
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic regioselective C–H bond functionalizations in arenes
Jun
Hu†
 ,
Suman
Pradhan†
,
Satyadeep
Waiba†
and
Shoubhik
Das
*
,
Suman
Pradhan†
,
Satyadeep
Waiba†
and
Shoubhik
Das
*
Department of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: shoubhik.das@uni-bayreuth.de
First published on 9th December 2024
Abstract
The direct functionalization of C–H bonds has revolutionized the field of synthetic organic chemistry by enabling efficient and atom-economical modification of arenes by avoiding prefunctionalization. However, the inherent challenges of inertness and regioselectivity in different C–H bonds, particularly for distal positions, necessitate innovative approaches. In this aspect, photoredox catalysis by utilizing both transition metal and organic photocatalysts has emerged as a powerful tool for addressing these challenges under mild reaction conditions. This review provides a comprehensive overview of recent progress in regioselective C–H functionalization in arenes via photocatalysis. Emphasizing the strategies for achieving ortho-, meta-, and para-selectivity, we explore the mechanistic insights, catalyst designs, and the novel methodologies that have expanded the scope of C–H bond functionalization. This discussion aims to offer valuable perspectives for advancing the field and developing more efficient and sustainable synthetic methodologies.
 Jun Hu | Mr Jun Hu earned his Bachelor of Science in Chemistry from the University of Antwerp in 2021, followed by a Master of Science in Chemistry from the same institution in 2023. He is currently pursuing his PhD under the supervision of Prof. Shoubhik Das at the University of Bayreuth. His research focuses on the development of innovative strategies for C–H bond activation, photocatalytic organic synthesis, and CO2 utilization. |
 Suman Pradhan | Dr Suman Pradhan received his MSc. from the University of Calcutta in 2016. In November 2017, he joined the Indian Institute of Technology (IIT) Ropar, India, as a PhD student under the guidance of Prof. Indranil Chatterjee. After earning his PhD, he began his postdoctoral research in October 2022 at the University of Antwerp, Belgium, under the supervision of Prof. Shoubhik Das. Subsequently, he moved with Prof. Das to the University of Bayreuth, Germany, to continue his postdoctoral studies. His current research focuses on photocatalytic organic synthesis, plastic valorization, and CO2 reduction. |
 Satyadeep Waiba | Dr Satyadeep Waiba received his MSc. in Chemistry from the University of North Bengal, India, in 2017. He then pursued his doctoral studies under the guidance of Prof. Biplab Maji in the Department of Chemical Sciences at the Indian Institute of Science Education and Research (IISER) Kolkata and earned his PhD degree in June 2022. In July 2023, he joined the University of Bayreuth in Germany as a postdoctoral researcher under the supervision of Prof. Shoubhik Das. His current research focuses on photocatalytic organic transformations, plastic valorizations, and (de)hydrogenation reactions. |
 Shoubhik Das | Prof. Dr Shoubhik Das received his PhD under the supervision of Prof. Matthias Beller from the Leibniz-Institut für Katalyse in 2012. Subsequently, he joined the research group of Prof. Matthew Gaunt at Cambridge University as a postdoctoral research associate. In 2013, he moved to the research group of Prof. Paul J. Dyson at the EPFL, Switzerland. In August 2015, he started his independent research career at Georg-August-Universität Göttingen. In November 2019, he joined the Department of Chemistry at the University of Antwerp. Later in August 2023, he moved to the University of Bayreuth, Germany, as a chair professor in the organic chemistry division. His main research interests are photocatalytic organic synthesis, heterogeneous photocatalysis, activation of small molecules and their applications in organic synthesis and fuel generation. |
1. Introduction
The direct functionalization of C–H bonds in arenes is a pivotal technique in contemporary organic synthesis, presenting an attractive and efficient strategy for incorporating diverse functionalities into arenes.1–7 These approaches generate derivatives with potentially enhanced biological activity, making them highly valuable in the fields of drug discovery and biochemistry. Additionally, direct C–H bond functionalization strategies offer an opportunity to increase molecular complexity using readily available starting materials, which obviates the necessity for intricate prefunctionalization steps.8–11 This approach is particularly advantageous since it minimizes the environmental and economic impact of organic synthesis and promotes the principles of green chemistry. In this respect, it is worth mentioning that direct C–H functionalization offers substantial advantages; however, it is tempered by two fundamental challenges: selective activation of the C–H bond and attaining high regioselectivity.12 The inert nature of most of the C–H bonds makes them poorly reactive, which often requires stringent reaction conditions such as elevated temperature and the use of strong oxidants. At the same time, the prevalence of C–H bonds in organic molecules and the subtle reactivity differences among multiple C–H bonds present significant challenges to achieving high regioselectivity in direct C–H bond functionalization. In this aspect, transition metal catalysis provides a highly desirable approach for proximal C–H bond activation through the formation of conformationally rigid five- or six-membered metallacycle intermediates, which are both kinetically and thermodynamically favored.13,14 However, this strategy is less applicable for selectively targeting C–H bonds that are distant from existing functional groups, since it requires the formation of much larger and less feasible metallacycles. Nevertheless, various research groups have developed methods to achieve regioselective functionalization at remote meta- and para-positions of benzene rings. These techniques often employ directing groups or exploit weak interactions to guide the functionalization process.15–17 Recently, there have been reports of undirected C–H bond functionalization at remote sites where the choice of reagents, ligands and catalysts plays a crucial role in determining the regioselectivity.18Although transition metal-catalyzed C–H bond functionalization has effectively addressed the issue of regioselectivity, there remains a strong demand for achieving highly regioselective C–H functionalization reactions under mild reaction conditions.19 In this regard, visible light-promoted photoredox catalysis has emerged as a promising solution due to its environmentally benign nature and efficiency in chemical transformations.20–22 In general, photo-induced organic reactions are catalyzed by either transition metal complexes or organic photocatalysts.23,24 Among transition metal photocatalysts, iridium and ruthenium complexes are extensively studied. These metal complexes benefit from the lower energy of the ligand's π* orbital, facilitating the metal–ligand charge transfer process.25 Alternatively, metal-free organic photocatalysts are gaining significant attention due to their low price and easy availability.26–30 Notable organic photocatalysts include benzophenones, acridinium salts, rhodamines and cyanoarenes. Both types of photocatalysts (PCs) harness sunlight as an inexpensive and renewable energy source. Upon irradiation of light, PCs form long-lived photoexcited states (PC*), which exhibit increased reactivity in electron-transfer events compared to their ground states. In reductive quenching, PC* accepts an electron from a substrate or reductant, forming a radical anion, which subsequently undergoes oxidation. Conversely, oxidative quenching is commonly involved in C–H bond functionalization via photoredox reactions. Here, PC* donates an electron to a substrate or an oxidant, generating the radical cation that is later reduced (Fig. 1A). These single electron transfer (SET) processes enhance radical generation, significantly advancing the radical-based transformations in synthetic organic chemistry (Fig. 1B). In addition, the tunability of light as an energy source in photoredox catalysis provides precise control over reaction conditions, enabling enhanced selectivity for functionalization in aromatic systems.
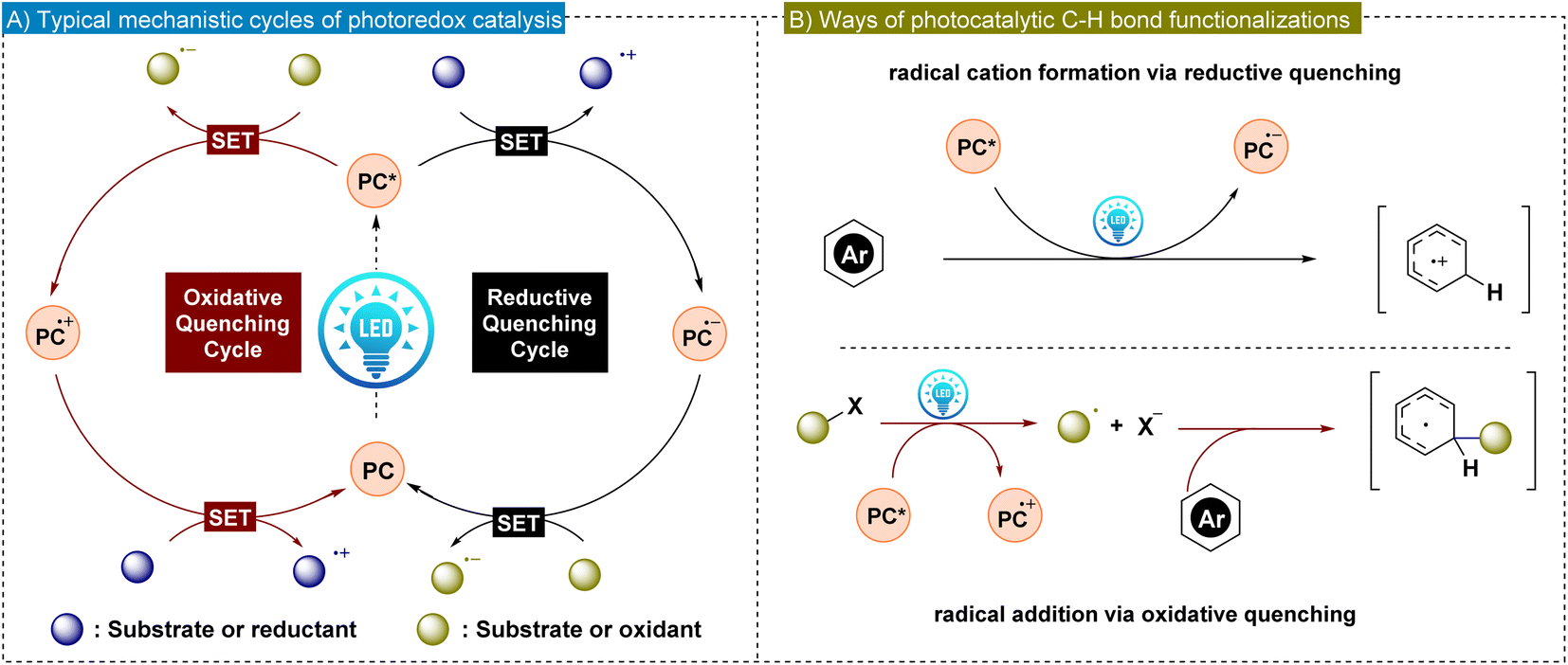 | ||
| Fig. 1 (A) General mechanism of photoredox catalysis. (B) Photoredox catalysis mediated arene C–H bond activation strategies. | ||
Several strategies have been developed to achieve regioselective functionalization of arenes via photocatalytic pathways (Fig. 2). One prominent approach is σ-bond activation-assisted C–H functionalization, where initial ortho-cycloruthenation plays a pivotal role in guiding selective radical coupling at the meta- or para-position. Another method employs templates or directing groups, which position the reactive metal center near the targeted C–H bond using a molecular bridge to control the geometry between the substrate and the catalyst. An alternative strategy involves the oxidation of aromatic compounds, followed by nucleophilic attack by the reagent to functionalize the phenyl ring. Moreover, regioselective functionalization of arenes can also be achieved by first generating a radical via SET, which then inserts into the phenyl ring in a regioselective manner. However, these latter methods often face challenges in achieving high regioselectivity, as the outcome is highly substrate-dependent.
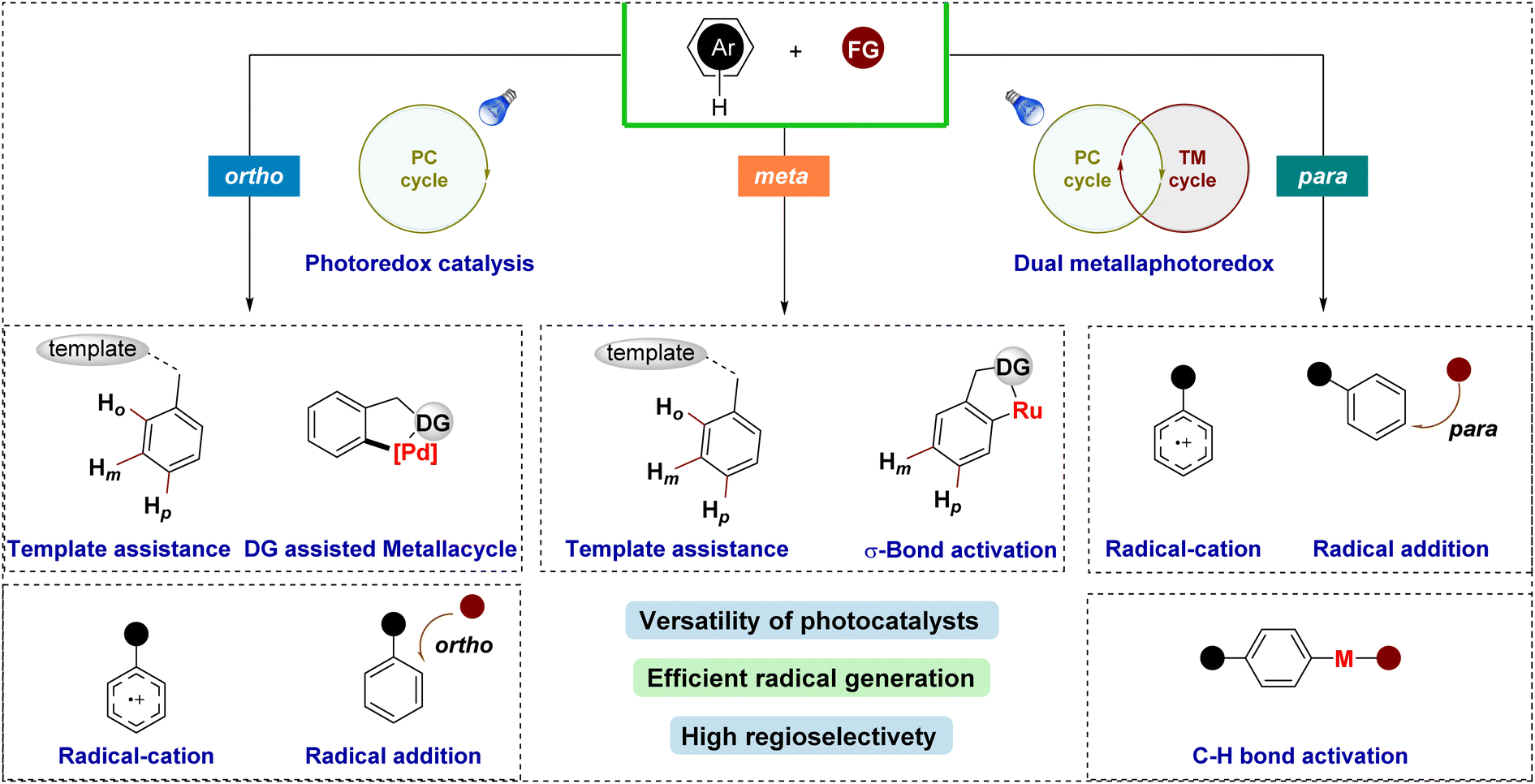 | ||
| Fig. 2 Summary of this review. | ||
Recent literature has extensively reviewed photocatalyzed aromatic C–H bond functionalization11,31–33 and regioselective C–H bond functionalization of arenes.1,3,15,16 However, comprehensive articles specifically addressing photocatalytic regioselective C–H bond functionalization are relatively scarce. Existing reviews predominantly focus on specific topics such as visible light-induced C–H functionalization34 with transition metal complexes or photoredox-catalyzed remote C–H bond functionalization.35 Despite significant advancements and detailed insights in these areas, there remains a need for a comprehensive review that systematically explores regioselectivity in photocatalytic phenyl C–H bond functionalization. This review aims to bridge this gap by offering an in-depth analysis of recent developments in the field, focusing on the regioselective photocatalytic C–H bond functionalization of benzene derivatives. It will assess the advantages, limitations, scopes, and reaction mechanisms of these transformations, providing critical insights to guide future research. The content is organized according to the site of functionalization on the benzene ring: ortho-, meta-, or para-positions, thus delivering a structured and detailed overview of regioselectivity in these processes.
2. Ortho-selective C–H functionalization
Ortho-C–H functionalization represents a pivotal transformation in organic synthesis, offering a direct approach to modify aromatic compounds without the need for pre-functionalization steps. This approach significantly streamlines synthetic pathways and enhances overall efficiency. Traditionally, these transformations required high temperatures and specific reagents, but recent advancements, including palladium-catalyzed methodologies and photoredox catalysis, have introduced milder conditions and expanded substrate compatibility.10,34,35 These innovations not only increase the versatility of C–H functionalization but also facilitate the incorporation of a wide range of functional groups into complex molecular architectures, thereby proving indispensable in drug discovery and materials science. Such ortho-regioselectivity can be achieved by using arenes containing heteroatom-based directing groups to facilitate ortho-selective reagent attack by coordinating with the catalyst or reagent. Alternatively, substrate control relies on carefully modulating electronic and steric factors inherent to the molecule, enabling site-selective functionalization predominantly at the ortho-position.2.1. Directing group-mediated ortho-C–H functionalization
In this aspect, the group of Sanford reported a room-temperature ligand-directed C–H arylation that synergistically combined palladium-catalyzed C–H functionalization with visible-light photoredox catalysis (Scheme 1, top left).44 This protocol employed aryldiazonium salts as aryl radical precursors under ambient temperature, facilitating the efficient arylation of a wide range of substrates, whereas the previously reported conditions required a harsh reaction temperature of higher than 100 °C.45–47 These included pyridine derivatives with varied electronic properties as well as other directing groups such as amides, pyrazoles, pyrimidines, oxime ethers, and even free oximes. Although this report presented a significant advancement in the field of C–H arylation, the use of expensive Ru-based photocatalysts somewhat limited its application.
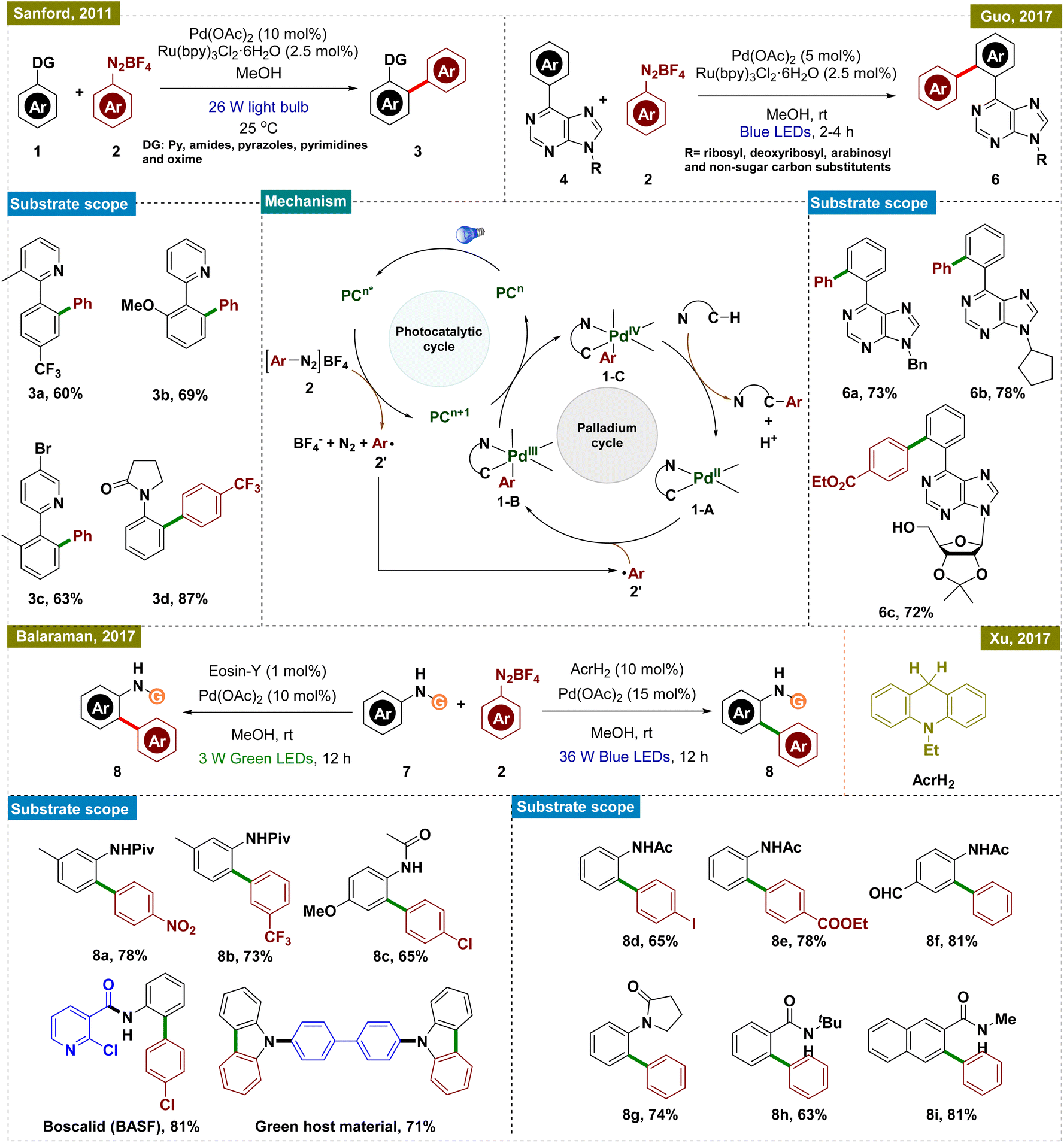 | ||
| Scheme 1 Photoinduced ortho-C–H arylation with aryldiazonium salts. | ||
To address this drawback, research groups of Xu and Balaraman independently reported similar protocols for C(sp2)–H functionalization replacing Ru-photocatalysts with organo-photocatalysts, AcrH2 (9,10-dihydroacridine) and Eosin-Y, respectively (Scheme 1, bottom right and left).48,49 While their approach paralleled Sanford's work, they primarily employed amides as the directing group. Both protocols demonstrated a broad substrate scope, accommodating functional groups such as halides, esters, amides, and ketones, thereby extending their applicability to complex synthetic contexts. Notably, Balaraman's protocol was successfully utilized in the synthesis of boscalid, a potent fungicide and a green host material for phosphorescent OLEDs, highlighting its practical significance in both pharmaceuticals and advanced materials.
