
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal- and light-induced valence tautomerism with a concerted spin transition in an iron tris(diimine) complex†
Jett T.
Janetzki
 a,
Dominic S.
Brown
a,
Florian
Daumann
b,
I. Haseena
Ismail
a,
Robert W.
Gable
a,
Moya A.
Hay
a,
Roger J.
Mulder
c,
Alyona A.
Starikova
d,
Birgit
Weber
b,
Marcus J.
Giansiracusa
a and
Colette
Boskovic
*a
a,
Dominic S.
Brown
a,
Florian
Daumann
b,
I. Haseena
Ismail
a,
Robert W.
Gable
a,
Moya A.
Hay
a,
Roger J.
Mulder
c,
Alyona A.
Starikova
d,
Birgit
Weber
b,
Marcus J.
Giansiracusa
a and
Colette
Boskovic
*a
aSchool of Chemistry, University of Melbourne, Victoria 3010, Australia. E-mail: c.boskovic@unimelb.edu.au
bInstitute for Inorganic and Analytical Chemistry, Friedrich Schiller University Jena, Humboldtstraße 8, 07743 Jena, Germany
cCSIRO Manufacturing, Clayton, Victoria 3168, Australia
dInstitute of Physical and Organic Chemistry, Southern Federal University, Rostov-on-Don, 344090, Russian Federation
First published on 24th February 2025
Abstract
The switching phenomena of spin crossover (SCO) and valence tautomerism (VT) are respectively dominated by iron(II) and cobalt-dioxolene systems. To explore new possibilities for SCO or VT, the redox-active α-diimine ligand bis((phenyl)imino)acenaphthene (Ph-BIAN), which can adopt neutral (L0), monoanionic (L˙−), and dianionic (L2−) states, was paired with zinc, cobalt, manganese and iron to give [M(Ph-BIAN)3](BPh4)2 (M = Zn (1), Co (2), Mn (3), Fe (4)). Compounds 1, 2 and 3 adopt a temperature invariant MII-(L0)3 state, (2 and 3 are high spin (HS)) in the solid- and solution-states. Electrochemical measurements show the metal controls the degree of electronic communication between the Ph-BIAN ligands. In stark contrast to 1, 2 and 3, compound 4 adopts the LS-FeIII-(L˙−)(L0)2 (LS = low spin) tautomeric form as the ground state in both the solid-state and in solution. Combined variable temperature solid- and solution-state structural, Mössbauer and electronic spectroscopy, and magnetic measurements, show that 4 undergoes the thermally-induced VT process LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3, the only example of VT accompanied by a concerted spin transition in an iron complex with a redox-active ligand. Solid-state photomagnetic measurements suggest that the VT interconversion is also induced by light. Light-induced VT has not been previously observed for complexes other than cobalt-dioxolene, and is potentially afforded here by the unique spin-state change that results in large differences in the Fe–N bond lengths for the two valence tautomers. This study introduces a new example of VT, and suggests that optically-induced VT can be displayed by iron systems, opening alternate pathways toward molecular switches that can be controlled with multiple stimuli.
Introduction
The capability of earth abundant 3d transition metals to access multiple oxidation- and spin-states has led to the synthesis of complexes that interconvert between distinct electronic states. These switchable molecules are key synthetic targets in efforts to miniaturize and improve current technologies, with applications in sensors, displays, actuators, and electronics and spintronics.1–7 A classic example of molecular switching is spin crossover (SCO), in which a metal complex interconverts between the low spin (LS) and high spin (HS) states.8 Incorporation of a redox-active ligand opens additional possibilities for modulating the electronic and magnetic properties of a metal complex. In certain circumstances, metal complexes with a redox-active ligand can undergo valence tautomerism (VT), in which a reversible intramolecular electron transfer occurs between the redox-active metal and redox-active ligand.9,10 Valence tautomerism is akin to metal-to-metal electron transfer, commonly termed charge-transfer-induced spin transition (CTIST), which is observed in Prussian blue analogues.11 In this study, VT refers solely to electron transfer between a metal and organic ligand. Both SCO and VT are induced by stimuli such as temperature, light, or pressure.8,9,12–14Spin crossover is most commonly observed for octahedral Fe(II) compounds with an N6 coordination sphere.8 More recently, SCO in Fe(III) complexes, usually evident with a N4O2-coordination, has received increased attention.15 In certain ligand environments, SCO is observed for Co(II) and Mn(III).16 Valence tautomerism is most common for cobalt complexes with dioxolene (diox) redox-active ligands,9,10 in which a LS-Co(III) centre coordinated to a catecholate ligand (LS-CoIII-cat) undergoes an intramolecular electron transfer and concerted spin transition to afford HS-Co(II)-semiquinonate (HS-CoII-SQ). In contrast, VT involving a redox-active ligand in a discrete complex is significantly more unusual for iron (Table 1)17–22 and manganese species.23–25
| Compounds | Interconversion | Phase | Ref. |
|---|---|---|---|
| a Chart S1 displays representations of these complexes. b Four different porphyrin/thiolate ligands were utilized.18 Bispicen = N,N′-bis(2-pyridylmethyl)-1,2-ethanediamine, Cl4-SQ˙− = 3,4,5,6-tetrachlorosemiquinonate, Cl4-diox = 3,4,5,6-tetrachlorodioxolene, paapH2 = 2-(2-phenylazo)-anilino-4,6-di-tert-butylphenol, tpa = tris(2-pyridylmethyl)amine, 3,6-dbdiox = 3,6-di-tert-butyldioxolene, cth = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetra-azacyclotetradecane, DHBQH2 = 2,5-dihydroxy-1,4-benzoquinone, 4-MeO-bpiH = 4,4′-di-methoxy-bis(pyridyl-imino)isoindoline. cat2− = catecholate, SQ˙− = semiquinonate, AP2− = amidophenolate, ISQ˙− = iminobenzosemiquinonate, IBQ = iminobenzoquinonate. | |||
| [Fe(bispicen)(Cl4-diox)(Cl4-SQ)] | HS-FeIII-(cat)(SQ) ⇌ HS-FeII-(SQ)2 | Solution | 17 |
| [Fe(porphyrin)(thiolate)]b | HS-FeIII-thiolate− ⇌ HS-FeII-thiyl˙ | Solution | 18 |
| [Fe(paap)2]BF4 | LS-FeIII-(ISQ)2 ⇌ LS-FeII-(IBQ)(ISQ) | Solid & solution | 19 |
| [CoIII(η5-C10H15)2][Fe(paap)2] | LS-FeIII-(AP)2 ⇌ LS-FeII-(ISQ)(AP) | Solid & solution | 19 |
| [Fe(tpa)(3,6-dbdiox)]BF4 | LS-FeIII-cat ⇌ HS-FeIII-cat ⇌ HS-FeIII-SQ | Solid | 20 |
| [(Fe(RR-cth))(Co(SS-cth))(DHBQ)](PF6)3 | LS-FeIII-cat ⇌ HS-FeIII-cat ⇌ HS-FeIII-SQ | Solid | 21 |
| [Fe(4-MeO-bpi)2] | LS-FeII-(bpi−)2 ⇌ LS-FeIII-(bpi˙2−)(bpi−) ⇌ HS-FeIII-(bpi˙2−)(bpi−) | Solid | 22 |
Unlike Co-dioxolene complexes, there are no discrete iron complexes that undergo VT with a spin transition occurring at the same time as electron transfer; either there is no spin-state change,17–19 or electron transfer and spin transition occur sequentially.20–22 Compounds [Fe(tpa)(3,6-dbdiox)]BF4 and [(Fe(RR-cth))(Co(SS-cth))(DHBQ)](PF6)3 (Chart S1†) (tpa = tris(2-pyridylmethyl)amine, 3,6-dbdiox = 3,6-di-tert-butyldioxolene, cth = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetra-azacyclotetradecane, DHBQH2 = 2,5-dihydroxy-1,4-benzoquinone) were found to display a LS-FeIII-cat ⇌ HS-FeIII-cat SCO interconversion at low temperature, followed by a separate VT interconversion to HS-FeII-SQ at higher temperatures.20,21 Alternative isolation of [Fe(tpa)(3,6-dbdiox)]+ as the PF6−, BPh4− and ClO4− and [(Fe(RR-cth))(Co(SS-cth))(DHBQ)]3+ as the AsF6− salts results in only Fe(III) SCO.20,26 The complex [Fe(4-MeO-bpi)2] (4-MeO-bpiH = 4,4′-di-methoxy-bis(pyridyl-imino)isoindoline) undergoes a VT transition in the solid-state from LS-FeII-bpi− to LS-FeIII-bpi2˙−, triggering SCO to HS-FeIII-bpi2˙−.22 However, in solution [Fe(4-MeO-bpi)2] only adopts a HS-FeII-bpi− state. As the observation of VT in these reported iron compounds is highly dependent on the solid-state environment, the VT process is unlikely to be molecular in origin.
In certain cases, in addition to the common use of temperature, light can induce SCO and VT.12,27,28 For SCO, light stimulation is known as light-induced excited spin-state trapping (LIESST),12,27 in which light irradiation generates a metastable HS state. Trapping of the HS form is promoted by a large change in the metal–ligand bond distances between the LS and HS state. For Fe(II), the change is significant (∼0.2 Å), resulting in efficient LIESST.12,27 For Fe(III), the smaller change (∼0.13 Å) commonly results in rapid relaxation of the HS-Fe(III) metastable state, making Fe(III) LIESST rarer.12 The analogous process for VT complexes, light-induced valence tautomerism (LIVT), is observed for many Co-dioxolene complexes.13,28–30 However, to the best of our knowledge, no other metal complex has been shown to display LIVT, limiting the available chemical building blocks to engineer light-induced VT materials. Of the discrete iron complexes that display thermally-induced VT, none have been reported to undergo LIVT.17–22,31 We hypothesize that is due to the lack of a concerted spin transition in the VT process, minimizing the differences in the iron–ligand bond lengths between the tautomers, resulting in rapid relaxation of the metastable state.
