
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble-core nanothread formation from α-furil via a pressure-induced planarization pathway†
Samuel G.
Dunning‡
 *a,
Anirudh
Hari
a,
Li
Zhu
b,
Bo
Chen
cd,
George D.
Cody
a,
Sebastiano
Romi
a,
Dongzhou
Zhang
e and
Timothy A.
Strobel
*a
*a,
Anirudh
Hari
a,
Li
Zhu
b,
Bo
Chen
cd,
George D.
Cody
a,
Sebastiano
Romi
a,
Dongzhou
Zhang
e and
Timothy A.
Strobel
*a
aEarth and Planets Laboratory, Carnegie Institution for Science, Washington, District of Columbia 20015, USA. E-mail: sdunning@carnegiescience.edu; tstrobel@carnegiescience.edu
bPhysics Department, Rutgers University-Newark, 101 Warren Street, Newark, NJ 07102, USA
cDonostia International Physics Center, Paseo Manuel de Lardizabal, 4, 20018 Donostia-San Sebastian, Spain
dIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Spain
eHawaii Institute of Geophysics and Planetology, School of Ocean and Earth Science and Technology, University of Hawaii at Manoa, Honolulu, HI 96822, USA
First published on 3rd February 2025
Abstract
The packing and geometry of compressed small molecule precursors largely dictate the kinetically controlled formation processes of carbon nanothread materials. Structural ordering and chemical homogeneity of nanothread products may deteriorate through competing reaction pathways, and molecular phase transitions can disrupt precursor stacking geometries. Here, we report the formation of well-ordered, double-core nanothreads from compressed α-furil via a unique polymorphic transition pathway that serves to facilitate a pressure-induced reaction. At ∼1.6 GPa, α-furil transforms to the photoactive trans-planar conformation, which was previously theorized but not observed. Crystalline packing of the trans-planar structure provides closely overlapping molecular stacks that result in topochemical-like Diels–Alder cycloaddition reactions between furan rings upon further compression. The controlled reaction pathways on both sides of the molecule produce two linked “cores” of chemically homogenous nanothreads, and successive nucleophilic addition reactions crosslink a large fraction of the diketone bridges between monomers.
Introduction
Diamond nanothreads represent a novel class of one-dimensional sp3 carbon-rich nanomaterials, formed from the compression of small molecules to high (GPa) pressures.1–15 The initial discovery of crystalline nanothreads from benzene1,2 suggested synthetic design principles for controlled high-pressure reactions, in contrast to previous work that showed the formation of disordered products.16–23 While crystalline one-dimensional nanothreads are in stark contrast to the previous paradigm of disorder, this crystallinity is typically the result of the macromolecular packing of individual chemically inhomogeneous threads.1–3,14,24–26 That is, atomically precise nanothreads with full three-dimensional long-range order still represent a major synthetic challenge, and most threads show a range of local chemical environments due to competing reaction pathways.27,28The initial discovery of nanothreads in 2015 sparked new interest in high-pressure solid-state organic reactions and nanothreads with a diverse range of chemical functionalities and structural motifs have since been produced.3–15 Several strategies to improve thread homogeneity and 3D order have been employed, and different approaches can significantly constrain reaction paths to produce highly ordered materials.5,6,9 Solution phase reactions29,30 and theoretical studies31 have shown that certain reaction pathways, such as [4 + 2] Diels–Alder cycloaddition reactions, are accelerated by increasing pressure due to their negative activation volumes. Such reactivity can also be translated to solid-state reactions, and accordingly, precursors containing five-membered ring systems (e.g., furan,4–6 thiophene7) have been shown to promote the formation of nanothreads with greatly increased chemical homogeneity, largely due to the incorporation of heteroatoms that constrain reactivity along a single [4 + 2] Diels–Alder cycloaddition pathway.4–6,32,33 While this reaction pathway may seem unorthodox in the context of traditional organic chemistry, recent theoretical studies have shown that small molecules such as furan can act as both diene and dienophile at extreme pressures.33 Indeed, the first “perfect” threads were synthesized from furans4–6,32 and pyridazine9 (a 6-membered diazine ring system). In addition, reduced aromaticity caused by heteroatom incorporation can contribute to lower reaction pressures for nanothread formation, e.g., 10–13 GPa4–6,9,32 for furan/pyridazine vs. ∼20 GPa for benzene.1,34 Interestingly, the formation of “perfect” nanothreads via sequential [4 + 2] cycloaddition reactions from carbonyl-substituted furans (e.g., 2,5-furandicarboxylic acid)5,6 suggest that high-pressure reactions may be a route to increase the reactivity of typically electron-poor dienes. It is also important to note that the most chemically homogeneous threads have been synthesized from planar systems which crystallize with appropriate π-stacking for topochemical-like nanothread formation, allowing nanothreads to be formed under rapid, quasi-hydrostatic compression conditions in a diamond anvil cell. Precursors that crystallize without a significant degree of π-stacking overlap (e.g., furan, benzene) may undergo non-topochemical nanothread formation induced by anisotropic stress, typically under slow (GPa hour−1) uniaxial compression. This is important to keep in mind as we move towards synthetic strategies that incorporate precursors with increasing chemical diversity, in which functional groups will impact π-stacking geometries.
Recent research has investigated nanothread formation in larger molecular precursors with different functionalities and increased structural complexity.5,6 One interesting class of more complex nanothreads are the so-called “double-core” nanothreads.11–14,35 First reported by Romi and coworkers, double-core nanothreads are formed from the compression of molecules containing linked aromatic rings (e.g., stilbene,14 azobenzene11,12), resulting in the formation of two 1D nanothread polymers connected together by a bridging motif. Double/multi-core nanothreads provide an opportunity to extend nanothread systems to pseudo-2D structures with an expanded range of chemical properties. For example, double-core nanothreads produced from diphenylacetylene represent tunable low-bandgap polyacetylene-like polymers, protected on the sides by sp3 nanothread cores.13,35 To date, double-core threads have only been produced from precursors containing six-membered aromatic ring systems, and the resulting nanothread products contain a large degree of disorder and coexist with an amorphous sp2–sp3 carbon matrix due to the presence of multiple competing reaction pathways (cf. benzene). This disorder has made precise structural determination a significant challenge and has led to debate over the final structure of the high-pressure reaction products for these systems. For example, recent publications on the pressure-induced polymerization of azobenzene have proposed both double-core nanothread,11,24 and pseudo-two dimensional nanoribbon36 structures for the recovered polymeric materials.
