
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAffinity-based covalent sialyltransferase probes enabled by ligand-directed chemistry†
Jun Yang
Ong
 a,
Erianna I.
Alvarado-Melendez
a,
Joshua C. L.
Maliepaard
b,
Karli R.
Reiding
b and
Tom
Wennekes
*a
a,
Erianna I.
Alvarado-Melendez
a,
Joshua C. L.
Maliepaard
b,
Karli R.
Reiding
b and
Tom
Wennekes
*a
aChemical Biology and Drug Discovery, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: t.wennekes@uu.nl
bBiomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute of Pharmaceutical Sciences, Utrecht University, Netherlands Proteomics Center, 3584 CH Utrecht, The Netherlands
First published on 13th January 2025
Abstract
Sialyltransferases (ST) are key enzymes found in, among others, mammals and bacteria that are responsible for producing sialylated glycans, which play critical roles in human health and disease. However, chemical tools to study sialyltransferases have been limited to non-covalent inhibitors and probes that do not allow isolation and profiling of these important enzymes. Here we report a new class of covalent affinity-based probes (AfBP) for ST by using ligand-directed chemistry (LDchem). Our affinity-based probes are armed with a simple to synthesise but robust O-nitrobenzoxadiazole (O-NBD) warhead, which is a lysine-specific SNAr electrophilic warhead with an advantageous turn-on fluorescence property. We chemoenzymatically synthesised a series of CMP-Neu5Ac based probes and demonstrated their high specificity in labelling a range of recombinant STs with submicromolar sensitivity. Importantly, with our LDchem ST probe, we successfully labelled the endogenous lipooligosaccharide ST (Lst) in live Neisseria gonorrhoeae, a clinically relevant human pathogen. Our results demonstrated that this new class of covalent ST probes offer a robust platform for ST profiling and future studies of STs in their native environments.
Introduction
Sialyltransferases (ST) are enzymes that catalyse sialylation, which is the transfer of sialic acids onto glycan chains. Sialylation plays many important roles in healthy human physiology, but has also been implicated as a key cancer and bacterial pathogenicity marker.1–6 Various sialic acid mimics have been developed as reversible inhibitors and fluorescent probes to aid in the study of ST,7–9 but their use is limited by the lack of a stable covalent linkage to the enzyme. While activity-based covalent probes (ABPs) have been successfully developed and proved invaluable in profiling and studying many glycosidases,10–12 such probes have not been reported for ST or glycosyltransferases in general. This effort is in part hindered by the non-covalent, single displacement SN2 catalytic mechanism by which all ST and many inverting glycosyltransferases operate13–15 as supported by crystal structures and mechanistic studies.16–25 In this mechanism, no covalent intermediate between the CMP-Neu5Ac substrate and the enzyme is formed (Scheme 1), therefore a stable covalent adduct cannot be trapped by stabilising such an intermediate, unlike what has been commonly done for the retaining glycosyl hydrolase ABPs.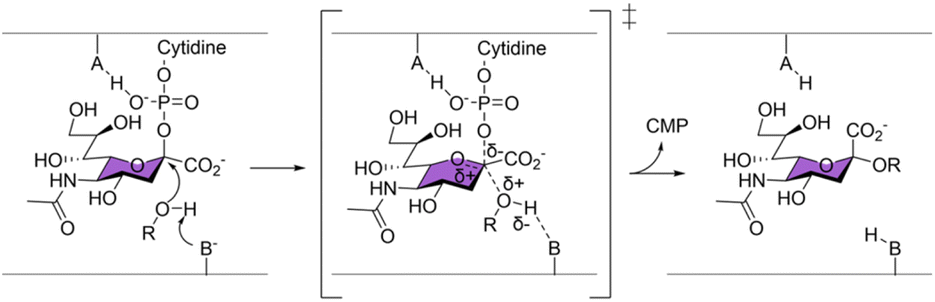 | ||
| Scheme 1 All sialyltransferases characterised so far have been proposed to operate by a single displacement SN2 mechanism. | ||
On the other hand, an affinity-based probe (AfBP) overcomes this limitation because it does not depend on a covalent catalytic intermediate. A typical AfBP consists of three parts: (1) a ligand with high affinity to the target protein (the directing group); (2) a linker of appropriate length; (3) a reactive group (the warhead) that is capable of forming a covalent bond with an amino acid residue close by (Fig. 1A). One major type of AfBP is a photoaffinity probe, where the warhead is activated for chemical crosslinking upon photo-irradiation. A photoaffinity-based sialyltransferase probe was recently reported to selectively photocrosslink to purified recombinant human sialyltransferase ST6Gal1 over ST3Gal1 and ST3Gal4.26 This probe, based on photoaffinity labeling (PAL), has the potential to become a valuable tool for studying STs, offering high spatiotemporal control and the ability to crosslink with various amino acid residues. However, its high reactivity when activated often results in significant background interference.