In this study, we aimed to discover new examples of SCO or VT complexes. The family of N-donor redox-active bis((aryl)imino)acenaphthene (Ar-BIAN) ligands are well suited for this goal. The Ar-BIAN ligand has comparable redox chemistry to dioxolene ligands, with accessible neutral Ar-BIAN0 (L0), monoanionic radical Ar-BIAN˙− (L˙−), and dianionic Ar-BIAN2− (L2−) states.32,33 Unlike dioxolene ligands in their neutral quinone form, Ar-BIAN0 will readily coordinate with a metal centre,33 allowing for two possible redox-couples to achieve VT: L2−/L˙− and L˙−/L0. In addition, the electronic properties of Ar-BIAN can be easily tuned via variation of the aryl groups.34
We have recently reported the use of Ar-BIAN ligands to generate a new example of cobalt VT; the complex [Co(4-MeO-BIAN)3]0 undergoes a LS-CoIII-(L˙−)3 ⇌ HS-CoII-(L0)(L˙−)2 interconversion in solution (4-MeO-BIAN = bis[(4-methoxy-phenyl)imino]acenaphthene) (Chart S2†).35 In that same study, the compound [CoII(Ph-BIAN)3](PF6)2 (Ph-BIAN = bis((phenyl)imino)acenaphthene) remains as HS-CoII-(L0)3, with no SCO to LS-CoII-(L0)3 or VT to LS-CoIII-(L˙−)(L0)2.35 For this work, we hypothesized that replacement of cobalt with manganese or iron could result in SCO and/or VT in several possible ways. The lower Mn(II) and Fe(II) oxidation potential compared to Co(II) could stabilize a MIII-(L˙−)(L0)2 ground state (M = Fe, Mn), such that a MIII-(L˙−)(L0)2 ⇌ MII-(L0)3 VT interconversion occurs. Alternatively, resultant FeII-(L0)3, FeIII-(L˙−)(L0)2, and MnIII-(L˙−)(L0)2 states could show SCO.16,36 Previously, the syntheses of [Fe(Ph-BIAN)3][FeCl4]2 and [Fe(Ph-BIAN)3](BF4)2 were reported, although the electronic structure and magnetic properties were not investigated.37
We report the synthesis and analysis of a family of [M(Ph-BIAN)3](BPh4)2 compounds, where M = Zn (1), Co (2), Mn (3) and Fe (4) (Fig. 1), with a view towards possible SCO or VT. Compound 1 was used as a reference for the MII-(L0)3 state, and to study the spectroscopic and electrochemical properties of coordinated Ph-BIAN isolated from a redox-active metal. The detailed multi-technique experimental studies of this family of complexes have allowed us to deduce the electronic structure of 1, 2, 3 and 4. Compound 4 undergoes the first example of thermally-induced iron valence tautomerism that occurs with a concerted spin-state change. In addition, 4 displays a photoinduced interconversion that is hypothesised to be LIVT.
 | ||
| Fig. 1 Representation of the [M(Ph-BIAN)3]2+ metal complexes (M = Zn (1), Co (2), Mn (3), Fe (4), with numbering scheme. All complexes isolated as the BPh4− salt. Photographs of MeCN solutions of 1, 2, 3 and 4 at room temperature. | ||
Results and discussion
Synthesis
The homoleptic complexes [M(Ph-BIAN)3]2+ (M = Zn, Co, Mn, and Fe, Fig. 1) were synthesized (ESI) by combining three equivalents of Ph-BIAN with the corresponding M(II) salt in methanol (in air for 1, 2 and 3, under N2 for 4). The combination of the metal salts and Ph-BIAN yielded a yellow solution of 1, orange solutions for 2 and 3, and a dark green solution for 4. Addition of two equivalents of BPh4− resulted in 1, 2 and 3, forming as orange solids, and 4 as a green solid. Crystals suitable for X-ray diffraction were grown by layering a saturated dichloromethane (DCM) solution with diethylether (Et2O) for 1, DCM solution with diisopropylether (iPr2O) for 2, 1,2-dichloroethane (DCE) with iPr2O for 3, and an acetonitrile (MeCN) solution with iPr2O for 4. Bulk samples for 1, 2 and 3 were recrystallized using DCE/iPr2O, and 4 using MeCN/iPr2O. Elemental analysis (EA) indicates solvation of compounds as 1·1.5DCE·1.5iPr2O, 2·DCE·1.6iPr2O, 3·0.4iPr2O and 4·1.7iPr2O. Thermogravimetric analysis (Fig. S1†) supports the formulation of 1, 2, 3 and 4 as solvates; deviation between the solvation determined from EA and thermogravimetric analysis for 3 is attributed to partial desolvation during sample preparation for EA, and 4 exhibits concurrent desolvation and thermal-decomposition. The experimental powder X-ray diffraction (PXRD) patterns match with the associated simulated pattern, indicating phase purity for all four compounds (Fig. S2†). The dramatic colour difference of 4 as a solid and in solution compared to the similar colours of 1, 2 and 3 provided an initial indication that complex 4 adopts a significantly different electronic structure compared to 1, 2 and 3.Solid-state analysis
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with a whole complex and two BPh4− anions in the asymmetric unit. The presence of two BPh4− anions per complex indicates an overall dicationic complex for 1, 2, 3 and 4.
space group with a whole complex and two BPh4− anions in the asymmetric unit. The presence of two BPh4− anions per complex indicates an overall dicationic complex for 1, 2, 3 and 4.
In the solid-state structures, all four complexes share a M(Ph-BIAN)3 motif with a distorted octahedral geometry and approximate D3 point symmetry (Fig. 2 and S4†). The oxidation and spin state of the metal centres can be determined by the M–N bond lengths, bond valence sum (BVS) parameters,38 and octahedral distortion parameters (SHAPE index, Σ, and Θ).39–41 The oxidation state of the Ph-BIAN ligands can be assigned via the α-diimine C–N and C–C bonds distances.32,35,42 Neutral Ph-BIAN0 has shorter C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1.28–1.31 Å) and longer C–C (1.46–1.53 Å) distances compared to the monoanionic Ph-BIAN˙− CN (1.32–1.35 Å) and C–C (1.41–1.44 Å) bond lengths.32,43
N (1.28–1.31 Å) and longer C–C (1.46–1.53 Å) distances compared to the monoanionic Ph-BIAN˙− CN (1.32–1.35 Å) and C–C (1.41–1.44 Å) bond lengths.32,43
 | ||
| Fig. 2 Cationic structure of 4·2iPr2O at 100 K with relevant atoms labelled. Hydrogen atoms, solvent molecules, and BPh4− anions have been omitted for clarity. Colour code: C (dark grey), N (blue), Fe (orange). | ||
Compound 1·3DCM can only adopt a ZnII-(L0)3 charge distribution. The average CN and C–C bond distances of the three Ph-BIAN0 ligands of 1.279(5) and 1.522(2) Å (Table 2 and Fig. S5†), respectively, deviate only slightly from the bond lengths of uncoordinated Ph-BIAN0 (CN: 1.275(2) Å, C–C 1.526(2) Å).43 In both 2·DCM·2iPr2O and 3·1.5iPr2O, the long Co/Mn–N distances, large octahedral distortion, lack of Jahn–Teller distortion, and BVS values (Table 2 and Fig. S5†) are all consistent with HS-Co(II) and HS-Mn(II), respectively.35,45 The CN and C–C bond lengths for 2·DCM·2iPr2O and 3·1.5iPr2O are similar to those in 1 (Table 2 and Fig. S5†), indicating three Ph-BIAN0 ligands. The lack of deviation in the CN/C–C bond distances in 2 and 3 compared with 1 discount significant π-backbonding. Therefore, 1, 2, and 3 all adopt the same MII-(L0)3 (2 and 3 are HS) electronic form in the solid-state at 100 K.