One factor that has hindered the formation of larger, chemically homogeneous systems is their molecular geometry. Planar molecules are regularly used as precursors for nanothread formation as their molecular geometries at ambient pressure often already occupy the smallest volume, and will therefore be favored under high-pressure conditions. While high-pressure phase changes may result in alteration of the overall molecular packing, such phase changes are unlikely to result in significant structural rearrangements of planar molecules. Therefore, the crystal structures of these systems collected at ambient may be used to predict high-pressure reaction pathways. While this approach has been successful, it has naturally excluded studies of non-planar systems. Nevertheless, many bridged systems are non-planar, yet possess aromatic rings that are well positioned for nanothread formation and structural motifs that would be interesting to incorporate into nanothread materials. We are therefore interested in investigating the high-pressure behavior of a non-planar system that also incorporates five-membered ring systems. Such a precursor would allow us to test if the design principles that have allowed for the formation of “perfect” nanothreads from small molecule precursors can also be applied to more chemically and structurally complex precursors in order to extend reaction control to larger systems with diverse future applications.
Herein, we report the high-pressure behavior of the 1,2-diketone system α-furil. Compression of α-furil leads to the formation of high-pressure trans-planar phase with excellent π-stacking overlap, which had previously been theorized but not validated experimentally. Additional compression results in the formation of an extended, double-core nanothread polymer through sequential [4 + 2] Diels–Alder cycloaddition reactions between overlapping furan rings.
Results and discussion
In situ characterization of high-pressure phase changes in α-furil
α-Furil (2,2′-furil, C10H6O4) is a potential candidate for the formation of double-core nanothreads with improved order and homogeneity due to the presence of overlapping five-membered furan rings. These furan rings, linked by a 1,2-diketone bridge, show parallel-displaced π-stacking and short intermolecular C⋯C distances (i.e., <4 Å), compatible with previous reports of temperature37 and pressure-induced3–6,9,10,32 [4 + 2] Diels–Alder cycloaddition reactions. We hypothesize that a cascade Diels–Alder polymerization reaction would proceed along the stacking axis with high selectivity3 to produce a double-core nanothread consisting of two connected degree-4 threads, each forming in the syn conformation (i.e., all oxygen atoms aligned along the same side). Double-core threads linked by a 1,2-diketone bridge could enable photoreactions such as those observed for structurally analogous systems such as benzil,38 phenanthrenequinone,38 retenequinone,38 diacetyl,39 and pyruvic acid,40 which have all been found to react with alkenes, alkynes and water in the presence of sunlight. Nevertheless, unlike the majority of nanothread-forming precursors, α-furil is non-planar; the furan ring planes of furil are canted by ∼48° from one another and the molecule exists in a buckled conformation such that simultaneous nanothread formation for both ring stacks would induce significant bridge strain. Furthermore, intermolecular ketone interactions could potentially impact reactivity of the rings.To investigate reactivity under high-pressure conditions, powder samples of α-furil were loaded into diamond anvil cells (DACs), and the structure and behavior was monitored with in situ X-ray diffraction (XRD) and vibrational spectroscopy. PXRD patterns of starting α-furil at 1 atm readily index to the previously reported orthorhombic Fdd2 phase.41,42 Upon compression, several new diffraction peaks emerge, indicating a pressure-induced phase transition (Fig. 1A and S1†). Single-crystal diffraction of α-furil collected between 0.5 and 4.4 GPa was used to solve the structure of this high-pressure phase (Fig. 1B and Table S1†). At 0.5 GPa, α-furil remains in the ambient-pressure Fdd2 phase, albeit with a 6.7% reduction in cell volume. Upon further compression to 1.6 GPa, α-furil planarizes, resulting in a transition to the monoclinic space group P21/n with cell parameters a = 14.586(5) Å, b = 7.0535(14) Å, c = 3.4512(6) Å, β = 93.132(6)° (see ESI†). This new cell is in good agreement with the analogous thiophene structure, α-thenil, which crystallizes in the trans-planar conformation, occupying a P21/n lattice (a = 14.73, b = 7.27, c = 4.75, β = 112.41°) albeit with minimal stacking overlap between aromatic rings.43,44 Planar 1,2-diketone systems are generally uncommon due to steric interactions between carbonyl oxygens and attached ring systems.41 However, the dihedral angle between rings in α-furil at ambient conditions is notably larger than analogous complexes (138.4° vs. 114.9° in benzil45) indicating that the smaller ring size may reduce steric clashes and therefore the barrier to planarization. Indeed, our theoretical calculations indicate a minimal (+0.2 kcal mol−1) energy difference between the ambient-pressure and trans-planar phases of α-furil. This is consistent with previous theoretical studies that indicate that a free molecule of α-furil with a dihedral angle of 180° is very close in energy to the global minimum (156.1°).46 It is important to note that the isolated molecular global minimum does not account for stabilization effects caused by crystal packing, which result in the smaller, crystallographically observed dihedral angle. The reduced volume of the high-pressure phase is a result of improved packing efficiency of the trans-planar structure. Confirmation of the existence of the trans-planar phase of α-furil validates previous studies into the photophysics of 1,2-dicarbonyl systems that theorized the existence of a short-lived trans-planar excited state for α-furil, from which photoemission occurs.46–48 A second structure of α-furil collected at 4.4 GPa (see ESI†) indicates that phase II persists upon further compression. Again, the furan rings are well placed to undergo a pressure-induced [4 + 2] Diels–Alder cycloaddition reaction, with an average intermolecular C⋯C distance of 2.98 Å (see Fig. 1B).
