
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProximity-enhanced cysteine–histidine crosslinking for elucidating intrinsically disordered and other protein complexes†
Qi
Wu
a,
Sebastian A. H.
van den Wildenberg
a,
Jeroen C. R.
Brzoskowski
 a,
Maxime C. M.
van den Oetelaar
a,
Carlo J. A.
Verhoef
a,
Sylvia A. A. M.
Genet
a,
Christian
Ottmann
a,
Albert J.
Markvoort
b,
Luc
Brunsveld
*a and
Peter J.
Cossar
*a
a,
Maxime C. M.
van den Oetelaar
a,
Carlo J. A.
Verhoef
a,
Sylvia A. A. M.
Genet
a,
Christian
Ottmann
a,
Albert J.
Markvoort
b,
Luc
Brunsveld
*a and
Peter J.
Cossar
*a
aLaboratory of Chemical Biology, Department of Biomedical Engineering and Institute for Complex Molecular Systems, Eindhoven University of Technology, Netherlands. E-mail: l.brunsveld@tue.nl; p.cossar@tue.nl
bSynthetic Biology Group, Department of Biomedical Engineering and Institute for Complex Molecular Systems, Eindhoven University of Technology, Netherlands
First published on 7th January 2025
Abstract
Disordered proteins and domains are ubiquitous throughout the proteome of human cell types, yet the biomolecular sciences lack effective tool compounds and chemical strategies to study this class of proteins. In this context, we introduce a novel covalent tool compound approach that combines proximity-enhanced crosslinking with histidine trapping. Utilizing a maleimide–cyclohexenone crosslinker for efficient cysteine–histidine crosslinking, we elucidated the mechanism of this dual-reactive tool compound class. This tool compound concept was then applied to profile the full-length complex of 14-3-3 and hyperphosphorylated Tau (hpTau), relevant to Alzheimer's. This approach identified a cryptic binding interaction between 14-3-3 and hpTau via its phosphorylated Ser356, overlooked by the majority of 14-3-3/Tau literature. Utilizing a mutational study and an equilibrium model, this cryptic binding interaction is revealed to play a prominent biomolecular role at cellularly relevant concentrations. This finding necessitates a re-evaluation of the mechanism of the 14-3-3/Tau interaction. The histidine-trap crosslinker approach reported here not only advances our understanding of the 14-3-3/Tau interaction but also demonstrates the potential of dual-covalent tool compounds in studying complex interactions involving IDPs and IDDs.
Introduction
The biomolecular understanding of the function and interactome of intrinsically disordered proteins (IDPs) and intrinsically disordered domains (IDDs) of proteins remains in its infancy.1 Unlike structured proteins, where the application of non-covalent tool compounds and drugs have significantly advanced our biomolecular understanding of protein structure and function,2,3 such non-covalent tool compounds have been largely ineffective against IDPs and IDDs. This inefficacy originates from the absence of a stable tertiary protein structure, preventing recognition-driven small molecule binding.1 Therefore, novel chemical strategies and tool compound approaches not only contingent on molecular recognition are needed to circumvent the challenge of targeting unfolded proteins and domains, enablinggreater molecular insights into protein function.The application of covalent chemistry, next to non-covalent approaches, has greatly improved our biomolecular understanding of the functions of structured proteins, DNAs/RNAs and lipid membranes. For example, protein complex ligation4 and spatiotemporal photo-release of caged-peptides5 have enabled a deeper understanding of transcription factors and signalling protein function. As an example, the application of reversible covalent crosslinkers to study RNA has enabled characterization of ternary RNA structures6 and transient biomolecular complexes7 not otherwise detectable. Covalent tool compounds have also led to significant advances in our ability to functionalize proteins, for small molecule biosensing,8 ligand-directed cargo release,9 and peptide based covalent probes.10
Covalent small molecule strategies have shown initial promise for probing the function of IDPs and IDDs. Specifically, covalent small molecules EN4,11 which blocks MYC DNA binding, and EPI-001, that covalently binds the disordered transactivation domain of the androgen receptor, have proven effective in studying oncogenic IDPs.12,13 Notwithstanding these attractive examples, the scarcity of chemical matter targeting IDPs and the absence of broadly applicable general approaches underscore the existing challenge of targeting an IDP. Given that IDPs and IDDs typically form complexes to execute their functions, developing covalent tool compounds that go beyond the individual protein or protein–protein interaction (PPI) inhibition will be essential to studying IDP complexes.
Chemical crosslinking and proximity ligation represent an attractive approach to study IDP complexes. The two reactive groups of chemical crosslinkers allow for the capture and immobilization,14 identification of protein complexes,15 and probing of binding motifs.16 Chemical crosslinking permanently freezes IDP complexes in the interaction state.17 This is particularly important as IDP complexes are typically weak and highly transient.18
Significant efforts over the last decade have been dedicated to the development of novel chemical crosslinkers.19,20 Breakthroughs have brought forth mass-spectrometry (MS)-cleavable linkers (DSSO)21 and affinity tags (PhoX).22,23 However, there has been limited innovation in the electrophilic reactive groups. Commonly used crosslinking chemistries include lysine–lysine (Lys–Lys) crosslinking using homobifunctional NHS esters such as DSS and BS3 crosslinkers,24 and acid-amine (amide) crosslinking using carbodiimide reagents like EDC.25 However, these chemistries are highly reactive and typically target residues with high abundance across the protein surface, leading to off-target reactivity and a lack of selectivity.26 Hence, there is a need for developing diverse crosslinking chemistries with enhanced selectivity and differentiated applicability.
Phosphorylation mediated PPIs play a central role in regulating IDPs, such as those with the prototypical hub protein 14-3-3.27,28 This highly structured dimeric protein typically binds phosphorylated motifs within disordered regions of proteins.28–30 14-3-3 exhibits remarkable versatility, binding and modulating a large number of proteins, including c-Myc,31 Tau,32 and the cystic fibrosis transmembrane conductance regulator (CFTR) protein,33,34 many of which are intimately linked to debilitating diseases like cancer, neurodegenerative disorders and cystic fibrosis.35,36 Therefore, decoding the interactions of IDPs and IDDs with 14-3-3 holds great potential for the development of novel diagnostic and therapeutic strategies to combat these diseases.
In this study, we developed a tool compound concept merging proximity-enhanced ligation and histidine trapping to study interactions between IDPs and 14-3-3 (Fig. 1A), using a maleimide–cyclohexenone crosslinker for efficient cysteine–histidine crosslinking. The crosslinking mechanism and the critical role of the cyclohexenone electrophile were elucidated using a focused library of eleven analogues and a 14-3-3/peptide interaction model. The reactivity of these histidine-trap crosslinkers was profiled with two peptide panels: the first assessing reactivity of the crosslinker strategy and position of the nucleophilic histidine within the 14-3-3 binding sequence, and the second comprising nine native 14-3-3 binding peptides to explore sequence influence on crosslinking efficiency. The strategy selectively targets cysteine and histidine and is effective across a range of 14-3-3 partners. Focusing in depth on the PPI of 14-3-3 and Tau, a cryptic binding interaction was identified between 14-3-3 and Tau at phosphorylation site Ser356, an interaction that has been overlooked by the majority of literature on the 14-3-3/Tau complex. By utilizing a mutational study and an equilibrium model, this cryptic binding interaction was demonstrated to be of relevant affinity to modulate the 14-3-3/Tau PPI.
 | ||
| Fig. 1 Structure-based design of the 14-3-3/B-Raf pS365 crosslinker. (A) Schematic illustration of crosslinking protein complexes of 14-3-3 and IDPs/IDDs. Amino acids Cys, His and pSer are represented by orange, green and red dots, respectively. (B) The co-crystal structure of the B-Raf (Ser365 phosphorylated) peptide in complex with 14-3-3σ monomer (PDB ID: 9F35). The B-Raf peptide is shown as blue spheres with the His370 residue as green spheres. The 14-3-3σ protein is shown as grey surface with the Cys38 residue as orange sticks. (C) The measured distance (black dashes) between Cys38 (orange sticks) of 14-3-3σ and His370 (green sticks) of B-Raf. The 2Fo–Fc electron density map is shown as blue mesh contoured at 1.5 σ. (D) The synthesis scheme and the structure of crosslinker 1. | ||
Results and discussion
Structure-based design of a histidine-trap crosslinker
The design of a chemical crosslinker to study 14-3-3/IDP PPIs started with electrophile selection. 14-3-3σ contains a single surface-exposed cysteine (Cys38) located at the interface of the 14-3-3/phospho-partner PPI interface (Fig. 1B and C).37 Cys38 has previously been targeted in drug discovery campaigns as an anchoring point for covalent fragment screening.38,39 The low proteogenic abundance of cysteine, its close location to the protein complex interface and its chemical tractability made this amino acid a logical starting point to develop a covalent tool compound to study 14-3-3 interactions with IDPs. To target Cys38 a maleimide electrophile was selected, based on the fast reaction kinetics and high thiol selectivity.40–42 IDPs and IDDs are typically characterized by their sequences featuring a higher charge density relative to those of structured proteins and domains.1,43–45 With this in mind, we sought a positively charged, reactive and relatively abundant amino acid present in the disordered polypeptide chains as the complementary nucleophilic amino acid. Recently, 2-cyclohexenone electrophile has been reported to react with histidine residue via an aza-Michael addition to the imidazole side chain.46 We selected the histidine–cyclohexenone pair, as histidine is highly represented in IDPs, such as Tau.47 Unlike NHS- and EDC-based crosslinkers, which are highly reactive, the cyclohexenone electrophile presents an interesting case study due to its lower reactivity, which has the potential to distinguish bona fide IDP interactions from highly transient and less biologically relevant PPIs.Next, we sought a model system to represent a 14-3-3/IDP complex. The protein–peptide interaction between 14-3-3 and the binding pSer365 motif of B-Raf was selected.48 The pSer365 peptide (sequence: 360-RDRSS(pS)APNVH-370, KD = 7.0 ± 0.2 μM, Fig. S1A†) contains a histidine (His370) at the C-terminus of the peptide. To gain detailed spatial information regarding the distance between the His370 and the Cys38, we solved the co-crystal structure of the 14-3-3σ/B-Raf (pSer365) protein–peptide complex. Specifically, we employed a soaking method previously developed by Ottmann and colleagues.49 This approach afforded 14-3-3σ/B-Raf crystals with a resolution of 2.3 Å. Analysis of the 14-3-3σ/B-Raf (pSer365) structure showed the electron density of the B-Raf peptide to be well resolved (Fig. 1B), with His370 of B-RAF is in close proximity to Cys38 of 14-3-3σ. However, the precise positioning of the imidazole sidechain of His370 remained unresolved. As such, two potential conformations of the imidazole sidechain were modelled and the distances to Cys38 of 14-3-3σ were measured at approximately 10.2–12.3 Å (Fig. 1C). Utilizing this information crosslinker 1 with a butyrate linker was designed with a distance of 10.4 Å spanning the C2 (3) of the maleimide electrophile and C19 of the cyclohexenone (Fig. 1D; MM2-minimalized maximum, Chem3D).