| 1·3DCM | 2·DCM·2iPr2O | 3·1.5iPr2O | 4·2iPr2O | |
|---|---|---|---|---|
| a Average distance (Å) between the three N–C–C–N centroids. b SHAPE index for octahedral geometry in SHAPE 2.1.39,40 A value of 0 represents a perfect octahedron. c ∑ = sum of the deviation of the 12 N/O–Co–N/O angles from 90°. Θ = sum of the deviation of 24 unique torsional angles between the N/O atoms on opposite triangular faces of the octahedron from 60°, providing the degree of trigonal distortion from an octahedron to trigonal prism. These were calculated using OctaDist39–41 – a program for determining the structural distortion of the octahedral complexes. For a perfect octahedron, ∑ and Θ are zero. d Bond valence sum.38,44 | ||||
| M–N bond lengths/Å | ||||
| M–N1 | 2.1734(9) | 2.131(1) | 2.262(1) | 1.991(2) |
| M–N2 | 2.193(1) | 2.149(1) | 2.242(1) | 2.008(2) |
| M–N3 | 2.1519(9) | 2.124(1) | 2.263(1) | 1.991(2) |
| M–N4 | 2.1519(9) | 2.124(1) | 2.283(1) | 2.008(2) |
| M–N5 | 2.193(1) | 2.149(1) | 2.268(1) | 1.991(2) |
| M–N6 | 2.1734(9) | 2.131(1) | 2.275(1) | 1.991(2) |
| M–Nav | 2.173(2) | 2.135(2) | 2.266(2) | 1.997(5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| C–N/C–C bond lengths and interatomic distances/Å | ||||
| C1–N1 | 1.280(2) | 1.274(2) | 1.287(2) | 1.292(3) |
| C2–N2 | 1.281(2) | 1.285(2) | 1.286(2) | 1.295(3) |
| C3–N3 | 1.276(2) | 1.275(2) | 1.286(2) | 1.292(3) |
| C4–N4 | 1.276(2) | 1.275(2) | 1.285(2) | 1.295(3) |
| C5–N5 | 1.281(2) | 1.285(2) | 1.284(2) | 1.292(3) |
| C6–N6 | 1.280(2) | 1.274(2) | 1.285(2) | 1.292(3) |
| C–Nav | 1.279(5) | 1.278(5) | 1.286(5) | 1.293(7) |
| C1–C2 | 1.524(1) | 1.507(2) | 1.516(2) | 1.482(3) |
| C3–C4 | 1.519(2) | 1.500(2) | 1.520(2) | 1.482(3) |
| C5–C6 | 1.524(1) | 1.507(2) | 1.522(2) | 1.480(4) |
| C–Cav | 1.522(2) | 1.505(3) | 1.519(3) | 1.481(6) |
| r AB | 3.895(2) | 3.843(2) | 4.075(2) | 3.632(3) |
|
||||
| Distortion parameters | ||||
| SHAPE (Oh)b | 1.271 | 1.175 | 2.015 | 0.665 |
| Σ/°c | 76.7 | 74.7 | 97.2 | 54.4 |
| Θ/°c | 242.1 | 237.0 | 296.1 | 177.6 |
| BVSd | 1.77 | 1.96 | 1.95 | 3.67 |
In contrast, 4·2iPr2O (Fig. 2) exhibits significantly shorter Fe–N bond lengths and reduced octahedral distortion compared with 1, 2 and 3 (Table 2). The Fe–N bond distances for LS-Fe(II) (1.95–2.05 Å) and LS-Fe(III) (1.95–2.09 Å) are shorter compared with HS-Fe(II) (2.15–2.21 Å) and HS-Fe(III) (2.09–2.15 Å).36,46,47 Therefore, 4 is likely characterized by a LS-Fe(II) or LS-Fe(III) centre at 100 K, with the BVS value of 3.67 supporting the latter. The average Ph-BIAN CN and C–C bonds are elongated and contracted, respectively, compared with 1, 2 and 3 (Table 2 and Fig. S5†), and match with the distances observed for [CoII(Ph-BIAN˙−)(Ph-BIAN0)2]+ (CN: 1.295(3) Å, C–C: 1.485(3) Å, Fig. S6†).35 The bond lengths indicate one Ph-BIAN˙− and two Ph-BIAN0 ligands, which are either crystallographically disordered or electronically delocalized. An overall dicationic charge is therefore consistent with 4·2iPr2O adopting the LS-FeIII-(L˙−)(L0)2 electromer at 100 K in the solid-state. Overall, structural analysis shows that at 100 K, 1, 2 and 3 adopt the MII-(L0)3 electromeric state (2 and 3 are HS), whereas 4 exists in the alternate LS-MIII-(L˙−)(L0)2 form. The different charge distribution of 4 explains the drastic colour variance of 4 compared to 1, 2 and 3.
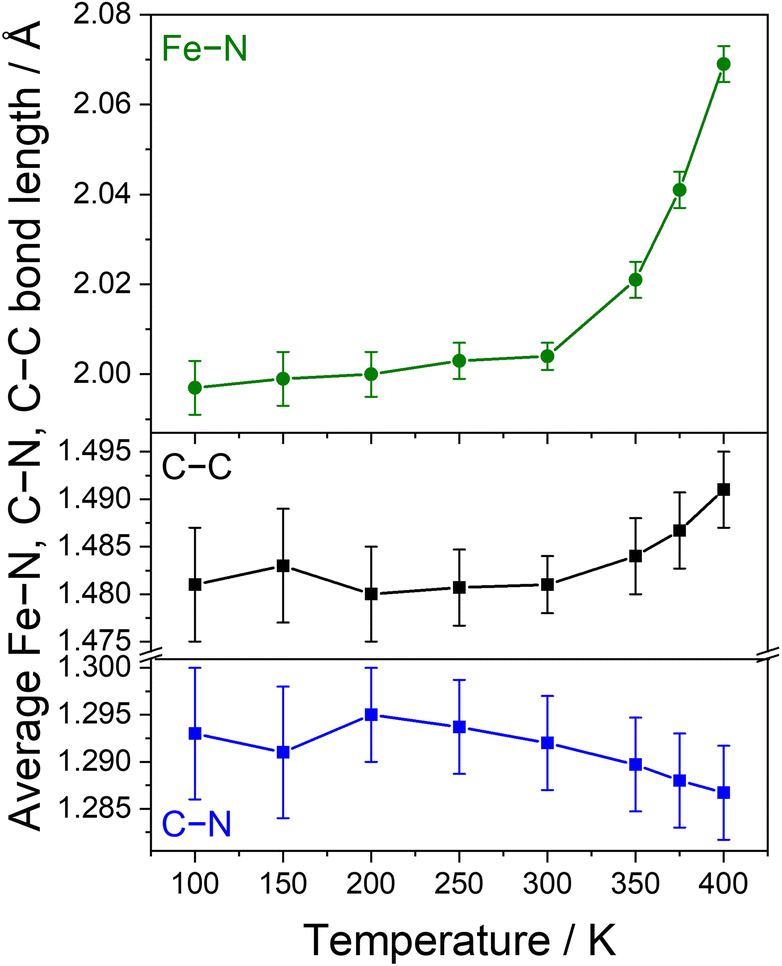 | ||
| Fig. 3 Temperature dependence of the average Fe–N bond distances (top) and C–C and C–N bond distances of the Ph-BIAN ligands (bottom) for 4·2iPr2O. | ||
To aid differentiation between a SCO or VT transition, we analysed the changes to the CN and C–C bond lengths of the Ph-BIAN ligands; VT would result in contraction and elongation, respectively, whereas no change is expected for SCO. The Ph-BIAN˙− ligand remains disordered across the three ligands at all temperatures (Table S4†). Between 300 and 400 K, the average C–C bond distances increase from 1.481(6) to 1.491(4) Å, and the average CN lengths decrease from 1.293(7) to 1.287(5) Å (Fig. 3, S9 and Table S4†), in conjunction with the changes to the Fe–N bond lengths. Unfortunately, the errors of the averaged ligand bond lengths are too high to be able to determine if there is a ligand oxidation state change. The individual C–C and CN bond distances increase and decrease with increasing temperature, respectively, some within the margin of error (Fig S9 and Table S4†). From the present structural data, we cannot definitively say if 4·2iPr2O undergoes SCO or VT; we tentatively suggest the VT transition LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 in the solid-state.
N stretches of neutral Ph-BAN0 in the range 1700–1550 cm−1.35,42 The intensities (normalized to the most intense peak at 695 cm−1) of these CN stretches are comparable for 1, 2 and 3 (Fig. 4), consistent with the coordination of three neutral Ph-BIAN0 ligands and adoption of a MII-(L0)3 state. In contrast, the intensity of these CN stretches is markedly reduced in 4 (Fig. 4), which suggests less Ph-BIAN0. A reduced amount of Ph-BIAN0 is consistent with 4 adopting a LS-FeIII-(L˙−)(L0)2 electronic form in the solid-state at room temperature.
 | ||
| Fig. 4 Top: ATR-IR spectra of 1·1.5DCE·1.5iPr2O, 2·DCE·1.6iPr2O, 3·0.4iPr2O and 4·1.7iPr2O measured at room temperature in the CN region between 1700–1500 cm−1, normalized to the most intense peak at 695 cm−1. Bottom: solid-state UV-vis spectra of 1·1.5DCE·1.5iPr2O, 2·DCE·1.6iPr2O, 3·0.4iPr2O and 4·1.7iPr2O measured as diffuse reflectance at room temperature, diluted ∼5% in KBr. | ||
Solid-state diffuse reflectance ultraviolet-visible (UV-vis) spectra were recorded for 1·1.5DCE·1.5iPr2O, 2·DCE·1.6iPr2O, 3·0.4iPr2O and 4·1.7iPr2O at room temperature (Fig. 4). The spectra for 1, 2 and 3 are dominated by intraligand (IL) Ph-BIAN0 processes centred at ∼420 nm (resulting in the observed orange colour) and ∼340 nm (Fig. 4).35,42 No other features are observed that can be attributed to the presence of Ph-BIAN˙− or metal-to-ligand-charge transfer (MLCT) processes.35,48,49 The presence of only Ph-BIAN0 processes supports 1, 2 and 3 adopting a MII-(L0)3 tautomer in the solid-state at room temperature. Compound 4 also exhibits IL Ph-BIAN0 processes in the solid-state (430, 340 nm, Fig. 4). In addition, 4 also features an intense peak at 700 nm, giving rise to the green colour. This process is not observed for free Ar-BIAN˙−,48 discounting a Ph-BIAN˙− ligand process. Instead, this feature is assigned as a Ph-BIAN˙− → Fe(III) ligand-to-metal charge transfer (LMCT), which would arise from the LS-FeIII-(L˙−)(L0)2 state adopted by 4 at room temperature.
 | ||
| Fig. 5 Variable temperature, zero-field 57Fe Mössbauer spectra of 4·1.7iPr2O. The squares are the raw data, and the solid lines are the least-square Lorentzian fits presented in Table 3. | ||
| T/K | Oxidation & spin state | δ/mm s−1 | ΔEQ/mm s−1 | Γ HWHM/mm s−1 |
|---|---|---|---|---|
| a δ denotes the isomer shift, ΔEQ is the quadrupole splitting, ΓHWHM is the half width at half maximum. The ratio of relative intensities in doublets (A2/A1) is expected to be A2/A1 = 1. The value has therefore been fixed in the fit. Deviations from an ideal ratio may be attributed to e.g. texture effects due to preferred orientation of larger crystals relative to the γ-ray.50 | ||||
| 80 | LS-Fe(III) | 0.393(10) | 0.456(19) | 0.111(14) |
| 300 | LS-Fe(III) | 0.336(9) | 0.399(15) | 0.150(11) |
| 350 | LS/HS-Fe(III) | 0.358(20) | — | 0.231(29) |
| 400 | HS-Fe(II) | 0.329(25) | 0.919(46) | 0.262(33) |
Upon increasing to 350 K, the doublet collapses to a broad singlet with δ = 0.348 mm s−1 (Fig. 5 and Table 3), implying HS-Fe(III).50 The broad shape is likely due to dynamic effects between LS-Fe(III) and HS-Fe(III) which occur faster than, or on, the timescale of the Mössbauer experiment (107–108 s−1).51 At 400 K, the spectrum is now characterized by a doublet at δ = 0.329 mm s−1 (Fig. 5 and Table 3). The large quadrupole splitting of ΔEQ = 0.919 mm s−1 is not consistent with a symmetric HS-Fe(III) (ΔEQ < 0.5 mm s−1) structure, and is therefore assigned as HS-Fe(II)15 which would arise from formation of HS-FeII-(L0)3. Due to pronounced Doppler broadening of the resonance line at 400 K, identification of additional unresolved contributions (e.g. HS-Fe(III)) is impaired. However, given that the transition to HS-Fe(II) is not yet complete, this cannot be ruled out.