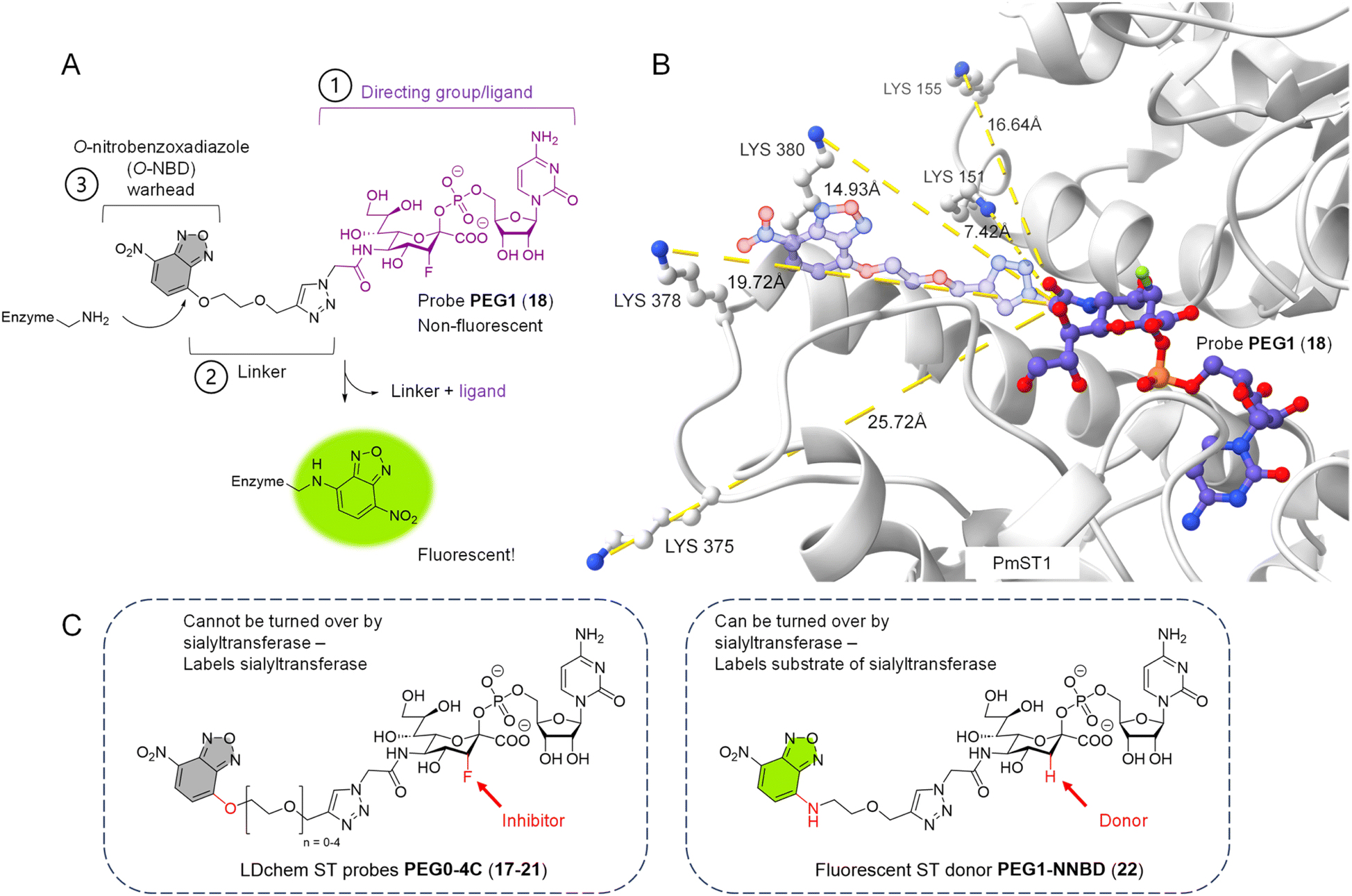 | ||
| Fig. 1 Design of O-NBD armed sialyltransferase probes. (A) Turn-on fluorescence property of O-NBD warhead upon reacting with a lysine residue is highly advantageous to allow wash-free labelling and reduce background signals. Linker length of probe PEG1 (18) is presented here. (B) Multiple lysines in the crystal structure of PmST1 WT (PDB: 2IHZ) are positioned close to the N-acetyl group of the bound CMP-3F(axial)-Neu5Ac ligand. The triazole-PEG1 linker and O-NBD group of probe PEG1 (18) are modelled onto the ligand structure (represented in 50% transparency), showing the O-NBD warhead to be in close proximity to the lysine residues for possible directed reactivity. (C) Chemical structures of probes with different linker lengths PEG0-4C (17–21) and donor 22. The differences (O/N-NBD, 3-F/H) between the two groups of reagents are highlighted in red. Linker lengths PEG0-4C in Å can be found in Table S2.† | ||
In this paper, we report the facile chemical synthesis and biological validation of a new class of highly selective covalent AfBP for sialyltransferases using the principle of ligand-directed chemistry (LDchem). LDchem employs cleavable warheads whose activities are driven solely by proximity effect upon ligand binding, and which upon reaction, cleave and release the ligand to label the protein-of-interest with a reporter moiety in a traceless manner. The activation of an LDchem warhead only upon binding and long residence time of the ligand on the target protein also allows repeated equilibrium between the probe and the target protein over a longer period of time, thereby increases the signal-to-noise ratio (eqn (1), ESI†). This approach has been applied successfully in endogenous protein labelling and profiling.27 However, its application in chemical glycobiology research is scarce, with only one example reported on isolated glycosidases.28
By screening for seven STs across four glycosyltransferase (GT) families, we show here that our LDchem ST probes are not only highly specific for a range of recombinant sialyltransferases, but also are capable of labelling the endogenous lipooligosaccharides (LOS) sialyltransferase Lst in live Neisseria gonorrhoeae, a clinically relevant human pathogen. We envision that our probes, with an efficient one-step labelling protocol, could serve as a valuable tool for endogenous ST profiling, complimentary to the photoaffinity technology.
Results and discussion
Rational design and synthesis of the covalent ST probes
Our design rationale for each part of the LDchem ST probes (Fig. 1A) is as follows:(1) The ligand/directing group was selected based on the known universal sialyltransferase inhibitor CMP-3F(axial)-Neu5Ac (7), which is itself a non-hydrolysable mimic of CMP-Neu5Ac, the natural universal substrate of all sialyltransferases.
(2) Linkers were designed to span a distance from ∼7 Å to ∼23 Å (PEG0-4C) to systematically study the effect of the linker length on labelling efficiency. As we aimed to target non-catalytic lysines residues, these residues are not conserved and can lie at different distances from the active sites in different sialyltransferases. Furthermore, the linker is connected to the C5 position of the sialic acid in the ligand, as it has been shown that the C5 N-acetyl group is solvent-exposed and can tolerate modifications without affecting binding to the sialyltransferases.
(3) O-Nitrobenzoxadiazole (O-NBD) is a lysine-specific SNAr electrophilic warhead with a desirable turn-on fluorescence property.29 The use of the O-NBD warhead in LDChem probes has been reported in several studies and has proven to be robust, notably for its stability in aqueous solution.29–32
The three parts of the probe were connected through a facile modular synthesis, namely (Part 2 + Part 3) + Part 1 (Schemes S1 and S2†) to give a panel of LDchem ST probes PEG0-4C (17–21). The crucial final step was facilitated by copper-catalysed azide–alkyne cycloaddition (CuAAC) between the chemoenzymatically synthesised azide-modified CMP-Neu5Ac-based ligand and the set of five alkyne-appended O-NBD warheads with varying linker length. Here, the remarkable aqueous stability of the O-NBD warhead, despite its high reactivity upon ligand binding, was fundamental to the successful assembly of the probe molecules, which due to their highly hydrophilic properties necessitate the use of water as the reaction solvent.
At the same time, we designed and synthesised a new fluorescently labelled CMP-sialic acid donor PEG1-N-NBD (22), which is isosteric to probe PEG1 (18) but can act as a substrate to the sialyltransferases (Fig. 1C). This donor allows us to rapidly determine the activity of the sialyltransferases by tracking the fluorescently labelled products directly on a TLC plate or in a SDS-PAGE gel.