Crosslinker 1 was synthesized through a four-step synthetic route, starting from the commercially available 1,4-cyclohexanedione monoethylene acetal (Fig. 1D). First, the ketone functionality was transformed into a silyl enol ether using TMSOTf under basic conditions. The silyl enol ether underwent a Saegusa reaction, leading to the formation of an α,β-unsaturated ketone. A Luchi reduction was then employed to selectively reduce the ketone to an allyl alcohol. Immediately after this step, the acetal protection was removed in a single step using oxalic acid. The 4-hydroxyl cyclohexenone was later merged with 4-maleimidobutyric acid under the Steglich esterification condition, resulting in the production of crosslinker 1.
The histidine-trap crosslinking reaction proceeds via a proximity-enhanced mechanism
To assess 14-3-3/B-Raf crosslinking efficiency, 1 (10 eq., Fig. 1D) was incubated with either 14-3-3σ alone or the 14-3-3σ/B-Raf complex (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) overnight at pH 7, 10 °C and monitored using intact Mass Spectrometry (MS) (Fig. 2A). Analysis of the 14-3-3σ/1 mixture showed a single peak (26787 Da) corresponding to a single covalent addition of crosslinker 1 (277 Da) to 14-3-3σ (26509 Da). In contrast, two peaks were observed for the 14-3-3σ/B-Raf/1 mixture. The major peak (28133 Da, >90%) corresponds to the crosslinked 14-3-3σ/B-Raf complex and the minor peak overlaps with the single addition of 1 to 14-3-3σ (26787 Da). This result indicates successful and selective crosslinking of 14-3-3σ/B-Raf complex induced by 1. The kinetics of the crosslinking reaction was then investigated using time-dependent intact MS measurements of the 14-3-3σ/B-Raf/1 mixture (Fig. 2B and C). At 5 min, apo-14-3-3σ was completely consumed, resulting in a major peak assigned to the 14-3-3σ–1 conjugate (Fig. 2B). The 14-3-3σ–1 conjugate further reacted with B-Raf peptide over the following 6 h to furnish the crosslinked complex of 14-3-3σ and B-Raf (Fig. 2C). Further analysis of the kinetic intact experiment showed a crosslinking yield of ∼80% was observed after 6 h. Raycroft et al. have reported a reaction rate (k2) of (17.6 ± 0.9) × 103 M−1 s−1 for L-cysteine–maleimide conjugation.50 Further, Sauerland et al. compared cysteine conjugation to α,β-unsaturated carbonyl-containing compounds and reported reaction rates (k2) of approximately 0.25–0.53 M−1 s−1 for cysteine–cyclohexanone conjugations across a panel of proteins, and glutathione.51 Given the three log-fold difference in reactivity between cysteine–maleimide and cysteine–cyclohexanone conjugations, this suggest that the intermediate 14-3-3–crosslinker 1 conjugate species is likely the cysteine–maleimide species.
:2) overnight at pH 7, 10 °C and monitored using intact Mass Spectrometry (MS) (Fig. 2A). Analysis of the 14-3-3σ/1 mixture showed a single peak (26787 Da) corresponding to a single covalent addition of crosslinker 1 (277 Da) to 14-3-3σ (26509 Da). In contrast, two peaks were observed for the 14-3-3σ/B-Raf/1 mixture. The major peak (28133 Da, >90%) corresponds to the crosslinked 14-3-3σ/B-Raf complex and the minor peak overlaps with the single addition of 1 to 14-3-3σ (26787 Da). This result indicates successful and selective crosslinking of 14-3-3σ/B-Raf complex induced by 1. The kinetics of the crosslinking reaction was then investigated using time-dependent intact MS measurements of the 14-3-3σ/B-Raf/1 mixture (Fig. 2B and C). At 5 min, apo-14-3-3σ was completely consumed, resulting in a major peak assigned to the 14-3-3σ–1 conjugate (Fig. 2B). The 14-3-3σ–1 conjugate further reacted with B-Raf peptide over the following 6 h to furnish the crosslinked complex of 14-3-3σ and B-Raf (Fig. 2C). Further analysis of the kinetic intact experiment showed a crosslinking yield of ∼80% was observed after 6 h. Raycroft et al. have reported a reaction rate (k2) of (17.6 ± 0.9) × 103 M−1 s−1 for L-cysteine–maleimide conjugation.50 Further, Sauerland et al. compared cysteine conjugation to α,β-unsaturated carbonyl-containing compounds and reported reaction rates (k2) of approximately 0.25–0.53 M−1 s−1 for cysteine–cyclohexanone conjugations across a panel of proteins, and glutathione.51 Given the three log-fold difference in reactivity between cysteine–maleimide and cysteine–cyclohexanone conjugations, this suggest that the intermediate 14-3-3–crosslinker 1 conjugate species is likely the cysteine–maleimide species.
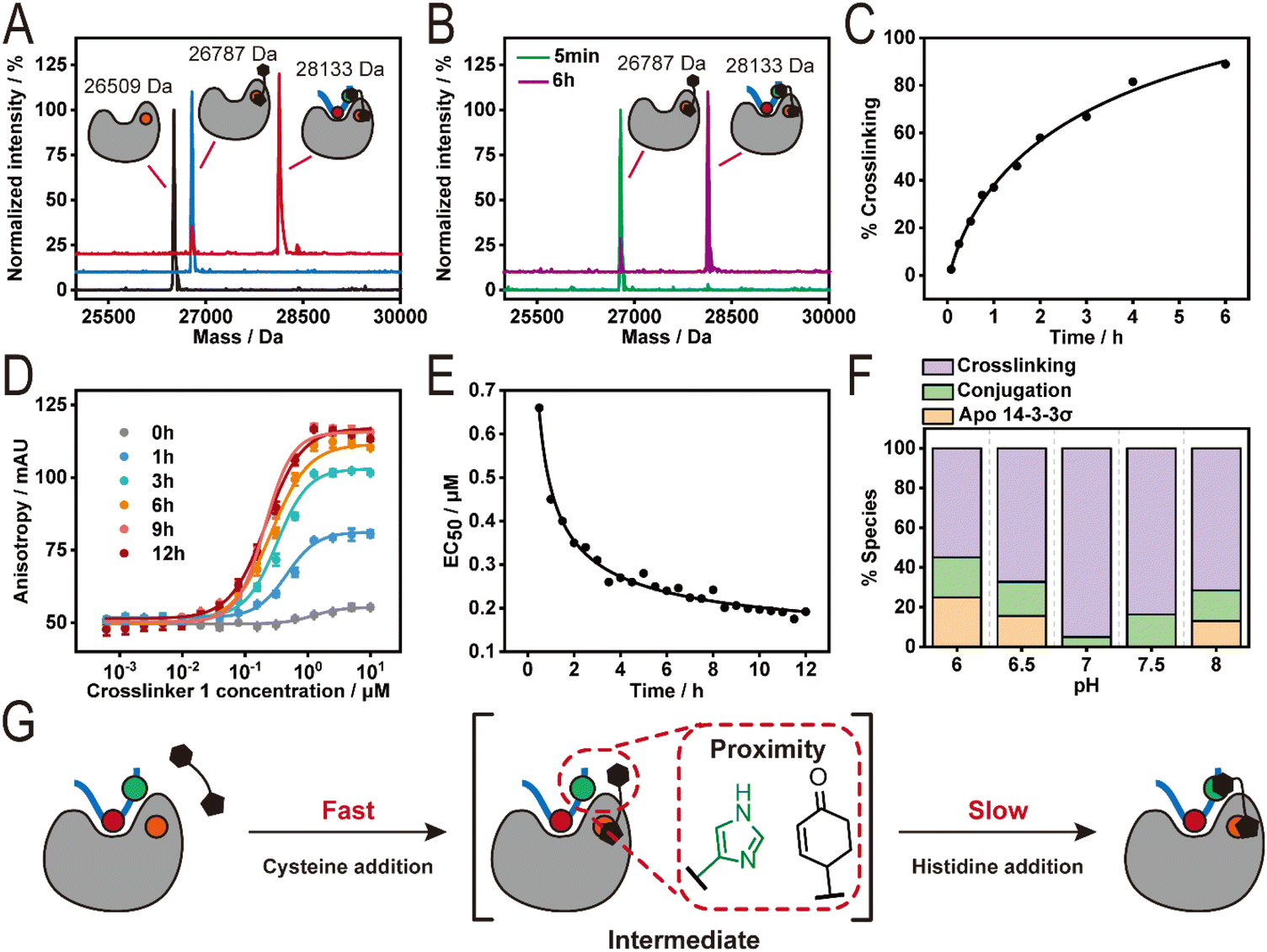 | ||
| Fig. 2 Investigation of the crosslinking reaction by intact mass analysis and FA assay. (A) The deconvoluted mass spectra of 14-3-3σ (8 μM) incubated with DMSO (black), 80 μM crosslinker 1 (blue) and 80 μM crosslinker 1 plus 16 μM B-Raf pS365 (red) in a 20 mM BTP buffer at pH 7, 10 °C. (B) The deconvoluted mass spectra of samples incubated with 8 μM 14-3-3σ, 80 μM crosslinker 1, and 16 μM B-Raf pS365 for 5 min (green) and 6 h (purple). (C) The percentage of crosslinking at 8 μM 14-3-3σ and 16 μM B-Raf pS365 by 80 μM crosslinker 1 over 6 h. (D) Time-dependent fluorescence anisotropy (FA) measurements were crosslinker 1 was titrated to a fixed concentration of 14-3-3σ and FITC-labelled B-Raf pS365 over 12 h (n = 3). (E) A plot of the EC50 values of of crosslinker 1 over time determined by FA assay. (F) A plot of 14-3-3/B-Raf pS365 crosslinking showing the effect of varying pH. (G) A schematic illustration of the proximity-driven crosslinking reaction between 14-3-3σ, B-Raf pS365 and crosslinker 1. | ||
We further validated the kinetics of 14-3-3σ/B-Raf crosslinking from the perspective of the peptide using a fluorescence anisotropy (FA) assay. Crosslinker 1 was titrated to the mixture of 1.5 μM 14-3-3σ and 50 nM fluorescein labelled B-Raf peptide, and the polarized emission was measured over time (Fig. 2D). The half maximal effective concentration (EC50) of 1 required to recruit B-Raf to 14-3-3σ was plotted over time (Fig. 2E). As expected, the EC50 value reduced with time, reaching saturation at approximately 6 h, correlating with the time-dependent intact MS measurements.