It appears that the transition first occurs with SCO from LS-FeIII-(L˙−)(L0)2 to HS-FeIII-(L˙−)(L0)2 (broad dynamic singlet at 350 K) followed by VT to HS-FeII-(L0)3 (doublet at 400 K), with both steps resolved by Mössbauer spectroscopy. From the present spectra, it cannot be determined if the VT interconversion in the solid-state has to proceed via HS-FeIII-(L˙−)(L0)2 before electron transfer, or if both LS-FeIII-(L˙−)(L0)2 and HS-FeIII-(L˙−)(L0)2 are able to undergo VT. It is possible that both LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 and LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 are occurring, with the latter process possibly dominating at higher temperatures. Both SCXRD and solid-state magnetic measurements (see below) indicate that the process is not complete at 400 K. The observation of only one doublet at 400 K and the singlet at 350 K indicates fast dynamic effects and therefore averaged signals between the LS-FeIII-(L˙−)(L0)2 and the HS-FeII-(L0)3 species. Therefore, it is safe to assume that the quadrupole splitting of the pure HS-FeII-(L0)3 species will be even larger. In summary, Mössbauer spectroscopy confirms 4 has a LS-FeIII-(L˙−)(L0)2 ground state and undergoes an overall VT interconversion to HS-FeII-(L0)3.
 | ||
| Fig. 6 Plots of χMT vs. T for 2·DCE·1.6iPr2O (black), 3·0.4iPr2O (red) and 4·1.7iPr2O (green) in the solid-state, and plots of χMT vs. T for 4 in d3-MeCN (blue). | ||
Compound 4·1.7iPr2O exhibits a χMT value of 0.1 cm3 mol−1 K at 300 K, remaining essentially constant until 20 K, below which the χMT decreases to 0.05 cm3 mol−1 K at 1.8 K (Fig. 6). A diamagnetic LS-FeII-(L0)3 state (S = 0) is discounted based on the structural and spectroscopic data. Instead, 4 is best interpreted as adopting a LS-FeIII-(L˙−)(L0)2 state between 1.8 and 300 K with strong antiferromagnetic coupling between Ph-BIAN˙− and LS-Fe(III). The magnetic data in the range 20–250 K were fit to the isotropic Heisenberg exchange Hamiltonian Ĥex = −2J(Ŝ1·Ŝ2) (Fig. S13†) using PHI,53 where J is the exchange interaction between LS-Fe(III) and Ph-BIAN˙− (both S = 1/2) and Si are the corresponding spin operators, with g fixed to 2.0 for both spin centres. A good fit was achieved with J = −700(40) cm−1 with the inclusion of temperature independent paramagnetic (TIP) = 9.4(2) × 10−5 cm3 mol−1 and a paramagnetic impurity (ρ) = 2.4(1)% that corresponds to residual paramagnetism from possible trapped HS-Fe(II) (S = 2). The rapid decrease below 20 K could be due to depopulation of the spin–orbit coupled states of trapped HS-Fe(II) and/or intermolecular antiferromagnetic exchange interactions. The Mössbauer spectroscopy data discount a VT transition from LS-FeIII-(L˙−)(L0)2 to LS-FeII-(L0)3 occurring up to 300 K, which that would not be detected by magnetometry due to a lack of spin state change.
Above 300 K, the χMT profile of 4·1.7iPr2O rapidly increases to 1.2 cm3 mol−1 K at 400 K (Fig. 6). The feature is reversible (Fig. S14†). The reversible increase in χMT with temperature confirms the overall LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 VT interconversion process tentatively suggested by SCXRD and shown by Mössbauer spectroscopy. The theoretical spin-only χMT value of HS-FeII-(L0)3 is 3.0 cm3 mol−1 K, with experimental values for HS-Fe(II) ranging between 3.2–3.5 cm3 mol−1 K due to spin–orbit contributions.36,54 The VT transition is approximated as being ∼35–40% complete (Fig. S15†), matching the estimation from variable temperature SCXRD. Overall, magnetic data confirm that in the solid-state, compounds 2 and 3 do not display VT or SCO and adopt a temperature invariant HS-MII-(L0)3 state. In contrast, magnetic measurements confirms that 4 adopts a LS-FeIII-(L˙−)(L0)2 ground state and undergoes an overall incomplete thermally-induced VT transition to HS-FeII-(L0)3 above 300 K.
Photoirradiation of 4·1.7iPr2O at 10 K was performed using the setup represented in Fig. S16† by scanning the wavelengths between 280–500 nm (Fig. S17†) and 600–1000 nm, corresponding to the ligand-based and LMCT transitions, respectively. The largest response was observed for 420 nm, with photoirradiation for 12 hours resulting in an increase in χMT from 0.09 to 0.15 cm3 mol−1 K (Fig. 7 and S18†). No photo-stationary limit was reached even after this 12 hour period. Repeating the measurement with a smaller amount of sample (0.3 mg vs. 0.8 mg) produced a larger photomagnetic response (χMT = 0.18 cm3 mol−1 K) due to increased light penetration, but resulted in significantly noisier thermal data. The photomagnetic response is either the conversion of the LS-FeIII-(L˙−)(L0)2 ground state to a metastable HS-FeII-(L0)3 (LIVT, ∼5% conversion) or HS-FeIII-(L˙−)(L0)2 (LIESST, ∼3% conversion) state. Based on the thermal behaviour, we suggest 4 displays LIVT. On warming after 10 K irradiation, the curve rejoins the non-irradiated curve at ∼100 K (Fig. 7), resulting in a T(photoirradiation) value of 82 K. If this temperature corresponds to T(LIVT), it is in the upper range compared to values reported for Co-diox complexes (30–80 K).29,55–58
 | ||
| Fig. 7 Plot of χMT vs. T for 4·1.7iPr2O upon cooling (blue circles) response with irradiation with 420 nm for 12 hours at 10 K (green circles), and subsequent warming scan after the light is switched off (red circles). Inset figure shows δχMT/δT vs. T, with T(photoirradiation) indicated by the red line (82 K). * Bump due to molecular oxygen. | ||
To investigate the relaxation features of the photoinduced state of 4, isothermal relaxation profiles from the photoinduced state to the ground state were monitored by time-dependent magnetic susceptibility measurements at five temperatures upon irradiation at 420 nm (Fig. S19†). Unfortunately, the data are noisy due to the weakly paramagnetic nature of 4 and the limited photoconversion. The photoinduced fraction γ is defined as γ(t) = (μDC(t) − μDC-off)/(μDC-t=0 − μDC-off), where μDC(t) is the DC magnetic moment at time t, μDC-off is the DC magnetic moment without irradiation, and μDC-t=0 is the DC magnetic moment reached at the end of the photoirradiation period (i.e., at time t = 0 of the relaxation curve).55,59 Over nine hours, γ decays by ∼15% between 10–50 K, and ∼25% at 60 K (Fig. S19†), representing a relatively large temperature range at which the photoinduced state remains trapped.
The shape of the decay curves for 4 match the stretched exponential decays observed for Co-diox LIVT complexes,29,30,55–63 rather than the typical decay of LIESST compounds, which show self-acceleration processes best modelled by a sigmoidal function.64,65 We propose that the stretched exponential decay, in conjunction with the thermal behaviour, indicates a photoinduced LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 VT interconversion. The decay curves were fit with a stretched exponential function γ(t) = (1 − γHS)e(−(t/τ)β) + γHS (Fig. S19†), where γ(t) is the photoinduced fraction at time t, γHS is the equilibrium photoinduced fraction and 1 represents the photoinduced fraction at the end of the photoirradiation period (t = 0), τ is a decay constant (indicative of the time it takes the signal to have relaxed to 1/e of its initial value (at t = 0 s)), 0 < β ≤ 1 is a parameter accounting for the inhomogeneous distribution of spin-centres in the sample,60–62,66 and the γHS value has been fixed to zero as it represents the HS photoinduced fraction as t → ∞. The relaxation times (τ values) range 105–107 seconds (Table S5†). As the measurement time is 3.5 × 104 seconds, the extracted relaxation times have no physical meaning as they represent extrapolation of the measured relaxation times by 1–3 orders of magnitude.57 Despite this, the relaxation of the metastable state of 4 is clearly slow over a large temperature range (10–80 K). The relaxation data, coupled with the high T(photoirradiation) value, suggest a highly stable photoinduced state with respect to temperature.