Details on the synthesis of the probes with varying linker length PEG0-4C (17–21) and donor PEG1-N-NBD (22) are provided in the ESI.†
Evaluation of the specificity and promiscuity of the ST probes
We selected seven ST with different linkage specificities (α2,3/2,6/2,8) from four different glycosyltransferase (GT) families and six different organism origins to investigate the specificity and the promiscuity of our affinity-based probes (Table 1). Both X-ray diffraction crystal structures and AlphaFold structure prediction of these enzymes showed at least one lysine residue in the vicinity of the sugar nucleotide binding site. The proximity of the O-NBD warhead to these lysine residues upon binding of the ligand was further verified by docking of our designed probes into the crystal structure of PmST1 (PDB: 2IHZ) (Fig. 1B).| Sialyltransferase | Organism | GT family | Specificities | Crystal structure |
|---|---|---|---|---|
| a AlphaFold structure prediction.37,38 b NST from N. meningitidis L1 was used as homology for N. gono Lst. | ||||
| PmST1 WT | Pasteurella multocida | GT80 | 2,3/2,6 ST | 2IHZ (ref. 18) |
| PmST1 M144D | Pasteurella multocida | GT80 | 2,3 ST | 3S44 (ref. 33) |
| VJT-FAJ-16 (ref. 34) | Vibrio sp. JT-FAJ-16 | GT80 | 2,3 ST | A8R0Y0a |
| Pd2,6ST | Photobacterium damselae | GT80 | 2,6 ST | 4R84 (ref. 35) |
| ST6Gal1 | Homo sapiens | GT29 | 2,6 ST | 4JS1 (ref. 36) |
| Cst-II | Campylobacter jejuni | GT42 | 2,3/2,8 ST | 1RO7 (ref. 16) |
| N. gono Lst | Neisseria gonorrhoeae | GT52 | 2,3 ST | 2YK7 (ref. 22)b |
All ST enzymes were expressed recombinantly, purified and tested for their activity prior to labelling experiments using donor PEG1-N-NBD (22) and lactose (Fig. S1†). This result also confirmed that the triazole-PEG1-N-NBD (and by structural homology, O-NBD) modification on the C5 position of the sialic acid was well tolerated and did not prevent the binding of the CMP-sialic acid derivatives to the sialyltransferases.
Standard labelling experiments were carried out in the presence of 1 μM sialyltransferase, 1 μM of sialic acid aldolase (as a background non-binding control protein to detect potential non-specific labelling) and 1 μM of probe PEG0-4C (17–21) in 25 mM pH 7.2 phosphate buffer at 37 °C for 2 hours. For all sialyltransferases, we observed a linker length-dependent labelling (Fig. 2 and S2†), with a detection limit down to 0.0625 μM of VJT-FAJ-16 and 0.125 μM of Cst-II (Fig. S3†). Labelling was observed even at pH 6.0, but when the pH of the reaction buffer was increased to 7.7 and 8.5, the labelling intensity increased as expected, without significant loss of selectivity (Fig. S4A and B†). We also observed exceptional stability of the probes when incubating them for 4 h in phosphate/Tris buffer in this pH range at 37 °C (Fig. S4C†). However, under all conditions, PmST1 M144D and ST6Gal1 showed minimal labelling (Fig. 2A and B).
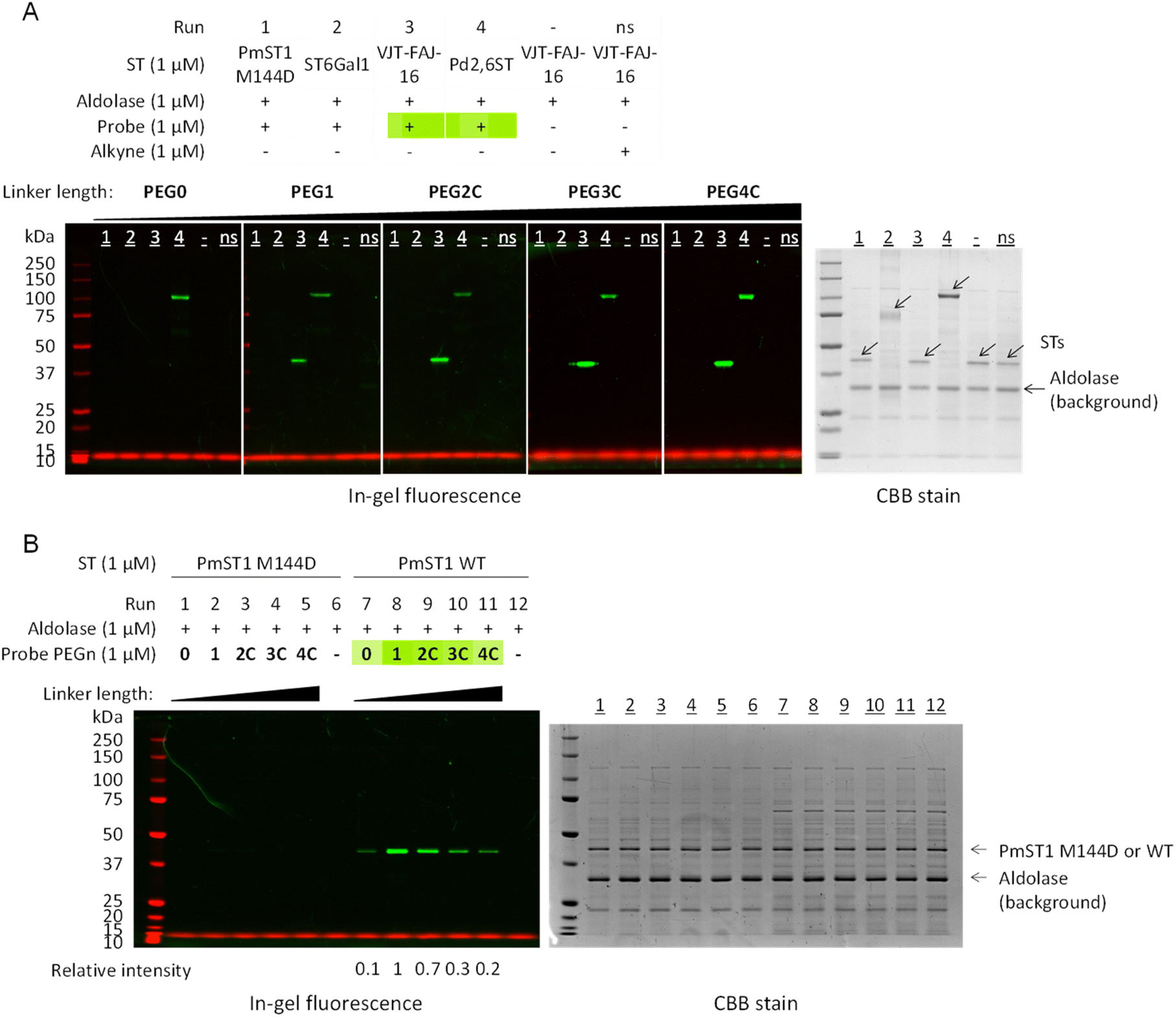 | ||
| Fig. 2 In-gel fluorescence was used to analyse the effect of linker length on the labelling efficiency of PmST1 WT, PmST1 M144D, ST6Gal1, VJT-FAJ-16 and Pd2,6ST by the affinity-based probes PEG0-4C (17–21). (A) VJT-FAJ-16 and Pd2,6ST were labelled to different degrees by probes with linker length ranging from PEG0 to PEG4C, while PmST1 M144D and ST6Gal1 showed no labelling. (B) Side-by-side comparison of PmST1 WT and M144D single mutant showed contrasting labelling efficiency, although both enzymes were active and able to transfer donor 22 onto lactose efficiently (Fig. S1†). ns: non-specific labelling control, alkynes with the corresponding linker length (8–9, 13–15) were used. In all experiments no off-target labelling was observed on the non-binding aldolase control (the aldolase is responsible for contributing the numerous background bands in the CBB). Relative intensity was measured by ImageJ. | ||
Investigation of the contrasting labelling efficiency between PmST1 WT and its M144D mutant