Interestingly, crosslinking of the 14-3-3σ/B-Raf complex was insensitive to protein:crosslinker stoichiometries of 1:1, 1:5 and 1:10 (Fig. S1B†). This result is likely due to the high reaction efficiency of the cysteine–maleimide addition, maintaining the local concentration of the cyclohexenone electrophile even at lower crosslinker concentration. Conjugation of 1 to 14-3-3σ and subsequent crosslinking proved sensitive to pH changes (Fig. 2F). The final proportion of the 14-3-3σ–1 conjugate varied from 20% to 0% over the pH range of 6–8 after overnight incubation while the fraction of the crosslinked complex ranged from approximately 55% at pH 6 to over 90% at pH 7. Notably, pH 7 was optimal with complete consumption of 14-3-3σ.
Mutation of Cys38 of 14-3-3 to asparagine abolished the conjugation of 1 to 14-3-3σ (Fig. S1C†), confirming the selectivity for this 14-3-3 cysteine. Further, incubation of the B-Raf peptide with crosslinker 1 in the absence of 14-3-3σ did not result in the formation of the B-Raf–1 conjugate (Fig. S2A–E†), revealing a low intrinsic reactivity between both the cyclohexenone and maleimide electrophiles with B-Raf His370. Incubation of 1 and B-Raf with the ligand binding domain of the Peroxisome proliferator-activated receptor gamma (PPARγ) or bovine serum albumin (BSA) which both have exposed cysteines but do not bind the B-Raf peptide, revealed exclusive crosslinker 1 conjugation to protein alone with no B-Raf crosslinking observed (Fig. S1D and E†). The 14-3-3σ/B-Raf crosslinking is thus dependent on PPI complex formation and proceeds via a proximity-enhanced mechanism, with the 14-3-3σ–crosslinker conjugate serving as the intermediate (Fig. 2G).
Maleimide and cyclohexenone electrophiles are critical to dual site-selective crosslinking
To further understand the structure–activity relationship (SAR) of the Cys–His crosslinker, a focused library based on 1 was developed (Fig. 3A). This SAR investigation focused on linker length and rigidity (1–4), as well as the histidine and cysteine reactive groups (5–11). Intact MS profiling showed that a shorter linker (2, n = 2, 8.8 Å) had no effect on crosslinking efficiency, while increasing the linker length (3, n = 5, 12.9 Å) or rigidifying the linker (4, 11.6 Å) resulted in a small (∼10%) decrease in crosslinking (Fig. 3B and C). Unsurprisingly, reduction of the histidine electrophile (5) or the maleimide (6) abolished crosslinking. Substituting the maleimide with chloro- or bromo-substituted acet/propionamide (7–9) also led to the loss of crosslinking. Substitution of the cyclohexenone with N-hydroxysuccinimide (NHS, 10) resulted in a complex mixture, likely due to promiscuous reactivity of the NHS ester with surface-exposed lysines on 14-3-3σ. Homodimeric maleimide crosslinker (11) elicited a 20% crosslinking efficiency, highlighting the lower reactivity of the maleimide electrophile with histidine. Analogues 6–9 that lacked the maleimide group or replaced the maleimide electrophile resulted in a loss of crosslinking (Fig. 3B, C, S3A and B†). However, 100% 14-3-3 conjugation was still observed indicating that in the absence of the maleimide the cyclohexenone reacts with Cys38. Given that Michael-addition reactions are reversible we next investigated the thermodynamic stability of the Michael-addition products. First, cyclohexenone was incubated with 14-3-3σ and MS analysis showed ∼80% conjugation (Fig. S3C and D†). We then co-incubated 1:1 cyclohexanone and N-ethylmaleimide (NEM) with 14-3-3σ. Experimental analysis showed exclusively the 14-3-3σ–NEM conjugate (Fig. S3E and F†). This result indicates the cysteine–maleimide conjugate is significantly more stable relative to cyclohexenone. In context to the literature, this SAR study emphasizes the significance of the maleimide for proximity enhanced crosslinking, the essential role of the cyclohexanone in targeting histidine, and cysteine–maleimide reaction is significantly faster and thermodynamically more stable relative to cyclohexanone.
 | ||
| Fig. 3 Structure–activity relationship of the crosslinker. (A) The chemical structures of analogues 1–11. (B) Schematic illustration of conjugated and crosslinked species observed by intact mass analysis. (C) A plot of the percentage of conjugation and crosslinking after overnight incubation at 8 μM 14-3-3σ, 80 μM compound, and 16 μM B-Raf pS365 peptide. (D) Schematic illustration summarising the key structure–activity relationship (SAR) insights. | ||
Histidine-trap crosslinkers show a wide substrate scope
The amino acid selectivity and relevance of histidine position were investigated using a panel of mutated B-Raf peptides (Fig. 4A). Specifically, three mutated B-Raf peptides (17-mer) were prepared, where the His residue was replaced by either an alanine (H+5A), lysine (H+5K), or cysteine (H+5C) in the +5 position and eight B-Raf peptides with the histidine residue shifted along the sequence, analogous to alanine scanning, from positions +1 to +8 (Fig. 4A). The amino acid numbering refers to the position of the residue relative to the phosphorylated amino acid (pSer). Notably, for the histidine scanning the native residue was replaced with an alanine if not at +5 position. | ||
| Fig. 4 Residue & position preference profile and target scope of the crosslinkers. The amino acid numbering (+1, +2, +3…) refers to the position of the residue relative to the phosphorylated amino acid. (A) Schematic representation of the +5 position mutation and histidine scanning peptide panels. (B) A plot of the percentage of crosslinking of 14-3-3σ and B-Raf mutants induced by crosslinker 1 and 3. (C) A table of the sequences of natural 14-3-3 binding motifs containing His or Cys residues. (D) A plot of the percentage of crosslinking of 14-3-3σ and natural 14-3-3 binding motifs induced by crosslinker 1 and 3. | ||
To confirm peptide binding with 14-3-3, the affinities of the peptide panel were first determined using a FA assay (Fig. S4A and B†). All peptides maintained their affinity to 14-3-3σ, with KD values ranging from 1.80–135 μM. His +1, +2, and +3 elicited the poorest affinities (KD > 50 μM). However, all peptides showed sufficient 14-3-3 affinity for subsequent intact MS study.