Guo and his group also utilized the same concept of dual catalysis for carrying out the ortho-C–H arylation of 6-arylpurine nucleosides, which are important motifs in medicinal chemistry due to their biological activities (Scheme 1, top right).50 By employing this dual catalytic protocol, they successfully arylated a variety of nucleosides by using aryl diazonium salts as arylating agents. Furthermore, the protocol was also applied to gram-scale synthesis, demonstrating its robustness and applicability for large-scale production.
To elucidate the reaction mechanism, a series of experimental studies were conducted, including radical trapping experiments and isotope effect analyses, independently in these reports. Based on these insights, a similar mechanism, as illustrated in Scheme 1, center, was proposed. The dual catalytic process was initiated by the photoexcitation of the photocatalyst (PC), which generated an aryl radical (2′) from the aryldiazonium salt (2) while concurrently getting oxidized to generate an oxidized PC (PCn+1). This aryl radical was then added to the palladacycle intermediate (1-A), which was formed via C–H activation of the substrate, leading to the formation of a Pd(III) species (1-B). The Pd(III) intermediate was further oxidized by the oxidized state of the photocatalyst (PCn+1), thereby regenerating the photocatalyst and producing a Pd(IV) intermediate (1-C). The catalytic cycle was concluded with a C–C bond-forming reductive elimination from the Pd(IV) intermediate, releasing the arylated product and regenerating the Pd(II) catalyst, thus ensuring the continuity of the catalytic cycle. While these protocols offered significant advancements, they all necessitated the use of a photocatalyst in conjunction with a Pd catalyst for C–H activation, increasing the overall cost and limiting their practical applicability.
In contrast, Ackermann and Greaney groups reported an exogenous photocatalyst-free Ru-catalyzed C–H arylation by using aryl halides as effective aryl radical sources, which reduced the traditionally high reaction temperatures required for such processes (Scheme 2).51,52 Both of the research groups utilized a Ru-cymene-based catalyst; however, Greaney's team employed [Ru(p-cymene)Cl2]2, while Ackermann's team used [Ru(OAc)2(p-cymene)] as the catalyst. In both cases, a stoichiometric amount of base was necessary, and the reactions were conducted under the irradiation of blue LEDs. Greaney's protocol utilized various aryl bromides and iodides to arylate arenes with directing groups like pyridines, quinolines, and isoquinolines. Conversely, Ackermann's method was selective for aryl iodides, leaving bromides and pseudohalides such as -OTf intact, and was applicable to a broader range of directing groups, including pyridines, pyrimidines, imidates, pyrazoles, ketimines, and triazoles. Moreover, Ackermann's group extended their protocol to the late-stage diversification of biorelevant molecules such as purines, nucleosides, and nucleotides. Both groups conducted mechanistic studies, ruling out radical formation via quenching experiments and highlighting the critical role of the carboxylate ligand. Additionally, p-cymene was found to dissociate during the reaction, initiating the catalytic cycle. Detailed DFT studies by Ackermann's group further elucidated the mechanism, as depicted in Scheme 2.
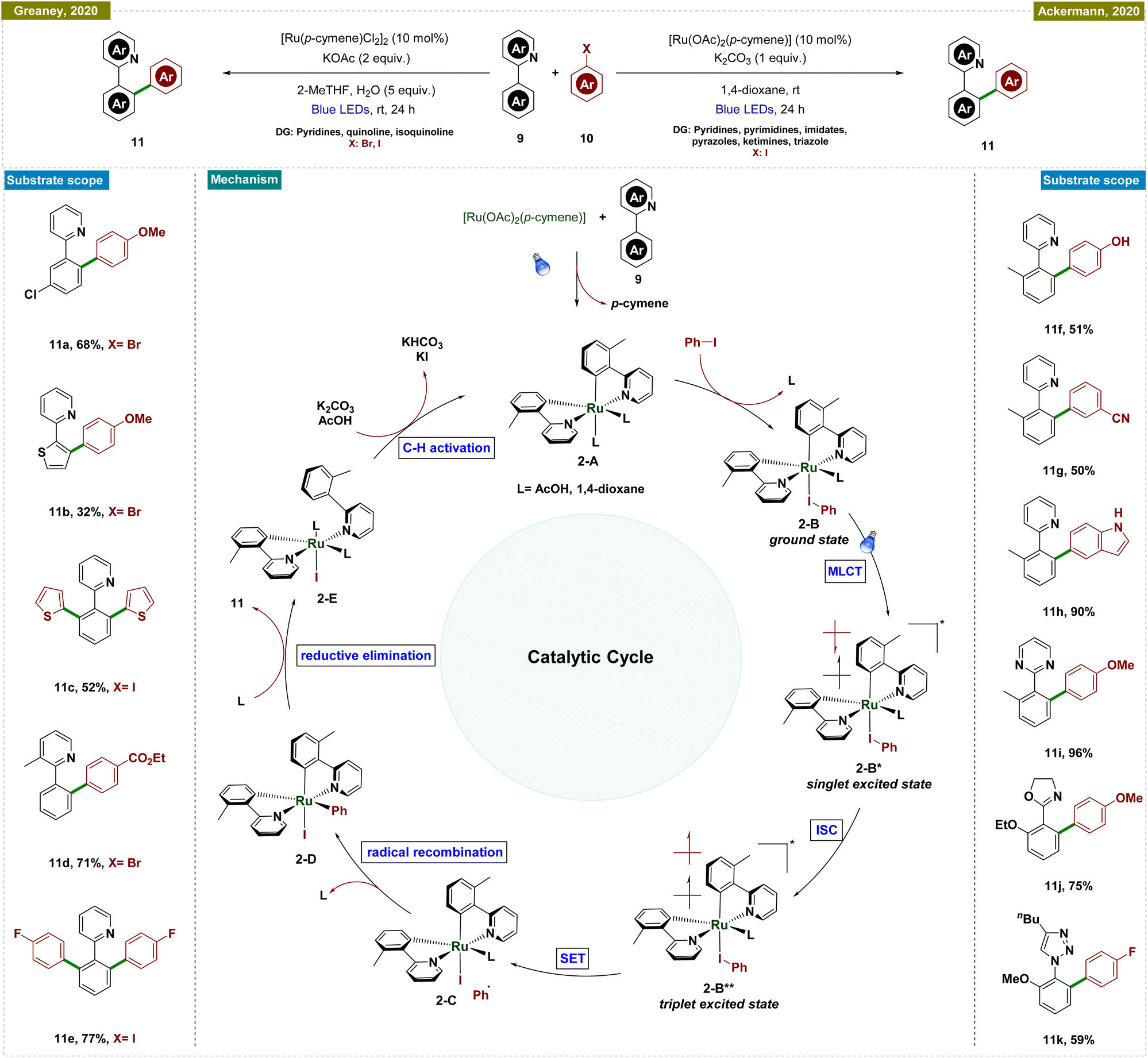 | ||
| Scheme 2 Photoinduced ortho-C–H arylation with aryl halides. | ||
The proposed catalytic cycle began with carboxylate-assisted C–H activation of 9 and p-cymene dissociation, forming the biscyclometalated complex 2-A (Scheme 2). Coordination of the haloarene to complex 2-A generated ruthenacycle 2-A, which, upon blue-light excitation, formed the singlet excited species 2-B*. After intersystem crossing (ISC) to a long-lived triplet state (2-B**), an inner-sphere electron transfer (ISET) occurred, producing a phenyl radical and a ruthenium(III) intermediate (2-C). These recombined to yield a stable ruthenium(IV) species (2-D). Reductive elimination followed by ligand exchange released the arylated product and regenerated the active ruthenium(II) complex (2-E), completing the cycle.
Recently, Ackermann and his team also reported a Ru-catalyzed C–H benzylation and allylation reaction, which followed a similar mechanistic pathway (Scheme 3).53 This exogenous photocatalyst-free ruthenium catalysis was performed via a Ru(II/III) cycle, effectively functionalizing a range of arenes, including those with pyridines, pyrazoles, oxazolines, and biorelevant purines as directing groups, using various substituted benzyl chlorides and allyl bromides. The protocol also extended to electrophiles bearing biorelevant groups and even utilized water as a non-toxic solvent. Mechanistic studies and control experiments were conducted to substantiate the proposed mechanism, similar to that illustrated in Scheme 2.
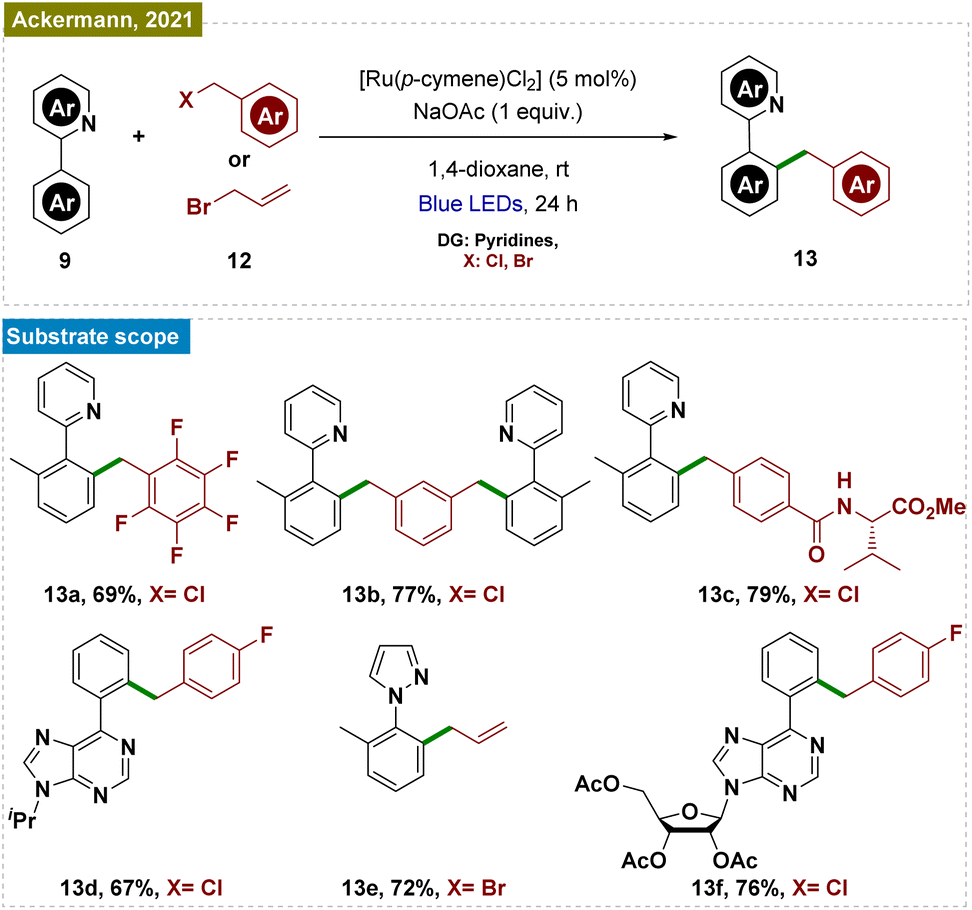 | ||
| Scheme 3 Photoinduced ortho-C–H benzylation and allylation. | ||
 | ||
| Scheme 4 Photoinduced directed ortho-C–H olefination. | ||
Recently, they also employed semiconductors as heterogeneous photocatalysts in dual metal-catalyzed C–H functionalization reactions, including the ortho-olefination of phenol derivatives, the olefination of Weinreb amides, and indole synthesis (Scheme 4, bottom).60 In these processes, various metal catalysts were combined with semiconductor photocatalysts [(BiVO4 (band gap: 2.4 eV), WO3 (band gap: 2.6–3.0 eV), and TiO2)] to achieve the desired transformations.
A general mechanism for the directing group-assisted photocatalytic ortho-C–H olefination is depicted in Scheme 4. Upon the formation of the active Ru(II) catalyst 4-A, which occurred by following the precipitation of chloride ligands from the Ru dimer, ortho-C–H activation of the substrate took place, leading to the formation of intermediate 4-B, facilitated by the coordinating directing group (shown with pyridine in Scheme 4). Subsequent olefin insertion produced intermediate 4-C, followed by reductive elimination to yield the desired product and generate the ruthenium complex 4-D. The photoredox catalyst, PCn, upon excitation by visible light, oxidized complex 4-Dvia electron transfer, thereby regenerating the catalytically active Ru species 4-A. The resulting PCn−1 was reoxidized to PCn by molecular oxygen, generating a superoxide anion. This superoxide anion further oxidized Ru complex 4-D, accepting an electron and thus maintaining the catalytic cycle.
 | ||
| Scheme 5 Photoinduced ortho-C–H acylation. | ||
Based on the mechanistic and control experiments, a probable mechanism was proposed (Scheme 5, center). Initially, the photocatalyst (Eosin Y, PC) was excited to its excited state (PC*) under the irradiation of visible light. This excited state of the PC underwent an oxidation process with phenylglyoxylic acid (20), producing a benzoyl radical (20′′). Concurrently, the reduction of PC* generated the radical anion PC˙−. Electron transfer from PC˙− to molecular oxygen regenerated the photocatalyst for the next catalytic cycle and formed a superoxide radical anion (O2˙−). Concurrently, the palladium catalytic cycle was initiated through C–H activation of acetanilide by Pd(OAc)2, forming a palladacycle intermediate (5-B). It is important to note that, in our view, the addition of substrate 19 to Pd(OAc)2 to form intermediate 5-B may have occurred through a photoinduced LMCT mechanism.62 This intermediate reacted with the in situ-generated benzoyl radical (20′′) to yield a Pd(III) species (5-C), which was further oxidized by the superoxide radical anion to a Pd(IV) intermediate (5-D), with the concomitant formation of O22− and H2O2. The final step involved reductive elimination from the Pd(IV) intermediate, producing the desired ortho-acylated product and regenerating the Pd(II) catalyst (5-A), thereby completing the catalytic cycle.
Building on a similar catalytic framework, Singh and colleagues recently developed a novel method for the regioselective decarboxylative acylation of N-methyl-3-phenylquinoxalin-2(1H)-ones (22) using dual palladium-photoredox catalysis under visible light (Scheme 5, top right).63 The method employed fluorescein dye as the photocatalyst and tert-butyl peroxybenzoate as the oxidant, though an oxidant was required in excess. While the reaction conditions tolerated a wide range of substituents, ortho-substituents on the aryl ring failed to yield the desired product (23b). To elucidate the mechanism, various control experiments, including radical trapping experiments, were conducted, indicating a mechanistic pathway similar to that proposed by Wang and co-workers.61
Continuing along similar lines, Ablajan, Zhu, and colleagues developed a novel method for synthesizing fluorenone derivatives (25) via dual photoredox-catalyzed deoxygenative intramolecular acylation reactions at room temperature (Scheme 5, bottom).64 Their protocol enabled the synthesis of a diverse array of fluorenones, featuring both electron-donating and electron-withdrawing substituents. This method represented a significant advancement over traditional fluorenone synthesis techniques, such as Friedel–Crafts acylations,65,66 fluorenes oxidations,67,68 and Diels–Alder reactions,69 which generated considerable waste. Through a series of mechanistic experiments and control studies, the researchers proposed a reaction mechanism aligned with previous reports, highlighting the crucial role of dimethyl dicarbonate (DMDC) and 2,6-lutidine in facilitating acyl radical formation. The study revealed that DMDC and 2,6-lutidine facilitate the formation of an anhydride intermediate (25′) from the biaryl carboxylic acid. This anhydride intermediate is essential for generating the acyl radical through a SET process. The resulting radical undergoes an intramolecular ortho-addition, ultimately yielding the desired product via a one-electron oxidation and deprotonation step.
 | ||
| Scheme 6 Photoinduced ortho-C–H borylation. | ||
Later, Tanaka and colleagues reported a method for photo-induced ortho-C–H borylation of arenes through the in situ generation of rhodium(II) ate complexes (Scheme 6, bottom).74 This study presented a novel approach to ortho-C–H borylation of arenes through the use of photoexcited rhodium complexes, specifically demonstrating the in situ generation of anionic rhodium(II) ate complexes from rhodium(III) precursors under blue LED irradiation. The catalyst optimization identified a CpRh(III) complex with an electron-rich N-phenylcarbamoyl moiety as the most effective, achieving a 65% yield with NaOtBu as the base. Mechanistic investigations suggested that the process involved the reduction of Rh(III) to Rh(I), followed by oxidative addition to the ortho-C–H bond, leading to the formation of an aryl Rh(III) hydride intermediate and ultimately yielding the borylated product. The method also expanded the scope of C–H functionalization by accommodating a broader range of directing groups. Computational studies using TDDFT calculations further corroborated the experimental observations, providing insight into the electronic transitions and photophysical properties of the rhodium complexes.
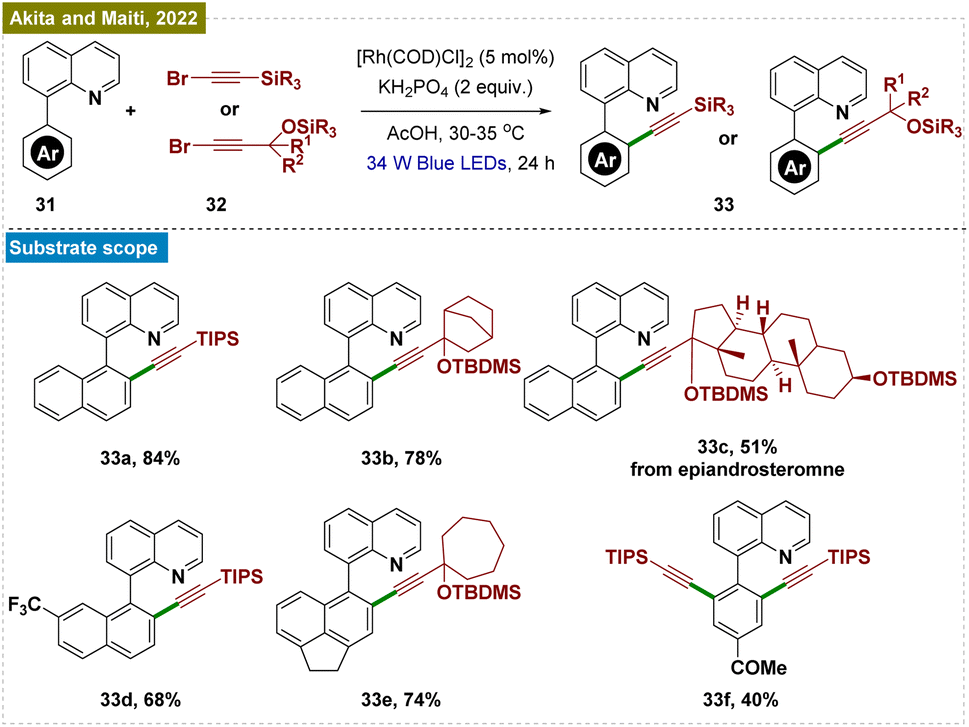 | ||
| Scheme 7 Photoinduced ortho-C–H alkynylation. | ||
2.2. Substrate-controlled ortho-C–H functionalization
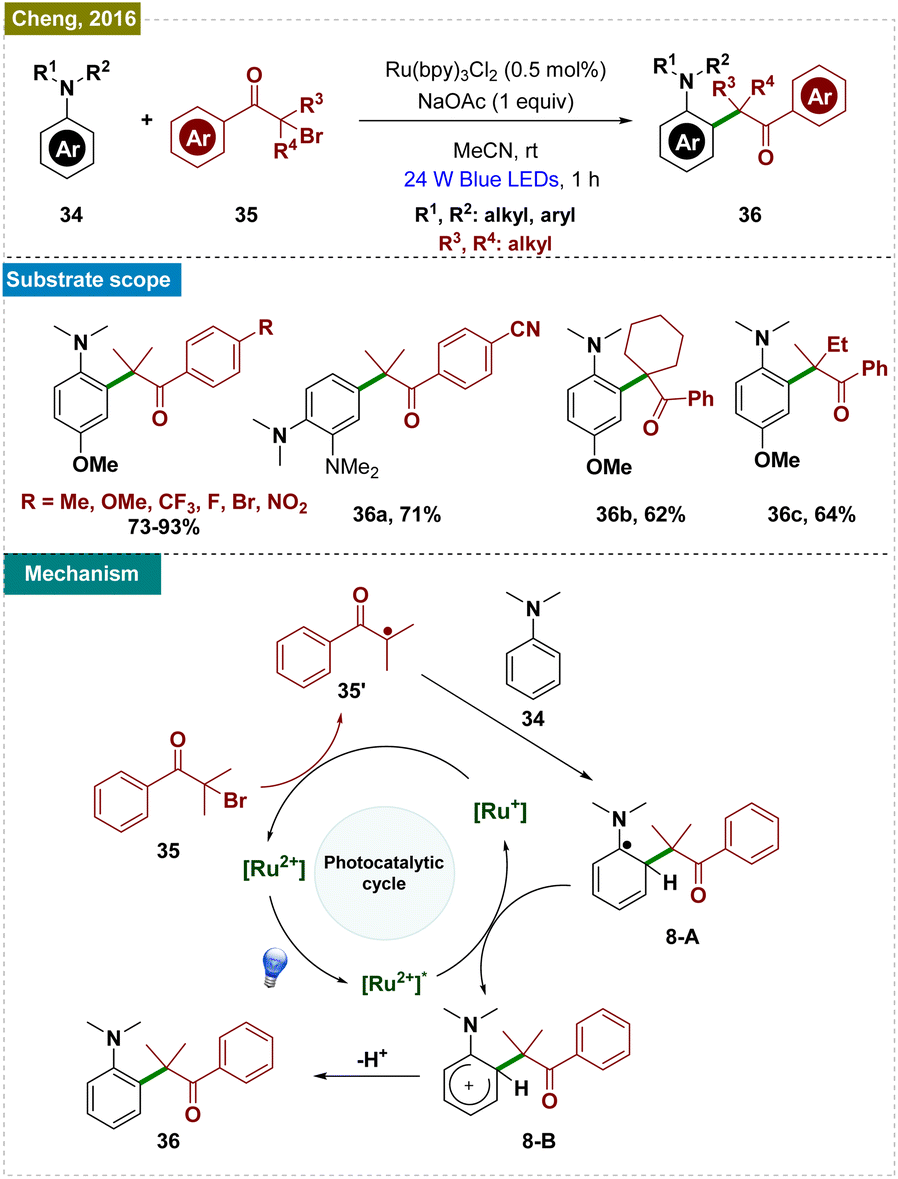 | ||
| Scheme 8 Photoinduced ortho-C–H alkylation from α-bromoketones. | ||
 | ||
| Scheme 9 Photoinduced ortho-C–H trifluoromethylation. | ||
Through quenching experiments and mechanistic studies, the authors proposed the mechanism, as depicted in Scheme 9. The reaction began with the photoexcitation of the photocatalyst (9-A), leading to its excited state (9-B). A single-electron transfer (SET) event from the excited state of the PC to triflyl chloride (38) generated the oxidized Ru3+ photocatalyst (9-C) and a –CF3 radical, which rapidly reacted with the arene (37) to form a cyclohexadienyl radical intermediate (9-D). This intermediate underwent a second SET with the oxidized Ru3+ photocatalyst, regenerating the active catalyst (9-A) and cyclohexadienyl cation (9-E). The final step involved the deprotonation of the cyclohexadienyl cation in the presence of a base, yielding the desired trifluoromethylated product. This method offered a milder, more accessible approach to trifluoromethylation, broadening its applicability in synthetic organic chemistry.