The combined solid-state analysis shows that 4 displays the thermally-induced VT interconversion LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3, which might also be accessible with light (Scheme 1). If the VT interconversion can be light-induced, this would be the first for an Fe-based VT system. Future work is required to confirm the photoinduced metastable state is HS-FeII-(L0)3.
 | ||
| Scheme 1 Valence tautomeric equilibrium LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 displayed by 4 in the solid- and solution-state. | ||
Solution-state analysis
The analysis in the solid state indicates that 1, 2 and 3 do not display SCO or VT, adopting a MII-(L0)3 electronic state (2 and 3 are HS). In contrast, 4 displays an overall LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 VT interconversion. As demonstrated by previous Fe-VT compounds, the VT behaviour can be highly dependent on the surrounding chemical environment.20–22 We therefore performed complementary solution studies to determine the electronic states exhibited by 1, 2, 3 and 4 in solution, with a focus on determining if the VT interconversion exhibited by 4 is molecular in origin or dependent on the extended chemical environment. | ||
| Fig. 8 Top: UV-vis-NIR spectra of 1, 2, 3 and 4 in MeCN at 298 K. Bottom: NIR spectra of 4 in MeCN, BuCN, DCE and THF at 298 K. | ||
The LS-FeIII-(L˙−)(L0)2 tautomeric state of 4 contains Ph-BIAN in two different oxidation states (neutral and monoanionic), which is an example of organic-based mixed-valence (MV).81–83 Organic MV systems share similarities with the more common instance of mixed-valency in which the metal ions in a multinuclear complex differ in oxidation state. If compound 4 has a LS-FeIII-(L˙−)(L0)2 ground state and electronic coupling between the Ph-BIAN ligands, a ligand-to-ligand Ph-BIAN˙− → Ph-BIAN0 intervalence charge transfer (IVCT) band should be observed in the near-infrared (NIR) region.81–83 In the NIR region, 4 displays an IVCT band across all four solvents (∼1550 nm, ∼6450 cm−1), confirming a LS-FeIII-(L˙−)(L0)2 state in solution at room temperature. Compounds 1, 2 and 3 do not feature this IVCT band (Fig. 8), consistent with the non-MV MII-(L0)3 state.
The degree of electronic coupling between the Ph-BIAN ligands in 4 can be described by the Robin and Day MV classification (class I, II, II–III. III, see ESI†)84–87 and the electronic coupling parameter (HAB), extracted from the energy and shape of the Ph-BIAN˙− → Ph-BIAN0 IVCT band in the framework of Marcus-Hush theory.88 These parameters have been used to define other metal complexes with MV ligands.81,83 The presence of an IVCT band indicates intramolecular ligand-to-ligand electron transfer, discounting class I for 4. The IVCT bands of 4 in MeCN, BuCN, DCE and THF were deconvoluted with two Gaussian functions (Fig. S27,† peak 1 at low energy, peak 2 at high energy), to extract νmax, half-height-width (Δν1/2) and molar absorptivity (εmax) values (Table S10†). The MV class can be determined based on solvent-dependency: class II is solvent-dependent with Δνmax > 200 cm−1 for a range of dielectric constant (Δκ) of 30, class II–III and III are solvent-independent with (Δνmax < 200 cm−1).84–86 Across all four solvents used, (Δκ = 30), peak 1 has a Δνmax of ∼120 cm−1 that suggests class II–III or III, and peak 2 has a Δνmax of 300 cm−1, suggesting class II (Table S10†). The cumulative fit has a Δνmax of 100 cm−1 across all four solvents, giving a tentative assignment of class II–III. To support a possible MV II–III assignment, we used the Γ parameter (eqn (S6), see ESI† for details), determined from the peak width: for class II, Γ < 0.5, class II and II, Γ ≈ 0.5, and class III, Γ > 0.5.89 Across all four solvents, peaks 1 and 2 have Γ values of 0.64–0.66 and 0.06–0.38 (Table S10†) that support assignment of class II–III and II, respectively. Marcus-Hush analysis estimates HAB values of 160–670 cm−1 for peak 1 and 230–920 cm−1 for peak 2 (Table S10 and eqn (S7)†) of 4. The 2HAB/νmax values of 0.05–0.22 for both bands suggest MV class II.84–86 The strength of electronic coupling is comparable to other organic MV systems in transition metal complexes; HAB values of 810–840 cm−1 for class II90,91 and 920–1100 cm−1 for class II–III.91,92 Overall, 4 is on the border of class II and II–III. Therefore, 4 has a localized electronic structure and solvent averaging, with a residual barrier to electron transfer arising from intramolecular structural changes.85,86 The average of the Ph-BIAN0 and Ph-BIAN˙− CN/C–C bond lengths observed in the structure of 4·2iPr2O must be due to crystallographic disorder, not electronic delocalization.
The same analysis of the previously reported35 IVCT band of [CoII(Ph-BIAN˙−)(Ph-BIAN0)2]+ in MeCN and DCM indicate class II–III (Fig. S28 and Table S11†), with HAB values of 1190–1380 cm−1 for peak 1 and 1190–1630 cm−1 for peak 2. The electronic coupling between the Ph-BIAN units in the LS-FeIII-(L˙−)(L0)2 state for 4 is weaker than for [CoII(Ph-BIAN˙−)(Ph-BIAN0)2]+. We initially expected the shorter metal–ligand bond lengths in 4 that decrease the distance between the Ph-BIAN ligands to promote increased electron transfer. Analysis using electrochemistry (see Electrochemistry section) shows electron transfer actually occurs through the metal, with Co(II) facilitating more efficient electron transfer in [CoII(Ph-BIAN˙−)(Ph-BIAN0)2]+ compared with Fe(III) in 4. Previously, Fe(III) has been shown to be less effective than other 3d metals at promoting intramolecular electron transfer between ligands.93 The class II/II–III IVCT behaviour for 4 indicates stronger coupling between the ligand centres compared to Fe(III) complexes with MV dioxolene ligands.93,94 The difference between 4 and these Fe-dioxolene complexes is that 4 shows VT,93,94 which would increase electron density on the iron centre, resulting in more efficient electron transfer between the ligands.
 | ||
| Fig. 9 Top: variable temperature UV-vis absorption spectra of 4 in BuCN between 268 (blue) and 373 K (red) in 5 K increments. Bottom: comparison of the UV-vis spectra of 4 in BuCN at 268 (blue) and 373 K (red) with 1 in BuCN at 373 K. | ||
The spectral profile of 4 upon heating (most evident in BuCN) approaches that of 1, 2 and 3 (in BuCN at 373 K for comparison) (Fig. 9, S33 and S34†); specifically the loss of the LMCT band, replacement of the IL Ph-BIAN˙− peaks at ∼420 nm with IL Ph-BIAN0 features, and the change in the peak shape at 320 nm. Therefore, the high temperature spectra of 4 resembles that expected for a HS-MII-(L0)3 state, supporting the interpretation that 4 undergoes LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 VT in solution. The difference in the spectra of the HS-MII-(L0)3 state in 4 compared with 1, 2 and 3 appears to be a peak at ∼620 nm, which we assign as a Fe(II) → Ph-BIAN0 metal-to-ligand charge transfer (MLCT). Deconvolution of the spectra of 4 in BuCN between 268–373 K using the spectra of 1 in BuCN at the same temperature, and plotting the relative spectral area (430–1100 nm) yields a transition temperature (T1/2) of ∼320 K (Fig. S35†). The observation of the same spectral change upon heating for 4 across all four solvents further supports that the observation of VT is not dependent on the surrounding chemical environment. The relative changes in the spectra of 4 upon heating (268–323 K, maximum collection temperature in THF), are greater in MeCN/BuCN compared with DCE/THF (Fig. S36†). The smaller change in the spectra with temperature in DCE and THF suggest these solvents stabilize the LS-FeIII-(L˙−)(L0)2 tautomer to a greater degree.
The transition remains incomplete up to 328 K, but the transition temperature (T1/2) in solution is shifted ∼50 K lower compared to the solid-state. To obtain thermodynamic quantities, we fit the solution susceptibility data with the regular solution model (eqn (S3)†). A fit was obtained with (χMT)max = 3.5(9) cm3 mol−1 K, ΔH = 30(2) kJ mol−1, ΔS = 80(10) J K−1 mol−1, and T1/2 of 370(30) K (Fig. S37†). The values of ΔH and ΔS for 4 are similar to those reported for the VT interconversion of Co-dioxolene complexes in solution (ΔH = 31–37 kJ mol−1, ΔS = 90–134 J K−1 mol−1).96–99 In Co-dioxolene VT complexes, the transition occurs either between spin = 1/2 and spin = 5/2 (ref. 97 and 99) (assume spin of 5/2 due to very weak magnetic exchange),100 or spin = 0 and spin = 2 (ref. 96 and 98) (assume spin of 2 due to weak magnetic exchange),52 resulting in Δspin = 2. For 4, the transition also occurs with Δspin = 2 (from spin = 0 due to strong antiferromagnetic exchange between LS-Fe(III) and Ph-BIAN˙−, and spin = 2 from HS-Fe(II)), resulting in similar thermodynamic parameters to Co-dioxolene VT. Overall, the charge distribution of 1, 2, 3 and 4 in the solution-state are the same as in the solid-state. Crucially, variable temperature solution UV-vis and magnetic measurements confirm that the LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 VT transition displayed by 4 is molecular in origin.