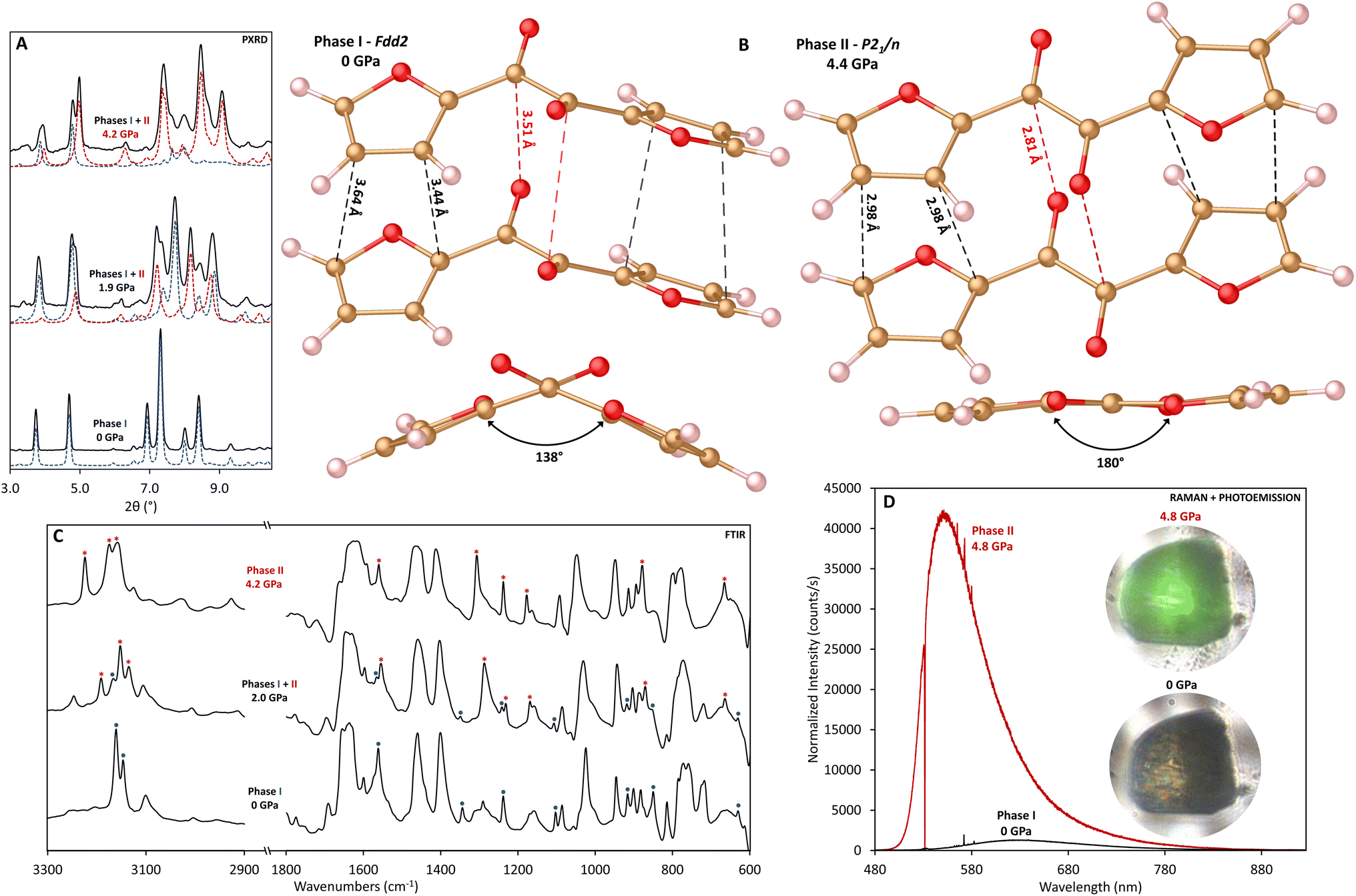 | ||
| Fig. 1 (A) Comparison of experimental XRD data collected at 0, 1.9, and 4.2 GPa (black lines) and phase profiles from Le Bail refinements at the same pressures calculated using phase I (blue dash) and phase II (red dash) structures of α-furil (λ = 0.4340 Å); (B) top: structures of phase I and phase II α-furil taken from crystal structure data collected at 0 GPa and 4.4 GPa respectively. The orientation of the leftmost furan ring is kept constant between structures to highlight the twisting of the 1,2-diketone bridge between phases. Black lines indicate the closest contact intermolecular C⋯C distances corresponding to a [4 + 2] cycloaddition pathway. Red lines represent the closest contact intermolecular C⋯O distances. Bottom: View of the same structures along the 1,2-diketone bridge, highlighting the torsion angle between the four carbons in the bridge motif; (C) comparison of the FTIR spectra for α-furil collected at 0, 2.0, and 4.2 GPa, between 600–1800 and 2900–3300 cm−1. The region between 1800 and 2900 cm−1 with diamond absorption has been omitted for clarity. Highlighted are vibrational modes corresponding with phase I (blue dots, decreasing in intensity as a function of pressure) and phase II (red asterisks, increasing in intensity as a function of pressure); (D) comparison of the Raman/photoemission spectra for phase I (black) and phase II (red) α-furil (excitation: 532 nm). Spectra have been normalized based on collection time. The inset shows photomicrographs of an α-furil crystal (∼50 × 50 μm) across the transition with laser-induced emission. | ||
The low-pressure FTIR spectra of α-furil are in good agreement with previously reported studies.49 However, above 1.1 GPa, several peaks disappear and new peaks emerge, at ca. 665, 874, 1167, 1234, 1287, 1552, 1629, 3131, and 3186 cm−1 (Fig. 1C and S2†), caused by the change to the trans-planar phase. Above 8.6 GPa, a new set of peaks begin to emerge at ca. 879, 1022, and 1141 cm−1, increasing in intensity as a function of pressure. These new peaks are generally consistent with changes seen upon the onset of nanothread formation in the case of 2,5-furandicarboxylic acid (cf. 1163, 899 cm−1) which polymerizes to form a syn-furan nanothread wherein all the furan atoms have the same orientation. At pressures above 15 GPa peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches above 1600 cm−1 begin to dramatically decrease in intensity, complimented by a corresponding increase in the new peak at ca. 1022 cm−1, which is generally consistent with the formation of sp3 C–O bonds. While this may indicate reaction of the carbonyl groups at pressure, it is important to note that furan nanothread formation will also result in the formation of sp3 C–O–C bonds.
O stretches above 1600 cm−1 begin to dramatically decrease in intensity, complimented by a corresponding increase in the new peak at ca. 1022 cm−1, which is generally consistent with the formation of sp3 C–O bonds. While this may indicate reaction of the carbonyl groups at pressure, it is important to note that furan nanothread formation will also result in the formation of sp3 C–O–C bonds.
Raman studies show behavior that is consistent with FTIR, and at 1.5 GPa several new vibrational modes appear at ca. 206, 616, 656, 1405, and 1562 cm−1 (Fig. S3†). The Raman spectra also show that α-furil exhibits a mechanochromic response to pressure. At ambient pressure a broad emission can be seen at ca. 623 nm, which may be related to trace impurities which can be seen in the 1H NMR of the α-furil starting material (see ESI†). Above 3.3 GPa a second emission centered at ca. 541 nm emerges, becoming more intense with increasing pressure and overwhelming Raman scattering above 4 GPa (Fig. 1D). Previous studies into the photoemission behavior of α-furil in solution have assigned emission in this region to the phosphorescence bands from the triplet state of a theorized trans-planar intermediate.46–48 However, this trans-planar phase has not been directly observed due to its transient nature. The vibrational changes observed in both Raman and FTIR studies were found to be reversible up to at least 4.6 GPa (see Fig. S2†), strongly indicating that they are related to the polymorphic molecular phase change and not the onset of an irreversible chemical reaction.