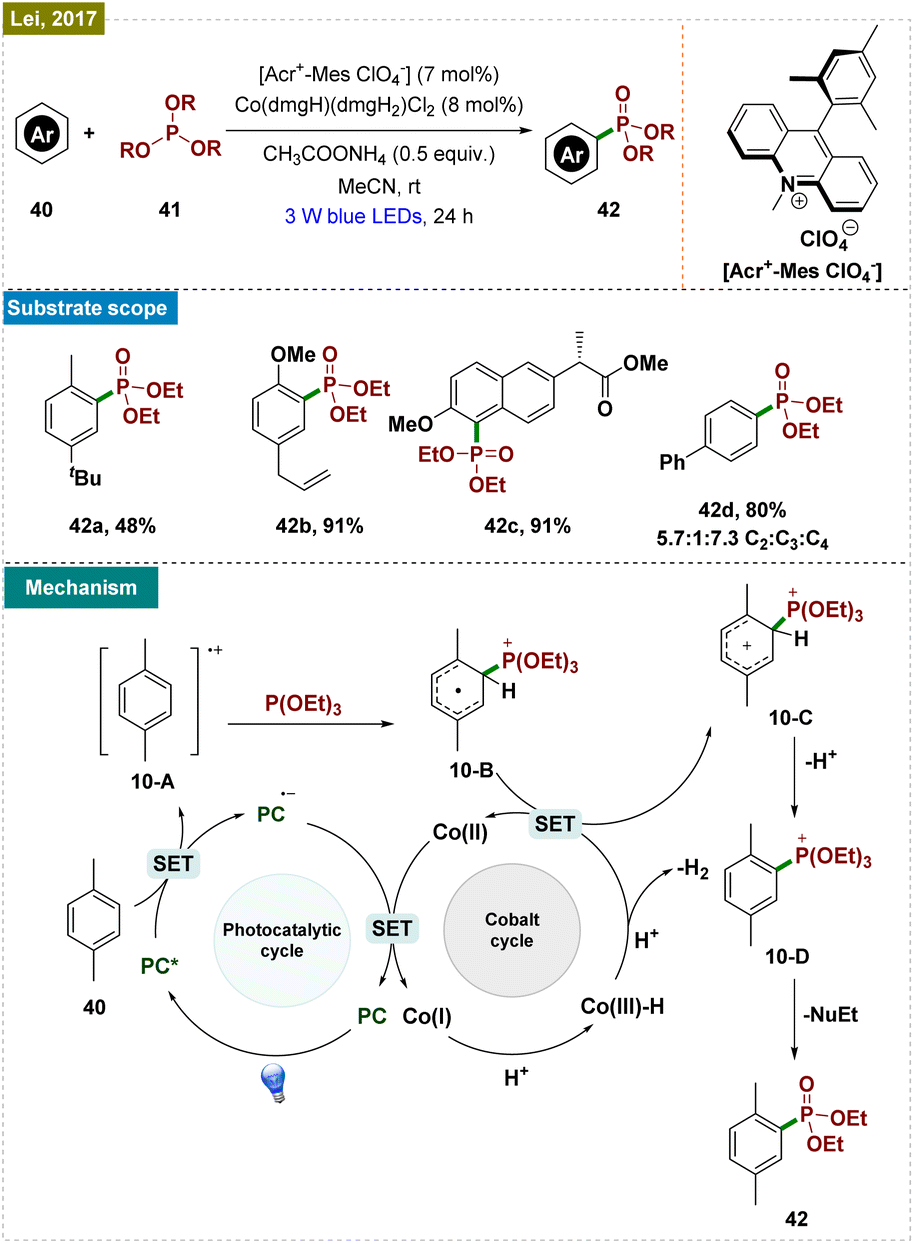 | ||
| Scheme 10 Photoinduced ortho-C–H phosphonylation. | ||
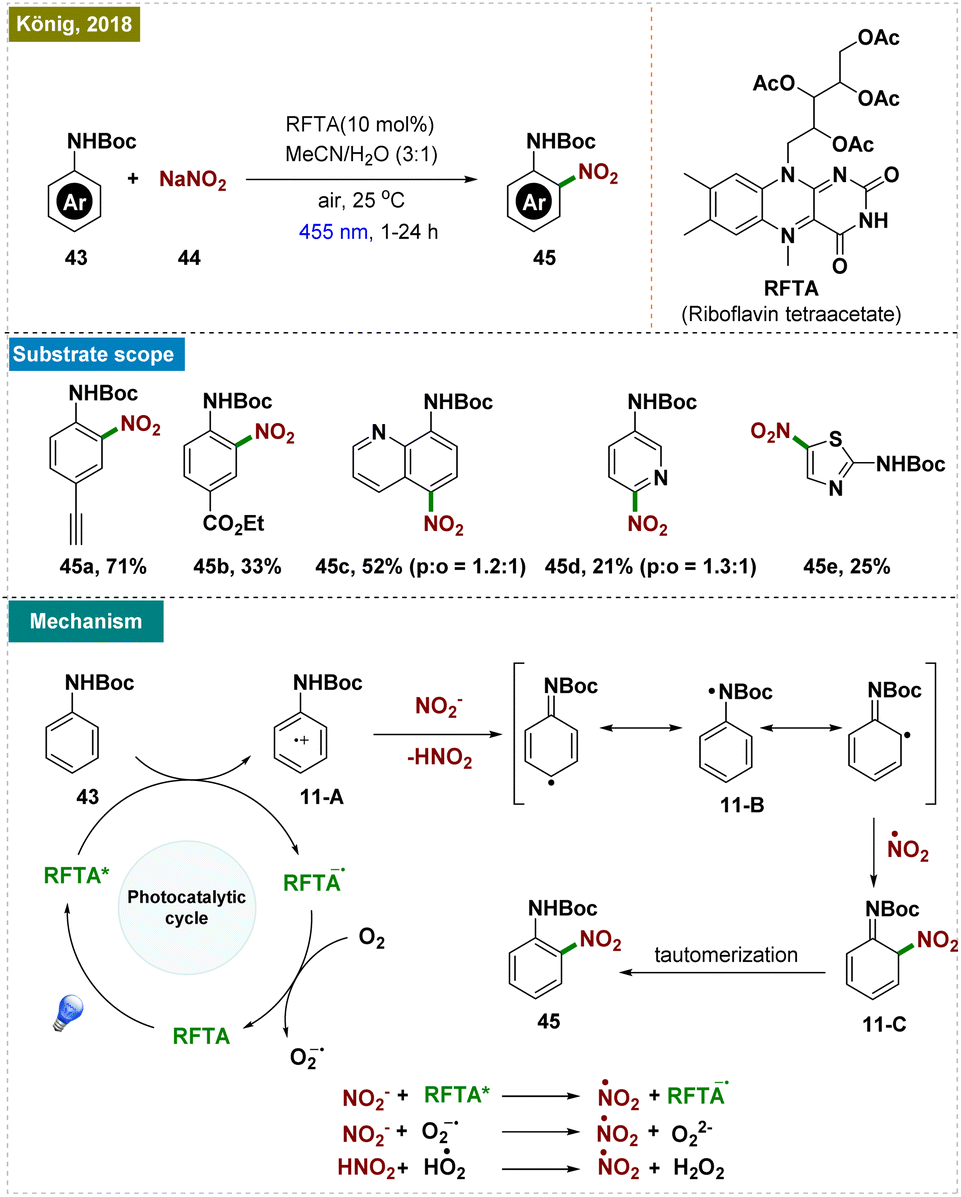 | ||
| Scheme 11 Photoinduced ortho-C–H nitration. | ||
As illustrated in Scheme 11, the reaction mechanism began with the excitation of the photocatalyst, which oxidized the aniline derivative (43), generating the arene radical cation 11-A. Subsequent deprotonation led to the formation of intermediate 11-B, which, upon reacting with a nitrate radical, yielded the desired product via the intermediate 11-C.
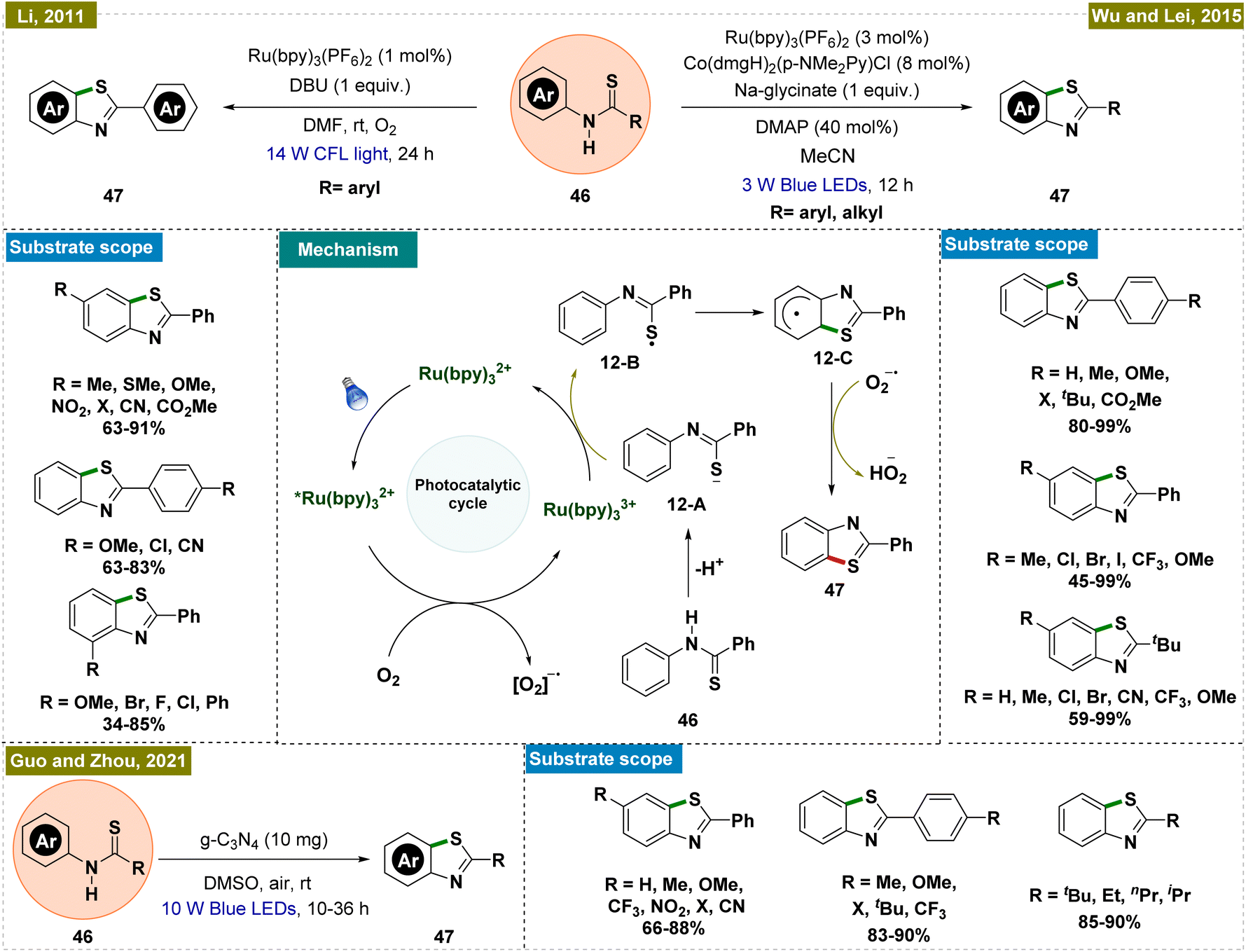
2.2.5.1. Synthesis of benzothiazoles by ortho-C–H thiolation. Benzothiazoles represent a crucial class of compounds with diverse biological and pharmaceutical applications.99,100 Considering this, in 2011, Li and colleagues introduced a mild and efficient methodology for synthesizing benzothiazoles through C–H functionalization, which was notable for its avoidance of direct metal catalysts and its use of visible light-mediated photoredox catalysis (Scheme 12, top left).101 This approach allowed for the effective synthesis of a variety of benzothiazole derivatives, with electron-donating groups showing enhanced reactivity. The reaction employed molecular oxygen as an oxidant and produced water as the sole byproduct, aligning with the principles of green chemistry. Mechanistic insights were gained through various control and kinetic experiments, leading to the proposed mechanism depicted in Scheme 12, center. Upon irradiation with light, the Ru(bpy)32+ complex was excited to the *Ru(bpy)32+ state. This excited photocatalyst was subsequently oxidized by molecular oxygen, producing the Ru(bpy)33+ species and a superoxide radical anion (O2˙−) via a single electron transfer (SET) process. Concurrently, the thioanilide substrate (46) was deprotonated to form an anionic intermediate (12-A), which was then reduced to a sulfur-centered radical (12-B) through SET with the oxidized Ru(bpy)33+, thereby regenerating the Ru(bpy)32+ catalyst and completing the photoredox cycle. The sulfur-centered radical underwent cyclization via a nucleophilic attack on the benzene ring, forming a key intermediate (12-C). This intermediate subsequently transferred a hydrogen atom to the O2˙− radical anion, facilitating rearomatization and resulting in the formation of the final benzothiazole product.
 | ||
| Scheme 12 Synthesis of benzothiazoles by photoinduced ortho-C–H thiolation. | ||
Wu and Lei subsequently further advanced the protocol by developing an external oxidant-free dual photoredox catalytic system for synthesizing benzothiazoles,102 marking a significant improvement over previous methods that often led to undesirable side products and necessitated the use of oxidants (Scheme 12, top right).103,104 Their approach was notably more efficient, producing only hydrogen as a byproduct and proceeding under oxidant-free conditions, although a base was required for the reaction. For the synthesis of 2-aryl-substituted benzothiazoles, a mild base such as sodium glycinate was sufficient, whereas the synthesis of 2-alkyl-substituted benzothiazoles required a stronger base like tetrabutylammonium hydroxide (TBAOH). Mechanistically, this method differed from Li's protocol,101 as it did not rely on molecular oxygen as the oxidant. Instead, the cobalt catalytic cycle played a crucial role in regenerating the photocatalyst and forming the cationic complex, facilitating the desired transformation.
Building on the principles established by Li's group,101 Guo and Zhou's team recently developed a novel heterogeneous catalytic system for intramolecular ortho-C–H thiolation to synthesize benzothiazoles (Scheme 12, bottom).105 This method employed a metal-free, heterogeneous photocatalyst based on graphitic carbon nitride (g-C3N4), enabling the efficient synthesis of a wide array of 2-substituted benzothiazoles, accommodating both aryl and alkyl substituents. The reaction proceeded under ambient air without the necessity of an additional base or oxidant. The simplicity of the system, coupled with the stability and reusability of the g-C3N4 catalyst—up to five cycles—made this approach highly practical and provided additional advantages over previous reports. Mechanistic and control studies suggested that substrate absorption onto g-C3N4 played a crucial role in facilitating the photocatalytic cycle and subsequent product formation, underscoring the catalyst's effectiveness in this transformation.
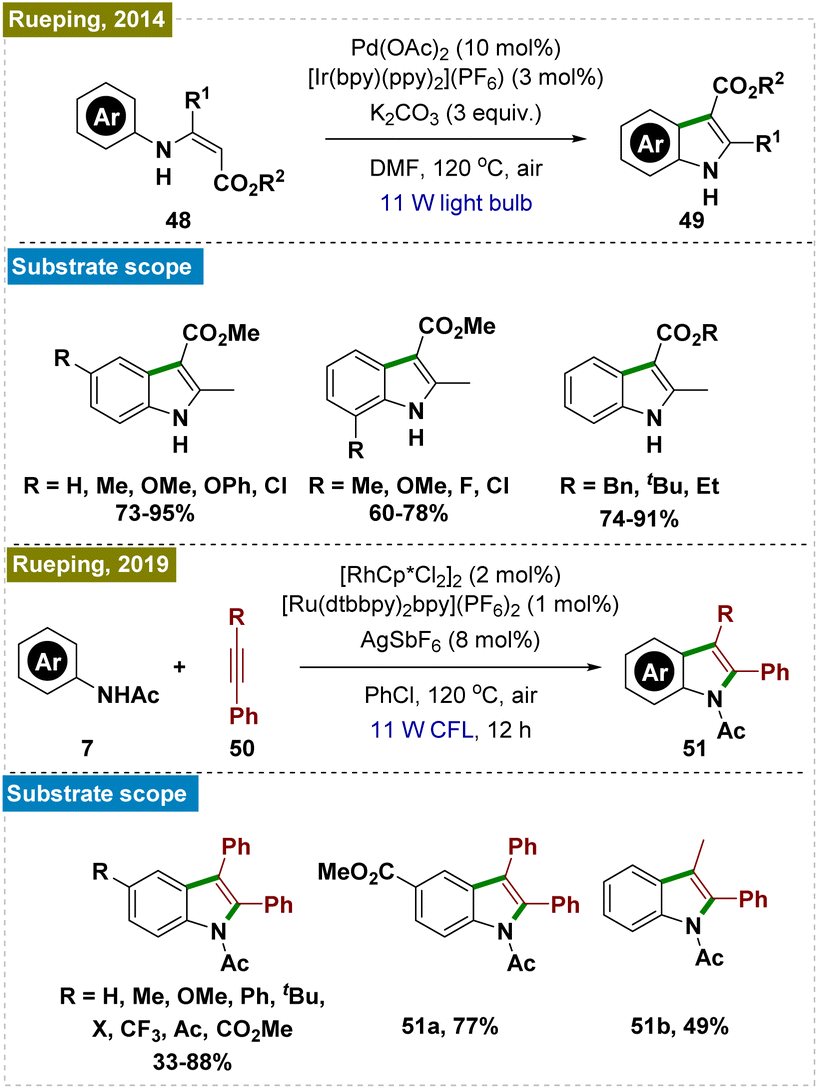
2.2.5.2. Synthesis of indoles and coumarins by ortho-C–H cyclization. Indoles and coumarins are pivotal compounds found in a variety of bioactive natural products, which has spurred significant research into the efficient construction of these important scaffolds.106–109 In this context, Rueping and his team developed a dual photocatalytic system for synthesizing a broad range of indoles through the intramolecular cyclization of aromatic enamines (Scheme 13, top).110 Although high temperature was still necessary, they successfully overcame the necessity of excess Cu(OAc)2—commonly used as an oxidant in earlier methods for regenerating active Pd catalysts—by employing a catalytic amount of an Ir-based photocatalyst.111–113
 | ||
| Scheme 13 Synthesis of indoles by ortho-C–H cyclization. | ||
Subsequently, the same group introduced an alternative protocol for the synthesis of indoles via the cyclization of substituted anilides with alkynes (Scheme 13, bottom).114 To avoid the use of copper salts as oxidants, they used a Ru photocatalyst in conjunction with a Rh catalyst.115,116 Furthermore, they demonstrated that the reaction could be performed by using a heterogeneous inorganic photocatalyst (BiVO4) instead of the Ru photocatalyst. Mechanistic investigations indicated that the photoredox catalysis process proceeded independently of C–H activation, underscoring the crucial role of photoredox catalysts in generating superoxide radicals, which drove the reaction forward. This work highlighted the advances in indole synthesis and the broader potential of photoredox catalysis in organic synthesis.