 | ||
| Fig. 10 Top: cyclic voltammograms of process I, II, III, IV, V and VI for compound 1, and processes I and II for compounds 2 and 3 (1.0 mM MeCN solutions with 0.25 M Bu4NPF6) obtained with a scan rate of 100 mV s−1. Arrow indicates direction of measurement, with all measurements started at position of zero current. Bottom: assignment of the redox processes I, II and III for complexes 1, 2 and 3. | ||
| Process | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | BPh4− | VII | VIII | |
| a Potentials reported vs. Ferrocene/Ferrocenium couple, measured as 65–75 mV with CV under the same conditions. Error in potentials is ±5 mV. b 1.0 mM in MeCN with 0.25 M Bu4NPF6, scan rate 100 mV s−1. c E p rather than Em. d Only an estimation as the process is obscured by IV. e Processes combined due to close spacing. | |||||||||
| Cyclic voltammetry E m or E pa /V (ΔE p /mV)b | |||||||||
| 1 | −0.91(80) | −1.07c | −1.25(50) | −1.73c | −2.19(100) | −2.61(140) | 0.47c | — | — |
| 2 | −0.59(65) | −1.24c | −1.43(68) | −1.71c | −2.17(85) | −2.67c | 0.46c | 0.68c | — |
| 3 | −0.89(75) | −1.17(65) | −1.78c | −2.27(115) | −2.75c | 0.43c | — | 1.04c | |
| 4 | −0.67(70) | −1.18(68) | −1.7c,d | −1.74c | −2.32(110) | −2.71c | 0.45c | — | 1.01(65) |
|
|||||||||
| Rotating disk electrode voltammetry E 1/2 /V(i L /μA)b | |||||||||
| 1 | −0.91(24) | −1.09(18) | −1.26(17) | −1.76(65) | — | — | 0.50(120) | — | — |
| 2 | −0.59(20) | −1.24(23) | −1.44(20) | −1.73(64) | — | — | 0.45(115) | — | — |
| 3 | −0.87(22) | −1.17(30) | −1.79(108)e | — | — | 0.43(145) | — | — | |
| 4 | −0.67(23) | −1.19(25) | −1.66(30) | −1.75(32) | — | — | 0.43(140) | — | 1.03(20) |
The voltammograms of 2 and 3 share features with 1 (Fig. S38†). In the reduction scan, 2 also exhibits six reductions (I–VI), whereas 3 features five reductions, with process III potentially combined with IV. As for 1, processes I, II and III in 2 and 3 are assigned as the sequential one-electron reduction of each of the three Ph-BIAN0 ligands (Fig. 10). The limiting current for processes IV, V and VI are different for 2 and 3 compared to 1, suggesting reduction of Ph-BIAN˙− and the Co(II)/Mn(II) centre. Upon oxidation, both 2 and 3 feature the BPh4− oxidation, and an additional oxidation process not observed for 1. In compound 2, process (VII) is assigned as a Co(II)/Co(III) oxidation.35 For 3, the oxidation (VIII) occurs at similar potential as the Mn(III)/Mn(IV) process observed for mononuclear manganese complexes with N6 coordination sphere formed with pyridyl ligands.45,102,103 The Mn(II)/Mn(III) oxidation of 3 is potentially hidden by the BPh4− process; the iL in the RDE for 3 (145 μA) is 25% larger than the four-electron BPh4− process of 1 (115 μA) and 2 (120 μA), suggesting a five-electron process.
The separation between the potentials of processes I and II (ΔL) is significantly different between 1 (160 mV), 2 (650 mV), and 3 (280 mV) (Fig. 10). The value of ΔL provides an indication of the strength of electronic communication between the three Ph-BIAN ligands in the MII-(L˙−)(L0)2 redox state, with increasing communication in the order 1 < 3 < 2. The comproportionation constants (Kc) for 1, 2 and 3, calculated using log10(Kc) = 17.2 × ΔL (ΔL in V),85 are 5.6 × 102, 1.5 × 1011, and 6.5 × 104, respectively. As the MV class for CoII-(L˙−)(L0)2 (see Solution-state electronic spectroscopy section) is II–III, the smaller ΔL for 1 and 3 suggests an MV class approaching/being II. Spectroelectrochemistry or isolation of [ZnII(Ph-BIAN˙−)(Ph-BIAN0)2]+ and [MnII(Ph-BIAN˙−)(Ph-BIAN0)2]+ would be required to confirm the MV class of the MII-(L˙−)(L0)2 state of 1 and 3. A through-space origin of the variation of electronic communication between the three complexes is discounted; Ph-BIAN N–C–C–N centroid distances (Table 2) are similar for all three complexes, and no significant structural changes upon reduction occurs.35 Instead, ligand communication must occur via the coordination bonds. We hypothesize that as the degree of metal–ligand covalency increases, together with the degree of metal character in these predominantly ligand-centred reductions, electronic delocalization also increases.91,104,105 The extent of covalency arises from the amount of overlap between the metal d-orbitals and the Ph-BIAN singly occupied molecular orbital (SOMO).104,105 For Co(II), the d-orbitals and SOMO have similar energies, increasing covalency and enhancing electron transfer. For Mn(II), the reduction in d-orbital energy relative to the SOMO reduces covalency, weakening electronic coupling. The redox-inactive Zn(II) has negligible metal–ligand covalency, minimising interligand electron transfer. Increased covalency between the metal centre and Ph-BIAN results in stabilisation of the [MII(Ph-BIAN˙−)(Ph-BIAN0)2]+ redox state; this manifests as an increase in the potential of process I in the order 1 < 3 < 2.
The voltammogram of 4 exhibits six reduction processes (I–VI), the BPh4− oxidation, and an additional oxidation process (VIII) (Fig. S38†). The assignment of the processes for 4 are complicated by the presence of concomitant VT in MeCN solution at room temperature, and the possibility for both ligand- and metal-based processes. Process I could be either reduction of the Fe(III) centre (FeII-(L˙−)(L0)2) or reduction of Ph-BIAN0 (FeIII-(L˙−)2(L0)), with subsequent processes II and III potentially involving either/both metal and ligand orbitals. If process I is indeed the reduction of Fe(III), 4 has a ΔL of 510 mV. The degree of ligand communication in the MII-(L˙−)(L0)2 redox state would therefore increase in the order 1 < 3 < 4 < 2. This trend is consistent with previous observations that Fe(II) and Co(II) enhance delocalisation compared to Zn(II) and Mn(II).104,105 Spectroelectrochemistry or isolation of a reduced derivative of 4 is required to confirm whether process I is metal- or ligand-based. The oxidation process VIII could be assigned as Ph-BIAN˙− oxidation; however, the potential appears too high for this oxidation process.106 The Ph-BIAN˙− oxidation is possibly obscured by the BPh4− oxidation, with the iL of 140 (compared with 120 for 1 and 115 for 2) suggesting a five-electron process.
Conclusions
The aim of this work at the outset was to find new examples of metal–ligand pairings that display molecular switchability in the form of VT and/or SCO. Building on past studies that identified VT in a Co-Ar-BIAN complex,35 we report the synthesis of [Zn(Ph-BIAN)3](BPh4)2 (1), [Co(Ph-BIAN)3](BPh4)2 (2), [Mn(Ph-BIAN)3](BPh4)2 (3), and [Fe(Ph-BIAN)3](BPh4)2 (4). The green colour of 4 compared to the orange coloured 1, 2 and 3 suggested immediately that 4 adopts a different charge distribution. Combined structural, IR and UV-vis-NIR spectroscopy, magnetic susceptibility and computational analysis shows 1, 2 and 3 adopt a temperature-invariant MII-(L0)3 state, with 2 and 3 being HS, in the solid- and solution-states. Electrochemical analysis demonstrated that the metal centre modulates the degree of communication between the three coordinated Ph-BIAN ligands; increased covalency between the metal and ligand enhances electronic communication.In contrast to 1, 2 and 3, comprehensive investigation unambiguously confirms that 4 exhibits a LS-FeIII-(L˙−)(L0)2 ground state in both the solid- and solution-states. Solid-state structural, magnetic, and Mössbauer spectroscopy measurements demonstrate that 4 undergoes a thermally-induced overall VT transition from LS-FeIII-(L˙−)(L0)2 to HS-FeII-(L0)3. Solution-state variable temperature UV-vis and magnetic measurements confirm this VT process is molecular in origin, and not dependent on solid-state packing effects. Therefore, this is the first instance of a thermally-induced VT transition in an iron complex with a redox-active ligand that occurs with a spin-state transition accompanying electron transfer. Solid-state photomagnetic measurements indicate a possible light-induced LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 VT interconversion. The photoinduced metastable state of 4 is highly stable with respect to temperature. Light-induced VT (LIVT) has not previously been observed for complexes other than cobalt-dioxolene. We attribute the possible LIVT for this Fe complex to the unique concerted spin-state transition accompanying electron transfer, which results in large changes to the Fe–N bond lengths. Future work would focus on confirming the identity of the metastable state.
This study indicates that an iron complex can undergo a thermally-induced VT interconversion that proceeds with a concerted spin-state change. This spin-state change is likely crucial to any LIVT. The key to achieving a VT process with a spin transition is therefore to utilize a ligand that can support both VT and a spin-state transition. The Ph-BIAN ligand is redox-active with an orbital energy that matches iron (VT), and has a free ligand N⋯N distance (2.84 Å) that falls within a range for diimine ligands that gives FeII(diimine)3 SCO (2.78–2.93 Å).35 Future work targeting Fe-LIVT should use ligands that satisfy these criteria.
In summary, all data are consistent with 4 undergoing LS-FeIII-(L˙−)(L0)2 ⇌ HS-FeII-(L0)3 valence tautomerism in both the solid- and solution-state, which can be induced thermally, and potentially with visible light (Scheme 1). This represents a new type of valence tautomerism, in which an iron complex displays both electron transfer and a concerted spin-state change. This work suggests that LIVT could be possible for systems other than Co-dioxolene, with LIVT potentially beneficial for applications in devices such as single-molecule junctions. As the percentage of photoconversion is low, future work should focus on decreasing the temperature of the thermally-induced VT transition in related iron complexes. A principal finding is that the metal centre with a conserved ligand set can modulate the magnetic switching behaviour. The system investigated, [M(Ph-BIAN)3]2+, is extremely electronically versatile, offering multiple avenues for VT and SCO. Incorporation of other metal centres, substituents on the aryl ring, and/or isolation of different oxidation states are all possible paths toward new spin transitions.