Powder X-ray diffraction patterns and IR spectra of α-furil collected above 1.9 GPa are in good agreement with the new planar phase. Interestingly, while single crystals of α-furil compressed under quasi-hydrostatic conditions show complete conversion to the trans-planar phase above 1.9 GPa, powder samples compressed without a pressure transmitting medium (i.e., anisotropic compression) show coexistence of both phases at similar pressures (see Fig. 1A). Further compression of α-furil results in increased conversion to the trans-planar phase and a uniform shift of Bragg peaks to higher angles, consistent with pressure-induced contraction of the unit cell. Above 8.5 GPa, significant changes are observed in the diffraction pattern with the emergence of new peaks at d = 6.30, 5.08, 4.00 and 3.37 Å (see Fig. S1†). These new reflections grow in intensity with increasing pressure and no further changes are noted upon additional compression to 25.2 GPa. These new reflections remain upon decompression to ambient pressure, suggesting that these new peaks are not due to another polymorphic pressure-induced phase change, and indicate that α-furil has undergone a pressure-induced chemical reaction. In situ IR data collected after the phase change are consistent with the changes observed in XRD. The intensity of the additional vibrational modes that emerge above 8.6 GPa increases upon further compression. At the same time the intensity of the peaks corresponding to phase I and II α-furil decreases significantly up to 19.6 GPa. Additionally, the large photoemission intensity observed after the transition to the trans-planar phase broadens and shifts to lower energy above 7 GPa, shifting to ca. 634 nm at 18 GPa. This broad emission remains ca. 634 nm upon decompression to ambient pressure (see Fig. S3†), and is consistent with the formation of other “perfect” nanothreads such as pyridazine-derived threads, which also shows broad fluorescence at ca. 630 nm.
Characterization of nanothread polymer
Given the relatively low reaction onset pressure, synthesis was readily scaled using a Paris–Edinburgh (PE) press at ∼20 GPa to produce bulk samples for advanced analysis. XRD and FTIR of recovered PE press and DAC samples are in good agreement (Fig. S4†), with the PE press sample exhibiting clearly defined diffraction out to d ≈ 2.13 Å. Unlike molecular furan which is a yellow powder, the recovered sample is a deep red color. The recovery of a highly colored sample is consistent with other nanothread systems, such as pyridazine,9 pyridine,25,26 and 2,5-furandicarboxylic acid5,6 which all possess an intense orange color. While the exact cause of this color change is likely to be system dependent, it is commonly believed to be due to the presence of defects along the thread backbone comparable to diamond, which can become highly colored by the inclusion of minor defects.Comparison of solid-state 13C NMR collected on α-furil, and the washed product (Fig. 2A) agrees with the formation of a nanothread material rich in sp3 carbon. Two new peaks can be seen at ca. 50 and 83 ppm, which are consistent with previous solid-state NMR studies carried out on syn-2,5-furandicarboxylic acid nanothreads (cf., 58 and 89 ppm).5 However, the peak widths in the recovered sample are notably larger than those seen in comparable syn-furan nanothreads, strongly indicating that the presence of a broader distribution of sp3 C environments. Accordingly, we can only generally assign the peak at ca. 50 ppm to sp3 C–C bonds, and the peak at ca. 83 ppm to sp3 C–O bonds. Interestingly, the recovered product shows no significant intensity associated with CO functionality at ca. 178 ppm in the molecular phase, which strongly indicates reaction of the 1,2-diketone bridge during polymerization. It is also important to note that the recovered sample exhibits significantly different relaxation dynamics compared to the starting material, resulting in a significant decrease in the observed signal-to-noise ratio under the same collection times, and it is possible that a small number of sp2 CO environments remain, but are not detected. Indeed, some weak intensity above the baseline can be seen up to approximately 200 ppm. While carbon environments in this region would be consistent with α,β-diketones attached to saturated, sp3 hybridized carbon,50,51 it is not possible to carry out meaningful interpretation of this data due to poor signal intensity. Although cross-polarization magic-angle spinning (CPMAS) NMR is not quantitative (see ESI†), the presence of smaller peaks at 148 ppm and 113 ppm potentially indicates retention of some amount of residual sp2 carbon after washing. 1H NMR studies collected on the same sample (Fig. 2B) confirm the presence of residual sp2 carbon environments. The resulting spectrum is described by two peaks at ca. 3.6 and 7.3 ppm, which we assign to sp3 and sp2 C–H bonds, respectively. Comparison of the area under each peak indicates a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of sp3:sp2 C–H bonds, consistent with the formation of a nanothread containing residual sp2 carbon defects and/or a disordered sp2 material. It is important to note that while the 1H spectrum can be used to estimate the relative proportion of hydrogen environments within the sample, the broad nature of the spectrum limits the certainty of quantification.
:1 ratio of sp3:sp2 C–H bonds, consistent with the formation of a nanothread containing residual sp2 carbon defects and/or a disordered sp2 material. It is important to note that while the 1H spectrum can be used to estimate the relative proportion of hydrogen environments within the sample, the broad nature of the spectrum limits the certainty of quantification.
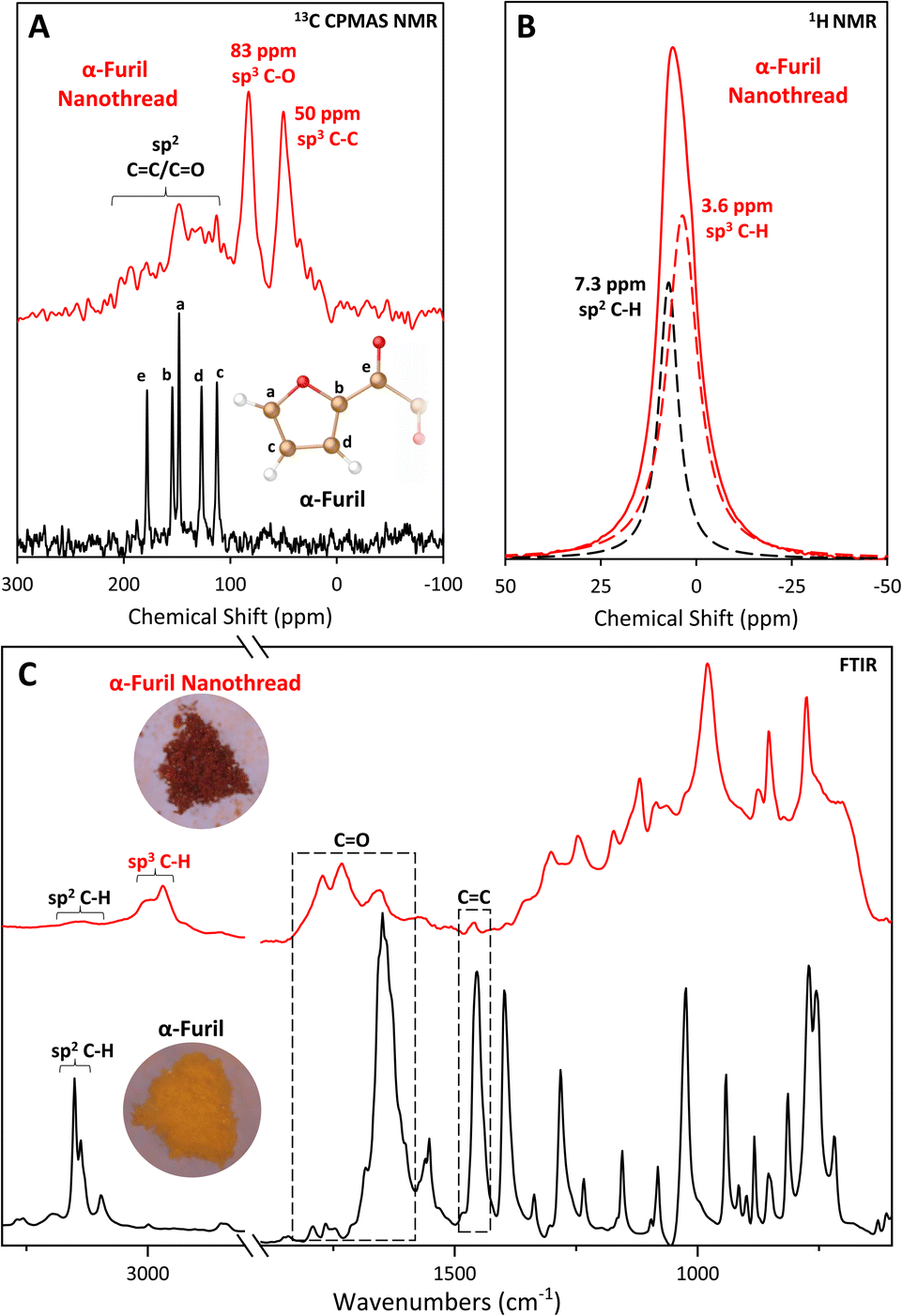 | ||