To explain why a single amino acid difference led to the contrasting labelling efficiency between PmST1 WT and its M144D single mutant, we looked into their kinetics characterisation and crystal structures. Here it has been shown that there are marked differences in the protein conformation between the two variants and about a five times higher KM for the donor for the M144D mutant (by comparison of the donor hydrolysis kinetics data).18,33 There are therefore two possible factors that could cause the difference in labelling efficiency. Firstly, the homologous lysine residue which was labelled in PmST1 WT is positioned out of reach in PmST1 M144D mutant because of its altered conformation and therefore prevented the labelling. Secondly, the equilibrium dissociation constant (Kd) of the ligand is higher for the M144D mutant (lower affinity) than the WT enzyme, which would diminish the rate of the labelling reaction as predicted by eqn (1), ESI.†39To test the first hypothesis, we carried out bottom-up liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify the labelling site in PmST1 WT by probe PEG1 (18) (Fig. S5, Table S3†). Rather surprisingly, proteomics revealed lysines K375 and K378 on the β12–α12a loop of PmST1 WT as the modification sites (SHIIFTSN![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) QVSKED). These lysines were not expected from initial docking experiments because the β12–α12a loop (residues 374–382) was missing from all the ligand-bound crystal structures solved by two independent research groups due to its highly disordered nature (PDB: 2IHZ and 2IY8),18,40 except for in the ligand-free apo structure (PDB: 2EX0).41 However, we were able to reconstruct this region by using the One-to-One Threading function of Phyre2 with PDB: 2IHZ as the model (Fig. 1B).42 On the other hand, sequence coverage on the N-terminal catalytic domain containing lysines K151 and K155 (DDGSMEYVDLEEENDIS) was limited, so they cannot be definitively excluded as potential labelling sites.
QVSKED). These lysines were not expected from initial docking experiments because the β12–α12a loop (residues 374–382) was missing from all the ligand-bound crystal structures solved by two independent research groups due to its highly disordered nature (PDB: 2IHZ and 2IY8),18,40 except for in the ligand-free apo structure (PDB: 2EX0).41 However, we were able to reconstruct this region by using the One-to-One Threading function of Phyre2 with PDB: 2IHZ as the model (Fig. 1B).42 On the other hand, sequence coverage on the N-terminal catalytic domain containing lysines K151 and K155 (DDGSMEYVDLEEENDIS) was limited, so they cannot be definitively excluded as potential labelling sites.
Dramatic conformational change upon binding to cytidine monophosphate (CMP) and CMP-3F(axial)-Neu5Ac (7) has been observed in PmST1 WT but not in its M144D mutant.18,33 These changes include closing of the N-terminal domain and movement of the β12–α12a loop together with the β8–α8 loop, which contains the important gatekeeper tryptophan residue W270. This movement may be essential to bring the lysines K375 and K378 into the reacting distance of the O-NBD warhead, as illustrated by Movie 1 (see ESI†). Serendipitously, our labelling experiment thus provided further evidence for this lack of conformational change in PmST1 M144D upon donor binding, corroborating the earlier crystal structure and NMR studies,33 and also shedding light on this disordered region that was unable to be observed by crystallography. However, we cannot dismiss the possibility that a higher Kd for the probes is also an equally important contributing factor for the lack of labelling in PmST1 M144D.
LC-MS/MS reveals anomalous labelling sites in VJT-FAJ-16
At the same time, LC-MS/MS was able to identify lysines K301 (FGHPSATF) and K353 (SIPEVKNKF) as the labelling sites in VJT-FAJ-16 (Fig. S5, Table S3†). This finding was highly interesting because K301 is located inside the binding pocket of CMP, and therefore its selectivity was not intuitive. We were confident that the modified CMP-sialic acids bind correctly in the catalytic pocket of VJT-FAJ-16 because the donor PEG1-N-NBD (22) could be successfully turned over by VJT-FAJ-16 (Fig. S1†). Subsequently, we carried out molecular docking of probe PEG1 (18) and found a second in silico binding mode (Fig. S6D†), where the NBD could bind in the place of cytidine. Our hypothesis is that the probe first binds in the correct conformation with the CMP-sialic acid ligand in the catalytic binding site (Fig. S6A–C†), and statistical rebinding flips the molecule to position the O-NBD in the CMP binding pocket (Fig. S6D†). It is followed by the attack of K301 on the electrophilic centre of the O-NBD. Molecular docking also showed a third binding mode (Fig. S6E†) which justified the labelling of K353. This finding serves to remind us that although rational design of affinity-based probes is a powerful technique, one should always investigate their molecular-level activity by multiple techniques to identify unexpected modes of action.