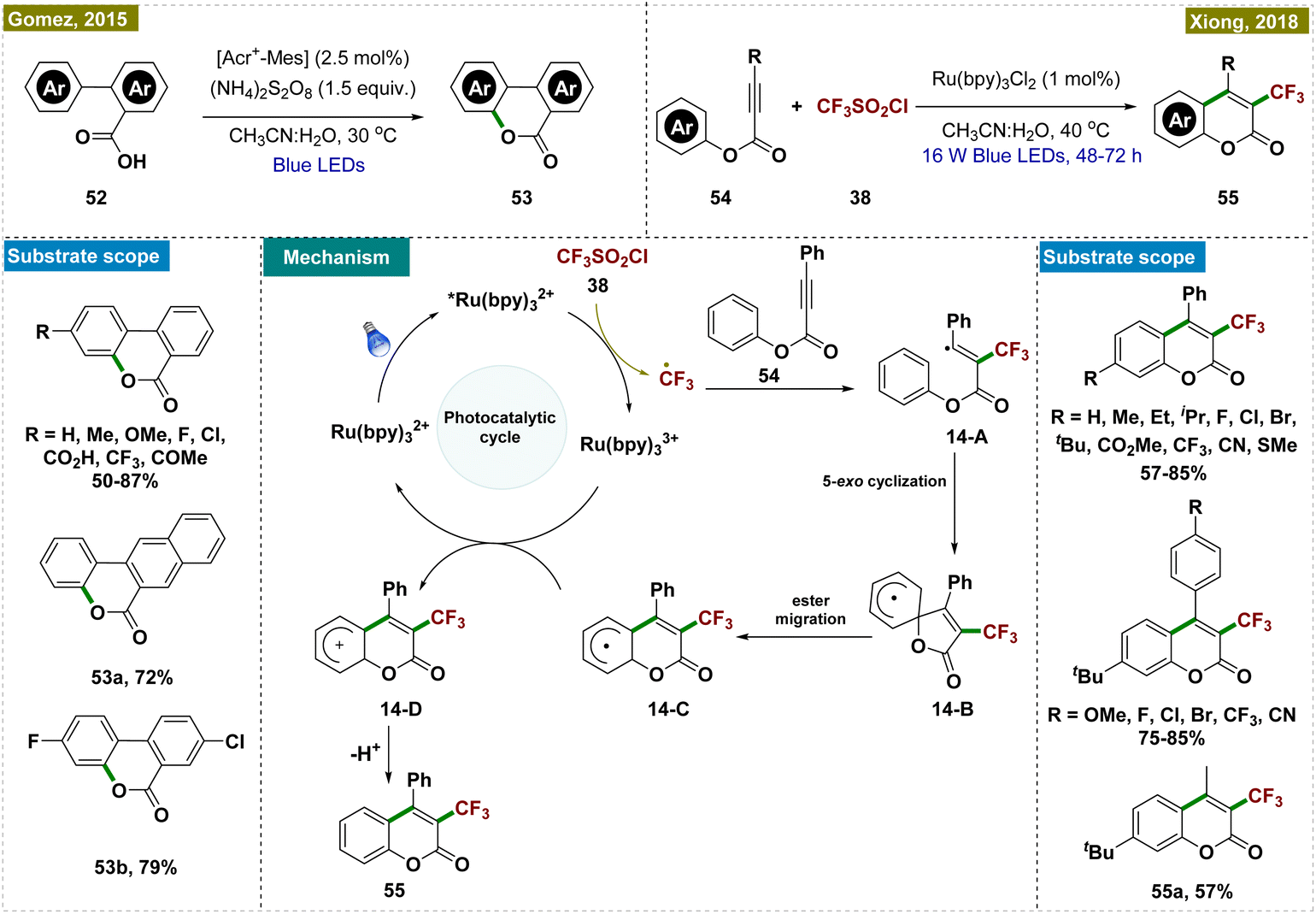
Gonzalez-Gomez and co-workers developed a novel protocol for synthesizing benzo-3,4-coumarins,117 a crucial intermediate in the synthesis of various natural products, which has been previously synthesized following harsh reaction conditions.24,118,119 Their approach utilized an organophotocatalyst, [Acr+-Mes], in combination with an excess of ammonium persulfate ((NH4)2S2O8) as the oxidant to facilitate the dehydrogenative lactonization of aryl benzoic acids (Scheme 14, top left). Crucially, no product formation was observed in the absence of either light or the photoredox catalyst, highlighting the indispensable role of photocatalysis in this transformation. Further mechanistic studies, including kinetic studies, were conducted to elucidate the mechanism.
 | ||
| Scheme 14 Synthesis of coumarins by ortho-C–H cyclization. | ||
Later, Xiong and co-workers developed an alternative protocol for the synthesis of 3-trifluoromethyl coumarins via a visible light-promoted cascade radical cyclization, employing CF3SO2Cl and Ru(bpy)3Cl2 as photocatalysts (Scheme 14, top right).120 This method offered milder reaction conditions compared to previous approaches that required more stringent conditions.121 Through various control experiments and mechanistic studies, the authors proposed a detailed reaction mechanism (depicted in Scheme 14, center). The process began with a single electron transfer (SET) reduction of triflyl chloride (38), coupled with the oxidation of *[Ru(bpy)3]2+ to [Ru(bpy)3]3+ under the irradiation of visible light, generating a trifluoromethyl radical. This radical then underwent electrophilic addition to p-methylphenyl 3-phenylpropiolate (54), forming intermediate 14-A, which subsequently transformed into radical 14-B through a 5-exocyclization. The next step involved the conversion of radical 14-B into radical 14-Cvia a 1,2-ester migration, a process previously observed in the literature.
While authors have documented the formation of 14-C through 5-exo-cyclization involving intermediate 14-B, an alternative phenomenon may occur, allowing the direct conversion of 14-A into 14-Cvia 6-exo-dig cyclization. Radical intermediate 14-C then underwent a second SET event with the strongly oxidizing Ru3+ photocatalyst, resulting in cationic intermediate 14-D. Finally, a proton was released and rearomatization yielded the desired product.
2.2.5.3. Carbazole synthesis by intramolecular C–H amination. The synthesis of carbazoles, which are biologically significant molecules, traditionally necessitates high temperatures and the use of stoichiometric amounts of oxidants.122–124 To address these limitations, Cho and his team developed a dual photoredox-catalyzed intramolecular C–H amination of N-substituted 2-amidobiaryls, utilizing a combination of an Ir-based photocatalyst and Pd(OAc)2 under aerobic conditions (Scheme 15).125 This method was proved to be effective for synthesizing a diverse array of carbazole derivatives with various substituents. Notably, one of the carbazoles synthesized through this protocol was further transformed into the alkaloid clausine C in a single deprotection step.
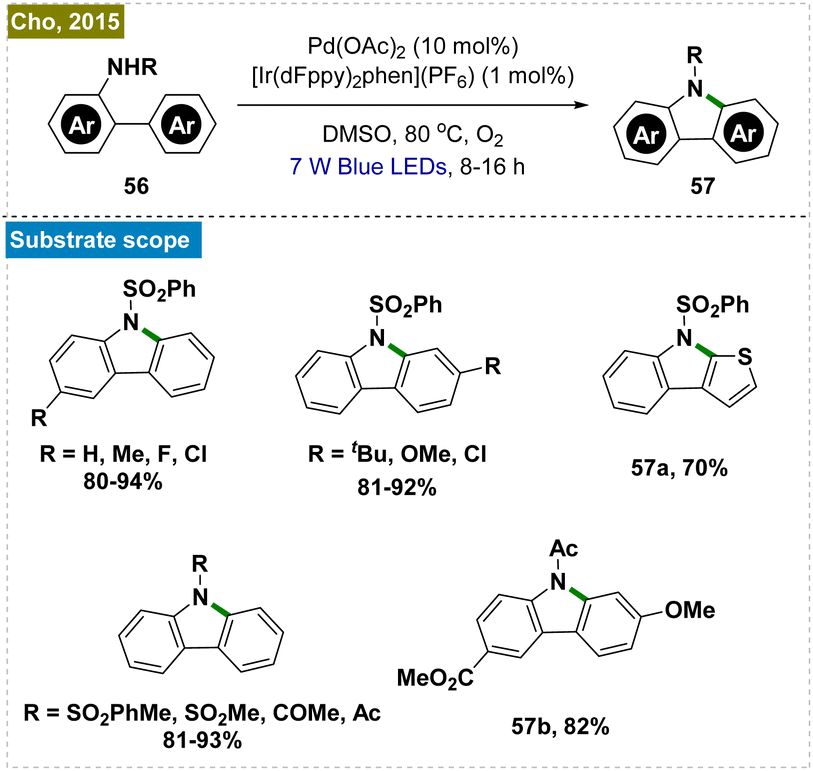 | ||
| Scheme 15 Synthesis of carbazoles by intramolecular ortho-C–H amination. | ||
3. Meta-selective C–H functionalization
Meta-selective C–H functionalization has been relatively underexplored compared to the well-established ortho- and para-functionalization strategies. This discrepancy arises from the intrinsic electronic properties of the benzene ring. In electron-deficient arenes, the meta-position becomes more reactive, but activating these electron-poor substrates remains challenging, particularly in the context of photocatalysis, where their lower reactivity presents a significant obstacle. Recent advances have sought to overcome these limitations through the development of novel methodologies for meta-selective C–H bond activation.Approaches such as template-directed functionalization and σ-activation have shown promise in achieving meta-selectivity.1 These methods exploit weak interactions and strategic catalyst design to direct reactivity to the meta-position, bypassing the inherent electronic preference for ortho- and para-functionalization. Despite the progress, the field of meta-selective C–H functionalization is still in its early stages and further innovation is required to broaden its applicability and efficiency.
3.1. σ-bond activation strategy
A promising approach for achieving meta-C–H functionalization involved the use of ruthenium catalysts through arene σ-activation, particularly in substrates with azine directing groups.126 The key mechanistic feature of this approach was the formation of a ruthenium metallacycle intermediate, which preferentially facilitated radical addition at the position para to the Ru–C bond, thereby enabling selective functionalization at the meta-position. The initial developments by Frost127 and Ackermann128 demonstrated the versatility of Ru-catalyzed meta-functionalization across a variety of transformations, including sulfonylation, alkylation, halogenation, nitration, and carboxylation.129–131 However, these processes typically required elevated temperatures, which, while effective, limited the compatibility with sensitive functional groups and thus emphasized the necessity of developing methods that can be performed under milder reaction conditions.To address this limitation, a novel photochemical strategy was independently developed by the groups of Ackermann and Greaney in 2019, enabling Ru-catalyzed meta-C–H alkylation of 2-aryl pyridines under photocatalytic conditions at room temperature (Scheme 16).132,133 Both groups employed the [RuCl2(p-cymene)]2 catalyst, which formed a cyclometalated ruthenium complex (16-B) through reversible C–H ruthenation of 2-phenylpyridine. Upon irradiation of light, the Ru(II) complex was photoexcited and acted as a single-electron donor, reducing the alkyl halide and forming an oxidized Ru(III) complex (16-C). The resulting alkyl radical then underwent a para-selective addition, followed by intramolecular ligand-to-metal charge transfer (LMCT), re-aromatization, and protodemetalation, yielding the meta-functionalized product and regenerating the Ru-catalyst. Unlike in thermal catalysis, where the ruthenium metallacycle functioned as a nucleophile, the oxidized form under photochemical conditions served as a radical acceptor, driving distinct reactivity and expanding the reaction scope. This photochemical approach accommodated a broader range of functional groups, including tertiary and secondary unactivated alkyl bromides and synthetically valuable α-bromo esters.
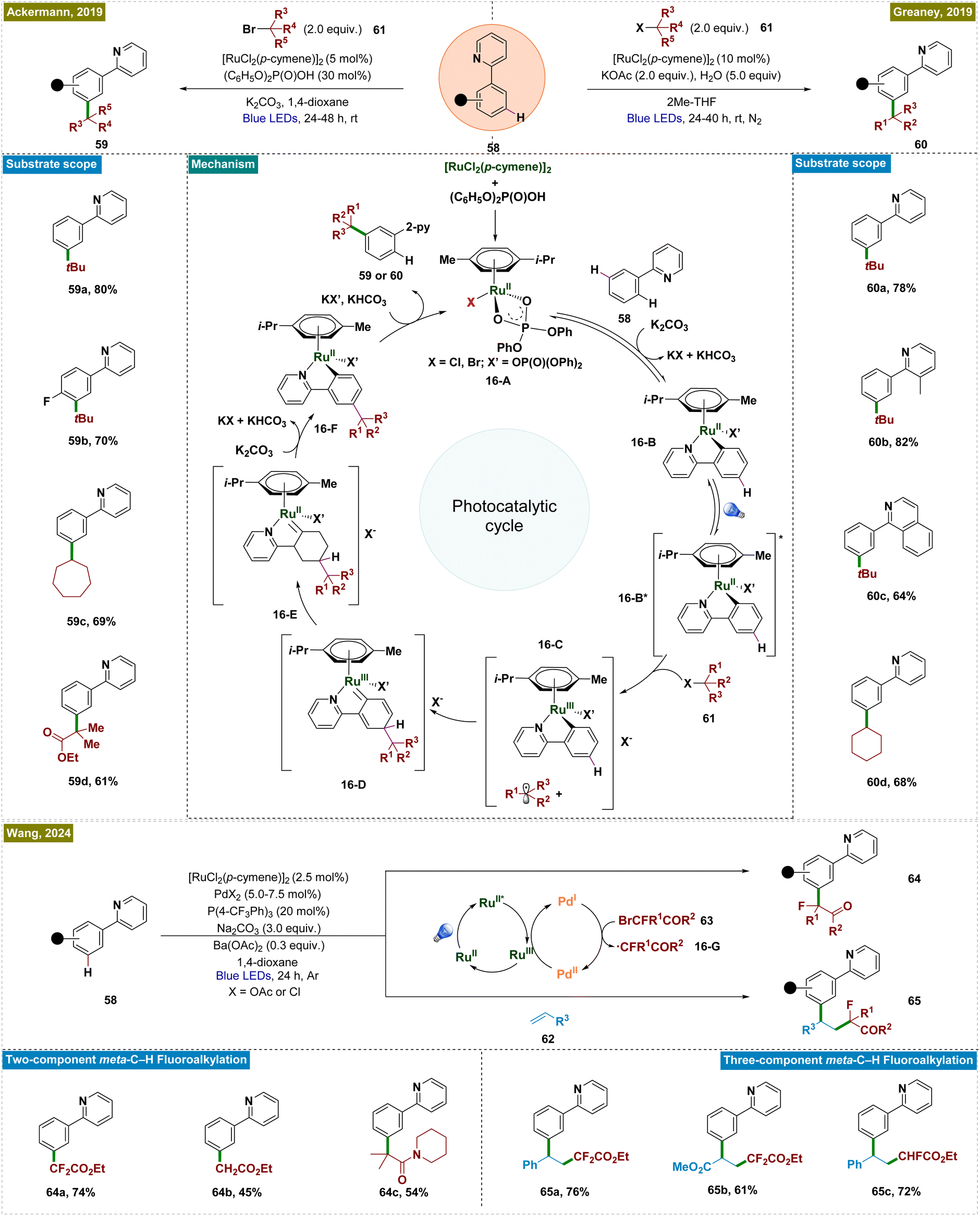 | ||
| Scheme 16 Photoinduced ruthenium-catalyzed meta-C–H alkylation with alkyl halides. | ||
Several mechanistic investigations were conducted by the groups of Ackermann133 and Greaney,132 including on/off experiments, cyclic voltammetry, and radical trapping, providing evidence that the photocatalytic reaction proceeded via single-electron transfer from the photoexcited ruthenium metallacycle, leading to the formation of alkyl radical intermediates. These investigations established the reaction's light dependency, with a relatively low quantum yield of 1.6%, and demonstrated that radical scavengers significantly inhibited the process. Cyclic voltammetry revealed a reversible oxidation event with an oxidation peak at 0.98 V in DCE, further supporting the proposed mechanism. Wang et al. recently extended this approach by developing a visible-light-mediated bimetallic catalysis system for meta-fluoroalkylation of arenes, utilizing a ruthenium–palladium dual catalyst strategy (Scheme 16).134 In this system, ruthenium served as both the directing group and the photoredox catalyst, while palladium was responsible for generating fluoroalkyl radicals. This methodology not only achieved high regioselectivity in alkylation but also exhibited broad functional group tolerance under mild reaction conditions. It is applicable to both two-component aromatic meta-fluoroalkylation reactions and three-component olefin difunctionalization, enabling the construction of complex molecular architectures. Mechanistic studies have clarified that ruthenium mediated both C–H activation and photoredox processes. Concurrently, the palladium cocatalyst facilitated the single-electron transfer (SET) process with bromofluoroalkanes (63), generating fluoroalkyl radicals (16-G) via Pd(0) or Pd(I) species. The distinct roles of ruthenium and palladium in this dual catalytic system worked efficiently to drive the catalytic cycle for meta-alkylation of arenes.
In general, meta-functionalization reactions have relied on substrates that generate radicals via SET with Ru catalysts, limiting the scope of meta-arene C–H bond functionalization. In the field of remote C(sp3)–H functionalization, the hydrogen atom transfer (HAT) process has emerged as a complementary and highly effective approach to the established cyclometallation and metal carbene pathways.135 This process is typically performed under mild reaction conditions, offering high efficiency and regioselectivity, with a preference for tertiary C–H sites over secondary and primary ones. In 2022, Liang's group introduced a novel strategy that integrated ruthenium-catalyzed meta-C–H functionalization with an aliphatic HAT process (Scheme 17).136 This method enabled the regioselective cross-dehydrogenative coupling of dual remote C–H bonds, specifically targeting inert γ-C(sp3)–H bonds in amides and meta-C(sp2)–H bonds in arenes, thereby facilitating the construction of meta-alkylated arenes. This new catalytic approach bypassed the reliance on SET with Ru catalysts, broadening the scope of meta-arene C–H bond functionalization reactions. In this process, Ir(ppy)3 acted as a photocatalyst, which upon irradiation, transitioned to its excited Ir(III)* state. This state initiated a SET event to the hydroxamide substrate (69), causing N–O bond cleavage and forming a nitrogen-centered radical alongside Ir(IV). This radical underwent intramolecular 1,5-HAT, generating a carbon-centered radical (17-E). Simultaneously, the Ru(II) complex (17-A), produced by C–H ruthenation of the arene, was oxidized by the Ir(IV) species, regenerating Ir(ppy)3 and forming a Ru(III) complex. The carbon-centered radical (17-E) then selectively added to the C–H bond para to the Ru(III) complex, finalizing the reaction. This process successfully functionalized primary, secondary, and tertiary C(sp3)–H bonds in amides, and it exhibited broad compatibility with a wide range of arenes, including natural products.