Data availability
Crystallographic data for all compounds have been deposited with the CCDC under deposition numbers 2393551–2393561 and can be obtained from https://www.ccdc.cam.ac.uk/. Other data are available from the authors upon reasonable request.Author contributions
Conceptualization: JTJ, CB; investigation and formal analysis: JTJ, DSB, FD, IHI, RWG, MAH, RJM, AAS; funding acquisition: CB; project administration: CB; resources: CB, RJM, BW; supervision: CB, MJG, BW; visualization: JTJ; writing – original draft: JTJ, CB; writing – review & editing: all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B. thanks the Australian Research Council for funding (FT190100293). We acknowledge the Australian Research Council for an equipment grant (LE210100009). J. T. J. acknowledges support from the Australian Government for a Research Training Stipend, and the Dr Albert Shimmins Fund for an Albert Shimmins Postgraduate Writing Up Award. M. J. G. thanks the University of Melbourne for a Melbourne Postdoctoral Fellowship. F. D. acknowledges financial support of the Deutsche Forschungsgemeinschaft (project no. 509879467). A. A. S. thanks the Ministry of Science and Higher Education of the Russian Federation (State assignment in the field of scientific activity, project no. FENW-2023-0017). We thank Stanley Bagio for assistance with the graphics. This work was performed in part at the Trace Analysis for Chemical, Earth and Environmental Sciences (TrACEES) Platform at the University of Melbourne. This research was undertaken in part using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, Australia and made use of the Australian Cancer Research Foundation (ACRF) detector (for MX2).References
- C. J. Yu, S. Von Kugelgen, D. W. Laorenza and D. E. Freedman, ACS Cent. Sci., 2021, 7, 712–723 CrossRef CAS PubMed.
- E. Resines-Urien, M. Á. G. García-Tuñón, M. García-Hernández, J. A. Rodríguez-Velamazán, A. Espinosa and J. S. Costa, Adv. Sci., 2022, 9, 1–7 Search PubMed.
- A. Cornia and P. Seneor, Nat. Mater., 2017, 16, 505–506 CrossRef CAS PubMed.
- J. Villalva, A. Develioglu, N. Montenegro-Pohlhammer, R. Sánchez-de-Armas, A. Gamonal, E. Rial, M. García-Hernández, L. Ruiz-Gonzalez, J. S. Costa, C. J. Calzado, E. M. Pérez and E. Burzurí, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- S. K. Karuppannan, A. Martín-Rodríguez, E. Ruiz, P. Harding, D. J. Harding, X. Yu, A. Tadich, B. Cowie, D. Qi and C. A. Nijhuis, Chem. Sci., 2021, 12, 2381–2388 RSC.
- H. J. Shepherd, I. A. Gural'Skiy, C. M. Quintero, S. Tricard, L. Salmon, G. Molnár and A. Bousseksou, Nat. Commun., 2013, 4, 1–9 RSC.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 1–47 Search PubMed.
- T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS.
- G. K. Gransbury and C. Boskovic, in Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley, 2021, pp. 1–24 Search PubMed.
- D. Aguilà, Y. Prado, E. S. Koumousi, C. Mathonière and R. Clérac, Chem. Soc. Rev., 2016, 45, 203–224 RSC.
- M. Nakaya, R. Ohtani, L. F. Lindoy and S. Hayami, Inorg. Chem. Front., 2021, 8, 484–498 RSC.
- O. Sato, A. Cui, R. Matsuda, J. Tao and S. Hayami, Acc. Chem. Res., 2007, 40, 361–369 CrossRef CAS PubMed.
- A. Summers, F. Zahra, M. Zahir, G. F. Turner, M. A. Hay, A. Riboldi-Tunnicliffe, R. Williamson, S. Boer, L. Goerigk, C. Boskovic and S. A. Moggach, Nat. Commun., 2024, 15, 8922 CrossRef CAS PubMed.
- D. J. Harding, P. Harding and W. Phonsri, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef CAS.
- J. Olguín, Coord. Chem. Rev., 2020, 407, 213148 CrossRef.
- N. Shaikh, S. Goswami, A. Panja, X. Y. Wang, S. Gao, R. J. Butcher and P. Banerjee, Inorg. Chem., 2004, 43, 5908–5918 CrossRef CAS PubMed.
- P. K. Das, S. Samanta, A. B. McQuarters, N. Lehnert and A. Dey, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6611–6616 CrossRef CAS PubMed.
- A. Rajput, A. K. Sharma, S. K. Barman, D. Koley, M. Steinert and R. Mukherjee, Inorg. Chem., 2014, 53, 36–48 CrossRef CAS PubMed.
- M. Chegerev, O. Demidov, P. Vasilyev, N. Efimov, S. Kubrin, A. Starikov, V. Vlasenko, A. Piskunov, S. Shapovalova, A. Guda, Y. Rusalev and A. Soldatov, Dalton Trans., 2022, 51, 10909–10919 RSC.
- P. Sadhukhan, S.-Q. Wu, J. I. Long, T. Nakanishi, S. Kanegawa, K. Gao, K. Yamamoto, H. Okajima, A. Sakamoto, M. L. Baker, T. Kroll, D. Sokaras, A. Okazawa, N. Kojima, Y. Shiota, K. Yoshizawa and O. Sato, Nat. Commun., 2021, 12, 3–8 CrossRef PubMed.
- A. Scheja, D. Baabe, D. Menzel, C. Pietzonka, P. Schweyen and M. Bröring, Chem.–Eur. J., 2015, 21, 14196–14204 CrossRef CAS PubMed.
- A. S. Attia and C. G. Pierpont, Inorg. Chem., 1995, 34, 1172–1179 CrossRef CAS.
- A. Caneschi and A. Dei, Angew. Chem., Int. Ed., 1998, 37, 3005–3007 CrossRef CAS PubMed.
- A. Panja, Inorg. Chem. Commun., 2012, 24, 140–143 CrossRef CAS.
- X. Zhang, W. H. Xu, W. Zheng, S. Q. Su, Y. B. Huang, Q. Shui, T. Ji, M. Uematsu, Q. Chen, M. Tokunaga, K. Gao, A. Okazawa, S. Kanegawa, S. Q. Wu and O. Sato, J. Am. Chem. Soc., 2023, 145, 15647–15651 CrossRef CAS PubMed.
- J. F. Létard, J. Mater. Chem., 2006, 16, 2550–2559 RSC.
- D. M. Adams, B. Li, J. D. Simon and D. N. Hendrickson, Angew. Chem., Int. Ed., 1995, 34, 1481–1483 CrossRef CAS.
- K. G. Alley, G. Poneti, P. S. D. Robinson, A. Nafady, B. Moubaraki, J. B. Aitken, S. C. Drew, C. Ritchie, B. F. Abrahams, R. K. Hocking, K. S. Murray, A. M. Bond, H. H. Harris, L. Sorace and C. Boskovic, J. Am. Chem. Soc., 2013, 135, 8304–8323 CrossRef CAS PubMed.
- W. H. Xu, Y. B. Huang, W. W. Zheng, S. Q. Su, S. Kanegawa, S. Q. Wu and O. Sato, Dalton Trans., 2024, 53, 2512–2516 RSC.
- J. Guasch, L. Grisanti, S. Jung, D. Morales, G. D'Avino, M. Souto, X. Fontrodona, A. Painelli, F. Renz, I. Ratera and J. Veciana, Chem. Mat., 2013, 25, 808–814 CrossRef CAS.
- I. L. Fedushkin, A. A. Skatova, V. A. Chudakova and G. K. Fukin, Angew. Chem., Int. Ed., 2003, 42, 3294–3298 CrossRef CAS PubMed.
- I. S. Fomenko, N. F. Romashev and A. L. Gushchin, Coord. Chem. Rev., 2024, 514, 215845 CrossRef CAS.
- M. Gasperini, F. Ragaini and S. Cenini, Organometallics, 2002, 21, 2950–2957 CrossRef CAS.
- M. A. Hay, J. T. Janetzki, V. J. Kumar, R. W. Gable, R. Clérac, A. A. Starikova, P. J. Low and C. Boskovic, Inorg. Chem., 2022, 61, 17609–17622 CrossRef CAS PubMed.
- H. Phan, J. J. Hrudka, D. Igimbayeva, L. M. Lawson Daku and M. Shatruk, J. Am. Chem. Soc., 2017, 139, 6437–6447 CrossRef CAS PubMed.
- M. Villa, D. Miesel, A. Hildebrandt, F. Ragaini, D. Schaarschmidt and A. Jacobi von Wangelin, ChemCatChem, 2017, 9, 3203–3209 CrossRef CAS.
- P. H. C. Camargo, J. Mater. Sci., 2017, 52, 9959–9962 CrossRef CAS.
- S. Llunell, M. Casanova, D. Cirera, J. Alemany and P. Alvarez, SHAPE 2.1, 2013 Search PubMed.
- S. Alvarez, D. Avnir, M. Llunell and M. Pinsky, New J. Chem., 2002, 26, 996–1009 RSC.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC.
- V. L. Nadurata, M. A. Hay, J. T. Janetzki, G. K. Gransbury and C. Boskovic, Dalton Trans., 2021, 50, 16631–16646 RSC.
- M. Schmitz, M. Seibel, H. Kelm, S. Demeshko, F. Meyer and H.-J. Krüger, Angew. Chem., Int. Ed., 2014, 53, 5988–5992 CrossRef CAS PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struc. Sci., Crys. Eng., Mat., 1985, 244, 244–247 CrossRef.
- N. R. East, C. Förster, L. M. Carrella, E. Rentschler and K. Heinze, Inorg. Chem., 2022, 61, 14616–14625 CrossRef CAS PubMed.