For the crosslinking experiment crosslinkers 1 (3-carbon linker) and 3 (5-carbon linker) were selected, and the previously established intact MS assay condition was applied. As expected, no crosslinking for H+5A and only trace amount of crosslinking was observed for H+5K (<5%) with 1 and 3 (Fig. 4B). Compounds 1 and 3 elicit 77% and 36% complex crosslinking for the H+5C peptide, respectively, aligning with previous observations that the cyclohexenone electrophile also reacts with cysteine. Notably, compounds 1 or 3 did not crosslink a non-phosphorylated H+5C peptide (Fig. S4C–E†) which lacks affinity for 14-3-3σ, further highlighting the proximity-enhanced nature of the crosslinking. Analysis of the histidine scanning on the crosslinking efficiency showed positions +1, +4, +5, +6, +7, and +8 were amendable to crosslinking (>50% crosslinking; Fig. 4A and B). In contrast, His+2 and His+3 showed minimal crosslinking. Increased peptide concentrations from 2 eq. to 16 eq. of His+2 and His+3 did not result in improved crosslinking (<10%, Fig. S4F†). We reasoned that affinity, amino acid conformation and distance from Cys38 collectively contribute to diminished crosslinking. His+1 clearly exhibited a distance effect, as 3 (five-carbon linker) showed ∼ two-fold higher crosslinking compared to 1 (three-carbon linker). These results demonstrated the broad suitability of our crosslinkers for Cys–His and Cys–Cys crosslinking at various positions (+1, +4, +5, +6, +7, and +8) in the peptide sequence.
We next sought to investigate how peptide sequence influenced histidine-trap crosslinking using a panel of other native 14-3-3 binding motifs. Each peptide contained either a His or Cys residue at different positions within the peptide sequence (Fig. 4C). These motifs included His at +1 (KSR1 and CDK11B), +5 (C-Raf and Cby), and remote +13 (p65), as well as Cys at +1 (PKR) and +3 (RGS18) positions. A bivalent Tau peptide with two phosphorylation sites (pSer214 and pSer324) that mimics the known 14-3-3 binding motifs of Tau was also used. Briefly, this 38-mer Tau peptide consists of the pSer214 and pSer324 binding motifs connected by a flexible, 11-amino-acid-long glycine–serine linker. Notably, there are two consecutive His residues at +5 and +6 positions relative to the pSer324. Most of the peptides are moderate binders of 14-3-3 with KD values in micro-molar range with the exception of p65 and PKR. p65 has a weak affinity (above micromolar) towards 14-3-3 while PKR binds 14-3-3 much stronger (sub-micromolar). Within this peptide panel varying degrees of crosslinking (10–98%) to 14-3-3σ were observed (Fig. 4D), indicating that our crosslinkers can target a diverse range of 14-3-3 binding partners. Peptides with His at +5 position (C-Raf and Cby) exhibited higher levels of crosslinking (58–81%) compared to those with His at +1 position (KSR1 and CDK11B) (10–46%). Crosslinkers 1 and 3 also showed crosslinking of the 14-3-3/p65 complex though with a lower yield (30%). Interestingly, the p65 peptide contains a His at the +13 position, considered outside the 14-3-3 binding groove. This result suggests that the Cα–Cα distance between His+13 of p65 and Cys38 of 14-3-3σ can be within 12.9 Å during binding. RGS18 with Cys at +3 position elicited the greatest crosslinking efficiency (98%), whereas the His residue in the B-Raf mutant at the same position exhibited negligible crosslinking. This result suggests the Cys residue at the +3 position of RGS18 peptide may adapt a more favourable conformation compared to the His residue at the +3 position of the B-Raf mutant. Despite relatively lower efficiency (18–23%), PKR with Cys at +1 position was also found to be crosslinked by both 1 and 3. Linker length had no significant effect on crosslinking, apart from the +1 His position (KSR1 and CDK11B) with 3 (five-carbon linker) eliciting a 3–4 fold increase in crosslinking relative to 1 (three-carbon linker), respectively. We postulated this to be a distance effect as observed earlier for the B-Raf His+1 mutant. The bivalent Tau peptide had a relatively low level of crosslinking (<30%, Fig. 4D) with both 1 and 3 even though two histidine residues are located at the empirically favorable position (+5 and +6) for crosslinking. We reasoned this is due to the presence of a cysteine residue at the −2 position which leads to intrapeptide crosslinking and competes with the 14-3-3σ/Tau crosslinking.
Taken together this SAR study shows that crosslinking efficiency is ultimately influenced by the accessibility of the reactive residue. Specifically, three parameters influence crosslinking efficiency: the distance between cyclohexanone and nucleophilic amino acid, spatial orientation of the nucleophilic amino acid, and steric hindrance caused by flanking residues. Having robustly characterized the reactivity of the histidine-trap crosslinker strategy, we shifted focus to probing a full-length IDP complex.
Probing the 14-3-3/Tau IDP complex using the histidine-trap crosslinkers
The interaction between 14-3-3 and hyperphosphorylated Tau is of particular interest. Research has shown that Tau in its dysregulated state is hyperphosphorylated.52,53 The hyperphosphorylation of Tau is promoted by cAMP-dependent protein kinase A (PKA).54,55 Further, emerging research has shown that hyperphosphorylated Tau accumulates in liquid–liquid phase separated droplets, leading to Tau aggregation and ultimately neurodegeneration. Interestingly, liquid–liquid phase separation (LLPS) experiments have shown that 14-3-3's binding specifically inhibits the aggregation of hyperphosphorylated Tau under phase separation conditions.56,57To explore potential 14-3-3 binding sites we sought to apply histidine-trapping to the 14-3-3/hyperphosphorylated Tau complex. First we optimized reaction conditions using the bivalent Tau peptide by; (1) reducing the crosslinker concentration to equimolar relative to 14-3-3σ, (2) crosslinker 1 was substituted for the rigid crosslinker 4—we rationalized the more rigid crosslinker would limit intra-peptide crosslinking, (3) the crosslinking reaction was performed in two steps (one-pot), and (4) methoxamine was employed to trap the formed ketone after crosslinking via an oxime ligation reaction (30659 Da) to prevent the retro-Michael reaction during the trypsin digestion (Fig. S5A–E†).46 The resulting optimization led to a favourable crosslinking of the 14-3-3σ/bivalent Tau peptide (74%, Fig. S5D†). Analysis of the MS1 spectrum of the digested sample showed peaks corresponding to triply (m/z = 852.7011), quadruply (m/z = 639.7750), and quintuply (m/z = 512.0248) charged αTau-β14-3-3 ions (Fig. S6A and B†) consistent with the peptide surrounding the pSer324 of the bivalent Tau peptide (αTau) and the peptide surrounding the Cys38 of the 14-3-3σ (β14-3-3).55 Having established a workflow for detection of the protein–peptide interaction, we proceeded with full-length Tau.
To biochemically investigate 14-3-3's binding to hyperphosphorylated Tau (hpTau), a full-length Tau construct (2N4R isoform) was recombinantly expressed and subjected to in vitro PKA phosphorylation to produce the hpTau construct.55 The phosphorylation sites were then mapped using trypsin digestion and mass spectrometry revealing up to five phosphorylated sites on Tau: Ser214, Ser324, Ser356, Ser409, and Ser416 (Fig. 5A and S7A–G†), consistent with previous reports.58,59 Whilst pSer214 and pSer324 have been studied extensively, limited biochemical and cellular data is available on 14-3-3's interaction with other phosphorylation sites on Tau, such as pSer356.60 The full-length hpTau was subjected to crosslinking with 14-3-3σ using the previously optimized crosslinking conditions and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis polyacrylamide gel electrophoresis (SDS-PAGE) to determine crosslinking species and molecular weights. Gel analysis revealed two new bands (lane 5, Fig. 5B, adapted from S8A†). Intact MS analysis of the data confirmed that the lower band corresponds to the crosslinking of hpTau with a 14-3-3σ monomer (single crosslinking, 73288 Da, Fig. S8B†) and the upper band aligned with the crosslinking of hpTau with two 14-3-3σ monomers (double crosslinking, 99804 Da, Fig. S8C†). Our team was intrigued by this result as the pSer214 site lacks a histidine or cysteine residue for crosslinking, suggesting that besides crosslinking with pSer324 site on hpTau one more crosslinking event took place either within two 14-3-3 monomers or 14-3-3/hpTau. Crosslinking of two 14-3-3 monomers was excluded based on gel experiments with 4 and apo-14-3-3 that showed no crosslinking (lane 2, Fig. 5B). Further, crosslinking experiment with the bivalent Tau peptide supported this reasoning (Fig. S5D†). Therefore, the presence of the upper band suggested an additional crosslinking event within the 14-3-3/hpTau. Unfortunately, attempts to elucidate the crosslinking sites with our MS workflow only afforded ions aligned with the crosslinking of the peptide surrounding pSer324 of full-length hpTau (Fig. S8D and E†). While this result is consistent with previously reported NMR studies that pSer324 in the microtubule binding domain binds 14-3-3 upon PKA phosphorylation,61 the other site was left unidentified. Given the challenges detecting the cryptic site on hpTau, likely due to the incomplete and/or an unknown mis-cleavage by trypsin, we shifted focus to mutation studies to elucidate it.
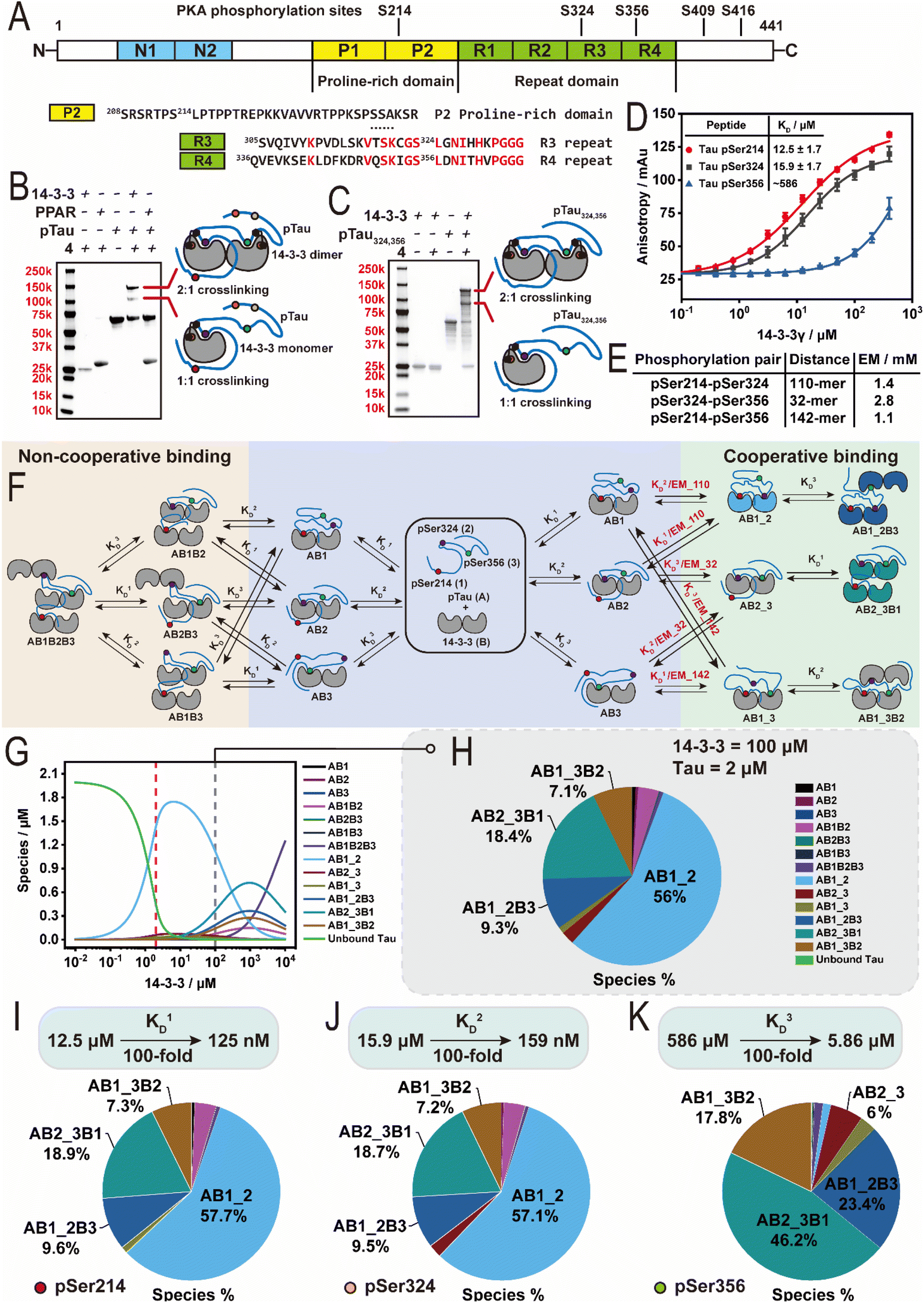 | ||
| Fig. 5 Probing interaction between 14-3-3 and full-length Tau. (A) Schematic representation of the domain structure of the Tau protein (2N4R isoform). (B) SDS-page analysis of the crosslinking reaction between 14-3-3σ and PKA phosphorylated Tau (hpTau). (C) SDS-page analysis of the crosslinking reaction between 14-3-3σ and hpTau324,356 mutant. (D) Fluorescence anisotropy (FA) measurement of titrating 14-3-3γ to 50 nM FITC-labelled Tau pSer214, pSer324 and pSer356 peptides respectively. (E) Overview of the effective molarity (EM) values of the phosphorylation pairs pSer214–pSer324, pSer324–pSer356 and pSer214–pSer356 on hpTau respectively. (F) Schematic representation of the binding model between 14-3-3 dimers and a multivalent hpTau. Only relevant binding sites pSer214, pSer324 and pSer356 are shown. (G) Simulation of the species distribution at a range of 14-3-3 concentration from 0.01 μM to 10 mM. A constant concentration of 2 μM hpTau (as reported in neuron) was applied. (H) Simulation of the fractions of different species in brain at a concentration of 100 μM 14-3-3 and 2 μM hpTau. Species below 4% are not labelled. (I–K) Simulation of the fractions of different species at a concentration of 100 μM 14-3-3 and 2 μM hpTau in a scenario where binding sites pSer214 (I), pSer324 (J) and pSer356 (K) get stabilized respectively (KD enhanced by 100-fold). Species below 4% are not labelled. | ||
Identification of pSer356 as a cryptic 14-3-3 binding site
Sequence inspection of the five Tau sites phosphorylated by PKA showed that, except for Ser324, only Ser356 contains a histidine in proximity to the phosphorylated amino acid, and no cysteine residues present (Fig. S7A†). Further, Ser324 and Ser356 contain 73% sequence homology in the C-terminal binding region (Fig. 5A). Sluchanko et al. first identified the Ser356 site as significant 14-3-3 binding site,60 however, this site has been largely overlooked in mechanistic models.32 Post-translational modification profiling of Alzheimer's patient brains has also shown Ser356 as highly phospho-enriched, relative to control brains,62 highlighting the significance of this phosphorylation site. To investigate this cryptic 14-3-3 binding site we expressed a Tau mutant protein where serine residues Ser214, Ser409, and Ser416 were mutated to alanine, whilst Ser324 and Ser356 were conserved (Tau324,356). The resulting Tau mutant was then subjected to PKA phosphorylation to afford a double phosphorylated Tau324,356 construct (hpTau324,356, 45706 Da, Fig. S9A†). Intact MS indicated a purity of around 60% double phosphorylated Tau out of a mixture of single and double phosphorylation. Next, the hpTau324,356 construct was then subjected to crosslinking experiments using 4 and 14-3-3σ. The resulting SDS-PAGE showed two protein bands corresponding to single and double crosslinking (lane 5, Fig. 5C, adapted from S9B†). Intact MS measurements also confirmed the two bands corresponded to a 1:1 and 1:2 Tau:14-3-3 crosslinking stoichiometry (72547 and 99389 Da, Fig. S9C and D†). These results suggested that the additional binding site indeed was pSer356. To validate this binding site a 16-mer Tau peptide representing the pSer356 binding site was prepared (Fig. S9E†). Crosslinking experiments with the pSer356 peptide confirmed that this motif binds 14-3-3 (Fig. S9F†). Binding studies with the established FA assay further confirmed that the pSer356 peptide binds 14-3-3 (both σ and γ isoforms), however does not reach binding saturation (Fig. 5D and S9G†).
14-3-3 binds pSer356 at cellularly relevant concentrations
To gain a greater thermodynamic understanding of 14-3-3/hpTau complex formation, given this new insight and the multivalent interaction between 14-3-3 and hpTau, we sought to develop a multicomponent equilibrium model. Critical for the development of the model was the determination of effective molarity (EM) values of hpTau binding to dimeric 14-3-3 for the phosphorylation pairs pSer214–pSer324 (110-mer), pSer324–pSer356 (32-mer), and pSer214–pSer356 (142-mer). To this end, we employed a Wormlike Chain Model (see ESI†), affording EM values of 2.8, 1.4, and 1.1 mM for the 32, 110, and 142-mer random-coil peptides, respectively (Fig. 5E). Utilizing the derived EM values and the mass balance equations, an equilibrium model was created that describe a system where a single hpTau (A) with three binding sites (1, 2 and 3) can bind multiple 14-3-3 dimers (B) with connected dissociation constants KD1–KD3 encoded (Fig. 5F). The model derived the fraction of protein complexes based on the total concentrations of 14-3-3 and hpTau used in the intact MS and gel experiments (hpTau = 16 μM; 14-3-3 = 8 μM), the previously calculated EM values, and peptide affinities with 14-3-3γ from the FA studies (Fig. 5D). Notably, the KD value for the pSer356 site (KD3) was extrapolated by confining the upper limit of the dose–response curve.Analysis of the equilibrium model under experimental conditions (Tau = 16 μM; 14-3-3 = 8 μM) revealed that complexes involving the bivalent binding of Tau to the 14-3-3 dimer are the dominant species at 8 μM 14-3-3 (Fig. S9H†). Among these, the bivalently bound pS214–pS324 (AB1_2) is the major species, while the bivalently bound pS324–pS356 (AB2_3) is the most prominent minor species. Bivalently bound pS214–pS324 (AB1_2), elicited a calculated KD value of 0.138 μM. Regarding bivalently bound pS324–pS356, this result highlights that even when only present at low fractions, our histidine trapping crosslinker can capture specific 14-3-3 interaction partners, suggesting this probe could also be useful to study other protein complexes.