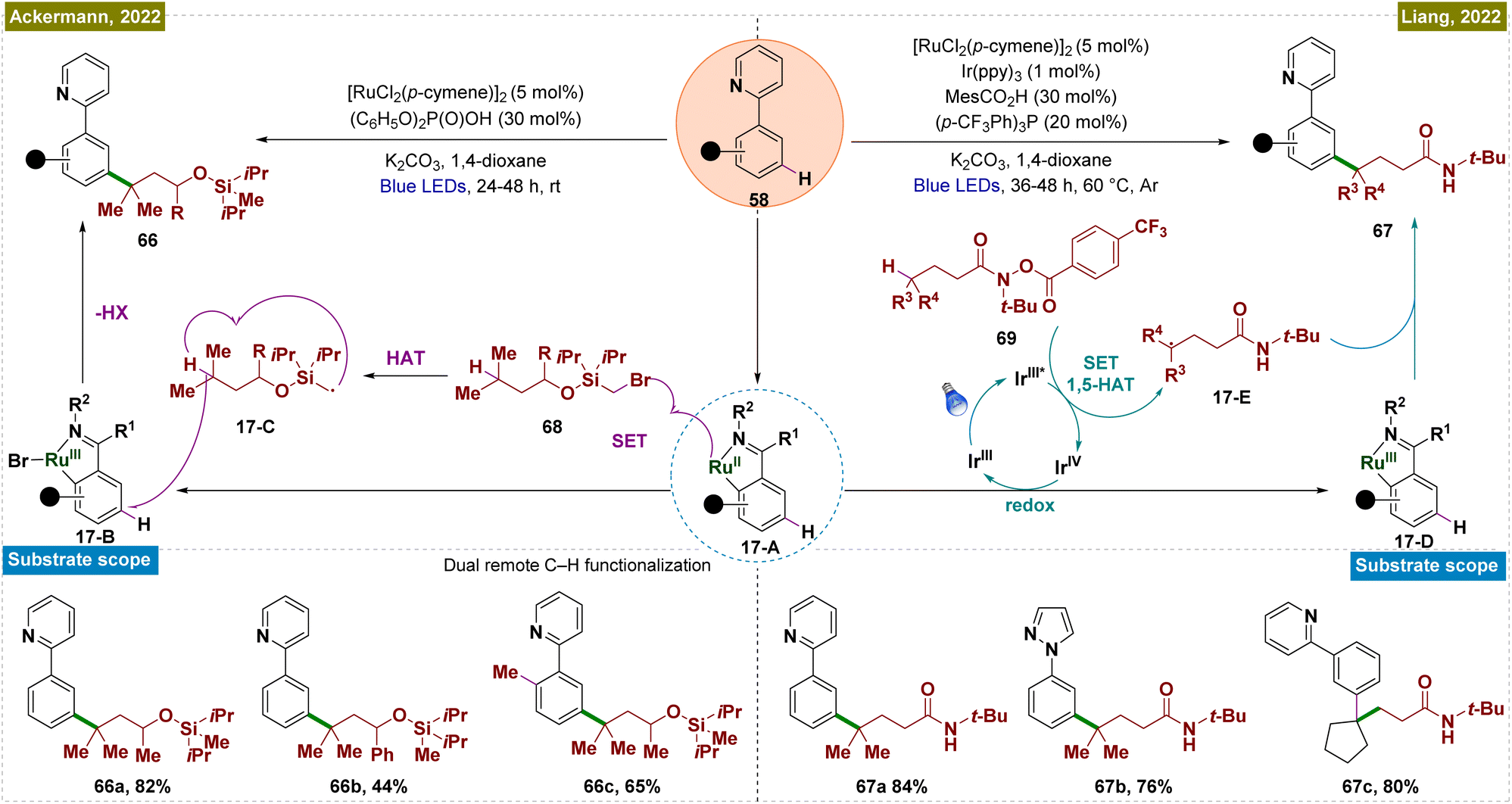 | ||
| Scheme 17 Site-selective coupling of remote C(sp3)–H/meta-C(sp2)–H bonds enabled by a Ru catalyst. | ||
In the same year, Ackermann and co-workers reported transition metal-catalyzed double remote C(sp2)–H/C(sp3)–H functionalization via a radical relay strategy, employing a [RuX2(p-cymene)]2 catalyst (Scheme 17).137 In this innovative approach, the Ru catalyst simultaneously activated the meta-C(sp2)–H bond of arenes and the remote C(sp3)–H bond of alkanes in a single operation, eliminating the necessity of an additional photocatalyst. The mechanism began with the formation of a Ru(II) metallacycle complex (17-A), followed by a SET between the metallacycle and the alkyl substrate (68), generating a Ru(III) intermediate (17-B) and a silyl methyl radical (17-C). This radical subsequently underwent a HAT process to form a more stable carbon-centered radical, which then added to the para-position relative to the C–Ru bond. Mechanistic studies further revealed that continuous irradiation of light was not required throughout the entire reaction but was essential in the initial phase to activate the [RuX2(p-cymene)]2 precatalyst, forming the catalytically active Ru(II) complex. The general applicability of this method was demonstrated through gram-scale synthesis, confirming its scalability. Additionally, the combination of dual remote C–H functionalization with Ru-catalyzed ortho-C–H functionalization was shown to be feasible in a one-pot process, underscoring the versatility and practicality of this approach.
3.2. Template assisted meta-C–H bond functionalization
Despite significant advancements in visible-light-mediated Ru-catalyzed meta-functionalization, these transformations consistently depended on the formation of a ruthenium metallacycle intermediate, which constrained the reaction scope. A potential solution involved direct releasing of the reactive metal catalyst near the targeted remote C–H bond. However, this strategy required the formation of a larger macrocyclic pretransition state, resulting in a thermodynamically unfavorable cyclometalated intermediate. To overcome these obstacles, Yu and co-workers introduced an end-on template with a linear nitrile functionality as the directing group.138 This design employed a long tethering fragment and a rigid, linear nitrile that effectively positioned the Pd-catalyst close to the meta-C–H bond, thereby minimizing the competing ortho-palladation, which was typically associated with significant ring strain due to the small cyclic pretransition state. Building on this concept and their prior success with nitrile-based templates for meta-C–H functionalization, Maiti et al. developed an efficient and selective photoinduced meta-oxygenation of substrates such as phenylacetic acids, biaryl acids, and alcohols through the synergistic combination of photoredox and Pd catalysis (Scheme 18).139 Unlike thermal catalysis, which demands elevated temperatures and excess acetylating agents for optimal selectivity, this catalytic system operated under mild conditions. The mechanism initiated with the weak Pd–nitrile interaction, positioning the Pd–ligand complex near the meta-C–H bond. Subsequently, photoexcitation activated the C–H bond, forming a palladacycle intermediate (18-A). The eosin Y-assisted reduction of PhI(OAc)2 (71) generated an acetoxy radical (18-B) that coordinated with this intermediate, leading to a Pd(III) complex. Subsequent oxidation by eosin Y produced a Pd(IV) intermediate, which underwent reductive elimination to yield the meta-acetoxylated product (72) and regenerated the Pd(II) catalyst. Radical quenching experiments and EPR analysis confirmed the radical nature of the process.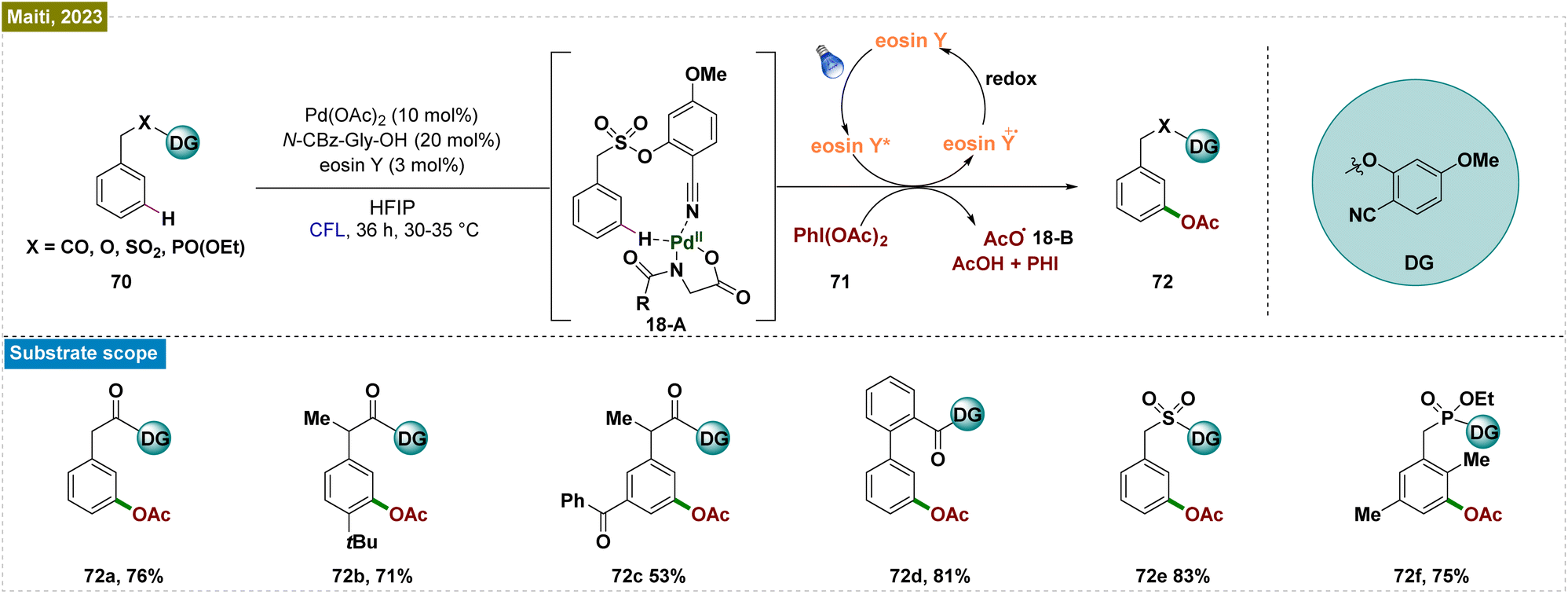 | ||
| Scheme 18 Photoinduced template mediated Pd-catalyzed meta-oxygenation of arenes. | ||
The use of directing groups is a widely adopted and critical strategy for achieving meta-selective functionalization of C–H bonds in arenes. However, the requirement for prefunctionalization and postfunctionalization steps restricted the substrate scope and rendered these processes step intensive. To overcome this limitation, Ohmiya et al. developed a method combining N-heterocyclic carbene (NHC) and organic photoredox catalysis for meta-selective acylation of electron-rich arenes, eliminating the need for directing groups (Scheme 19).140 The process began with SET oxidation of the arene (73), forming a radical cation. Simultaneously, the NHC catalyst (N) reacted with acyl imidazole (74) to generate an acyl azolium intermediate and an imidazolide anion (LG). This anion acted as a nucleophile, creating a cyclohexadienyl radical (19-A) that enabled meta-selective acylation. Subsequent SET reduction of acyl azolium gave the ketyl radical (19-B) and closed the photoredox cycle. The reaction was completed by radical–radical coupling of the cyclohexadienyl radical with the ketyl radical and rearomatization. This catalytic system efficiently functionalized a variety of mono- and disubstituted electron-rich arenes, including acenes, using a broad range of carboxylic acid derivatives.
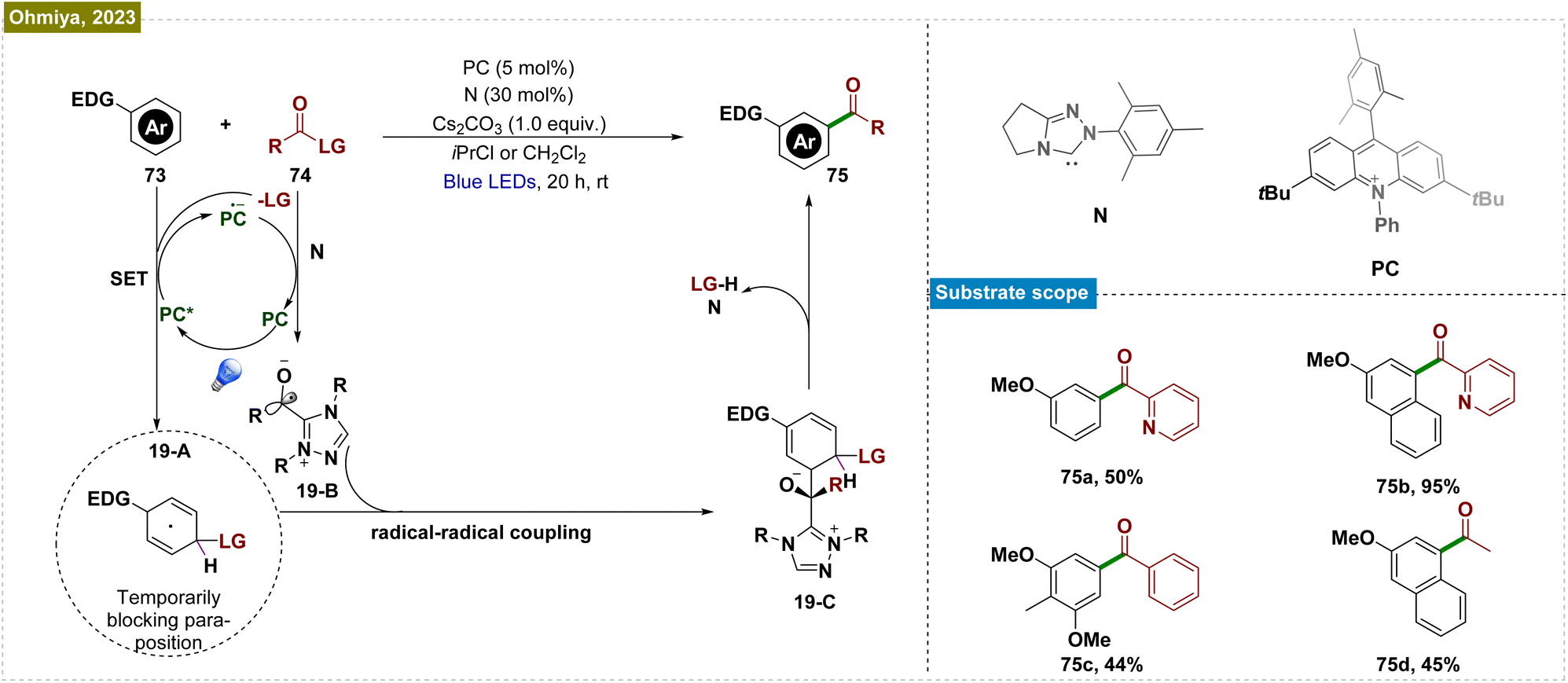 | ||
| Scheme 19 NHC and organic photoredox-catalyzed meta-selective acylation of electron-rich arenes without directing group assistance. | ||
4. Para-selective C–H functionalization
Para-selective C–H functionalization of aromatic compounds is a challenging task due to the difficulty of targeting specific C–H bonds in the symmetrical and reactive nature of the aromatic ring.6,141 The ortho- and meta-positions typically exhibit greater reactivity, making it hard to selectively functionalize the para-position. This challenge arises from a combination of electronic and steric effects, as the para-position is often less activated and harder to distinguish from other positions on the ring.142 However, recent advances in photoredox catalysis offer new strategies to address this issue. Photoredox catalysis uses light-activated catalysts to trigger selective radical or electrophilic processes, providing a gentler and more precise approach to C–H functionalization.143 By carefully designing photocatalysts, researchers can exploit their unique electronic properties to promote para-selectivity.143 This is achieved through catalyst-driven control over reaction mechanisms, which modulate reactivity and regioselectivity by stabilizing key intermediates.143,144 In addition, photoredox catalysis enables the use of mild oxidants and reductants, improving selectivity and tolerance to different functional groups compared to conventional methods.144 The tunability of light as an energy source allows for fine control over reaction conditions, resulting in enhanced selectivity for para-position functionalization in aromatic systems.1434.1. Radical-cation pathway
In recent decades, organometallic cross-coupling has become a cornerstone for constructing intricate aromatic compounds from pre-functionalized precursors.145 Photoredox catalysis offers a direct and efficient approach to arene functionalization, bypassing the necessity of pre-functionalization.23,143,144,146 One of the earliest strategies developed for the regioselective functionalization of phenyl rings involves the oxidation of aromatic compounds, followed by nucleophilic attack by the participating reagent to functionalize the phenyl ring. Fukuzumi is recognized as one of the pioneers in photocatalytic aromatic ring functionalization using this approach. High regioselectivity is typically attributed to: (a) specific interactions between the catalyst and substrate or reagent and substrate, (b) the electronic properties of the substrate, (c) the steric characteristics of the substrate or catalyst, or (d) particular mechanistic pathways.86,147–153In 2011, Fukuzumi and team disclosed photocatalytic bromination of aromatic hydrocarbons by using molecular oxygen and hydrogen bromide, which proceeded efficiently, yielding selectively monobrominated products with 9-mesityl-10-methylacridinium ion (Acr+-Mes) 77 as a photocatalyst under the irradiation of visible light (Scheme 20a).147 The photocatalytic turnover number reached 900, based on the initial concentration of Acr+-Mes. Reactive radical intermediates involved in the photocatalytic cycle were successfully detected by using laser flash photolysis. The reaction mechanism began with photoinduced electron transfer from the mesitylene moiety to the singlet excited state of the acridinium ion, forming the electron-transfer state (Acr˙-Mes˙+). This was followed by electron transfer from aromatic hydrocarbons to the mesitylene radical cation and from the acridinyl radical to O2. The radical cations of the aromatic hydrocarbons then reacted with Br−, selectively producing the monobrominated products.
 | ||
| Scheme 20 Para-selective C–H bond halogenation of arenes. | ||
However, the reaction conditions developed by Fukuzumi were harsh due to the use of corrosive HBr and a high-power xenon lamp, which limited the substrate scope. To overcome this, in 2018, König and colleagues utilized nucleophilic addition to an aromatic radical-cation intermediate for a bromination reaction under the irradiation of visible-light (Scheme 20b).151 In their study, they developed reaction conditions where the oxidation potential of the sodium anthraquinone-2-sulfonate (SAS) photocatalyst 78 increased from 1.8 V to approximately 2.3 V vs. SCE upon the addition of a Brønsted acid. This enhanced photooxidation ability of protonated anthraquinone was then applied for the regio-selective oxidative bromination of electron-rich (hetero)arenes and drugs, yielding good results. The reaction was carried out under mild conditions, which were compatible with various functional groups, including double and triple bonds, ketones, amides, amines, hydroxyl groups, carboxylic acids, and carbamates.
Later in 2020, the same group described a methodology where electron-rich arenes were oxidatively photochlorinated using catalytic amounts of bromide ions, visible light, and 4CzIPN 79 as an organic photoredox catalyst (Scheme 20c).153 From their previous report, they have successfully replaced the stoichiometric oxidant with new and efficient catalytic conditions. In this process, the substrates were first brominated in situ via a photoredox-catalyzed oxidation, followed by a photocatalyzed ipso-chlorination, resulting in the desired compounds with high ortho/para-regioselectivity. Dioxygen acted as a green and efficient terminal oxidant, while the use of aqueous hydrochloric acid as the chloride source minimized the formation of saline by-products.
Moving forward, Nicewicz and co-workers developed several photocatalytic strategies to functionalize the arene C–H bond for the formation of C–N,148 C–C and C–F150,152 bonds by applying this radical-cation pathway (Scheme 21). In all of these cases, the high regioselectivity was attributed to the unique catalytic cycle and the involvement of an electron-rich arene system.
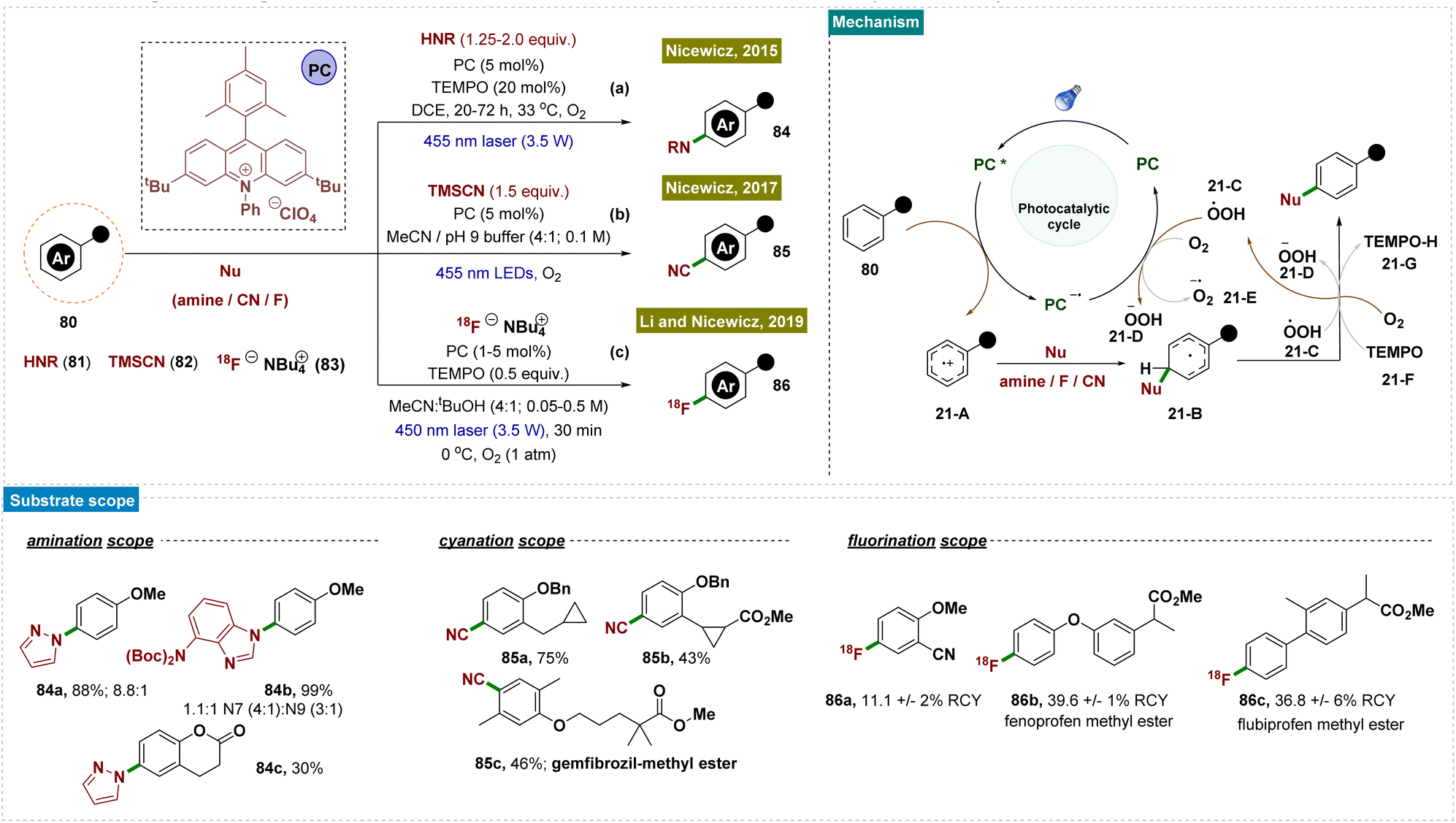 | ||
| Scheme 21 Para-selective C–H bond functionalization of arenes. | ||
In 2015, they developed a photoredox catalyst system comprising an acridinium photooxidant and a nitroxyl radical 21-F that enabled site-selective amination of diverse aromatics with heteroaromatic azoles (as nucleophiles), compounds of significant pharmaceutical interest (Scheme 21a).148 Furthermore, this approach utilized ammonia directly, providing an atom-economical route to anilines without requiring pre-functionalization of the aromatic substrate.