| Fig. 2 (A) Solid-state 13C CPMAS NMR spectra of α-furil (black, bottom) and the recovered nanothread (red, top) between 300 and −100 ppm. Inset: Assignment of 13C NMR peaks in phase I α-furil. Only chemically unique carbons are shown. No additional peaks were observed outside of the spectral range shown; (B) solid-state 1H NMR spectrum of the same recovered nanothread (solid red line) between 50 and −50 ppm. Dashed black and red lines indicate the fits corresponding to sp2 and sp3 C–H bonds respectively; (C) FTIR spectra of phase I α-furil (black) and the nanothread recovered to ambient pressure (red). The diamond absorption region between 1800 and 2900 cm−1 has been omitted for clarity. Inset: Optical photomicrographs of α-furil before (yellow) and after (red) compression, sample width is ∼1 mm. | ||
The sp2 carbon peaks in the 13C NMR spectrum of the recovered nanothread are in good agreement with the peaks assigned to the carbons in the a and c positions of the furan ring in unreacted α-furil, furthest away from the diketone bridge (Fig. 2A(a) and (c), ca. 148 and 112 ppm at ambient respectively), potentially indicating retention of this bond. Indeed, the FTIR spectrum of the washed nanothread also shows evidence for the retention of sp2 CC vibrations at ca. 1452 cm−1 and sp2 C–H stretching vibrations at ca. 3150 cm−1, although their intensities are significantly reduced (Fig. 2C). While the shortest intermolecular O⋯C separation distance between a carbonyl oxygen and a furan ring in phase II α-furil is longer than the closest contact [4 + 2] cycloaddition pathway (3.15 vs. 2.98 Å at 4.4 GPa, respectively), it is still possible that some reactivity occurs between ketone groups and the closest CC double bond in the furan ring. Such a reaction would therefore prevent [4 + 2] cycloaddition and would result in the formation of sp2 CC defects at the position furthest away from the diketone bridge.
The IR spectrum of the recovered PE press sample, washed to remove any residual starting phase, confirms the formation of a new material rich in sp3 carbon (Fig. 2C). The emergence of new sp3 C–H stretches at ca. 2968 and 3000 cm−1, clearly indicates the formation of a sp3-C rich material consistent with nanothread formation. Additionally, the presence of new peaks at ca. 1730 and 1770 cm−1 are consistent with α,β-diketones attached to sp3-hybridized carbons (cf., 1690–1769 cm−1 in diacetyl52) and analogous carbonyl-functionalized nanothreads (1742 cm−1 in 2,5-furandicarboxylic acid nanothreads5). Peaks at 775, 852, and 877 cm−1 are also consistent with ring vibrations of syn-furan nanothreads (cf., 765, 851, and 885 cm−1 in syn-2,5-furandicarboxylic acid nanothreads5). The formation of a double-core nanothread product, wherein the furan rings are arranged in the syn conformation (i.e., all oxygen atoms arranged on the same side of the nanothread core), is consistent with the orientation of the starting molecular structure. However, the presence of the carbonyl peak at ca. 1656 cm−1 (albeit with notably decreased intensity) suggests partial retention of the diketone bridge as seen in the molecular phase. Additionally, the relatively broad absorbance background indicates the presence of a disordered component coexisting with the resulting nanothread.
In order to determine if the carbonyl peak at 1656 cm−1 is due to the presence of residual trapped molecular precursor or polymerization defects, we carried out a combined thermogravimetric analysis/differential scanning calorimetry study on the same sample used for FTIR. While molecular α-furil exhibits an endothermic mass loss beginning at approximately 153 °C, consistent with sublimation, the recovered nanothread shows no mass loss in the same region (Fig. S5†). Instead, the recovered nanothread does not begin to exhibit any mass loss until the onset of an exothermic decomposition process at approximately 364 °C, indicating the formation of an extended, polymeric system.
To understand the potential side reactions which may result in loss of ketone functionality, it is necessary to reexamine the structure of phase II α-furil. In this planar phase the diketone bridge is indeed well positioned for potential reaction with only a 2.81 Å intermolecular O⋯C separation between carbonyl groups at 4.4 GPa, a shorter spacing than any of the intermolecular C⋯C separation distances observed in phase II α-furil (average = 2.98 Å). Indeed, studies in analogous systems have shown that carbonyl groups can readily act as dienes in Diels–Alder reactions,53,54 and can react with 5-membered ring systems via alternative reaction pathways such as electrophilic aromatic substitution.55 To investigate possible reaction pathways, including possible diketone reactions, we carried out first-principles molecular dynamics (MD) simulations on phase II α-furil (see Fig. S6†) at 50 GPa and 500 K (note that more extreme PT conditions were necessary to compensate for rare-event reaction observations on a computationally limited time scale). After 150 fs, a spontaneous [4 + 2] cycloaddition reaction occurs between furan rings, before the onset of a second reaction between ketones after 450 fs.
Based on these findings we optimized the packed crystal structures of four potential close-contact nanothread products for direct comparison to experimental data (see Table S2†). These structures represent the nanothread formed from [4 + 2] Diels Alder cycloaddition reactions between furan rings with (A) no reaction between ketones, (B) partial reaction between ketones (i.e., only one set of ketones react), (C) complete reaction between ketones, and (D) complete reaction between ketones with alternative diagonal bonding due to the close O⋯C contacts of 2.96 Å between ketone groups at 4.4 GPa. Initial comparison of the energies for the packed structures relative to structure A indicates that structure D is significantly less stable (+1.52 kcal mol−1) than the other candidates and can likely be discounted as a product.