Using turn-on fluorescence to measure the affinity of probe 18 for PmST1 WT and VJT-FAJ-16
We capitalised on the unique turn-on fluorescence property of the O-NBD probe to determine the binding affinity of probe 18 to the sialyltransferases. The fluorescence increase over time, as the labelled enzymes were formed, was measured and used to calculate the KM of the labelling reaction using an adapted version of previously published eqn (1), ESI.†43 The KM of probe PEG1 (18) was estimated to be 0.5 ± 0.1 μM for VJT-FAJ-16 and 0.6 ± 0.2 μM for PmST1 WT (Fig. 3). Since the estimated KM represents the upper limit value for the Kd of the probe, we concluded that the probe has a binding affinity for both enzymes in the submicromolar concentration.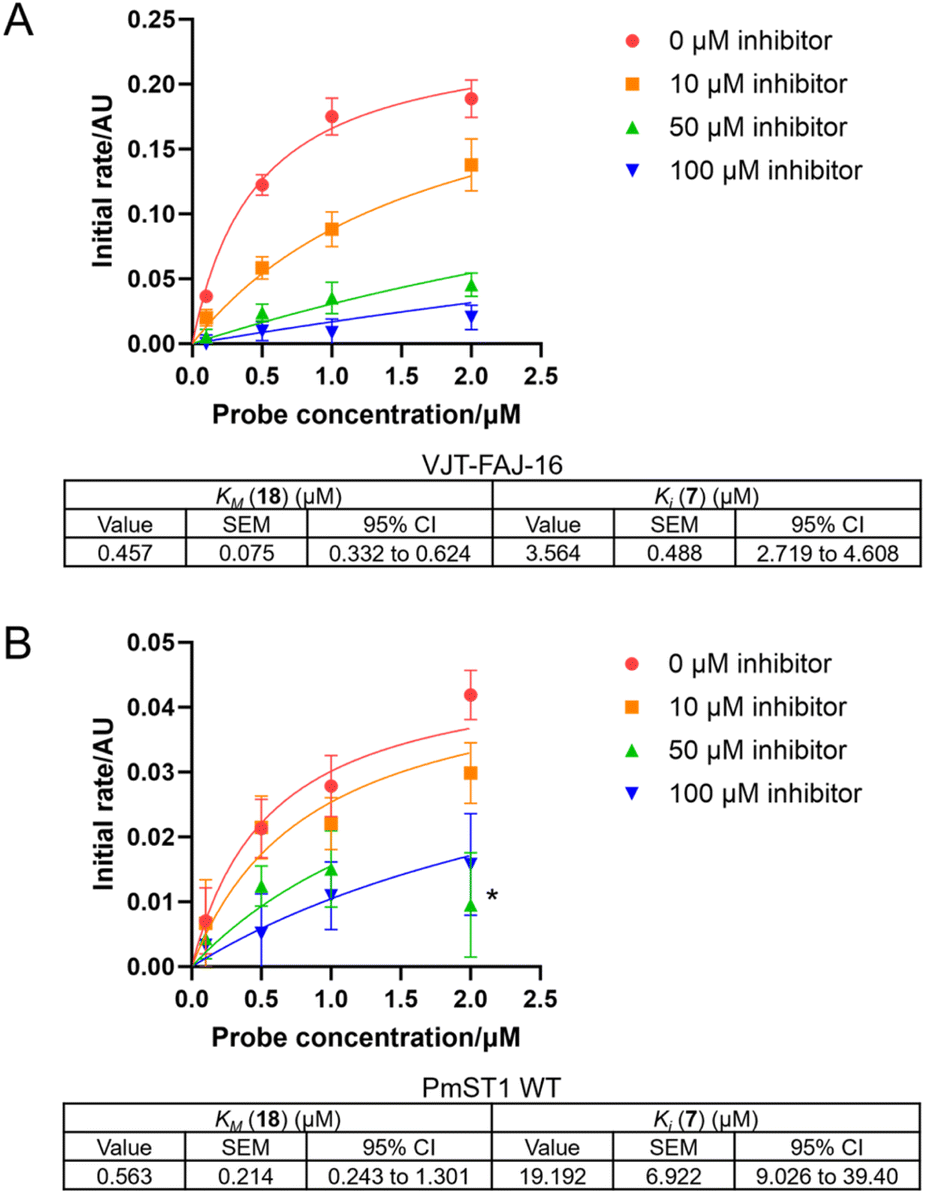 | ||
| Fig. 3 Kinetics curve fitting to eqn (2), ESI† was done to calculate KM of the probe PEG1 (18) and Ki of the competitive inhibitor CMP-3F(axial)-Neu5Ac (7) for (A) VJT-FAJ-16 and (B) PmST1 WT. *This outlier was omitted from curve fitting (2 μM probe, 50 μM inhibitor). n.b. Two independent experiments with duplicates were carried out for VJT-FAJ-16 with 0 μM and 100 μM inhibitor, and one experiment with duplicates was carried out for the rest. Error bars represent standard deviation (SD). Value of KM and Ki were reported with their standard error of the mean (SEM) and 95% confidence interval (CI). | ||
At the same time, we discovered that by taking into account a competitive inhibitor in the kinetics equation, we could establish a competition assay to measure the Ki of a competitive inhibitor. To evaluate our newly derived equation (eqn (2), full derivation available in the ESI†), we carried out a competition assay using various concentrations of the competitive inhibitor CMP-3F(axial)-Neu5Ac (7) (Fig. 3). The Ki for CMP-3F(axial)-Neu5Ac (7) was calculated to be 3.6 ± 0.5 μM for VJT-FAJ-16, while the Ki calculated for PmST1 WT was 19.2 ± 6.9 μM, which is close to the reported literature value of 25.7 μM.18 These preliminary results presented a possible use of our LDchem ST probes for high-throughput screening of ST inhibitors, as an alternative to activity assay or fluorescent polarisation (FP) assay.
Absence of labelling of STGal1
We attributed the absence of labelling of ST6Gal1 to a low binding affinity. The crystal structure of ST6Gal1 clearly shows lysine residues in the vicinity of the ligand binding site, but CMP-3F(axial)-Neu5Ac (7) has been shown to bind only weakly to ST6Gal1 with a calculated dissociation constant (Ki) of 0.65 mM,44 in contrast to 25.7 μM reported for PmST1 WT.18 ST6Gal1 was also not able to transfer donor PEG1-N-NBD (22) efficiently (Fig. S1†), which was surprising given that ST6Gal1 has been shown to be tolerant of donor substrates modified at the C5 position of the sialic acid with either a biotin or a fluorophore.45,46 All this suggested that the combination of modifications of the donor substrate analogues (probes 17–21, donor 22) led to a decrease in binding to ST6Gal1 and prevented efficient labelling or transfer.Evaluation of probe specificity under crude conditions
To further prove the fidelity of the LDchem ST probes and their potential use in a more complex biological setting, we carried out a heat inactivation and competition assay by spiking 1 μM of VJT-FAJ-16 into 1 mg mL−1 of E. coli cell lysate (Fig. 4). This experiment confirmed the labelling as ligand-directed and demonstrated its high specificity in E. coli cell lysate.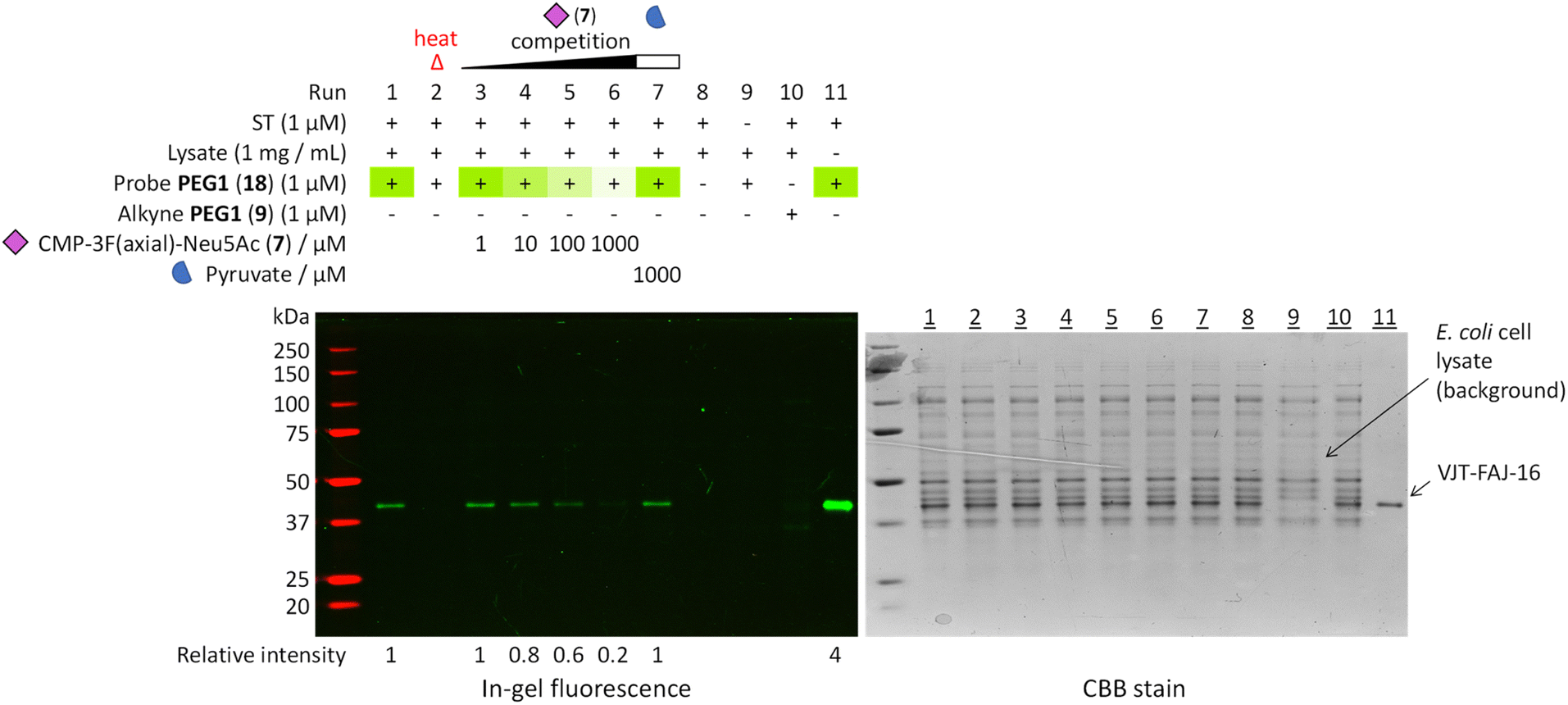 | ||
Fig. 4 Heat inactivation and a competition assay was used to further prove that the probes bind selectively to the active site of a properly folded sialyltransferase. Heat treatment denatured the enzyme and destroyed the binding pocket, resulting in no labelling. Increasing the concentration of active site competitor CMP-3F(axial)-Neu5Ac (7,  ) decreased the labelling intensity while the same highest concentration of non-competitor pyruvate ( ) decreased the labelling intensity while the same highest concentration of non-competitor pyruvate ( ) did not. All evidence pointed towards highly selective ligand-directed affinity-based labelling. Detailed procedure is described in the ESI.† Relative intensity was measured by ImageJ. ) did not. All evidence pointed towards highly selective ligand-directed affinity-based labelling. Detailed procedure is described in the ESI.† Relative intensity was measured by ImageJ. | ||
