This journal is © The Royal Society of Chemistry 2025 |