All of the considered DFT-optimized structural models exhibit calculated X-ray diffraction patterns that reproduce most of the experimentally observed features. Rietveld refinement of the recovered PE press sample indicates that the fully reacted structure (model C) provides the best description of the experimental data among the four basic models (Fig. 3, Tables S3 and S4†). While models A and B are poorer matches to the XRD of the recovered sample, some degree of partial reactivity must be present in the recovered structure, based on the experimental evidence for residual sp2 C–H and CO groups in the corresponding FTIR and NMR spectra. The actual polymer product clearly exhibits more complexity than the four base models, however, major structural aspects are well reproduced, and the prohibitively large number of all possible furan ring/diketone defect structures were not examined. Although the major features of the XRD of the recovered nanothreads are described by the simplified nanothread models, XRD patterns also contain small peaks at low angle that are not described by any of the model structures. Preliminary analysis shows that these peaks can be described by an expanded version of the monoclinic cell associated with phase II α-furil, and the impurity structure might be related to trapped molecular precursor or insoluble crystallized n-mer fragments. The intensity of these impurity peaks can be reduced, but not removed entirely, through washing the sample with dichloromethane.
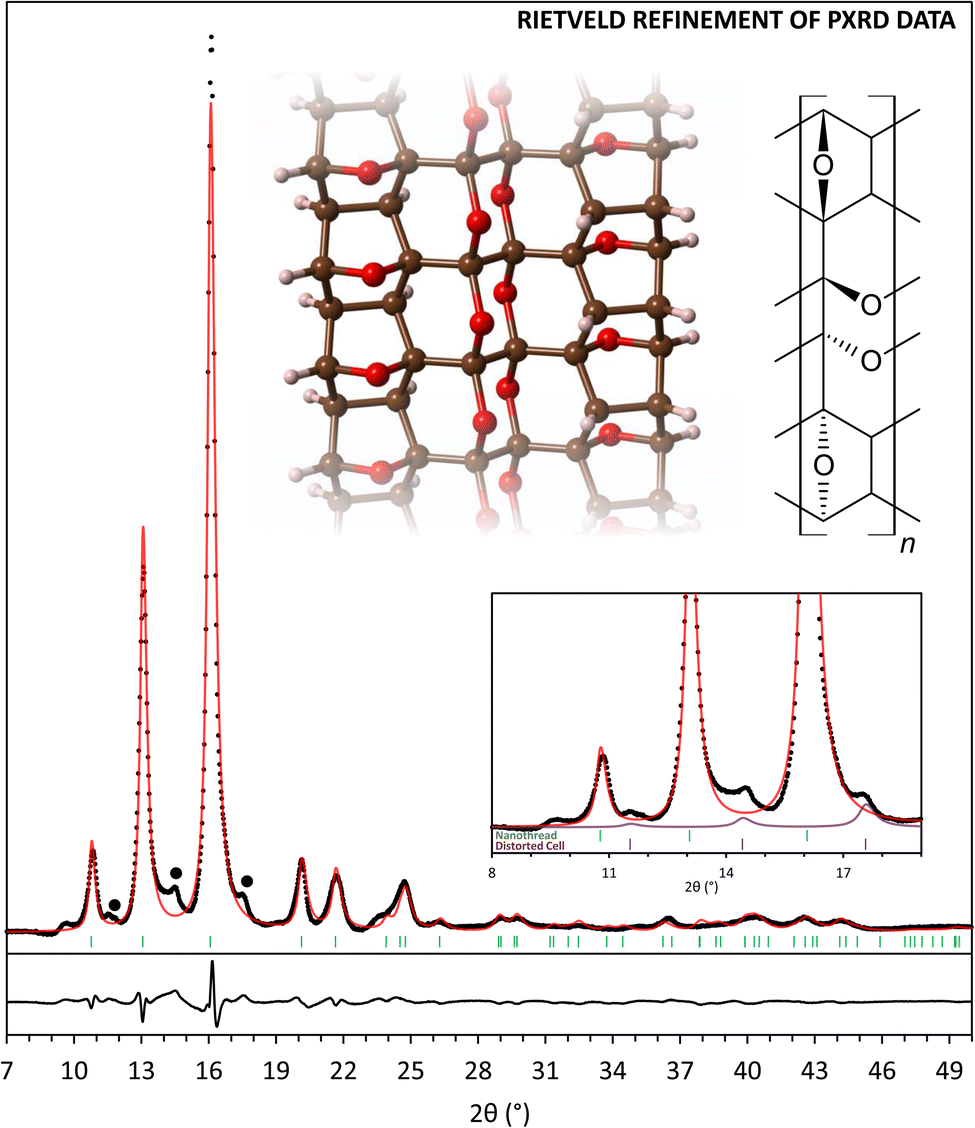 | ||
| Fig. 3 X-ray diffraction pattern (λ = 1.541 Å) of α-furil nanothreads synthesized in a Paris–Edinburgh press and recovered to 1 atm (black points) with Rietveld refinement using nanothread structure C (red line). Black dots indicate peaks corresponding to unidentified phases. Inset, top: A segment of refined nanothread structure C and associated structural formula. Inset, bottom: X-ray diffraction pattern of α-furil nanothreads between 8 and 19°. Green tick marks indicate the relative positions of the three low-angle reflections for the refined nanothread polymer. Purple pattern and tick marks indicate the relative positions of the three low-angle reflections for an expanded lattice of phase II α-furil. | ||
Comparison of calculated and experimental IR spectra provides some additional insights into the local structure of the recovered product (Fig. S7†). All model structures contain a syn-furan nanothread ring system and can therefore each generally describe the peaks seen between 700 and 1000 cm−1. While structure C is consistent with NMR studies (i.e., no significant evidence of carbonyl species) this structure alone cannot account for the peaks at 1656, 1730 and 1770 cm−1 seen in the FTIR spectrum of the recovered nanothread. Structure A is calculated to exhibit a single CO vibration at ca. 1654 cm−1, in excellent agreement with the experimentally observed peak at ca. 1656 cm−1. Structure B is also calculated to exhibit two, higher energy CO vibrations in reasonable agreement with the experimentally observed vibrations at ca. 1730 and 1770 cm−1.