Labelling endogenous sialyltransferase Lst in live N. gonorrhoeae and its Triton X-100 extract
Finally, we investigated the possibility to label the endogenous Lst in N. gonorrhoeae, a relevant human pathogen that causes the sexually transmitted disease gonorrhoea. We attempted labelling under two conditions: in live N. gonorrhoeae and its Triton X-100 extract, as it has been demonstrated that it is an efficient method to extract sialyltransferase activity from N. gonorrhoeae.47,48 Conveniently, a Lst-deficient mutant of N. gonorrhoeae F62 (strain JB1, referred to here as F62Δlst) was available49,50 for direct comparison with the wild type F62.Prior to affinity labelling, the presence of sialyltransferase activity in live bacteria and Triton X-100 extract was assessed using the native sialyltransferase (NST) bacteria labelling technique reported recently by our group.46,51 With this technique, native LOS was used as the sialyltransferase acceptor and precluded the need to synthesise bespoke complex glycan acceptors. The Triton X-100 extract has also been shown previously to contain sufficient amount of LOS to allow us to detect the NST sialylation products.47 Again we utilised the N-NBD fluorescently labelled CMP-sialic acid donor PEG1-N-NBD (22). With that, we were able to confirm sialyltransferase activity in N. gonorrhoeae F62 and the absence thereof in F62Δlst by in-gel fluorescence and silver staining of the LOS, both in live bacteria and in the Triton X-100 extract (Fig. S7†).
Subsequently, we carried out affinity labelling in live N. gonorrhoeae and its Triton X-100 extract using 1 μM probe PEG1 (18), which performed well with recombinant N. gonorrhoeae Lst. Similarly, we used competition with CMP-3F(axial)-Neu5Ac (7) to identify genuine ligand binding.
Excitingly, in both live bacteria and Triton X-100 extract, we could clearly see a labelled band at ∼41 kDa that corresponds to the molecular weight of Lst only in F62 but not F62Δlst (Fig. 5), which also matched the observed band size on western blot in the study by Shell et al. using the same strain of N. gonorrhoeae.52 The labelling could be competed off entirely by CMP-3F(axial)-Neu5Ac (7) and less efficiently by non-competitor ATP (although AMP can be present in the stock solution and is a weak competitive inhibitor for Lst).47
 | ||
| Fig. 5 Labelling of Lst was performed using probe PEG1 (18) in live N. gonorrhoeae and its Triton X-100 extract. A labelled band at ∼41 kDa that correspond to the molecular weight of Lst can be clearly seen in F62 and not F62Δlst, indicating successful labelling of the sialyltransferase in both live bacteria and Triton X-100 extract. Competition by CMP-3F(axial)-Neu5Ac completely abolished the labelling, providing additional evidence that the labelling was ligand-directed. An extra labelled protein (∼36 kDa) was observed in live bacteria but not in the Triton X-100 extract, and it was postulated to be the neisserial porin PorB. Full uncropped gel images are presented in Fig. S8.† | ||
Interestingly, there was another labelled band at ∼36 kDa that was observed only in live bacteria but not in the Triton X-100 extract. The fact that it was not extracted by 0.5% Triton X-100 suggested that it was likely integrally membrane bound. The identity of this protein is currently not known, but we speculate that it might be a major outer membrane protein (OMP), potentially the neisserial porin PorB that is known to bind nucleotides through lysine residues.53,54 This could hint at its involvement in CMP-sialic acid uptake in N. gonorrhoeae, although prior studies have shown Lst to be a surface-exposed outer membrane protein.52,55,56 Further study on this additional labelled protein is needed to determine if N. gonorrhoeae has another receptor protein for CMP-sialic acid.