In 2019, a heterogeneous version of photoredox catalysts was designed by the Wang and König groups for the formation of C–N bonds regioselectively with broad tolerance of substrates and high efficiency, by employing molecular oxygen as the terminal oxidant.154 This semiconductor-based photoredox system enabled C–H amination without the necessity of metals, ligands, strong oxidants, or additives, and was recyclable up to 6 cycles, offering a sustainable and efficient approach to C–H functionalization with broad synthetic utility. Mechanistic studies, supported by electron spin resonance (ESR) data, KI-starch tests, and control experiments, identified superoxide anion radicals (O2˙−) and H2O2 as the reactive oxygen species.
In 2017, as follow-up work, Nicewicz and team reported a novel method for synthesizing aromatic nitriles through direct C–H functionalization by using an acridinium photoredox catalyst and trimethylsilyl cyanide 82 (as a nucleophile) under ambient conditions (Scheme 21b).150 This reaction was performed at room temperature and tolerated a wide range of functional groups, including electron-donating and -withdrawing groups, halogens, nitrogen- and oxygen-containing heterocycles, and even aromatic moieties found in pharmaceutical compounds.
Two years later, in 2019, they disclosed a mild process for direct [18F] fluorination of aromatic C–H bonds using an [18F] fluoride salt 83 under photocatalytic conditions. This photoredox-catalyzed approach enabled the synthesis of a diverse range of [18F]-labeled arenes and heteroaromatics, including those relevant to pharmaceutical applications (Scheme 21c).152 These radiolabelled compounds held promise as diagnostic agents and provided valuable insights into the in vivo behaviour of their unlabelled counterparts, as demonstrated in preliminary animal studies. In this context, it is important to note that positron emission tomography (PET) plays a crucial role in drug discovery, development, and medical imaging. However, the lack of efficient and straightforward radiolabeling methods for aromatic C–H bonds presents a significant challenge, hindering progress in PET radiotracer development.
Mechanistically, all these reports followed similar reaction pathways as described in Scheme 21. According to the authors, the mechanism of this reaction was not fully understood and is currently under investigation. They proposed that TEMPO facilitated the aromatization of radical intermediate 21-B through direct H-atom abstraction (Scheme 21). The generation of radical intermediate 21-B was achieved through the trapping of radical-cation species 21-A with a suitable nucleophile (81/82/83). Alternatively, radical 21-B was trapped by O2, forming a 1,3-cyclohexadienyl peroxyl radical, which underwent internal elimination to yield products 84–86 and the hydroperoxyl radical  . As suggested in the literature, O2 oxidized the acridine radical Mes-Acr˙ (Mes, mesityl; Acr, acridinium), regenerating acridinium (Mes-Acr+) and superoxide (O2˙−), although other intermediates, such as HOO˙, might also contribute to catalyst turnover. The highly basic superoxide likely deprotonated intermediate 21-B, followed by hydrogen atom transfer with TEMPO-H, ultimately forming H2O2 and regenerating TEMPO. The observed reduction in unwanted byproducts when TEMPO was present supported the proposed role of TEMPO-H in scavenging reactive oxygen-centered radicals like
. As suggested in the literature, O2 oxidized the acridine radical Mes-Acr˙ (Mes, mesityl; Acr, acridinium), regenerating acridinium (Mes-Acr+) and superoxide (O2˙−), although other intermediates, such as HOO˙, might also contribute to catalyst turnover. The highly basic superoxide likely deprotonated intermediate 21-B, followed by hydrogen atom transfer with TEMPO-H, ultimately forming H2O2 and regenerating TEMPO. The observed reduction in unwanted byproducts when TEMPO was present supported the proposed role of TEMPO-H in scavenging reactive oxygen-centered radicals like  . Although the half-wave redox potential of TEMPO [E1/2 (TEMPO˙/TEMPO+) = +0.62 V vs. Ag/AgCl] suggested possible oxidation by cat+*, the use of 20 mol% TEMPOnium-BF4 yielded similar results to TEMPO in the aryl amination reaction, indicating a common mechanistic intermediate, presumably TEMPO, generated through electron transfer from cat˙ [E1/2 (cat+/cat˙) = −0.47 to −0.58 V vs. SCE] to TEMPOnium. In the absence of cat+, no aryl amine 84 was formed with 20 mol% TEMPO, though trace product formation was observed with 20 mol% TEMPOnium-BF4 when the acridinium photocatalyst was omitted.
. Although the half-wave redox potential of TEMPO [E1/2 (TEMPO˙/TEMPO+) = +0.62 V vs. Ag/AgCl] suggested possible oxidation by cat+*, the use of 20 mol% TEMPOnium-BF4 yielded similar results to TEMPO in the aryl amination reaction, indicating a common mechanistic intermediate, presumably TEMPO, generated through electron transfer from cat˙ [E1/2 (cat+/cat˙) = −0.47 to −0.58 V vs. SCE] to TEMPOnium. In the absence of cat+, no aryl amine 84 was formed with 20 mol% TEMPO, though trace product formation was observed with 20 mol% TEMPOnium-BF4 when the acridinium photocatalyst was omitted.
The direct incorporation of a trifluoromethyl group onto an aromatic ring via a radical pathway has been widely studied,155 but achieving highly para-selective C–H trifluoromethylation of specific arenes has remained challenging.
In 2021, Shi, Zhao, and their team reported a light-promoted, 4,5-dichlorofluorescein (DCFS, 90)-enabled para-selective C–H trifluoromethylation of arylcarbamates using the Langlois reagent 88 (Scheme 22).86 Preliminary mechanistic studies suggested that the activated organic photocatalyst, when coordinated with the arylcarbamate, facilitated the para-selective trifluoromethylation. The method's synthetic significance was underscored by its successful application on a ten-gram scale.
 | ||
| Scheme 22 Para-selective C–H bond trifluoromethylation of arenes. | ||
Mechanistically (Scheme 22), a trifluoromethyl radical 22-F was generated through a known process using 4,5-DCFS 90 as an organic photocatalyst and K2S2O822-C as the oxidant. The oxidation of DCFS formed the key intermediate 22-Avia a single-electron transfer (SET) process, which then underwent tautomerization to produce intermediate 22-B with the activated DCFS. The steric bulk of DCFS increased the hindrance at the ortho- and meta-positions on the phenyl ring, promoting highly para-selective trifluoromethylation. However, the possibility that the free trifluoromethyl radical 22-F reacted directly with the DCFS-activated phenyl ring could not be completely ruled out.
Apart from trifluoromethylation of secondary amines, synthesis of other fluorinated compounds, such as difluoromethylated aromatics, is also in high demand due to their synthetic and biological value.156 Recently in 2024, Guo, Zhao, Huang, and colleagues introduced a green and environmentally friendly modular platform for the sustainable para-difluoromethylation of readily available aromatic amines 95 using a metal-free photocatalytic system (Scheme 23).157 This reaction, for the first time, exhibited excellent functional group tolerance and a broad substrate scope, employing a radical–radical coupling C–H functionalization strategy to efficiently generate a range of valuable difluoromethylated products 97. Comparative experiments revealed that the activating group is crucial for electronic control of regioselectivity and for preventing the decomposition of arylamines. Moreover, the high atom economy achieved in the derivatization of complex molecules underscored the synthetic utility and practicality of this approach.
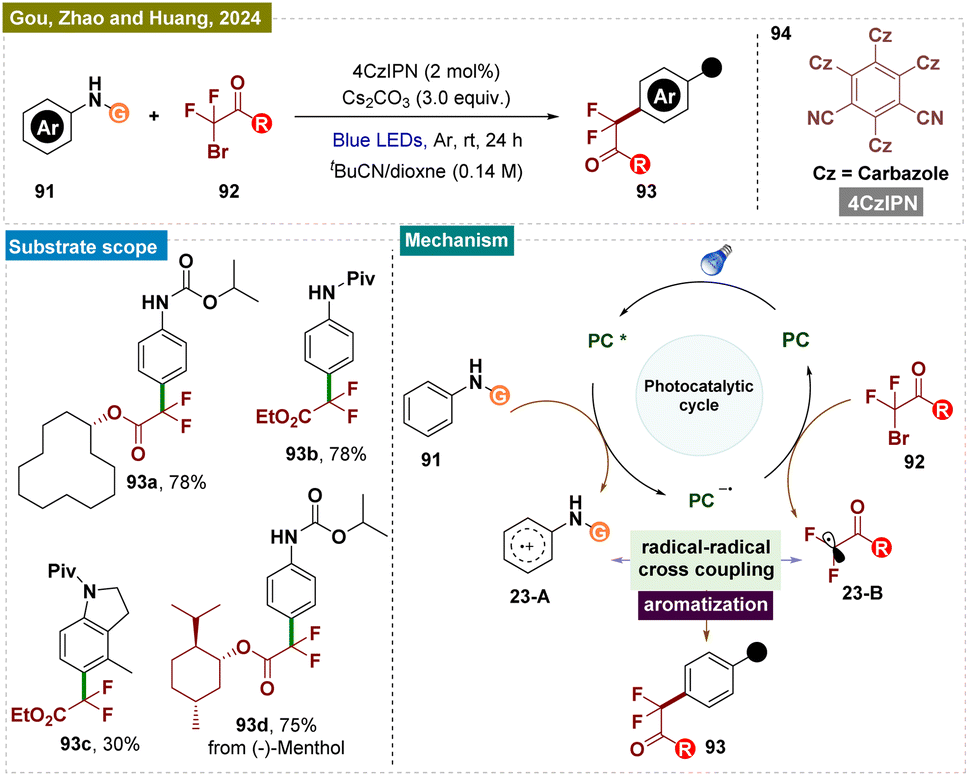 | ||
| Scheme 23 Para-selective C–H difluoromethylation of arenes. | ||
A plausible reaction mechanism was proposed by the authors and is illustrated in Scheme 23. Compound 95 underwent reductive quenching by the excited state 4CzIPN*, generated upon visible light irradiation, leading to the formation of 4CzIPN˙− and a key transient arene radical-cation 23-A (supported by Stern–Volmer emission quenching studies). Subsequently, the reduced photocatalyst 4CzIPN˙− transferred a single electron to alkyl bromide 96, generating the difluoroalkyl radical 23-B and restoring the photocatalyst 4CzIPN. The sp3–sp2 coupling between the ˙CF2CO2Et radical 23-B and arene cation radical 23-A then produced a cationic intermediate, which underwent deprotonation by a base to yield product 97.
4.2. Radical trifluoromethylation
Understanding the importance of aromatic trifluoromethylation158 regioselectively, Zhao and team presented a novel strategy for para-selective C–H trifluoromethylation of benzamide derivatives 91 through iminium activation (Scheme 24).159 This approach facilitated a radical nucleophilic substitution rather than a radical electrophilic substitution, driven by iminium activation, which lowered the lowest unoccupied molecular orbital (LUMO). This method was compatible with a wide range of substrates, producing predominantly para-trifluoromethylated products with high selectivity. | ||
| Scheme 24 Para-selective C–H bond trifluoromethylation of benzamide. | ||
A plausible reaction mechanism was proposed (Scheme 24). Initially, pyridinium complex 24-A and iminium intermediate 24-C are formed in situ.160 Upon irradiation with blue LEDs, the excited-state IrIII* was generated, which reduced complex 24-A to radical species 24-B through a single electron transfer (SET) process.160 The –CF3 radical 24-B, released from radical species 24-A, then underwent nucleophilic addition to iminium intermediate 24-C, forming intermediate 24-D. The selective activation at the para-position of the iminium salt resulted in high para-selectivity, avoiding meta-selectivity.149,161 Another SET event between radical 24-D and IrIV regenerated IrIII, continuing the catalytic cycle and leading to intermediate 24-E. Deprotonation of intermediate 24-E followed by hydrolysis of iminium intermediate 24-E yields the desired product 93.
However, a radical chain mechanism cannot be ruled out. Intermediate 24-D underwent deprotonation to form a radical anion, which directly reduced complex 24-A to radical species 24-B, initiating a radical chain process. While –CF3 radicals kinetically favored addition at the para-position, the exact direction of electron transfer in the transition state remained unconfirmed, requiring further investigation. Another plausible explanation was that cyclohexadienyl radical 24-D was thermodynamically stabilized by delocalization with the iminium group, favoring para-trifluoromethylation. Given the broad applicability of this protocol to various functionalized substrates, it was also likely that radical 24-D was both kinetically preferred and thermodynamically more stable.
4.3. Functionalization of unprotected functional groups bearing a phenyl ring for C–C bond formation
Aromatic substrates with unprotected functional groups present significant challenges due to their delicate nature. Free groups such as –OH and –NH2 often participate in side reactions, making selective functionalization difficult.162 Achieving regioselective functionalization of these substrates typically requires specialized catalytic conditions. In this context, Hwang and colleagues have developed a visible-light-mediated, copper-catalyzed photoredox process that enables direct oxidative coupling of phenols 99 with terminal alkynes 100 at room temperature (Scheme 25).163 This method facilitated para-selective formation of hydroxyl-functionalized aryl and alkyl ketones through the cleavage of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond.
C triple bond.
 | ||
| Scheme 25 Para-selective C–H bond functionalization of unprotected phenols. | ||
Visible-light irradiation of the in situ generated Cu(I)-phenylacetylide produced a long-lived excited state of Cu(I)-phenylacetylide 25-A. This excited state underwent a single-electron transfer (SET) process, donating an electron to O2 and forming the intermediate Cu(II)-phenylacetylide 25-B and a superoxide radical anion, as confirmed by EPR experiments. Given the redox potential of 25-A (−2.048 V vs. SCE in CH3CN) and the strong electron affinity of O2, it was likely that this SET process readily occurred between the photoexcited triplet Cu(I)-phenylacetylide and O2. Simultaneously, phenol 99 was oxidatively converted to benzoquinone (BQ) 25-C by the Cu(II)-superoxo intermediate. Next, the Paternò–Büchi-type [2 + 2] cycloaddition between Cu(II)-phenylacetylide 25-B and benzoquinone (BQ, 26-C) formed a labile Cu(II)-oxetene intermediate 26-D. This oxetene ring underwent rearrangement to produce the Cu(II)-coordinated quinone methide 26-E, which rapidly fragmented to generate an acyl radical intermediate 26-F and Cu(I). Subsequent dark reactions with molecular oxygen (O2) and the abstraction of a phenolic proton resulted in the formation of peracid 26-G. This peracid then underwent radical cleavage, releasing CO2 and producing the hydroxyl-substituted quinone methide 26-H. Finally, keto–enol tautomerism (via aromatization) led to the formation of the aryl ketone (101).
In addition to unprotected phenols, free anilines are also key structural motifs in organic and pharmaceutical chemistry.164 Therefore, functionalizing anilines while preserving the integrity of the –NH2 group is a challenging task, requiring selective reaction conditions to avoid undesired modifications. In 2023, An, Wang, and Li reported a visible light-mediated method for para-alkylation of unprotected anilines 102 using a Ru(II)–aniline complex, achieving remarkable efficiency in just 2 hours under mild conditions (Scheme 26).165 This protocol demonstrated excellent functional group tolerance, enabling late-stage functionalization of natural products and pharmaceuticals. Mechanistic studies indicated that the unique Ru(II)–aniline complex played a crucial role in both initiating the reaction and ensuring para-selectivity.
 | ||
| Scheme 26 Para-selective C–H bond alkylation of unprotected anilines. | ||
Mechanistically (Scheme 26), [RuCl2(p-cymene)]2 first coordinated with aniline 102 to form complex 26-B, which then reacted with alkyl halide 103 to produce intermediate 26-C. Upon exposure to visible light, 26-C was excited to its active state, 26-D. This excited state underwent single-electron transfer (SET) to yield Ru(III) species 26-E and an alkyl radical 26-F. The alkyl radical 26-F attacked the aromatic ring at the position para to the ruthenium, forming intermediate 26-G. Subsequently, a ligand-to-metal charge transfer (LMCT) occurred, generating complex 26-H. Finally, 26-H underwent rearomatization, released the para-alkylated product 104 and regenerated the ruthenium(II) species [RuCl2(p-cymene)]2.
In 2022, the Cho group developed a visible-light-induced method for para-selective C–H functionalization of anilines 105, avoiding N–H insertion, using diazomalonates 106 in the presence of an Ir(III) photocatalyst (Scheme 27).166 The para-selective radical–radical cross-coupling occurred via C-centered radical intermediates generated from both anilines 105 and diazomalonates 106. The reaction pathway for this selective C–C coupling was confirmed through electrochemical and photophysical experiments and computational studies.
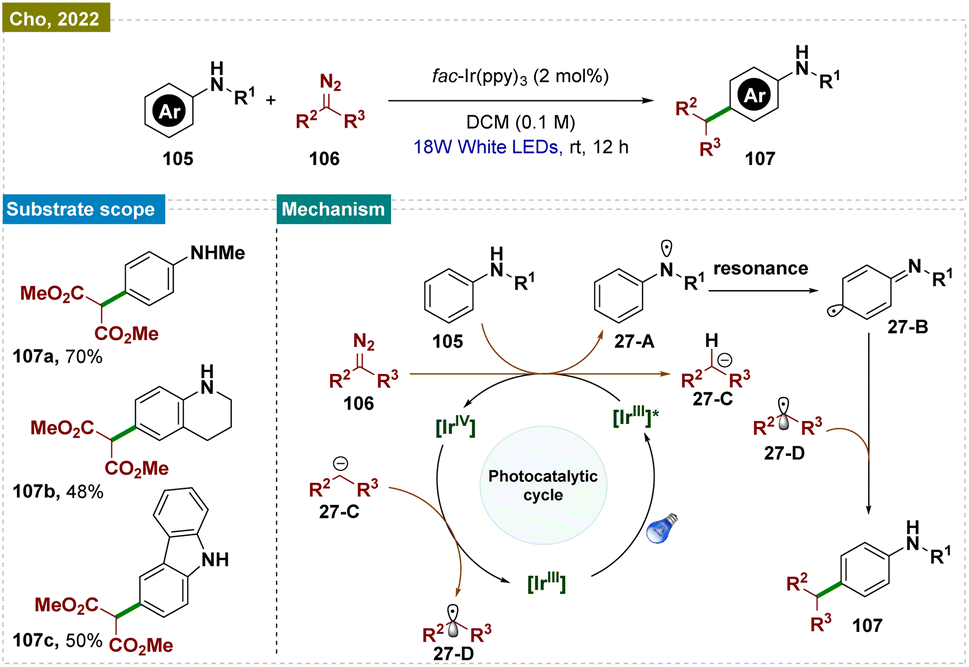 | ||
| Scheme 27 Para-selective C–H bond alkylation of arenes. | ||
Mechanistically (Scheme 27), upon visible-light irradiation, fac-Ir(ppy)3 underwent photoexcitation, resulting in a metal-to-ligand charge-transfer excited state [IrIV]* (E*(IrIV/*IrIII) = −1.58 V in DCM). This excited state was oxidatively quenched via single-electron transfer to 106 (Ered, −1.40 V vs. SCE), producing [IrIV] and the radical anion [106]˙−. Subsequent hydrogen atom abstraction (HAT) from 105 by [106]˙− generated intermediate 27-C, releasing N2 and forming the persistent radical intermediate 27-A, which was stabilized by resonance across the phenyl ring. Meanwhile, 27-C donated an electron to [IrIV], regenerating the ground state [IrIII] and creating the key C-centered radical species 27-D. The reaction was concluded with radical–radical coupling between 27-D·and 27-B, followed by rearomatization, yielding the para-C–C coupled product 107.
4.4. Dual-photocatalytic pathway
In addition to photoredox catalysis, the development of dual catalytic protocols further broadens the scope of reactivity available in organic synthesis.167 Dual-photocatalysis can be classified into two types:168 (a) the combination of photoredox and organocatalysis for mainly asymmetric transformations, and (b) metallaphotoredox, which enables access to odd-electron reactivity in transition metal catalysis, both involving radical species.In 2022, Maiti and colleagues developed a photoredox catalytic system combining palladium with an organo-photocatalyst (PC), enabling oxidative olefination with high regioselectivity across a broad range of arenes and heteroarenes (108) (Scheme 28).169 This system utilized visible light to conduct “regioresolved” Fujiwara–Moritani reactions without the need for silver salts or heat. The catalyst was versatile, accommodating both proximal and distal olefination through the use of specific directing groups (DGs), making it effective for C(sp2)–H olefination across a wide spectrum. The transformation underscored the dual function of visible light as both an oxidant and an activator, facilitating: (a) highly regioselective olefination of (hetero)arenes via a nondirected approach; (b) streamlined synthesis of phenolic natural products; (c) photoinduced atroposelective generation of axial chirality using a chiral ligand; (d) the first distal functionalization via photocatalysis; and (e) late-stage functionalization of biologically relevant molecules. Several control experiments were conducted by the authors to confirm key mechanistic aspects. To investigate whether a radical pathway was responsible for the formation of the dimerization product, the CDC reaction was performed in the presence of radical scavengers, as well as through a radical clock experiment using vinyl cyclopropane. In both cases, the yields remained unaffected, indicating a non-radical pathway. Additionally, the authors demonstrated that the C–H activation step was facilitated by the Pd catalyst and visible light, with the photocatalyst (PC) playing no significant role in this process. Based on the mechanistic investigations, a plausible mechanistic proposal was made (Scheme 28), suggesting that the direct excitation of the in situ generated Pd complex drove the C–H activation step. In line with previous studies, it was proposed that excitation of the photocatalyst allowed for the mild reoxidation of Pd(0) 28-B to Pd(II) 28-A in the presence of oxygen (Scheme 28). As an alternative to the proposed oxidation step, it was also possible that the regeneration of the photocatalyst from its reduced state led to the reduction of molecular oxygen (O2), forming [O2]˙−28-A. In the oxidation step involving Pd(0) 28-B to Pd(II) 28-A, O2 similarly participated in a single electron reduction process, generating [O2]˙−. This radical anion species, being highly short-lived, then rapidly underwent a protonation. The generated Pd(II) complex underwent a C–H insertion reaction at the para-position of the arene, forming intermediate 28-D. This intermediate then reacted with olefin substrates to produce species 28-E, followed by the formation of species 28-F. Subsequently, dehydrogenative elimination from 28-F led to the formation of product 110 and the intermediate LPd-H (28-G), which readily converted to Pd(0) (28-B) through the elimination of L-H.