Based on these studies, we can conclude that the structure of the resulting nanothread can largely be described by structure C, as two syn-furan nanothread cores containing a polymerized bridge. However, it is also evident from FTIR and NMR studies that the 1,2-diketone bridge is not fully reacted and the recovered nanothread contains some degree of unreacted carbonyl defects which can be described by the bridging motifs shown in structures A and B. This behavior is consistent with previous studies into double-core nanothreads that have shown uncertainty in the extent of reaction of bridging groups.11,14,35,36,56
Conclusions
In summary, α-furil undergoes pressure-induced planarization to the previously hypothesized trans-planar phase above 1 GPa, before undergoing a topochemical-like polymerization above 8.5 GPa. The discovery of the trans-planar phase validates previous photochemical studies of 1,2-dicarbonyl systems that theorized the existence of a trans-planar excited state from which photoemission occurs. Upon further compression, α-furil polymerizes to form a double-core nanothread material with a high degree of crystalline order. This work also emphasizes the importance of polymorphic phase transitions on the high-pressure behavior of small organic molecules and subsequent chemical reactions. The application of pressure may enable topochemical-like reaction pathways within complex molecular systems via structural rearrangement that might otherwise be overlooked based on initial crystalline geometries.Data availability
The data supporting this article have been included as part of the ESI.† Additional experimental details, plots of diffraction and spectroscopic data, as well as computational models are provided in PDF form. Crystal structures of α-furil at 0.5, 1.6, and 4.4 GPa are provided in cif form. CCDC 2343198–2343200 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.Author contributions
SGD, AH and TAS designed the research project and conducted experimental studies. BC and LZ carried out all theoretical calculations. GDC performed all NMR measurements. SR and DZ assisted with X-ray diffraction studies and structure solutions. All authors contributed to the writing of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Portions of this work were performed at GeoSoilEnviroCARS (The University of Chicago, Sector 13), Advanced Photon Source (APS), Argonne National Laboratory. GeoSoilEnviroCARS is supported by the National Science Foundation – Earth Sciences (EAR – 1634415). This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors acknowledge funding support from The Arnold and Mabel Beckman Foundation (Arnold O. Beckman Postdoctoral Fellowship in Chemical Sciences Program). TS acknowledges support from NSF-DMR under award No. 2226699. BC acknowledges financial support from MCIN/AEI/10.13039/501100011033/FEDER, UE (Project PID2021-123573NA-I00).Notes and references
- X. Li, M. Baldin, T. Wang, B. Chen, E. S. Xu, B. Vermilyea, V. H. Crespi, R. Hoffmann, J. J. Molaison, C. A. Tulk, M. Guthrie, S. Sinogeikin and J. V. Badding, J. Am. Chem. Soc., 2017, 139, 16343–16349 CrossRef CAS PubMed.
- T. C. Fitzgibbons, M. Guthrie, E. S. Xu, V. H. Crespi, S. K. Davidowski, G. D. Cody, N. Alem and J. V. Badding, Nat. Mater., 2015, 14, 43–47 CrossRef CAS PubMed.
- W. S. Tang and T. A. Strobel, J. Phys. Chem. C, 2020, 124, 25062–25070 CrossRef CAS.
- S. Huss, S. K. Wu, B. Chen, T. Wang, M. C. Gerthoffer, D. J. Ryan, S. E. Smith, V. H. Crespi, J. V. Badding and E. Elacqua, ACS Nano, 2021, 15, 4134–4143 CrossRef CAS PubMed.
- S. G. Dunning, B. Chen, L. Zhu, G. D. Cody, S. Chariton, V. B. Prakapenka, D. Zhang and T. A. Strobel, Angew. Chem., Int. Ed., 2023, 62, e202217023 CrossRef CAS.
- X. Wang, X. Yang, Y. Wang, X. Tang, H. Zheng, P. Zhang, D. Gao, G. Che, Z. Wang, A. Guan, J.-F. Xiang, M. Tang, X. Dong, K. Li and H.-k. Mao, J. Am. Chem. Soc., 2022, 144, 21837–21842 CrossRef CAS PubMed.
- A. Biswas, M. D. Ward, T. Wang, L. Zhu, H.-T. Huang, J. V. Badding, V. H. Crespi and T. A. Strobel, J. Phys. Chem. Lett., 2019, 10, 7164–7171 CrossRef CAS PubMed.
- H.-T. Huang, L. Zhu, M. D. Ward, T. Wang, B. Chen, B. L. Chaloux, Q. Wang, A. Biswas, J. L. Gray, B. Kuei, G. D. Cody, A. Epshteyn, V. H. Crespi, J. V. Badding and T. A. Strobel, J. Am. Chem. Soc., 2020, 142, 17944 CrossRef CAS PubMed.
- S. G. Dunning, L. Zhu, B. Chen, S. Chariton, V. B. Prakapenka, M. Somayazulu and T. A. Strobel, J. Am. Chem. Soc., 2022, 144, 2073–2078 CrossRef CAS PubMed.
- D. Gao, X. Tang, J. Xu, X. Yang, P. Zhang, G. Che, Y. Wang, Y. Chen, X. Gao, X. Dong, H. Zheng, K. Li and H.-k. Mao, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2201165119 CrossRef CAS PubMed.
- S. Romi, S. Fanetti, F. Alabarse, A. M. Mio and R. Bini, Chem. Sci., 2021, 12, 7048–7057 RSC.
- S. Romi, S. Fanetti, F. Alabarse and R. Bini, J. Phys. Chem. C, 2021, 125, 17174–17182 CrossRef CAS.
- S. Romi, S. Fanetti, F. G. Alabarse, R. Bini and M. Santoro, Chem. Mater., 2022, 34, 2422–2428 CrossRef CAS.
- S. Romi, S. Fanetti, F. Alabarse, A. M. Mio, J. Haines and R. Bini, Nanoscale, 2022, 14, 4614–4625 RSC.
- M. D. Ward, W. S. Tang, L. Zhu, D. Popov, G. D. Cody and T. A. Strobel, Macromolecules, 2019, 52, 7557–7563 CrossRef CAS.
- M. Citroni, R. Bini, P. Foggi and V. Schettino, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7658–7663 CrossRef CAS PubMed.
- L. Ciabini, M. Santoro, F. A. Gorelli, R. Bini, V. Schettino and S. Raugei, Nat. Mater., 2007, 6, 39–43 CrossRef CAS PubMed.
- P. Pruzan, J. C. Chervin, M. M. Thiery, J. P. Itie, J. M. Besson, J. P. Forgerit and M. Revault, J. Chem. Phys., 1990, 92, 6910–6915 CrossRef CAS.
- V. Schettino and R. Bini, Chem. Soc. Rev., 2007, 36, 869–880 RSC.
- M. D. Ward, H. T. Huang, L. Zhu, D. Popov and T. A. Strobel, J. Phys. Chem. C, 2019, 123, 11369–11377 CrossRef CAS.
- D. W. Keefer, H. Gou, Q. Wang, A. Purdy, A. Epshteyn, S. J. Juhl, G. D. Cody, J. Badding and T. A. Strobel, J. Phys. Chem. A, 2018, 122, 2858–2863 CrossRef CAS PubMed.
- H. Gou, L. Zhu, H.-T. Huang, A. Biswas, D. W. Keefer, B. L. Chaloux, C. Prescher, L. Yang, D. Y. Kim, M. D. Ward, J. Lerach, S. Wang, A. R. Oganov, A. Epshteyn, J. V. Badding and T. A. Strobel, Chem. Mater., 2017, 29, 6706–6718 CrossRef CAS.