Conclusions
In conclusion, we report here the utility of LDchem to develop highly specific covalent affinity-based probes for sialyltransferases. These O-NBD probes are straightforward to make, stable in water57 and easy to analyse by turn-on fluorescence. Since these affinity-based probes do not target conserved catalytic residues, they are not expected to be universal. Their effectiveness depends on the presence of lysine residues near the binding site and a certain affinity threshold of the protein for the ligand. However, we show that the probes that we designed by this principle are promiscuous for many sialyltransferases in this study and are highly specific even in a complex biological setting such as in live bacteria. This modular ligand-directed approach, combined with alternative warheads, has the potential to expand the toolbox of covalent probes for previously inaccessible glycosyltransferases, which we have started investigating.Data availability
Data associated with the LC-MS/MS experiments for this article are available at MassIVE repository at https://doi.org/10.25345/C5VH5CW11 and can additionally be accessed viahttps://MSV000095915@massive.ucsd.edu. Other data supporting this article have been included as part of the ESI.†Author contributions
J. Y. O. and T. W. conceived the research and designed the research approach. T. W. acquired the funding for this project. J. Y. O., E. I. A. and J. M. performed the experiments, collected and analyzed the raw data. T. W. and K. R. provided supervision. J. Y. O. wrote the initial manuscript with input from T. W. All authors contributed to the writing of the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Geert-Jan Boons (Utrecht University), Dr Gerlof P. Bosman (Utrecht University), Debóra van Ekeris (Utrecht University) and Dr Romane Breysse (Inbiose NV, Belgium) for kindly providing and assisting in the preparation of the recombinant enzymes. We thank Dr Javier Sastre Toraño for recording the high-resolution mass spectrometry data of synthesised chemical compounds. Research in this publication was supported by funding from the European Union's Horizon 2020 Marie Skłodowska-Curie Actions for the Innovative Training Network “Sweet Crosstalk” under the grant agreement No. 814102.Notes and references
- A. Varki, Trends Mol. Med., 2008, 14, 351–360 CrossRef CAS.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
- E. R. Vimr, K. A. Kalivoda, E. L. Deszo and S. M. Steenbergen, Microbiol. Mol. Biol. Rev., 2004, 68, 132–153 CrossRef CAS.
- W. H. D. Bowles and T. M. Gloster, Front. Mol. Biosci., 2021, 8, 705133 CrossRef CAS PubMed.
- H. De Jong, M. M. S. M. Wösten and T. Wennekes, Glycobiology, 2022, 32, 11–22 CrossRef CAS.
- A. J. Walklett, E. K. P. Flack, H. S. Chidwick, N. E. Hatton, T. Keenan, D. Budhadev, J. Walton, G. H. Thomas and M. A. Fascione, Angew. Chem., Int. Ed., 2024, 63, e202318523 CrossRef CAS PubMed.
- B. Müller, C. Schaub and R. R. Schmidt, Angew Chem. Int. Ed. Engl., 1998, 37, 2893–2897 CrossRef.
- J. J. Preidl, V. S. Gnanapragassam, M. Lisurek, J. Saupe, R. Horstkorte and J. Rademann, Angew. Chem., Int. Ed., 2014, 53, 5700–5705 CrossRef CAS PubMed.
- K. Suzuki, A. Ohtake, Y. Ito and O. Kanie, Chem. Commun., 2012, 48, 9744 RSC.
- M. D. Witte, G. A. Van Der Marel, J. M. F. G. Aerts and H. S. Overkleeft, Org. Biomol. Chem., 2011, 9, 5908–5926 RSC.
- L. Wu, Z. Armstrong, S. P. Schröder, C. de Boer, M. Artola, J. M. Aerts, H. S. Overkleeft and G. J. Davies, Curr. Opin. Chem. Biol., 2019, 53, 25–36 CrossRef CAS PubMed.
- Y. M. C. A. Luijkx, S. Jongkees, K. Strijbis and T. Wennekes, Org. Biomol. Chem., 2021, 19, 2968–2977 RSC.
- M. Audry, C. Jeanneau, A. Imberty, A. Harduin-Lepers, P. Delannoy and C. Breton, Glycobiology, 2011, 21, 716–726 CrossRef CAS PubMed.
- Y. Li and X. Chen, Appl. Microbiol. Biotechnol., 2012, 94, 887–905 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- C. P. C. Chiu, A. G. Watts, L. L. Lairson, M. Gilbert, D. Lim, W. W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, Nat. Struct. Mol. Biol., 2004, 11, 163–170 CrossRef CAS PubMed.
- C. P. C. Chiu, L. L. Lairson, M. Gilbert, W. W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, Biochemistry, 2007, 46, 7196–7204 CrossRef CAS.
- L. Ni, H. A. Chokhawala, H. Cao, R. Henning, L. Ng, S. Huang, H. Yu, X. Chen and A. J. Fisher, Biochemistry, 2007, 46, 6288–6298 CrossRef CAS.
- Y. Kakuta, N. Okino, H. Kajiwara, M. Ichikawa, Y. Takakura, M. Ito and T. Yamamoto, Glycobiology, 2008, 18, 66–73 CrossRef CAS PubMed.
- F. V. Rao, J. R. Rich, B. Raki, S. Buddai, M. F. Schwartz, K. Johnson, C. Bowe, W. W. Wakarchuk, S. Defrees, S. G. Withers and N. C. J. Strynadka, Nat. Struct. Mol. Biol., 2009, 16, 1186–1188 CrossRef CAS.
- H. J. Lee, L. L. Lairson, J. R. Rich, E. Lameignere, W. W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, J. Biol. Chem., 2011, 286, 35922–35932 CrossRef CAS.
- L. Y. C. Lin, B. Rakic, C. P. C. Chiu, E. Lameignere, W. W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, J. Biol. Chem., 2011, 286, 37237–37248 CrossRef CAS PubMed.
- L. Meng, F. Forouhar, D. Thieker, Z. Gao, A. Ramiah, H. Moniz, Y. Xiang, J. Seetharaman, S. Milaninia, M. Su, R. Bridger, L. Veillon, P. Azadi, G. Kornhaber, L. Wells, G. T. Montelione, R. J. Woods, L. Tong and K. W. Moremen, J. Biol. Chem., 2013, 288, 34680–34698 CrossRef CAS PubMed.