- J. T. Janetzki, M. G. Chegerev, G. K. Gransbury, R. W. Gable, J. K. Clegg, R. J. Mulder, G. N. L. Jameson, A. A. Starikova and C. Boskovic, Inorg. Chem., 2023, 62, 15719–15735 CrossRef CAS PubMed.
- J. A. Real, M. C. Muñoz, J. Faus and X. Solans, Inorg. Chem., 1997, 36, 3008–3013 CrossRef CAS PubMed.
- I. L. Fedushkin, A. A. Skatova, V. A. Chudakova, V. K. Cherkasov, G. K. Fukin and M. A. Lopatin, Eur. J. Inorg. Chem., 2004, 388–393 CrossRef CAS.
- I. L. Fedushkin, A. A. Skatova, M. Hummert and H. Schumann, Eur. J. Inorg. Chem., 2005, 1601–1608 CrossRef CAS.
- P. Gütlich; E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, Springer, 2011 Search PubMed.
- Y. Maeda, N. Tsutsumi and Y. Takashima, Inorg. Chem., 1984, 266, 2440–2447 CrossRef.
- G. K. Gransbury, M.-E. Boulon, R. A. Mole, R. W. Gable, B. Moubaraki, K. S. Murray, L. Sorace, A. Soncini and C. Boskovic, Chem. Sci., 2019, 10, 8855–8871 RSC.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55, 4327–4331 CrossRef CAS PubMed.
- F. Yu, M. Xiang, Q. G. Wu, H. He, S. Q. Cheng, X. Y. Cai, A. H. Li, Y. M. Zhang and B. Li, Inorg. Chim. Acta, 2015, 426, 146–149 CrossRef CAS.
- P. Dapporto, A. Dei, G. Poneti and L. Sorace, Chem.–Eur. J., 2008, 14, 10915–10918 CrossRef CAS PubMed.
- R. D. Schmidt, D. A. Shultz and J. D. Martin, Inorg. Chem., 2010, 49, 3162–3168 CrossRef CAS PubMed.
- G. Poneti, M. Mannini, B. Cortigiani, L. Poggini, L. Sorace, E. Otero, P. Sainctavit, R. Sessoli and A. Dei, Inorg. Chem., 2013, 52, 11798–11805 CrossRef CAS PubMed.
- Y. Mulyana, G. Poneti, B. Moubaraki, K. S. Murray, B. F. Abrahams, L. Sorace and C. Boskovic, Dalton Trans., 2010, 39, 4757–4767 RSC.
- A. Cui, K. Takahashi, A. Fujishima and O. Sato, J. Photochem. Photobiol., A, 2004, 167, 69–73 CrossRef CAS.
- C. Carbonera, A. Dei, C. Sangregorio and J. F. Létard, Chem. Phys. Lett., 2004, 396, 198–201 CrossRef CAS.
- A. Beni, C. Carbonera, A. Dei, J. F. Létard, R. Righini, C. Sangregorio and L. Sorace, J. Braz. Chem. Soc., 2006, 17, 1522–1533 CrossRef CAS.
- A. Beni, A. Dei, S. Laschi, M. Rizzitano and L. Sorace, Chem.–Eur. J., 2008, 14, 1804–1813 CrossRef CAS PubMed.
- A. Hauser, C. Enachescu, M. L. Daku, A. Vargas and N. Amstutz, Coord. Chem. Rev., 2006, 250, 1642–1652 CrossRef CAS.
- A. Hauser, Top. Curr. Chem., 2004, 55–198 Search PubMed.
- R. V. Chamberlin, G. Mozurkewich and R. Orbach, Phys. Rev. Lett., 1984, 52, 867–870 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovic, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS PubMed.
- T. Tezgerevska, E. Rousset, R. W. Gable, G. N. L. Jameson, E. C. Sañudo, A. Starikova and C. Boskovic, Dalton Trans., 2019, 48, 11674–11689 RSC.
- J. Cirera, M. Via-Nadal and E. Ruiz, Inorg. Chem., 2018, 57, 14097–14105 CrossRef CAS PubMed.
- O. S. Siig and K. P. Kepp, J. Phys. Chem. A, 2018, 122, 4208–4217 CrossRef CAS PubMed.
- T. E. Fischer, J. T. Janetzki, F. Z. M. Zahir, R. W. Gable, A. A. Starikova and C. Boskovic, Dalton Trans., 2024, 53, 3104–3117 RSC.
- J. T. Janetzki, G. K. Gransbury, R. W. Gable, M. J. Giansiracusa, A. A. Starikova and C. Boskovic, Eur. J. Inorg. Chem., 2024, 27, e202400227 CrossRef CAS.
- M. G. Chegerev and A. A. Starikova, Comput. Theor. Chem., 2022, 1211, 113693 CrossRef CAS.
- J. Sirirak, D. Sertphon, W. Phonsri, P. Harding and D. J. Harding, Int. J. Quantum Chem., 2017, 117, 1–8 CrossRef.
- M. Swart, J. Chem. Theory Comput., 2008, 4, 2057–2066 CrossRef CAS PubMed.
- Y. Hitomi, M. Higuchi, H. Minami, T. Tanaka and T. Funabiki, Chem. Commun., 2005, 1758–1760 RSC.
- F. Z. M. Zahir, M. A. Hay, J. T. Janetzki, R. W. Gable, L. Goerigk and C. Boskovic, Chem. Sci., 2024, 15, 5694–5710 RSC.
- V. L. Nadurata and C. Boskovic, Inorg. Chem. Front., 2021, 8, 1840–1864 RSC.
- A. Arnold, T. J. Sherbow, R. I. Sayler, R. D. Britt, E. J. Thompson, M. T. Muñoz, J. C. Fettinger and L. A. Berben, J. Am. Chem. Soc., 2019, 141, 15792–15803 CrossRef CAS PubMed.
- J. Hankache and O. S. Wenger, Chem. Rev., 2011, 111, 5138–5178 CrossRef CAS PubMed.
- B. J. O. Kessler, I. F. Mansoor, D. I. Wozniak, T. J. Emge and M. C. Lipke, J. Am. Chem. Soc., 2023, 145, 15924–15935 CrossRef CAS PubMed.
- D. M. D'Alessandro, A. C. Topley, M. S. Davies and F. R. Keene, Chem.–Eur. J., 2006, 12, 4873–4884 CrossRef PubMed.
- D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424–440 Search PubMed.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed.
- P. Robin and M. B. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef.
- C. Creutz, in Progress in Inorganic Chemistry, 2007, pp. 1–73 Search PubMed.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- T. Kojima, F. Ogishima, T. Nishibu, H. Kotani, T. Ishizuka, T. Okajima, S. Nozawa, Y. Shiota, K. Yoshizawa, H. Ohtsu, M. Kawano, T. Shiga and H. Oshio, Inorg. Chem., 2018, 57, 9683–9695 CrossRef CAS PubMed.
- C. H. Yu, X. Yang, X. Ji, C. H. Wang, Q. Lai, N. Bhuvanesh and O. V. Ozerov, Inorg. Chem., 2020, 59, 10153–10162 CrossRef CAS PubMed.
- I. F. Mansoor, D. I. Wozniak, Y. Wu and M. C. Lipke, Chem. Commun., 2020, 56, 13864–13867 RSC.
- C. G. Pierpont, Inorg. Chem., 2011, 50, 9766–9772 CrossRef CAS PubMed.
- A. S. Attia, S. Bhattacharya and C. G. Pierpont, Inorg. Chem., 1995, 34, 4427–4433 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- G. K. Gransbury, M. E. Boulon, S. Petrie, R. W. Gable, R. J. Mulder, L. Sorace, R. Stranger and C. Boskovic, Inorg. Chem., 2019, 58, 4230–4243 CrossRef CAS PubMed.
- D. M. Adams and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 11515–11528 CrossRef CAS.
- A. Dei, A. Feis, G. Poneti and L. Sorace, Inorg. Chim. Acta, 2008, 361, 3842–3846 CrossRef CAS.
- D. M. Adams, A. Dei, A. L. Rheingold and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 8221–8229 CrossRef CAS.
- M. W. Lynch, R. M. Buchanan, C. G. Pierpont and D. N. Hendrickson, Inorg. Chem., 1981, 20, 1038–1046 CrossRef CAS.
- P. K. Pal, S. Chowdhury, M. G. B. Drew and D. Datta, New J. Chem., 2002, 26, 367–371 RSC.
- S. Romain, C. Baffert, C. Duboc, J. C. Leprêtre, A. Deronzier and M. N. Collomb, Inorg. Chem., 2009, 48, 3125–3131 CrossRef CAS PubMed.
- S. Romain, C. Duboc, F. Neese, E. Rivière, L. R. Hanton, A. G. Blackman, C. Philouze, J. C. Leprêtre, A. Deronzier and M. N. Collomb, Chem.–Eur. J., 2009, 15, 980–988 CrossRef CAS PubMed.
- C. C. Lu, E. Bill, T. Weyhermüller, E. Bothe and K. Wieghardt, J. Am. Chem. Soc., 2008, 130, 3181–3197 CrossRef CAS PubMed.
- C. Fleming, S. Vu, D. J. R. Brook, S. Agrestini, E. Pellegrin and J. DaRos, Front. Chem., 2023, 11, 1–14 Search PubMed.
- R. A. Zarkesh, A. S. Ichimura, T. C. Monson, N. C. Tomson and M. R. Anstey, Dalton Trans., 2016, 45, 9962–9969 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic experimental details, TGA, powder X-ray diffraction, crystallographic data, structural analysis, IR, solid- and solution-state magnetic measurements, photomagnetism, UV-vis-NIR, mixed-valency analysis, electrochemistry, and DFT (PDF). DFT optimized XYZ coordinates of complexes (ZIP). CCDC 2393551–2393561. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07798a |
| This journal is © The Royal Society of Chemistry 2025 |