Based on the data of Sluchanko et al.60 and our new understanding of 14-3-3/hpTau complex formation, we then sought to gain a greater mechanistic understanding of 14-3-3 binding to hpTau at cellularly relevant concentrations. The concentration of Tau in a neuron is reported at ∼2 μM.63,64 Further, several studies have estimated the most abundant 14-3-3 isoform zeta in the brain has a concentration of 100 μM.65–68 Utilizing these protein concentrations (Tau = 2 μM; 14-3-3 = 100 μM) and our equilibrium model it was observed that hpTau binds one or two 14-3-3 dimers (Fig. 5G and H). At 100 μM of 14-3-3, AB1_2 remains the major species, however, it undergoes a significant reduction in concentration compared to at equimolar 14-3-3 and hpTau (Fig. 5G) – with the double bound 14-3-3, containing the bivalent binding pSer324–pSer356 (AB2_3B1) being the second most abundant species (Fig. 5H). Interestingly, at high concentrations of 14-3-3 effective molarity plays a more dominate role with AB2_3B1 out competing AB1_2B3 and AB1_3B2 (Fig. 5G). This key finding aligns with the data report by Sluchanko et al.60 and is highly significant to our current molecular understanding of 14-3-3's interaction. Most studies focus purely on binding affinities, however for this multivalent PPI, at concentrations higher than 10 μM of 14-3-3, affinity plays a less dominate role and effective molarity significantly influences protein complex species. This suggest the need for the Tau chemical biology and cellular biology communities to re-evaluate the molecular mechanism of 14-3-3/Tau binding.
Finally, we are particularly interested in the development of molecular glues that stabilize the native interaction between 14-3-3 and its interaction partners.37,69–72 14-3-3 binding to hpTau has been shown to suppress its aggregation that leads to neurodegeneration.56,57 Utilizing this equilibrium model we simulated 100-fold stabilization of 14-3-3 binding at each phosphorylation site (Fig. 5I–K). Simulated stabilization of the pSer214 and pSer324 sites had little effect on the overall fraction of 14-3-3 bound landscape compared to the non-stabilized system (Fig. 5H). Interestingly, stabilization of the weakest pSer356 site significantly modified the distribution landscape of 14-3-3/hpTau complexes (Fig. 5K). Specifically, the enhanced app. KD coupled with the higher effective molarity (30-mer vs. 110-mer) led to the complex containing the bivalent binding of pSer324–pSer356 (AB2_3B1) being the major species. This result suggests stabilizing the pSer356 site may provide an interesting line of research to mitigate hpTau aggregation enabling more 14-3-3 to be recruited to hpTau.
Conclusions
The development of tool compounds for studying the function of IDPs and IDDs remains a significant challenge. In this study, we have developed a novel histidine trapping crosslinking approach to study the interactions between IDPs/IDDs and the well-structured hub protein 14-3-3. Our histidine trapping crosslinker approach exhibited high crosslinking capacity (>90%) and selectivity for cysteine and histidine. Through kinetic, pH, and stoichiometry studies we identified key reaction intermediates, enabling the deconvolution of a two-step, proximity-driven crosslinking reaction pathway. SAR studies underscored the indispensable role of the maleimide electrophile for the subsequent histidine–cyclohexenone crosslinking. Further, replacement of either electrophile led to complete abrogation or drastic decrease of the crosslinking efficiency. In contrast, linker length was well tolerated as changes in linker length didn't elicit significant effect on the crosslinking efficiency. Mutational studies demonstrated that the crosslinker concept is also capable of crosslinking proximal Cys–Cys residues, but incapable of conjugating Lys. The cross-linking approach is accommodating to a variety of His positions in the IDPs/IDDs, except the +2 and +3 at the C-terminus of the 14-3-3 binding motifs. Further, we demonstrated the robustness of the approach on a panel of 14-3-3 binding peptides decorated with either a histidine or cysteine.Using the crosslinker to study the multivalent interaction between full-length proteins 14-3-3 and PKA phosphorylated Tau we identified a largely overlooked 14-3-3 binding site pSer356 on Tau. An equilibrium model provided a greater mechanistic understanding of the role of the pSer356 site in 14-3-3 binding. At cellularly relevant 14-3-3 and Tau concentrations a significant proportion of 14-3-3 is bound to the pSer356 site of Tau. Although first reported by Sluchanko et al.,60 this binding site has been less attended to in 14-3-3 mechanistic studies and poorly depicted in 14-3-3/Tau models. Interestingly, polypeptide regions R4 (354–369) and PRD (212–221) (Fig. 5A), which encompass Ser214 and Ser356, are significantly phospho-enriched in Alzheimer's patient brain samples.62 Given the phospho-enrichment of Ser356 in Alzheimer's, coupled with ours and Sluchanko's findings necessitate a re-evaluation of the mechanism of 14-3-3/Tau interactions in neurodegeneration.
Finally, we showed that stabilization of the pSer356 site has the greatest impact on the 14-3-3 binding landscape. This finding has significant impact to the molecular glue drug discovery field as it suggests that stabilization of weak interactions over high affinity interactions may have a greater biomolecular impact on the interactome landscape.
The histidine-trap crosslinker approach reported here not only advances our understanding of the 14-3-3/Tau interaction but also demonstrates the potential of covalent tool compounds in studying complex PPIs involving IDPs and IDDs.
Data availability
The data that support the findings of this study are available in the ESI† of this article. Specifically, the ESI† contains experimental procedures and additional data, as well as all the structures and characterization of the molecules described in this manuscript. Further, the details of the effective molarity and mass balance models including the python code can also be found within the ESI.† Crystallographic data presented in this manuscript has been deposited on the open access repository RSCB Protein Data Bank (https://www.rcsb.org/) via the PDB code 9F35.Author contributions
QW, JB, LB, and PC conceptualized and initiated the project. QW and JB designed, synthesized, purified and characterized all compounds. QW, JB, and MO expressed and purified full-length proteins. QW and CV synthesized and purified peptides. QW and JB performed the protein crystallization and solved the crystal structure. QW, JB, SW, and SG performed the mass spectrometry experiments. PC and AM wrote and developed Python models. CO, AM, LB, and PC supervised the project. QW and PC wrote the manuscript with input from all authors.Conflicts of interest
The authors declare the following competing financial interest: C. O. and L. B. are co-founders of Ambagon Therapeutics.Acknowledgements
This research was supported by the Netherlands Organization for Scientific Research via NWO Veni VI.Veni.212.27 and OCNW.KLEIN.300, a China Scholarship Council PhD fellowship (CSC 201906050031) and the European Union through ERC Advanced Grant PPI-Glue (101098234). We thank Auke A. Koops for providing PPARγ protein.Notes and references
- P. E. Wright and H. J. Dyson, Intrinsically disordered proteins in cellular signalling and regulation, Nat. Rev. Mol. Cell Biol., 2015, 16, 18–29 CrossRef CAS PubMed
.
- S. V. Frye, The art of the chemical probe, Nat. Chem. Biol., 2010, 6, 159–161 CrossRef CAS PubMed
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Muller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundstrom, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, The promise and peril of chemical probes, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed
- S. Pomplun, M. Jbara, C. K. Schissel, S. Wilson Hawken, A. Boija, C. Li, I. Klein and B. L. Pentelute, Parallel Automated Flow Synthesis of Covalent Protein Complexes That Can Inhibit MYC-Driven Transcription, ACS Cent. Sci., 2021, 7, 1408–1418 CrossRef CAS PubMed
- M. R. Banghart and B. L. Sabatini, Photoactivatable neuropeptides for spatiotemporally precise delivery of opioids in neural tissue, Neuron, 2012, 73, 249–259 CrossRef CAS PubMed
- R. Van Damme, K. Li, M. Zhang, J. Bai, W. H. Lee, J. D. Yesselman, Z. Lu and W. A. Velema, Chemical reversible crosslinking enables measurement of RNA 3D distances and alternative conformations in cells, Nat. Commun., 2022, 13, 911 CrossRef CAS PubMed
- W. A. Velema, H. S. Park, A. Kadina, L. Orbai and E. T. Kool, Trapping Transient RNA Complexes by Chemically Reversible Acylation, Angew. Chem., Int. Ed., 2020, 59, 22017–22022 CrossRef CAS PubMed
- L. Xue, P. Schnacke, M. S. Frei, B. Koch, J. Hiblot, R. Wombacher, S. Fabritz and K. Johnsson, Probing coenzyme A homeostasis with semisynthetic biosensors, Nat. Chem. Biol., 2023, 19, 346–355 CrossRef CAS PubMed
- R. N. Reddi, A. Rogel, E. Resnick, R. Gabizon, P. K. Prasad, N. Gurwicz, H. Barr, Z. Shulman and N. London, Site-Specific Labeling of Endogenous Proteins Using CoLDR Chemistry, J. Am. Chem. Soc., 2021, 143, 20095–20108 CrossRef CAS PubMed
- R. Gabizon, B. Tivon, R. N. Reddi, M. C. M. van den Oetelaar, H. Amartely, P. J. Cossar, C. Ottmann and N. London, A simple method for developing lysine targeted covalent protein reagents, Nat. Commun., 2023, 14, 7933 CrossRef CAS PubMed
- L. Boike, A. G. Cioffi, F. C. Majewski, J. Co, N. J. Henning, M. D. Jones, G. Liu, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC, Cell Chem. Biol., 2021, 28, 4–13e17 CrossRef CAS PubMed
- R. J. Andersen, N. R. Mawji, J. Wang, G. Wang, S. Haile, J. K. Myung, K. Watt, T. Tam, Y. C. Yang, C. A. Banuelos, D. E. Williams, I. J. McEwan, Y. Wang and M. D. Sadar, Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor, Cancer Cell, 2010, 17, 535–546 CrossRef CAS PubMed
- J. Zhu, X. Salvatella and P. Robustelli, Small molecules targeting the disordered transactivation domain of the androgen receptor induce the formation of collapsed helical states, Nat. Commun., 2022, 13, 6390 CrossRef CAS PubMed
- C. M. Joiner, M. E. Breen, J. Clayton and A. K. Mapp, A Bifunctional Amino Acid Enables Both Covalent Chemical Capture and Isolation of in Vivo Protein–Protein Interactions, ChemBioChem, 2017, 18, 181–184 CrossRef CAS PubMed
- S. Lenz, L. R. Sinn, F. J. O'Reilly, L. Fischer, F. Wegner and J. Rappsilber, Reliable identification of protein-protein interactions by crosslinking mass spectrometry, Nat. Commun., 2021, 12, 3564 CrossRef CAS PubMed
- M. A. Gonzalez-Lozano, F. Koopmans, P. F. Sullivan, J. Protze, G. Krause, M. Verhage, K. W. Li, F. Liu and A. B. Smit, Stitching the synapse: Cross-linking mass spectrometry into resolving synaptic protein interactions, Sci. Adv., 2020, 6, eaax5783 CrossRef CAS PubMed
- T. Berggard, S. Linse and P. James, Methods for the detection and analysis of protein-protein interactions, Proteomics, 2007, 7, 2833–2842 CrossRef PubMed
- J. R. Perkins, I. Diboun, B. H. Dessailly, J. G. Lees and C. Orengo, Transient protein-protein interactions: structural, functional, and network properties, Structure, 2010, 18, 1233–1243 CrossRef CAS PubMed
- B. Steigenberger, P. Albanese, A. J. R. Heck and R. A. Scheltema, To Cleave or Not To Cleave in XL-MS?, J. Am. Soc. Mass Spectrom., 2020, 31, 196–206 CrossRef CAS PubMed
- K. Yugandhar, Q. Zhao, S. Gupta, D. Xiong and H. Yu, Progress in methodologies and quality-control strategies in protein cross-linking mass spectrometry, Proteomics, 2021, 21, e2100145 CrossRef PubMed
- A. Kao, C. L. Chiu, D. Vellucci, Y. Yang, V. R. Patel, S. Guan, A. Randall, P. Baldi, S. D. Rychnovsky and L. Huang, Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes, Mol. Cell. Proteomics, 2011, 10, M110.002212 CrossRef PubMed
- P. L. Jiang, C. Wang, A. Diehl, R. Viner, C. Etienne, P. Nandhikonda, L. Foster, R. D. Bomgarden and F. Liu, A Membrane-Permeable and Immobilized Metal Affinity Chromatography (IMAC) Enrichable Cross-Linking Reagent to Advance In Vivo Cross-Linking Mass Spectrometry, Angew. Chem., Int. Ed., 2022, 61, e202113937 CrossRef CAS PubMed
- B. Steigenberger, R. J. Pieters, A. J. R. Heck and R. A. Scheltema, PhoX: An IMAC-Enrichable Cross-Linking Reagent, ACS Cent. Sci., 2019, 5, 1514–1522 CrossRef CAS PubMed
- R. Kluger and A. Alagic, Chemical cross-linking and protein-protein interactions-a review with illustrative protocols, Bioorg. Chem., 2004, 32, 451–472 CrossRef CAS PubMed
- K. Vyborny, J. Vallova, Z. Koci, K. Kekulova, K. Jirakova, P. Jendelova, J. Hodan and S. Kubinova, Genipin and EDC crosslinking of extracellular matrix hydrogel derived from human umbilical cord for neural tissue repair, Sci. Rep., 2019, 9, 10674 CrossRef PubMed
- Y. Jin Lee, Mass spectrometric analysis of cross-linking sites for the structure of proteins and protein complexes, Mol. BioSyst., 2008, 4, 816–823 RSC
- K. L. Pennington, T. Y. Chan, M. P. Torres and J. L. Andersen, The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein-protein interactions, Oncogene, 2018, 37, 5587–5604 CrossRef CAS PubMed
- D. M. Bustos and A. A. Iglesias, Intrinsic disorder is a key characteristic in partners that bind 14-3-3 proteins, Proteins, 2006, 63, 35–42 CrossRef CAS PubMed
- X. Yang, W. H. Lee, F. Sobott, E. Papagrigoriou, C. V. Robinson, J. G. Grossmann, M. Sundström, D. A. Doyle and J. M. Elkins, Structural basis for protein–protein interactions in the 14-3-3 protein family, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17237–17242 CrossRef CAS PubMed
- N. N. Sluchanko, K. V. Tugaeva, S. J. Greive and A. A. Antson, Chimeric 14-3-3 proteins for unraveling interactions with intrinsically disordered partners, Sci. Rep., 2017, 7, 12014 CrossRef PubMed
- L. Phan, P. C. Chou, G. Velazquez-Torres, I. Samudio, K. Parreno, Y. Huang, C. Tseng, T. Vu, C. Gully, C. H. Su, E. Wang, J. Chen, H. H. Choi, E. Fuentes-Mattei, J. H. Shin, C. Shiang, B. Grabiner, M. Blonska, S. Skerl, Y. Shao, D. Cody, J. Delacerda, C. Kingsley, D. Webb, C. Carlock, Z. Zhou, Y. C. Hsieh, J. Lee, A. Elliott, M. Ramirez, J. Bankson, J. Hazle, Y. Wang, L. Li, S. Weng, N. Rizk, Y. Y. Wen, X. Lin, H. Wang, H. Wang, A. Zhang, X. Xia, Y. Wu, M. Habra, W. Yang, L. Pusztai, S. C. Yeung and M. H. Lee, The cell cycle regulator 14-3-3sigma opposes and reverses cancer metabolic reprogramming, Nat. Commun., 2015, 6, 7530 CrossRef CAS PubMed
- Y. Chen, X. Chen, Z. Yao, Y. Shi, J. Xiong, J. Zhou, Z. Su and Y. Huang, 14-3-3/Tau Interaction and Tau Amyloidogenesis, J. Mol. Neurosci., 2019, 68, 620–630 CrossRef CAS PubMed
- Z. Bozoky, M. Krzeminski, R. Muhandiram, J. R. Birtley, A. Al-Zahrani, P. J. Thomas, R. A. Frizzell, R. C. Ford and J. D. Forman-Kay, Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4427–E4436 CrossRef CAS PubMed
- X. Liang, A. C. D. Paula, Z. Bozóky, H. Zhang, C. A. Bertrand, K. W. Peters, J. D. Forman-Kay and R. A. Frizzell, Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis, Mol. Biol. Cell, 2012, 23, 996–1009 CrossRef CAS PubMed
- E. Giusto, T. A. Yacoubian, E. Greggio and L. Civiero, Pathways to Parkinson's disease: a spotlight on 14-3-3 proteins, npj Parkinson's Dis., 2021, 7, 85 CrossRef CAS PubMed
- H. Hermeking, The 14-3-3 cancer connection, Nat. Rev. Cancer, 2003, 3, 931–943 CrossRef CAS PubMed
- E. Sijbesma, K. K. Hallenbeck, S. Leysen, P. J. de Vink, L. Skora, W. Jahnke, L. Brunsveld, M. R. Arkin and C. Ottmann, Site-Directed Fragment-Based Screening for the Discovery of Protein-Protein Interaction Stabilizers, J. Am. Chem. Soc., 2019, 141, 3524–3531 CrossRef CAS PubMed
- D. N. Kenanova, E. J. Visser, J. M. Virta, E. Sijbesma, F. Centorrino, H. R. Vickery, M. Zhong, R. J. Neitz, L. Brunsveld, C. Ottmann and M. R. Arkin, A Systematic Approach to the Discovery of Protein-Protein Interaction Stabilizers, ACS Cent. Sci., 2023, 9, 937–946 CrossRef CAS PubMed