- C. Lizak, L. J. Worrall, L. Baumann, M. M. Pfleiderer, G. Volkers, T. Sun, L. Sim, W. Wakarchuk, S. G. Withers and N. C. J. Strynadka, Sci. Rep., 2017, 7, 1–13 CrossRef CAS.
- D. Harrus, A. Harduin-Lepers and T. Glumoff, J. Struct. Biol., 2020, 212, 107628 CrossRef CAS.
- D. Kumawat, T. E. Gray, C. R. Garnier, D. T. Bui, Z. Li, Z. Jame-Chenarboo, J. Jerasi, W. O. Wong, J. S. Klassen, C. J. Capicciotti and M. S. Macauley, J. Am. Chem. Soc., 2024, 146, 28630–28634 CrossRef CAS.
- K. Shiraiwa, R. Cheng, H. Nonaka, T. Tamura and I. Hamachi, Cell Chem. Biol., 2020, 27, 970–985 CrossRef CAS.
- H. Prasch, A. Wolfsgruber, M. Thonhofer, A. Culum, C. Mandl, P. Weber, M. Zündel, S. A. Nasseri, A. Gonzalez Santana, G. Tegl, B. Nidetzky, K. Gruber, A. E. Stütz, S. G. Withers and T. M. Wrodnigg, ChemBioChem, 2023, 24, e202300480 CrossRef CAS.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- M. Chen, M. Asanuma, M. Takahashi, Y. Shichino, M. Mito, K. Fujiwara, H. Saito, S. N. Floor, N. T. Ingolia, M. Sodeoka, K. Dodo, T. Ito and S. Iwasaki, Cell Chem. Biol., 2021, 28, 475.e8–486.e8 Search PubMed.
- K. R. Barnes, J. Blois, A. Smith, H. Yuan, F. Reynolds, R. Weissleder, L. C. Cantley and L. Josephson, Bioconjugate Chem., 2008, 19, 130–137 CrossRef CAS PubMed.
- D. Xue, L. Ye, J. Zheng, Y. Wu, X. Zhang, Y. Xu, T. Li, R. C. Stevens, F. Xu, M. Zhuang, S. Zhao, F. Zhao and H. Tao, Org. Biomol. Chem., 2019, 17, 6136–6142 RSC.
- G. Sugiarto, K. Lau, J. Qu, Y. Li, S. Lim, S. Mu, J. B. Ames, A. J. Fisher and X. Chen, ACS Chem. Biol., 2012, 7, 1232–1240 CrossRef CAS PubMed.
- Y. Takakura, H. Tsukamoto and T. Yamamoto, J. Biochem., 2007, 142, 403–412 CrossRef CAS.
- N. Huynh, Y. Li, H. Yu, S. Huang, K. Lau, X. Chen and A. J. Fisher, FEBS Lett., 2014, 588, 4720–4729 CrossRef CAS PubMed.
- B. Kuhn, J. Benz, M. Greif, A. M. Engel, H. Sobek and M. G. Rudolph, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1826–1838 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Zídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS PubMed.
- D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997–1000 CrossRef CAS.
- D.-U. Kim, J.-H. Yoo, Y.-J. Lee, K.-S. Kim and H.-S. Cho, BMB Rep., 2008, 41, 48–54 CrossRef CAS PubMed.
- L. Ni, M. Sun, H. Yu, H. Chokhawala, X. Chen and A. J. Fisher, Biochemistry, 2006, 45, 2139–2148 CrossRef CAS PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 106(10), 845–858 CrossRef PubMed.
- D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997–1000 CrossRef CAS.
- S. Liu, NMR active substrates for structure and function studies of sialyltransferases, PhD Thesis, University of Georgia, Athens, US, 2008.
- T. Sun, S.-H. Yu, P. Zhao, L. Meng, K. W. Moremen, L. Wells, R. Steet and G.-J. Boons, J. Am. Chem. Soc., 2016, 138, 11575–11582 CrossRef CAS PubMed.
- H. de Jong, M. J. Moure, J. E. M. Hartman, G. P. Bosman, J. Y. Ong, B. W. Bardoel, G. J. Boons, M. M. S. M. Wösten and T. Wennekes, ChemBioChem, 2022, 23, e202200340 CrossRef CAS PubMed.
- R. E. Mandrell, H. Smith, G. A. Jarvis, J. McLeod Griffiss and J. A. Cole, Microb. Pathog., 1993, 14, 307–313 CrossRef CAS.
- M. Packiam, D. M. Shell, S. V. Liu, Y.-B. Liu, D. J. McGee, R. Srivastava, S. Seal and R. F. Rest, Infect. Immun., 2006, 74, 2637–2650 CrossRef CAS PubMed.
- J. Bramley, R. D. de Hormaeche, C. Constantinidou, X. Nassif, N. Parsons, P. Jones, H. Smith and J. Cole, Microb. Pathog., 1995, 18, 187–195 CrossRef CAS PubMed.
- M. J. Gill, D. P. Mcquillen, J. P. M. Van Putten, L. M. Wetzler, J. Bramley, H. Crooke, N. J. Parsons, J. A. Cole and H. Smith, Infect. Immun., 1996, 64, 3374–3378 CrossRef CAS.
- E. I. Alvarado-Melendez, H. Jong, J. E. M. Hartman, J. Y. Ong, M. M. S. M. Wösten and T. Wennekes, Glycobiology, 2024, 34, cwae071 CrossRef CAS.
- D. M. Shell, L. Chiles, R. C. Judd, S. Seal and R. F. Rest, Infect. Immun., 2002, 70, 3744–3751 CrossRef CAS.
- T. Rudel, A. Schmid, R. Benz, H. A. Kolb, F. Lang and T. F. Meyer, Cell, 1996, 85, 391–402 CrossRef CAS.
- M. Tanabe, C. M. Nimigean and T. M. Iverson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6811–6816 CrossRef CAS PubMed.
- L. Gao, L. Linden, N. J. Parsons, J. A. Cole and H. Smith, Microb. Pathog., 2000, 28, 257–266 CrossRef CAS.
- F. E. C. Jen, M. R. Ketterer, E. A. Semchenko, C. J. Day, K. L. Seib, M. A. Apicella and M. P. Jennings, mBio, 2021, 12, 1–11 CrossRef PubMed.
- M. Kosar, D. A. Sykes, A. E. G. Viray, R. M. Vitale, R. C. Sarott, R. L. Ganzoni, D. Onion, J. M. Tobias, P. Leippe, C. Ullmer, E. A. Zirwes, W. Guba, U. Grether, J. A. Frank, D. B. Veprintsev and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 15094–15108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07184k |
| This journal is © The Royal Society of Chemistry 2025 |