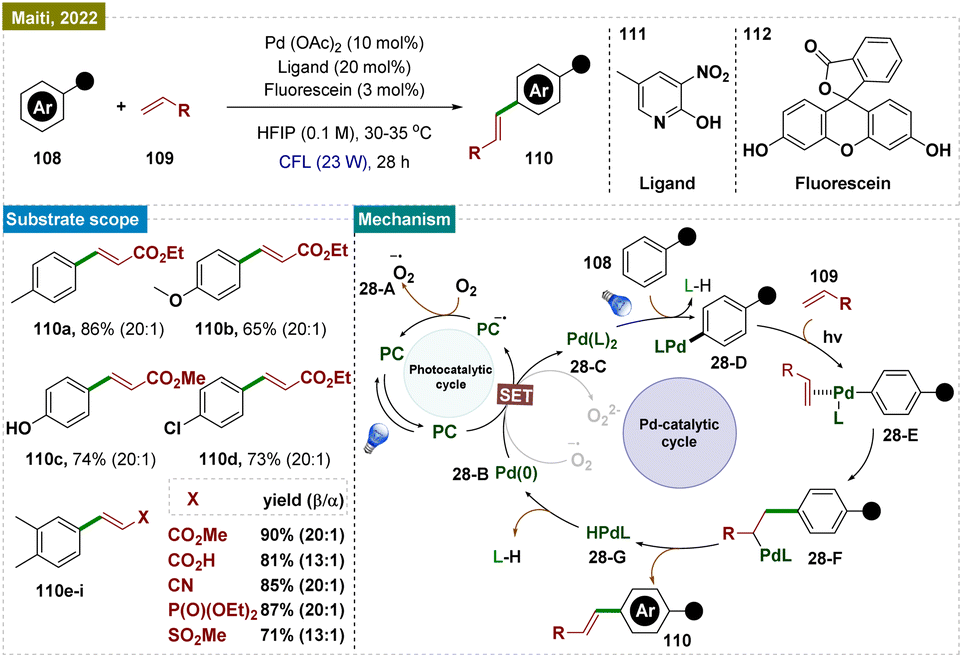 | ||
| Scheme 28 Para-selective C–H bond alkenylation of arenes. | ||
Distinct from Nicewicz's report,148 in 2023 Huang, Zhao and their team reported a novel method for para-selective C–H amination of aryl oximes 113 using transient steric control (Scheme 29).170 A preliminary mechanistic investigation identified an interaction between 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) 116 and the oxime 113, which activates the aromatic ring. Simultaneously, TPT 116 creates steric hindrance at the ortho- and meta-C–H bonds, directing the pyrazole radical to selectively react at the less hindered para-position of the phenyl ring. This approach offers a new strategy for achieving para-selective C–H functionalization.
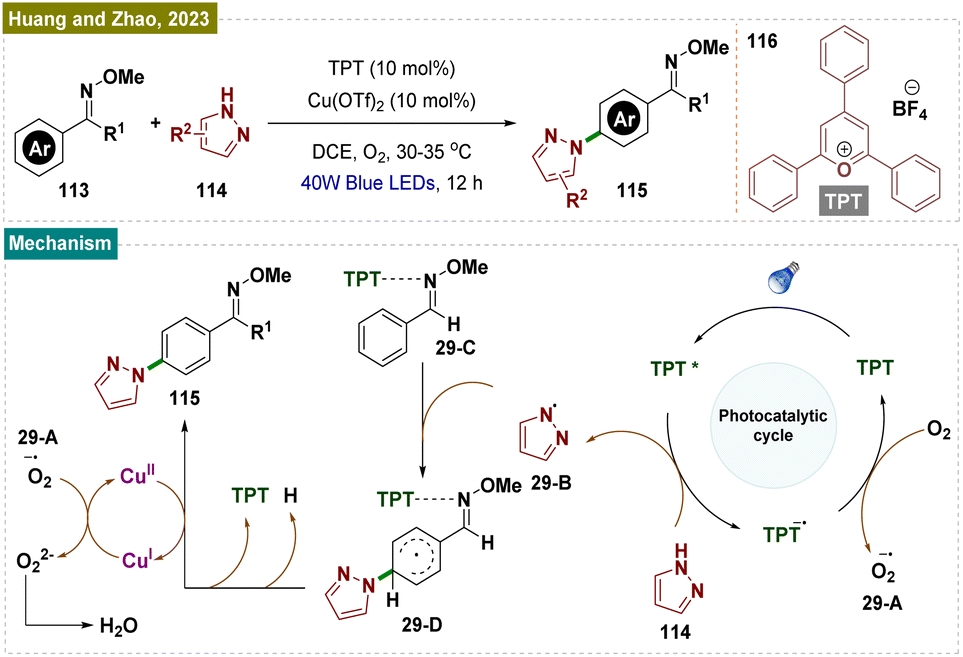 | ||
| Scheme 29 Para-selective C–H bond amination of arenes. | ||
Based on these results and prior studies, a plausible reaction mechanism proposed by the authors has been outlined here in Scheme 29. Upon irradiation, the photocatalyst TPT 116 generates an excited state (TPT*) (E1/2 (TPT*/TPT˙−) = +2.55 V vs. SCE), which facilitates a single electron transfer, producing a pyrazole radical 29-B (E1/2 (114/29-B˙) = +2.21 V vs. SCE). The pyrazole radical then reacted with intermediate 29-C at the less hindered para-position, forming intermediate 29-D. A subsequent single electron transfer to Cu(II), followed by aromatization, resulted in the formation of the amination product 115. The role of O2 was to oxidize back the Cu(I) to Cu(II) while generating [O2]˙−29-Avia SET.
5. Conclusions
In this review, we have provided a comprehensive examination of the recent advancements in photocatalytic regioselective C–H bond functionalization of benzene derivatives, with a focus on the strategies and mechanisms that enable high regioselectivity under mild conditions. By systematically analyzing both transition metal and organic photocatalysts, we have highlighted how SET processes and radical intermediates have been harnessed to achieve selective functionalization at the ortho-, meta-, and para-positions of aromatic rings. These methodologies not only enhance the efficiency of C–H activation but also align with the principles of green chemistry, minimizing the need for prefunctionalization and reducing the environmental impact of synthetic processes.Despite these successes, challenges remain, particularly in enhancing functional group compatibility, reaction scalability, and improving overall efficiency. Future efforts should focus on the design of more sustainable photocatalytic systems that avoid the use of rare metals, while maintaining or even improving selectivity. Moreover, combining photocatalysis with other catalytic strategies, such as HAT and dual-metal catalysis, offers promising avenues for further expanding the reaction scope and addressing current limitations in substrate generality. In this regard, advances in computational modeling and mechanistic understanding will be crucial for guiding the design of next generation photocatalysts. Furthermore, applying these catalytic strategies in the late-stage functionalization of complex, bioactive molecules and in industrial settings will be key to unlocking the full potential of photocatalytic C–H functionalization.
Overall, this review serves as a foundation for the continued development of regioselective photocatalytic C–H functionalization. The combination of detailed mechanistic understanding and innovative catalyst design will undoubtedly lead to new synthetic methods and offer valuable tools for fields such as drug discovery and materials science.
Data availability
No primary research results have been included and no new data were generated or analyzed as part of this review.Author contributions
J. H., S. P. and S. W. developed the concept, wrote the review, and contributed equally. S. D. developed the concept and revised and edited this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to all the reviewers for their critical evaluation. S. D. acknowledges the UBT start-up funding and FWO Research project grant for the financial support. J. H. thanks UBT for PhD funding. S. P. thanks DTU for the postdoctoral funding. S. W. thanks UBT for the postdoctoral funding.Notes and references
- U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Arene diversification through distal C(sp2)–H functionalization, Science, 2021, 372, eabd5992 CrossRef PubMed.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, Electrocatalytic C–H Activation, ACS Catal., 2018, 8, 7086–7103 CrossRef.
- J. Li, S. De Sarkar and L. Ackermann, in C–H Bond Activation and Catalytic Functionalization I, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 217–257 Search PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Mild metal-catalyzed C–H activation: examples and concepts, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- J. A. Labinger, Platinum-Catalyzed C–H Functionalization, Chem. Rev., 2017, 117, 8483–8496 CrossRef PubMed.
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Toolbox for Distal C–H Bond Functionalizations in Organic Molecules, Chem. Rev., 2022, 122, 5682–5841 CrossRef.
- S. Pradhan and S. Das, Recent advances on the carboxylations of C (sp3)–H bonds using CO2 as the carbon source, Synlett, 2023, 34, 1327–1342 CrossRef.
- W. R. Gutekunst and P. S. Baran, C–H functionalization logic in total synthesis, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, C–H Functionalization of Commodity Polymers, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef PubMed.
- L. Zhang and T. Ritter, A Perspective on Late-Stage Aromatic C–H Bond Functionalization, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds, Chem. Rev., 2018, 118, 7532–7585 CrossRef PubMed.
- T. W. Lyons and M. S. Sanford, Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions, Chem. Rev., 2010, 110, 1147–1169 CrossRef PubMed.
- B. Ye and N. Cramer, Chiral Cyclopentadienyls: Enabling Ligands for Asymmetric Rh(III)-Catalyzed C–H Functionalizations, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef PubMed.
- J. Dupont, C. S. Consorti and J. Spencer, The Potential of Palladacycles: More Than Just Precatalysts, Chem. Rev., 2005, 105, 2527–2572 CrossRef PubMed.
- M. T. Mihai, G. R. Genov and R. J. Phipps, Access to the meta position of arenes through transition metal catalysed C–H bond functionalisation: a focus on metals other than palladium, Chem. Soc. Rev., 2018, 47, 149–171 RSC.
- A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Accessing Remote meta- and para-C(sp2)–H Bonds with Covalently Attached Directing Groups, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, Achieving Site-Selectivity for C–H Activation Processes Based on Distance and Geometry: A Carpenter's Approach, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS.
- J. F. Hartwig and M. A. Larsen, Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed.
- Y. Zhang, T. Zhang and S. Das, Selective functionalization of benzylic C(sp3)-H bonds to synthesize complex molecules, Chem, 2022, 8, 3175–3201 CAS.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Photocatalytic Late-Stage C–H Functionalization, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- J. Vanderghinste and S. Das, Applications of Photoredox Catalysis for the Radical-Induced Cleavage of C–C Bonds, Synthesis, 2022, 54, 3383–3398 CrossRef CAS.
- R. Cauwenbergh and S. Das, Photocatalysis: a green tool for redox reactions, Synlett, 2022, 33, 129–149 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- V. Balzani and A. Juris, Photochemistry and photophysics of Ru(II) polypyridine complexes in the Bologna group. From early studies to recent developments, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS.
- Y. Zhang, S. Qin, N. Claes, W. Schilling, P. K. Sahoo, H. Y. V. Ching, A. Jaworski, F. Lemière, A. Slabon, S. Van Doorslaer, S. Bals and S. Das, Direct Solar Energy-Mediated Synthesis of Tertiary Benzylic Alcohols Using a Metal-Free Heterogeneous Photocatalyst, ACS Sustain. Chem. Eng., 2022, 10, 530–540 CrossRef CAS.
- Y. Zhang, W. Schilling and S. Das, Metal-Free Photocatalysts for C–H Bond Oxygenation Reactions with Oxygen as the Oxidant, ChemSusChem, 2019, 12, 2898–2910 CrossRef PubMed.
- P. K. Sahoo, Y. Zhang, Y. Qin, P. Ren, R. Cauwenbergh, G. Siva Raman and S. Das, Robust late-stage benzylic C(sp3)–H aminations by using transition metal-free photoredox catalysis, J. Catal., 2023, 425, 80–88 CrossRef.
- Y. Zhang, P. K. Sahoo, P. Ren, Y. Qin, R. Cauwenbergh, P. Nimmegeers, G. SivaRaman, S. Van Passel, A. Guidetti and S. Das, Transition metal-free approach for late-stage benzylic C(sp3)–H etherifications and esterifications, Chem. Commun., 2022, 58, 11454–11457 RSC.
- Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, Visible-Light-Mediated Efficient Metal-Free Catalyst for α-Oxygenation of Tertiary Amines to Amides, ACS Catal., 2018, 8, 6659–6664 CrossRef.
- N. Holmberg-Douglas and D. A. Nicewicz, Photoredox-Catalyzed C–H Functionalization Reactions, Chem. Rev., 2022, 122, 1925–2016 CrossRef PubMed.
- S. Pradhan, S. Patel and I. Chatterjee, Nitrosoarene-catalyzed regioselective aromatic C–H sulfinylation with thiols under aerobic conditions, Chem. Commun., 2020, 56, 5054–5057 RSC.
- S. Pradhan, S. Roy, S. Ghosh and I. Chatterjee, Regiodivergent Aromatic Electrophilic Substitution Using Nitrosoarenes in Hexafluoroisopropanol: A Gateway for Diarylamines and p-Iminoquinones Synthesis, Adv. Synth. Catal., 2019, 361, 4294–4301 CrossRef.
- C. Pei, C. Empel and R. M. Koenigs, Visible-Light-Induced, Single-Metal-Catalyzed, Directed C–H Functionalization: Metal-Substrate-Bound Complexes as Light-Harvesting Agents, Angew. Chem., Int. Ed., 2022, 61, e202201743 CrossRef PubMed.
- R. Siddiqui and R. Ali, Recent developments in photoredox-catalyzed remote ortho and para C–H bond functionalizations, Beilstein J. Org. Chem., 2020, 16, 248–280 CrossRef.
- L. Ackermann, Robust Ruthenium(II)-Catalyzed C–H Arylations: Carboxylate Assistance for the Efficient Synthesis of Angiotensin-II-Receptor Blockers, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef.
- D. A. Horton, G. T. Bourne and M. L. Smythe, The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Substructures, Chem. Rev., 2003, 103, 893–930 CrossRef.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Aryl–Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed.
- S. G. Ouellet, A. Roy, C. Molinaro, R. Angelaud, J.-F. Marcoux, P. D. O'Shea and I. W. Davies, Preparative Scale Synthesis of the Biaryl Core of Anacetrapib via a Ruthenium-Catalyzed Direct Arylation Reaction: Unexpected Effect of Solvent Impurity on the Arylation Reaction, J. Org. Chem., 2011, 76, 1436–1439 CrossRef CAS.
- J. Hubrich and L. Ackermann, Amino Acid Ligands for Ruthenium(II)-Catalyzed C–H Arylation of Aryltetrazoles with Chlorides: Expedient Access to Antihypertension Drugs, Eur. J. Org Chem., 2016, 2016, 3700–3704 CrossRef CAS.
- M. Seki, Efficient Catalytic System for Ru-Catalyzed C–H Arylation and Application to a Practical Synthesis of a Pharmaceutical, ACS Catal., 2014, 4, 4047–4050 CrossRef CAS.
- A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Bioorthogonal Diversification of Peptides through Selective Ruthenium(II)-Catalyzed C–H Activation, Angew. Chem., Int. Ed., 2017, 56, 1576–1580 CrossRef CAS.
- V. Gayakhe, Y. S. Sanghvi, I. J. S. Fairlamb and A. R. Kapdi, Catalytic C–H bond functionalisation of purine and pyrimidine nucleosides: a synthetic and mechanistic perspective, Chem. Commun., 2015, 51, 11944–11960 RSC.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef.
- W.-Y. Yu, W. N. Sit, Z. Zhou and A. S. C. Chan, Palladium-Catalyzed Decarboxylative Arylation of C–H Bonds by Aryl Acylperoxides, Org. Lett., 2009, 11, 3174–3177 CrossRef.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, Oxidative C–H Activation/C–C Bond Forming Reactions: Synthetic Scope and Mechanistic Insights, J. Am. Chem. Soc., 2005, 127, 7330–7331 CrossRef.
- M. K. Lakshman, A. C. Deb, R. R. Chamala, P. Pradhan and R. Pratap, Direct Arylation of 6-Phenylpurine and 6-Arylpurine Nucleosides by Ruthenium-Catalyzed C–H Bond Activation, Angew. Chem., Int. Ed., 2011, 50, 11400–11404 CrossRef.
- J. Jiang, W.-M. Zhang, J.-J. Dai, J. Xu and H.-J. Xu, Visible-Light-Promoted C–H Arylation by Merging Palladium Catalysis with Organic Photoredox Catalysis, J. Org. Chem., 2017, 82, 3622–3630 CrossRef PubMed.
- M. K. Sahoo, S. P. Midya, V. G. Landge and E. Balaraman, A unified strategy for silver-, base-, and oxidant-free direct arylation of C–H bonds, Green Chem., 2017, 19, 2111–2117 RSC.
- L. Liang, M.-S. Xie, H.-X. Wang, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Visible-Light-Mediated Monoselective Ortho C–H Arylation of 6-Arylpurine Nucleosides with Diazonium Salts, J. Org. Chem., 2017, 82, 5966–5973 CrossRef PubMed.
- K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C–H Arylations at Ambient Temperature, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef PubMed.
- A. Sagadevan, A. Charitou, F. Wang, M. Ivanova, M. Vuagnat and M. F. Greaney, Ortho C–H arylation of arenes at room temperature using visible light ruthenium C–H activation, Chem. Sci., 2020, 11, 4439–4443 RSC.
- J. Struwe, K. Korvorapun, A. Zangarelli and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C–H Benzylations and Allylations at Room Temperature, Chem.–Eur. J., 2021, 27, 16237–16241 CrossRef.
- Y. Wang, C. Li, Y. Li, F. Yin and X.-S. Wang, Rhodium-Catalyzed C–H Olefination of Aryl Weinreb Amides, Adv. Synth. Catal., 2013, 355, 1724–1728 CrossRef CAS.
- D. C. Fabry, J. Zoller, S. Raja and M. Rueping, Combining Rhodium and Photoredox Catalysis for C–H Functionalizations of Arenes: Oxidative Heck Reactions with Visible Light, Angew. Chem., Int. Ed., 2014, 53, 10228–10231 CrossRef CAS.
- F. W. Patureau and F. Glorius, Rh Catalyzed Olefination and Vinylation of Unactivated Acetanilides, J. Am. Chem. Soc., 2010, 132, 9982–9983 CrossRef CAS PubMed.
- F. Yang and L. Ackermann, Ruthenium-Catalyzed C–H Oxygenation on Aryl Weinreb Amides, Org. Lett., 2013, 15, 718–720 CrossRef CAS.
- W. Ma and L. Ackermann, Ruthenium(II)-Catalyzed C–H Alkenylations of Phenols with Removable Directing Groups, Chem.–Eur. J., 2013, 19, 13925–13928 CrossRef CAS PubMed.
- D. C. Fabry, M. A. Ronge, J. Zoller and M. Rueping, C–H Functionalization of Phenols Using Combined Ruthenium and Photoredox Catalysis: In Situ Generation of the Oxidant, Angew. Chem., Int. Ed., 2015, 54, 2801–2805 CrossRef CAS PubMed.
- D. C. Fabry, J. Zoller and M. Rueping, Semiconductors as heterogeneous visible light photoredox catalysts in combined dual metal catalyzed C–H functionalizations, Org. Chem. Front., 2019, 6, 2635–2639 RSC.
- C. Zhou, P. Li, X. Zhu and L. Wang, Merging Photoredox with Palladium Catalysis: Decarboxylative ortho-Acylation of Acetanilides with α-Oxocarboxylic Acids under Mild Reaction Conditions, Org. Lett., 2015, 17, 6198–6201 CrossRef.
- A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, Photoinduced Regioselective Olefination of Arenes at Proximal and Distal Sites, J. Am. Chem. Soc., 2022, 144, 1929–1940 CrossRef.
- M. Prince, P. Kumar and B. K. Singh, Visible-Light-Driven Regioselective Decarboxylative Acylation of N-Methyl-3-phenylquinoxalin-2(1H)-one by Dual Palladium–Photoredox Catalysis Through C–H Activation, ACS Omega, 2024, 9, 651–657 CrossRef PubMed.
- R. Ruzi, M. Zhang, K. Ablajan and C. Zhu, Photoredox-Catalyzed Deoxygenative Intramolecular Acylation of Biarylcarboxylic Acids: Access to Fluorenones, J. Org. Chem., 2017, 82, 12834–12839 CrossRef PubMed.
- L. G. Wade Jr, K. J. Acker, R. A. Earl and R. A. Osteryoung, Cyclodehydration reactions using molten sodium tetrachloroaluminate, J. Org. Chem., 1979, 44, 3724–3725 CrossRef.
- P. J. Coelho, L. M. Carvalho, S. Rodrigues, A. M. F. Oliveira-Campos, R. Dubest, J. Aubard, A. Samat and R. Guglielmetti, Synthesis and photochromic behaviour of novel 2H-chromenes derived from fluorenone, Tetrahedron, 2002, 58, 925–931 CrossRef.
- D. Rodríguez, M. F. Martínez-Esperón, A. Navarro-Vázquez, L. Castedo, D. Domínguez and C. Saá, Intramolecular Dehydro Diels-Alder Reactions of Diarylacetylenes: Switching between Benzo[b]- and Benzo[c]fluorenones as Products by Controlling the Rearrangement of Cyclic Allene Intermediates, J. Org. Chem., 2004, 69, 3842–3848 CrossRef.