- M. Konstantinidou, E. J. Visser, E. Vandenboorn, S. Chen, P. Jaishankar, M. Overmans, S. Dutta, R. J. Neitz, A. R. Renslo, C. Ottmann, L. Brunsveld and M. R. Arkin, Structure-Based Optimization of Covalent, Small-Molecule Stabilizers of the 14-3-3sigma/ERalpha Protein-Protein Interaction from Nonselective Fragments, J. Am. Chem. Soc., 2023, 145, 20328–20343 CrossRef CAS PubMed
- S. B. Gunnoo and A. Madder, Chemical Protein Modification through Cysteine, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed
- B. H. Northrop, S. H. Frayne and U. Choudhary, Thiol–maleimide “click” chemistry: evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity, Polym. Chem., 2015, 6, 3415–3430 RSC
- E. W. McConnell, A. L. Smythers and L. M. Hicks, Maleimide-Based Chemical Proteomics for Quantitative Analysis of Cysteine Reactivity, J. Am. Soc. Mass Spectrom., 2020, 31, 1697–1705 CrossRef CAS PubMed
- A. H. Mao, S. L. Crick, A. Vitalis, C. L. Chicoine and R. V. Pappu, Net charge per residue modulates conformational ensembles of intrinsically disordered proteins, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8183–8188 CrossRef CAS PubMed
- S. Muller-Spath, A. Soranno, V. Hirschfeld, H. Hofmann, S. Ruegger, L. Reymond, D. Nettels and B. Schuler, From the Cover: Charge interactions can dominate the dimensions of intrinsically disordered proteins, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14609–14614 CrossRef CAS PubMed
- R. van der Lee, M. Buljan, B. Lang, R. J. Weatheritt, G. W. Daughdrill, A. K. Dunker, M. Fuxreiter, J. Gough, J. Gsponer, D. T. Jones, P. M. Kim, R. W. Kriwacki, C. J. Oldfield, R. V. Pappu, P. Tompa, V. N. Uversky, P. E. Wright and M. M. Babu, Classification of intrinsically disordered regions and proteins, Chem. Rev., 2014, 114, 6589–6631 CrossRef CAS PubMed
- P. N. Joshi and V. Rai, Single-site labeling of histidine in proteins, on-demand reversibility, and traceless metal-free protein purification, Chem. Commun., 2019, 55, 1100–1103 RSC
- S. Vucetic, C. J. Brown, A. K. Dunker and Z. Obradovic, Flavors of protein disorder, Proteins, 2003, 52, 573–584 CrossRef CAS PubMed
- A. Fischer, A. Baljuls, J. Reinders, E. Nekhoroshkova, C. Sibilski, R. Metz, S. Albert, K. Rajalingam, M. Hekman and U. R. Rapp, Regulation of RAF activity by 14-3-3 proteins: RAF kinases associate functionally with both homo- and heterodimeric forms of 14-3-3 proteins, J. Biol. Chem., 2009, 284, 3183–3194 CrossRef CAS PubMed
- A. Ballone, R. A. Lau, F. P. A. Zweipfenning and C. Ottmann, A new soaking procedure for X-ray crystallographic structural determination of protein-peptide complexes, Acta Crystallogr., Sect. F:Struct. Biol. Commun., 2020, 76, 501–507 CrossRef CAS PubMed
- M. A. R. Raycroft, K. E. Racine, C. N. Rowley and J. W. Keillor, Mechanisms of Alkyl and Aryl Thiol Addition to N-Methylmaleimide, J. Org. Chem., 2018, 83, 11674–11685 CrossRef CAS PubMed
- M. Sauerland, R. Mertes, C. Morozzi, A. L. Eggler, L. F. Gamon and M. J. Davies, Kinetic assessment of Michael addition reactions of alpha, beta-unsaturated carbonyl compounds to amino acid and protein thiols, Free Radical Biol. Med., 2021, 169, 1–11 CrossRef CAS PubMed
- J.-Z. Wang, Y.-Y. Xia, I. Grundke-Iqbal and K. Iqbal, Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration, J. Alzheimer's Dis., 2013, 33, S123–S139 Search PubMed
- M. P. Mazanetz and P. M. Fischer, Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases, Nat. Rev. Drug Discovery, 2007, 6, 464–479 CrossRef CAS PubMed
- F. Liu, Z. Liang, J. Shi, D. Yin, E. El-Akkad, I. Grundke-Iqbal, K. Iqbal and C. X. Gong, PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners, FEBS Lett., 2006, 580, 6269–6274 CrossRef CAS PubMed
- J. X. Meng, Y. Zhang, D. Saman, A. M. Haider, S. De, J. C. Sang, K. Brown, K. Jiang, J. Humphrey, L. Julian, E. Hidari, S. F. Lee, G. Balmus, R. A. Floto, C. E. Bryant, J. L. P. Benesch, Y. Ye and D. Klenerman, Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses, Nat. Commun., 2022, 13, 2692 CrossRef CAS PubMed
- Y. Q. Liu, C. Q. Liang, Z. W. Chen, J. Hu, J. J. Hu, Y. Y. Luo, Y. X. Chen and Y. M. Li, 14-3-3zeta Participates in the Phase Separation of Phosphorylated and Glycated Tau and Modulates the Physiological and Pathological Functions of Tau, ACS Chem. Neurosci., 2023, 14, 1220–1225 CrossRef CAS PubMed
- J. Hochmair, M. C. M. v. d. Oetelaar, L. Diez, L. J. M. Lemmens, R. Ponce, L. Ravatt, M. W. Franck, E. Semenova, S. Mohapatra, C. Ottmann, L. Brunsveld and S. Wegmann, 14-3-3 binding regulates Tau assembly and microtubule association, bioRxiv, 2024, preprint, DOI:10.1101/2024.03.15.585148.
- C. W. Scott, R. C. Spreen, J. L. Herman, F. P. Chow, M. D. Davison, J. Young and C. B. Caputo, Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly, J. Biol. Chem., 1993, 268, 1166–1173 CrossRef CAS PubMed
- I. Landrieu, L. Lacosse, A. Leroy, J.-M. Wieruszeski, X. Trivelli, A. Sillen, N. Sibille, H. Schwalbe, K. Saxena, T. Langer and G. Lippens, NMR Analysis of a Tau Phosphorylation Pattern, J. Am. Chem. Soc., 2006, 128, 3575–3583 CrossRef CAS PubMed
- N. N. Sluchanko, A. S. Seit-Nebi and N. B. Gusev, Phosphorylation of more than one site is required for tight interaction of human tau protein with 14-3-3zeta, FEBS Lett., 2009, 583, 2739–2742 CrossRef CAS PubMed
- Y. Joo, B. Schumacher, I. Landrieu, M. Bartel, C. Smet-Nocca, A. Jang, H. S. Choi, N. L. Jeon, K. A. Chang, H. S. Kim, C. Ottmann and Y. H. Suh, Involvement of 14-3-3 in tubulin instability and impaired axon development is mediated by Tau, FASEB J., 2015, 29, 4133–4144 CrossRef CAS PubMed
- H. Wesseling, W. Mair, M. Kumar, C. N. Schlaffner, S. Tang, P. Beerepoot, B. Fatou, A. J. Guise, L. Cheng, S. Takeda, J. Muntel, M. S. Rotunno, S. Dujardin, P. Davies, K. S. Kosik, B. L. Miller, S. Berretta, J. C. Hedreen, L. T. Grinberg, W. W. Seeley, B. T. Hyman, H. Steen and J. A. Steen, Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer's Disease, Cell, 2020, 183, 1699–1713 CrossRef CAS PubMed
- A. d. C. Alonso, B. Li, I. Grundke-Iqbal and K. Iqbal, Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8864–8869 CrossRef CAS PubMed
- T. Arendt, J. Stieler, A. M. Strijkstra, R. A. Hut, J. Rüdiger, E. A. V. d. Zee, T. Harkany, M. Holzer and W. Härtig, Reversible Paired Helical Filament-Like Phosphorylation of Tau Is an Adaptive Process Associated with Neuronal Plasticity in Hibernating Animals, J. Neurosci., 2003, 23, 6972–6981 CrossRef CAS PubMed
- M. Wang, C. J. Herrmann, M. Simonovic, D. Szklarczyk and C. von Mering, Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines, Proteomics, 2015, 15, 3163–3168 CrossRef CAS PubMed
- P. F. Boston, P. Jackson and R. J. Thompson, Human 14-3-3 Protein: Radioimmunoassay, Tissue Distribution, and Cerebrospinal Fluid Levels in Patients with Neurological Disorders, J. Neurochem., 1982, 38, 1475–1482 CrossRef CAS PubMed
- R. J. Ellis, Macromolecular crowding: an important but neglected aspect of the intracellular environment, Curr. Opin. Struct. Biol., 2001, 11, 114–119 CrossRef CAS PubMed
- Z. Trosanova, P. Lousa, A. Kozelekova, T. Brom, N. Gasparik, J. Tungli, V. Weisova, E. Zupa, G. Zoldak and J. Hritz, Quantitation of Human 14-3-3zeta Dimerization and the Effect of Phosphorylation on Dimer-monomer Equilibria, J. Mol. Biol., 2022, 434, 167479 CrossRef CAS PubMed
- S. A. Andrei, P. de Vink, E. Sijbesma, L. Han, L. Brunsveld, N. Kato, C. Ottmann and Y. Higuchi, Rationally Designed Semisynthetic Natural Product Analogues for Stabilization of 14-3-3 Protein-Protein Interactions, Angew. Chem., Int. Ed., 2018, 57, 13470–13474 CrossRef CAS PubMed
- P. J. Cossar, M. Wolter, L. van Dijck, D. Valenti, L. M. Levy, C. Ottmann and L. Brunsveld, Reversible Covalent Imine-Tethering for Selective Stabilization of 14-3-3 Hub Protein Interactions, J. Am. Chem. Soc., 2021, 143, 8454–8464 CrossRef CAS PubMed
- E. Sijbesma, E. Visser, K. Plitzko, P. Thiel, L. G. Milroy, M. Kaiser, L. Brunsveld and C. Ottmann, Structure-based evolution of a promiscuous inhibitor to a selective stabilizer of protein-protein interactions, Nat. Commun., 2020, 11, 3954 CrossRef CAS PubMed
- E. J. Visser, P. Jaishankar, E. Sijbesma, M. A. M. Pennings, E. M. F. Vandenboorn, X. Guillory, R. J. Neitz, J. Morrow, S. Dutta, A. R. Renslo, L. Brunsveld, M. R. Arkin and C. Ottmann, From Tethered to Freestanding Stabilizers of 14-3-3 Protein–Protein Interactions through Fragment Linking, Angew. Chem., Int. Ed., 2023, 62, e202308004 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07419j |
| This journal is © The Royal Society of Chemistry 2025 |