- H. Gou, B. L. Yonke, A. Epshteyn, D. Y. Kim, J. S. Smith and T. A. Strobel, J. Chem. Phys., 2015, 142, 194503 CrossRef PubMed.
- S. Romi, S. Fanetti, F. Alabarse and R. Bini, J. Phys. Chem. C, 2021, 125, 17174–17182 CrossRef CAS.
- X. Li, T. Wang, P. Duan, M. Baldini, H. T. Huang, B. Chen, S. J. Juhl, D. Koeplinger, V. H. Crespi, K. Schmidt-Rohr, R. Hoffmann, N. Alem, M. Guthrie, X. Zhang and J. V. Badding, J. Am. Chem. Soc., 2018, 140, 4969–4972 CrossRef CAS.
- S. M. Oburn, S. Huss, J. Cox, M. C. Gerthoffer, S. Wu, A. Biswas, M. Murphy, V. H. Crespi, J. V. Badding, S. A. Lopez and E. Elacqua, J. Am. Chem. Soc., 2022, 144, 22026–22034 CrossRef CAS.
- P. Duan, X. Li, T. Wang, B. Chen, S. J. Juhl, D. Koeplinger, V. H. Crespi, J. V. Badding and K. Schmidt-Rohr, J. Am. Chem. Soc., 2018, 140, 7658–7666 CrossRef CAS PubMed.
- B. Chen, R. Hoffmann, N. W. Ashcroft, J. Badding, E. S. Xu and V. Crespi, J. Am. Chem. Soc., 2015, 137, 14373–14386 CrossRef CAS PubMed.
- H. Kotsuki, H. Nishizawa, S. Kitagawa, M. Ochi, N. Yamasaki, K. Matsuoka and T. Tokoroyama, Bull. Chem. Soc. Jpn., 1979, 52, 544–548 Search PubMed.
- W. G. Dauben, J. M. Gerdes and D. B. Smith, J. Org. Chem., 1985, 50, 2576–2578 CrossRef CAS.
- B. Chen, R. Hoffmann and R. Cammi, Angew. Chem., Int. Ed., 2017, 56, 11126–11142 CrossRef CAS PubMed.
- B. S. Matsuura, S. Huss, Z. X. Zheng, S. C. Yuan, T. Wang, B. Chen, J. V. Badding, D. Trauner, E. Elacqua, A. C. T. van Duin, V. H. Crespi and K. Schmidt-Rohr, J. Am. Chem. Soc., 2021, 143, 9529–9542 CrossRef CAS PubMed.
- B. Chen, V. H. Crespi and R. Hoffmann, J. Am. Chem. Soc., 2022, 144, 9044–9056 CrossRef CAS PubMed.
- T. C. Fitzgibbons, M. Guthrie, E.-s. Xu, V. H. Crespi, S. K. Davidowski, G. D. Cody, N. Alem and J. V. Badding, Nat. Mater., 2015, 14, 43–47 CrossRef CAS PubMed.
- S. Romi, M. Santoro, S. Fanetti, T. A. Strobel, S. G. Dunning and R. Bini, J. Phys. Chem. C, 2024, 128(46) DOI:10.1021/acs.jpcc.4c06125.
- P. Zhang, D. Gao, X. Tang, X. Yang, H. Zheng, Y. Wang, X. Wang, J. Xu, Z. Wang, J. Liu, X. Wang, J. Ju, M. Tang, X. Dong, K. Li and H.-k. Mao, J. Am. Chem. Soc., 2023, 145, 6845–6852 Search PubMed.
- T. A. Lau, S. Khorasani and M. A. Fernandes, CrystEngComm, 2023, 25, 3988–3997 RSC.
- A. Schonberg and A. Mustafa, Chem. Rev., 1947, 40, 181–200 CrossRef CAS PubMed.
- Y. Masuda, D. Ikeshita and M. Murakami, Helv. Chim. Acta, 2021, 104, e2000228 CrossRef CAS.
- K. J. Kappes, A. M. Deal, M. F. Jespersen, S. L. Blair, J.-F. Doussin, M. Cazaunau, E. Pangui, B. N. Hopper, M. S. Johnson and V. Vaida, J. Phys. Chem. A, 2021, 125, 1036–1049 CrossRef CAS PubMed.
- S. C. Biswas, S. Ray and A. Podder, Chem. Phys. Lett., 1987, 134, 541–544 Search PubMed.
- F.-F. Jian, K.-F. Wang and R.-R. Zhuang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o196 CrossRef CAS PubMed.
- G. Crundwell, J. Sullivan, R. Pelto and K. Kantardjieff, J. Chem. Crystallogr., 2003, 33, 239–244 CrossRef CAS.
- W. Zhou, K. Hernández-Burgos, S. E. Burkhardt, H. Qian and H. D. Abruña, J. Phys. Chem. C, 2013, 117, 6022–6032 CrossRef CAS.
- C. J. Brown and R. Sadanaga, Acta Crystallogr., 1965, 18, 158–164 CrossRef CAS.
- P. Kundu and N. Chattopadhyay, J. Chem. Phys., 2016, 144, 234317 CrossRef PubMed.
- A. K. Singh and D. K. Palit, Chem. Phys. Lett., 2002, 357, 173–180 CrossRef CAS.
- C. J. Sandroff and I. Y. Chan, Chem. Phys. Lett., 1983, 97, 60–65 CrossRef CAS.
- S. Lopes, A. Gómez-Zavaglia and R. Fausto, Phys. Chem. Chem. Phys., 2006, 8, 1794–1806 RSC.
- H. Matsuzawa, T. Nakagaki and M. Iwahashi, J. Oleo Sci., 2007, 56, 653–658 CrossRef CAS PubMed.
- N. Sawwan and A. Greer, J. Org. Chem., 2006, 71, 5796–5799 CrossRef CAS PubMed.
- L. T. M. Profeta, R. L. Sams, T. J. Johnson and S. D. Williams, J. Phys. Chem. A, 2011, 115, 9886–9900 CrossRef CAS PubMed.
- H. Kotsuki, H. Nishizawa, M. Ochi and K. Matsuoka, Bull. Chem. Soc. Jpn., 1982, 55, 496–499 CrossRef CAS.
- W. G. Dauben and H. O. Krabbenhoft, J. Org. Chem., 1977, 42, 282–287 CrossRef CAS.
- G. Jenner, N. Papadopoulos, J. Jurczak and T. Koźluk, Tetrahedron Lett., 1984, 25, 5747–5750 CrossRef CAS.
- C. M. Miliante, J. P. D. de Matos and A. R. Muniz, Carbon, 2023, 215, 118387 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2343198–2343200. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07412b |
| ‡ Current address: Division of Sciences and Mathematics, University of the District of Columbia, 4200 Connecticut Avenue NW, Washington, District of Columbia 20008, USA. E-mail: E-mail: samuel.dunning@udc.edu |
| This journal is © The Royal Society of Chemistry 2025 |