- C. Atienza, C. Mateo, Ó. de Frutos and A. M. Echavarren, Formation of Benzo[b]fluorenes and the Benzo[a]fluorene Core of the Fluostatins by Cyclization of Diaryldiynones, Org. Lett., 2001, 3, 153–155 CrossRef PubMed.
- A. J. Catino, J. M. Nichols, H. Choi, S. Gottipamula and M. P. Doyle, Benzylic Oxidation Catalyzed by Dirhodium(II,III) Caprolactamate, Org. Lett., 2005, 7, 5167–5170 CrossRef PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, C–H Activation for the Construction of C–B Bonds, Chem. Rev., 2010, 110, 890–931 CrossRef PubMed.
- J. F. Hartwig, Regioselectivity of the borylation of alkanes and arenes, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- J. F. Hartwig, Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations, Acc. Chem. Res., 2012, 45, 864–873 CrossRef.
- J. Thongpaen, R. Manguin, V. Dorcet, T. Vives, C. Duhayon, M. Mauduit and O. Baslé, Visible Light Induced Rhodium(I)-Catalyzed C–H Borylation, Angew. Chem., Int. Ed., 2019, 58, 15244–15248 CrossRef.
- J. Tanaka, Y. Nagashima, A. J. Araujo Dias and K. Tanaka, Photo-Induced ortho-C–H Borylation of Arenes through In Situ Generation of Rhodium(II) Ate Complexes, J. Am. Chem. Soc., 2021, 143, 11325–11331 CrossRef PubMed.
- F. Diederich, P. J. Stang and R. R. Tykwinski, in Acetylene Chemistry, WILEY-VCH, Weinheim, 2005, DOI:10.1002/3527605487.fmatter.
- A. Saha, A. Ghosh, S. Guin, S. Panda, D. K. Mal, A. Majumdar, M. Akita and D. Maiti, Substrate-Rhodium Cooperativity in Photoinduced ortho-Alkynylation of Arenes, Angew. Chem., Int. Ed., 2022, 61, e202210492 CrossRef.
- F. Xie, Z. Qi, S. Yu and X. Li, Rh(III)- and Ir(III)-Catalyzed C–H Alkynylation of Arenes under Chelation Assistance, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef PubMed.
- C. Feng and T.-P. Loh, Rhodium-Catalyzed C–H Alkynylation of Arenes at Room Temperature, Angew. Chem., Int. Ed., 2014, 53, 2722–2726 CrossRef.
- E. Tan, O. Quinonero, M. Elena de Orbe and A. M. Echavarren, Broad-Scope Rh-Catalyzed Inverse-Sonogashira Reaction Directed by Weakly Coordinating Groups, ACS Catal., 2018, 8, 2166–2172 CrossRef PubMed.
- E. Tan, M. Montesinos-Magraner, C. García-Morales, J. G. Mayans and A. M. Echavarren, Rhodium-catalysed ortho-alkynylation of nitroarenes, Chem. Sci., 2021, 12, 14731–14739 RSC.
- J. Cheng, X. Deng, G. Wang, Y. Li, X. Cheng and G. Li, Intermolecular C–H Quaternary Alkylation of Aniline Derivatives Induced by Visible-Light Photoredox Catalysis, Org. Lett., 2016, 18, 4538–4541 CrossRef PubMed.
- K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- W. K. Hagmann, The Many Roles for Fluorine in Medicinal Chemistry, J. Med. Chem., 2008, 51, 4359–4369 CrossRef.
- D. A. Nagib and D. W. C. MacMillan, Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis, Nature, 2011, 480, 224–228 CrossRef PubMed.
- Y. Jiang, B. Li, N. Ma, S. Shu, Y. Chen, S. Yang, Z. Huang, D. Shi and Y. Zhao, Photoredox-Catalyst-Enabled para-Selective Trifluoromethylation of tert-Butyl Arylcarbamates, Angew. Chem., Int. Ed., 2021, 60, 19030–19034 CrossRef.
- K. Moonen, I. Laureyn and C. V. Stevens, Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity, Chem. Rev., 2004, 104, 6177–6216 CrossRef.
- S. Branion and V. Benin, Preparation of Some Substituted Terephthalic Acids, Synth. Commun., 2006, 36, 2121–2127 CrossRef.
- G. P. Horsman and D. L. Zechel, Phosphonate Biochemistry, Chem. Rev., 2017, 117, 5704–5783 CrossRef.
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Review on Modern Advances of Chemical Methods for the Introduction of a Phosphonic Acid Group, Chem. Rev., 2011, 111, 7981–8006 CrossRef.
- L. Niu, J. Liu, H. Yi, S. Wang, X.-A. Liang, A. K. Singh, C.-W. Chiang and A. Lei, Visible-Light-Induced External Oxidant-Free Oxidative Phosphonylation of C(sp2)–H Bonds, ACS Catal., 2017, 7, 7412–7416 CrossRef.
- J. Huang, Z. Chen, J. Yuan and Y. Peng, Recent Advances in Highly Selective Applications of Nitroso Compounds, Asian J. Org. Chem., 2016, 5, 951–960 CrossRef.
- G. Yan and M. Yang, Recent advances in the synthesis of aromatic nitro compounds, Org. Biomol. Chem., 2013, 11, 2554–2566 RSC.
- B. Kilpatrick, M. Heller and S. Arns, Chemoselective nitration of aromatic sulfonamides with tert-butyl nitrite, Chem. Commun., 2013, 49, 514–516 RSC.
- Y.-X. Li, L.-H. Li, Y.-F. Yang, H.-L. Hua, X.-B. Yan, L.-B. Zhao, J.-B. Zhang, F.-J. Ji and Y.-M. Liang, Direct oxidative nitration of aromatic sulfonamides under mild conditions, Chem. Commun., 2014, 50, 9936–9938 RSC.
- G. G. Pawar, A. Brahmanandan and M. Kapur, Palladium(II)-Catalyzed, Heteroatom-Directed, Regioselective C–H Nitration of Anilines Using Pyrimidine as a Removable Directing Group, Org. Lett., 2016, 18, 448–451 CrossRef CAS PubMed.
- E. Hernando, R. R. Castillo, N. Rodríguez, R. Gómez Arrayás and J. C. Carretero, Copper-Catalyzed Mild Nitration of Protected Anilines, Chem.–Eur. J., 2014, 20, 13854–13859 CrossRef CAS.
- S. J. S. Düsel and B. König, Visible-Light-Mediated Nitration of Protected Anilines, J. Org. Chem., 2018, 83, 2802–2807 CrossRef.
- A. A. Weekes and A. D. Westwell, 2-Arylbenzothiazole as a privileged scaffold in drug discovery, Curr. Med. Chem., 2009, 16, 2430–2440 CrossRef CAS PubMed.
- R. S. Keri, M. R. Patil, S. A. Patil and S. Budagumpi, A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry, Eur. J. Med. Chem., 2015, 89, 207–251 CrossRef CAS PubMed.
- Y. Cheng, J. Yang, Y. Qu and P. Li, Aerobic Visible-Light Photoredox Radical C–H Functionalization: Catalytic Synthesis of 2-Substituted Benzothiazoles, Org. Lett., 2012, 14, 98–101 CrossRef CAS PubMed.
- G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C–H Thiolation for Constructing C–S Bonds, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS.
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- C. Liu, D. Liu and A. Lei, Recent Advances of Transition-Metal Catalyzed Radical Oxidative Cross-Couplings, Acc. Chem. Res., 2014, 47, 3459–3470 CrossRef CAS PubMed.
- J. Bai, S. Yan, Z. Zhang, Z. Guo and C.-Y. Zhou, Visible-Light Carbon Nitride-Catalyzed Aerobic Cyclization of Thiobenzanilides under Ambient Air Conditions, Org. Lett., 2021, 23, 4843–4848 CrossRef CAS.
- S. Kotha, R. Ali, V. Srinivas and N. G. Krishna, Diversity-oriented approach to spirocycles with indole moiety via Fischer indole cyclization, olefin metathesis and Suzuki–Miyaura cross-coupling reactions, Tetrahedron, 2015, 71, 129–138 CrossRef CAS.
- S. Feng, X. Xie, W. Zhang, L. Liu, Z. Zhong, D. Xu and X. She, Visible-Light-Promoted Dual C–C Bond Formations of Alkynoates via a Domino Radical Addition/Cyclization Reaction: A Synthesis of Coumarins, Org. Lett., 2016, 18, 3846–3849 CrossRef PubMed.
- S. Kotha, R. Ali and M. Saifuddin, Diversity-oriented approach to natural product inspired pyrano-carbazole derivatives: strategic utilization of hetero-Diels–Alder reaction, Fischer indolization and the Suzuki–Miyaura cross-coupling reaction, Tetrahedron, 2015, 71, 9003–9011 CrossRef.
- S. Kotha and R. Ali, Diversity-oriented approach to spirooxindoles: application of a green reagent ‘rongalite’, Tetrahedron Lett., 2015, 56, 3992–3995 CrossRef.
- J. Zoller, D. C. Fabry, M. A. Ronge and M. Rueping, Synthesis of Indoles Using Visible Light: Photoredox Catalysis for Palladium-Catalyzed C–H Activation, Angew. Chem., Int. Ed., 2014, 53, 13264–13268 CrossRef PubMed.
- T. Itahara, M. Ikeda and T. Sakakibara, Alkenylation of 1-acylindoles with olefins bearing electron-withdrawing substituents and palladium acetate, J. Chem. Soc., Perkin Trans. 1, 1983, 1361–1363 RSC.
- C. Jia, T. Kitamura and Y. Fujiwara, Catalytic Functionalization of Arenes and Alkanes via C–H Bond Activation, Acc. Chem. Res., 2001, 34, 633–639 CrossRef PubMed.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Towards mild metal-catalyzed C–H bond activation, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- H. J. Kim, D. C. Fabry, S. Mader and M. Rueping, Photoredox/rhodium catalysis in C–H activation for the synthesis of nitrogen containing heterocycles, Org. Chem. Front., 2019, 6, 2319–2323 RSC.
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, Indole Synthesis via Rhodium Catalyzed Oxidative Coupling of Acetanilides and Internal Alkynes, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef PubMed.
- D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, Rhodium(III)-Catalyzed Arene and Alkene C–H Bond Functionalization Leading to Indoles and Pyrroles, J. Am. Chem. Soc., 2010, 132, 18326–18339 CrossRef PubMed.
- N. P. Ramirez, I. Bosque and J. C. Gonzalez-Gomez, Photocatalytic Dehydrogenative Lactonization of 2-Arylbenzoic Acids, Org. Lett., 2015, 17, 4550–4553 CrossRef PubMed.
- X. Wang, J. Gallardo-Donaire and R. Martin, Mild ArI-Catalyzed C(sp2)–H or C(sp3)–H Functionalization/C–O Formation: An Intriguing Catalyst-Controlled Selectivity Switch, Angew. Chem., Int. Ed., 2014, 53, 11084–11087 CrossRef PubMed.
- Y. Wang, A. V. Gulevich and V. Gevorgyan, General and Practical Carboxyl-Group-Directed Remote C–H Oxygenation Reactions of Arenes, Chem.–Eur. J., 2013, 19, 15836–15840 CrossRef PubMed.
- L. Chen, L. Wu, W. Duan, T. Wang, L. Li, K. Zhang, J. Zhu, Z. Peng and F. Xiong, Photoredox-Catalyzed Cascade Radical Cyclization of Ester Arylpropiolates with CF3SO2Cl To Construct 3-Trifluoromethyl Coumarin Derivatives, J. Org. Chem., 2018, 83, 8607–8614 CrossRef PubMed.
- Y. Li, Y. Lu, G. Qiu and Q. Ding, Copper-Catalyzed Direct Trifluoromethylation of Propiolates: Construction of Trifluoromethylated Coumarins, Org. Lett., 2014, 16, 4240–4243 CrossRef.
- S. W. Youn, J. H. Bihn and B. S. Kim, Pd-Catalyzed Intramolecular Oxidative C–H Amination: Synthesis of Carbazoles, Org. Lett., 2011, 13, 3738–3741 CrossRef.
- S. H. Cho, J. Yoon and S. Chang, Intramolecular Oxidative C–N Bond Formation for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-Free Conditions, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef.
- C. Suzuki, K. Hirano, T. Satoh and M. Miura, Direct Synthesis of N-H Carbazoles via Iridium(III)-Catalyzed Intramolecular C–H Amination, Org. Lett., 2015, 17, 1597–1600 CrossRef.
- S. Choi, T. Chatterjee, W. J. Choi, Y. You and E. J. Cho, Synthesis of Carbazoles by a Merged Visible Light Photoredox and Palladium-Catalyzed Process, ACS Catal., 2015, 5, 4796–4802 CrossRef.
- J. A. Leitch and C. G. Frost, Ruthenium-catalysed σ-activation for remote meta-selective C–H functionalisation, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, Ruthenium-Catalyzed Meta Sulfonation of 2-Phenylpyridines, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef PubMed.
- N. Hofmann and L. Ackermann, meta-Selective C–H Bond Alkylation with Secondary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef.
- Q. Yu, L. a. Hu, Y. Wang, S. Zheng and J. Huang, Directed meta-Selective Bromination of Arenes with Ruthenium Catalysts, Angew. Chem., Int. Ed., 2015, 54, 15284–15288 CrossRef.
- Z. Fan, J. Ni and A. Zhang, Meta-Selective CAr–H Nitration of Arenes through a Ru3(CO)12-Catalyzed Ortho-Metalation Strategy, J. Am. Chem. Soc., 2016, 138, 8470–8475 CrossRef PubMed.
- H. L. Barlow, C. J. Teskey and M. F. Greaney, Ruthenium-Catalyzed meta-Carboxylation, Org. Lett., 2017, 19, 6662–6665 CrossRef.
- A. Sagadevan and M. F. Greaney, meta-Selective C–H Activation of Arenes at Room Temperature Using Visible Light: Dual-Function Ruthenium Catalysis, Angew. Chem., Int. Ed., 2019, 58, 9826–9830 CrossRef PubMed.
- P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Visible-Light-Enabled Ruthenium-Catalyzed meta-C–H Alkylation at Room Temperature, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS.
- P.-C. Cui and G.-W. Wang, Visible-Light-Mediated Bimetal-Catalyzed meta-Alkylation of Arenes, Org. Lett., 2024, 26, 427–432 CrossRef CAS PubMed.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Integrated strategy for the synthesis of aromatic building blocks via upcycling of real-life plastic wastes, Chem, 2022, 8, 2472–2484 CAS.
- H.-C. Liu, X. Kong, X.-P. Gong, Y. Li, Z.-J. Niu, X.-Y. Gou, X.-S. Li, Y.-Z. Wang, W.-Y. Shi, Y.-C. Huang, X.-Y. Liu and Y.-M. Liang, Site-selective coupling of remote C(sp3)–H/meta-C(sp2)–H bonds enabled by Ru/photoredox dual catalysis and mechanistic studies, Chem. Sci., 2022, 13, 5382–5389 RSC.
- Y. Wang, S. Chen, X. Chen, A. Zangarelli and L. Ackermann, Photo-Induced Ruthenium-Catalyzed Double Remote C(sp2)–H/C(sp3)–H Functionalizations by Radical Relay, Angew. Chem., Int. Ed., 2022, 61, e202205562 CrossRef CAS PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Activation of remote meta-C–H bonds assisted by an end-on template, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- W. Ali, A. Saha, H. Ge and D. Maiti, Photoinduced meta-Selective C–H Oxygenation of Arenes, JACS Au, 2023, 3, 1790–1799 CrossRef CAS PubMed.
- Y. Goto, M. Sano, Y. Sumida and H. Ohmiya, N-heterocyclic carbene- and organic photoredox-catalysed meta-selective acylation of electron-rich arenes, Nat. Synth., 2023, 2, 1037–1045 CrossRef CAS.
- D. Maiti and S. Guin, Remote CH Bond Functionalizations: Methods and Strategies in Organic Synthesis, John Wiley & Sons, 2021 Search PubMed.
- R. Siddiqui and R. Ali, Recent developments in photoredox-catalyzed remote ortho and para C–H bond functionalizations, Beilstein J. Org. Chem., 2020, 16, 248–280 CrossRef CAS.
- N. Holmberg-Douglas and D. A. Nicewicz, Photoredox-catalyzed C–H functionalization reactions, Chem. Rev., 2021, 122, 1925–2016 CrossRef PubMed.
- J. M. Narayanam and C. R. Stephenson, Visible light photoredox catalysis: applications in organic synthesis, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- L.-C. Campeau and N. Hazari, Cross-coupling and related reactions: connecting past success to the development of new reactions for the future, Organometallics, 2018, 38, 3–35 CrossRef.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li and Y. Liang, Metallaphotoredox: the merger of photoredox and transition metal catalysis, Chem. Rev., 2021, 122, 1485–1542 CrossRef PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Selective photocatalytic aerobic bromination with hydrogen bromidevia an electron-transfer state of 9-mesityl-10-methylacridinium ion, Chem. Sci., 2011, 2, 715–722 RSC.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Site-selective arene C-H amination via photoredox catalysis, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, para-Selective Alkylation of Benzamides and Aromatic Ketones by Cooperative Nickel/Aluminum Catalysis, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- J. B. McManus and D. A. Nicewicz, Direct C–H cyanation of arenes via organic photoredox catalysis, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- D. Petzold and B. König, Photocatalytic Oxidative Bromination of Electron-Rich Arenes and Heteroarenes by Anthraquinone, Adv. Synth. Catal., 2018, 360, 626–630 CrossRef CAS.
- W. Chen, Z. Huang, N. E. S. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Direct arene C-H fluorination with 18F via organic photoredox catalysis, Science, 2019, 364, 1170–1174 CrossRef CAS PubMed.
- S. J. S. Düsel and B. König, Oxidative Photochlorination of Electron-Rich Arenes via in situ Bromination, Eur. J. Org Chem., 2020, 1491–1495 CrossRef.
- M. Zheng, I. Ghosh, B. König and X. Wang, Metal-free Semiconductor Photocatalysis for sp2 C–H Functionalization with Molecular Oxygen, ChemCatChem, 2019, 11, 703–706 CrossRef CAS.
- W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, Recent advances in the trifluoromethylation methodology and new CF3-containing drugs, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS.
- S. Dehnen, L. L. Schafer, T. Lectka and A. Togni, Fluorine: A Very Special Element and Its Very Special Impacts on Chemistry, Inorg. Chem., 2021, 60, 17419–17425 CrossRef CAS.
- Q. Gou, M. Yu, Q. Chen, C. Gu, Q. Zhu, R. Ding, M. Tang, Q. Zhao, J. Shi and H. Huang, Direct remote Csp2–H transformation of aromatic amines enabled by organophotoredox catalysis, Green Chem., 2024, 26, 7007–7012 RSC.
- H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Radical trifluoromethylation, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC.
- Z. Tang, K. Mo, X. Ma, J. Huang and D. Zhao, para-Selective Radical Trifluoromethylation of Benzamide Derivatives via Iminium Intermediates, Angew. Chem. Int. Ed., 2022, 61, e202208089 CrossRef CAS.
- Y. Ouyang, X. H. Xu and F. L. Qing, Trifluoromethanesulfonic Anhydride as a Low-Cost and Versatile Trifluoromethylation Reagent, Angew. Chem. Int. Ed., 2018, 57, 6926–6929 CrossRef CAS.
- P. Liu, C. Chen, X. Cong, J. Tang and X. Zeng, Chromium-catalyzed para-selective formation of quaternary carbon centers by alkylation of benzamide derivatives, Nat. Commun., 2018, 9, 4637 CrossRef PubMed.
- J. F. Hartwig, Evolution of a fourth generation catalyst for the amination and thioetherification of aryl halides, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS.
- A. Sagadevan, V. P. Charpe, A. Ragupathi and K. C. Hwang, Visible Light Copper Photoredox-Catalyzed Aerobic Oxidative Coupling of Phenols and Terminal Alkynes: Regioselective Synthesis of Functionalized Ketones via C–C Triple Bond Cleavage, J. Am. Chem. Soc., 2017, 139, 2896–2899 CrossRef CAS PubMed.
- B. Amini and S. Lowenkron, Aniline and its derivatives, Kirk-Othmer Encyclopedia of Chemical Technology, 2000 Search PubMed.
- X. Lv, Y. Cheng, Y. Zong, Q. Wang, G. An, J. Wang and G. Li, Visible-light-mediated ruthenium-catalyzed para-selective alkylation of unprotected anilines, ACS Catal., 2023, 13, 7310–7321 CrossRef CAS.
- U. Karmakar, H. S. Hwang, Y. Lee and E. J. Cho, Photocatalytic para-Selective C–H Functionalization of Anilines with Diazomalonates, Org. Lett., 2022, 24, 6137–6141 CrossRef CAS.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS.
- M. M. Mastandrea and M. A. Pericàs, Photoredox dual catalysis: A fertile playground for the discovery of new reactivities, Eur. J. Inorg. Chem., 2021, 2021, 3421–3431 CrossRef CAS.
- A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, Photoinduced regioselective olefination of arenes at proximal and distal sites, J. Am. Chem. Soc., 2022, 144, 1929–1940 CrossRef CAS.
- C. Wang, X.-J. Zhang, Y.-W. Zhao, Q. Liu, H. Cheng, Z. Huang and Y. Zhao, An organic photoredox catalyst promoted para-selective C–H amination of aryl oximes, Org. Chem. Front., 2023, 10, 335–341 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |