
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering perfluoroarenes for enhanced molecular barrier effect and chirality transfer in solutions†
Tianhao
Wang
,
Zeyuan
Zhang
,
Aiyou
Hao
 * and
Pengyao
Xing
*
* and
Pengyao
Xing
*
School of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, PR China. E-mail: xingpengyao@sdu.edu.cn
First published on 10th January 2025
Abstract
Noncovalent forces have a significant impact on photophysical properties, and the flexible employment of weak forces facilitates the design of novel luminescent materials with a variety of applications. The arene-perfluoroarene (AP) force, as one type of π-hole/π interaction, shows unique directionality, involving an electron-deficient π-hole interacting with a π-electron-rich region, facilitating precise orientation and stabilization in supramolecular structures. Here we present an amination engineering protocol to build a perfluoroarene library based on an octafluoronaphthalene skeleton with various steric and electronic properties. In diluted solution-based assemblies, the perfluoroarenes perform as efficient molecular barriers to perylene building units, lighting up the luminescence. Enhanced steric effects, hydrophobicity and appended aromatic pendants are pivotal structural factors to boost the molecular barrier effect. Highly affinitive AP coassemblies transfer chirality from perfluoroarenes to achiral perylene moieties, inducing the appearance of chiral microstructures with tailored circularly polarized luminescence. Application as luminescent ink for enhanced water-resistance in displays and anti-counterfeiting is successfully realized. This work greatly extends the potential of molecular engineering in noncovalently bonded luminescent materials, and clearly reveals structure–property correlations.
Introduction
Noncovalent weak interactions such as van der Waals forces,1–5 π–π stacking,6–8 halogen bonds,9–13 and π-hole/π interactions14–18 drive the spontaneous formation of ordered supramolecular structures. The tunability of these forces enables precise control over relative orientation and aggregation. By modulating these weak intermolecular interactions, the luminescent properties of molecules can be effectively adjusted.19–23 This principle provides a robust framework and theoretical basis for designing intelligent, responsive, or multifunctional luminescent materials, with significant applications in sensing,24–27 imaging, and display technologies.28–31 Among these interactions, the π-hole/π interaction, with unique mode of action and strong regulatory capabilities, shows significant potential for wide-ranging applications. The π-hole/π interaction is a unique non-covalent interaction that differs from its analogue, π–π stacking. It involves a π-hole—a region of electron deficiency on an aromatic ring or an unsaturated molecule acting as an electron acceptor—interacting attractively with the π-electron-rich area of another molecule. Compared to conventional π–π interactions, the π-hole/π interaction offers greater directionality, as it depends on the spatial alignment of the electron-accepting and donating regions. This interaction is widely utilized in controlling supramolecular self-assembly,32–35 molecular recognition,36 and crystal engineering,37–41 where it stabilizes molecular structures by finely tuning the electronic characteristics and orientation of the interacting molecules.The role of arene-perfluoroarene (AP) interaction as a quintessential π-hole/π interaction is exemplified by the representative octafluoronaphthalene (OFN), which has been developed as a spatial barrier molecule to disrupt the aggregation-caused quenching (ACQ) of polycyclic aromatic hydrocarbons (PAHs), thereby enhancing luminescence.42,43 Compared to other π-hole/π interactions, like conventional charge transfer (CT) interactions,44,45 OFN can leverage the AP interaction to exploit the energy barrier effect by interrupting the conversion pathway between the singlet and triplet states of PAHs, thereby suppressing triplet-state annihilation and ultimately increasing the quantum yield. However, it still faces limitations in application and lacks structural diversity or universality in design. State-of-the-art studies have focused primarily on solid-state cocrystals, with limited exploration of assembled solutions and minimal investigation into diluted solution-based coassemblies, which suffer prominent solvation effects.36,46
In this work, a series of fluorinated compounds (guests) were synthesized through amination based on an OFN skeleton, and two model molecules (one flexible and one rigid) were employed as hosts to provide distinct assembly environments. The library encompasses a wide variety of perfluorinated compounds, differing in aromatic pendants, the presence of acidic hydrogen atoms, and varying degrees of steric hindrance (Scheme 1A). The structural diversity provides a robust foundation for investigating the universality of the barrier effect and how various factors influence AP interactions. It reveals the previously unexamined universality of modified OFN as an energy barrier in AP interactions and demonstrates precise control over the luminescent properties of materials through AP interactions. The results show that pristine OFN failed to light up the coassemblies in solution, while modification with specific steric and hydrophobic pendants would boost the molecular barrier effects to efficiently turn on the luminescence (Scheme 1B). It also facilitates chirality expression and transfer, generating chiral microstructures with emerged circularly polarized luminescence (CPL) (Scheme 1C). Leveraging this effect, luminescent materials were developed in which the host molecules were used to impregnate filter paper substrates, creating a highly luminescent canvas. The guest molecules were formulated into ink, enabling applications in digital displays and painting. Additionally, this ink demonstrated a certain level of water-resistance. This work clearly unveils the role of amination engineering in boosting the molecular barrier effects of AP coassemblies in solution, leading to the future design of supramolecular luminescent materials and devices.
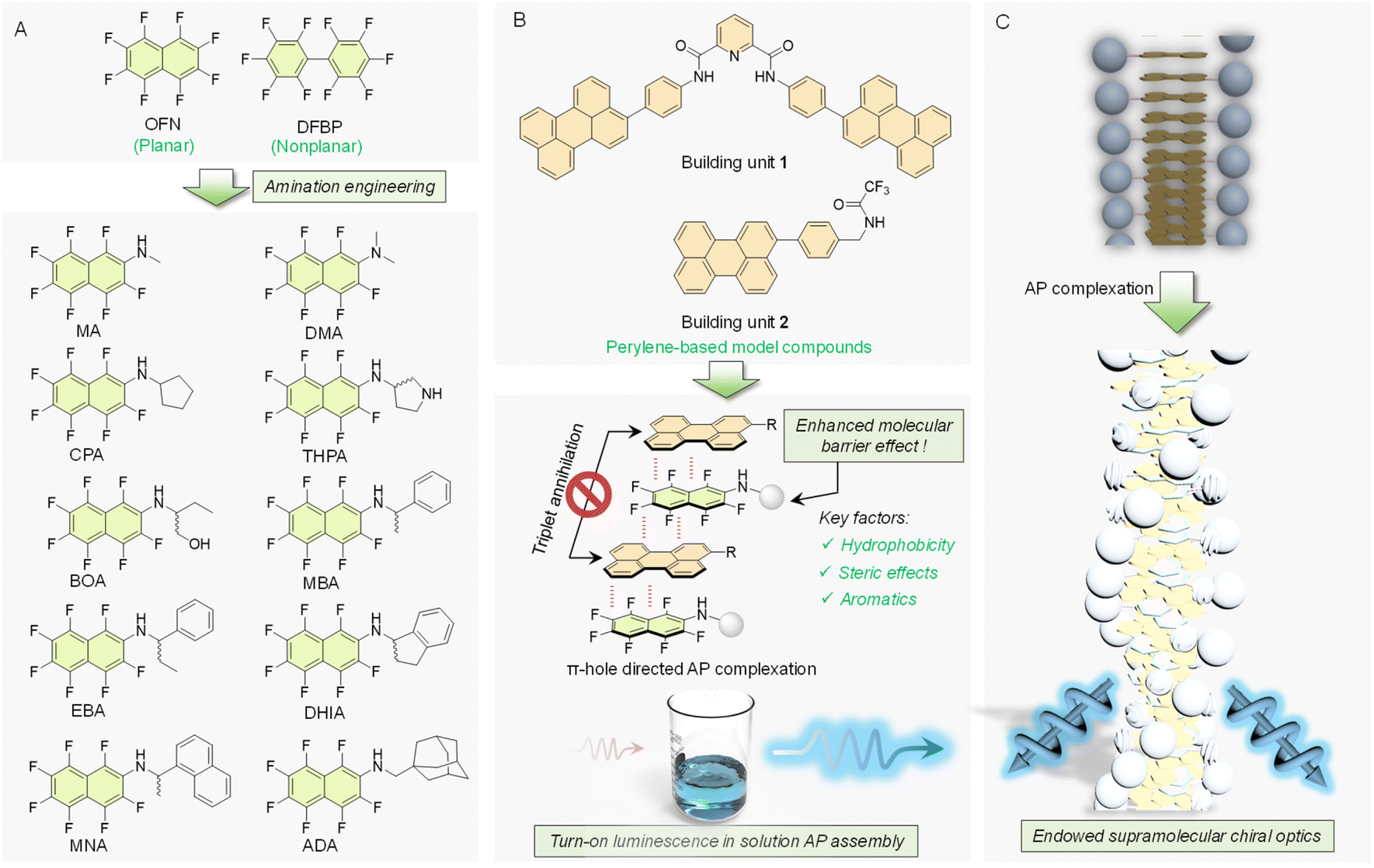 | ||
| Scheme 1 (A) A series of OFN derivatives was developed as guest molecules using a molecular engineering strategy. (B) Two perylene-based building units served as host molecules, enabling the modulation of certain properties through AP interactions. (C) An illustration depicting the chiral effects achieved by adding OFN-based compounds as barrier molecules. | ||
Results and discussion
Two host molecules were constructed using Suzuki coupling and condensation reactions, while the guest molecules were synthesized through aromatic nucleophilic substitution of OFN with various amines. The successful synthesis was confirmed through nuclear magnetic resonance (1H, 19F and 13C NMR) and mass spectrometry. Perylene-based compounds, as typical ACQ-type species,47–50 exhibit strong π–π stacking in aggregated states—such as in solid states and assembled solution phases—leading to pronounced fluorescence quenching. An energy barrier molecule, OFN, has been developed to interact with perylene through AP interactions, disrupting perylene aggregation. Through a periodic arrangement of π-conjugated molecular arrays, this molecule effectively isolates interactions between luminophores. We employed this strategy to enhance the luminescence of the two host molecules in diluted solutions (0.05 mmol L−1 in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 2:8, v/v); however, no turn-on luminescence was observed, highlighting the limitations of this barrier molecule (OFN) in solution. Zhang et al. observed the effective barrier effect of OFN in cocrystals with a series of PAHs, including perylene, pyrene, coronene and anthracene.42 The failed barrier effect of OFN in the present case might be caused by the strong solvation effect. The basic principle of the molecular barrier is based on a higher energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), referred to as the energy gap. By periodically inserting OFN between polycyclic aromatic hydrocarbons (PAHs), the increased energy gap effectively prevents the exchange of energy and electrons between the chromophores. An attempt was also made with the analogue, decafluorobiphenyl (DFBP), which has a larger energy gap (5.6 eV; the energy gap of OFN is 4.6 eV). However, it did not exhibit an effective shielding performance. Therefore, a series of modifications were made to OFN, aiming to enhance its barrier function through spatial optimization to overcome its limitations.
:H2O = 2:8, v/v); however, no turn-on luminescence was observed, highlighting the limitations of this barrier molecule (OFN) in solution. Zhang et al. observed the effective barrier effect of OFN in cocrystals with a series of PAHs, including perylene, pyrene, coronene and anthracene.42 The failed barrier effect of OFN in the present case might be caused by the strong solvation effect. The basic principle of the molecular barrier is based on a higher energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), referred to as the energy gap. By periodically inserting OFN between polycyclic aromatic hydrocarbons (PAHs), the increased energy gap effectively prevents the exchange of energy and electrons between the chromophores. An attempt was also made with the analogue, decafluorobiphenyl (DFBP), which has a larger energy gap (5.6 eV; the energy gap of OFN is 4.6 eV). However, it did not exhibit an effective shielding performance. Therefore, a series of modifications were made to OFN, aiming to enhance its barrier function through spatial optimization to overcome its limitations.
Initially, the feasibility of the design of aminated OFN derivatives was validated through a series of density functional theory (DFT) calculations. At the B3LYP-D3(BJ)/6-311G-(d,p) level, the electrostatic potential maps (ESPs) of each molecule, were calculated (Fig. S1†). The electron-deficient center of OFN exhibits an electrostatic potential as high as 21 kcal mol−1, while the electron-rich center of compound 2 reaches −37 kcal mol−1. Subsequently, the binding energies between each host–guest pair were calculated, and the interaction region indicator (IRI) method was employed to visualize the chemical bonds and weak interactions within the system (Fig. S2 and S3†). In the IRI, the green regions between the two segments indicate the presence of strong interactions, further supporting the feasibility of our proposed approach. Simultaneously, we calculated the binding energies between the host and guest molecules under the same basis set conditions. The corresponding data are summarized in Fig. 1D, indicating that the binding energies range approximately from 15 to 27 kcal mol−1. This is clearly stronger than a typical π–π stacking interactions, providing additional evidence in support of our design.
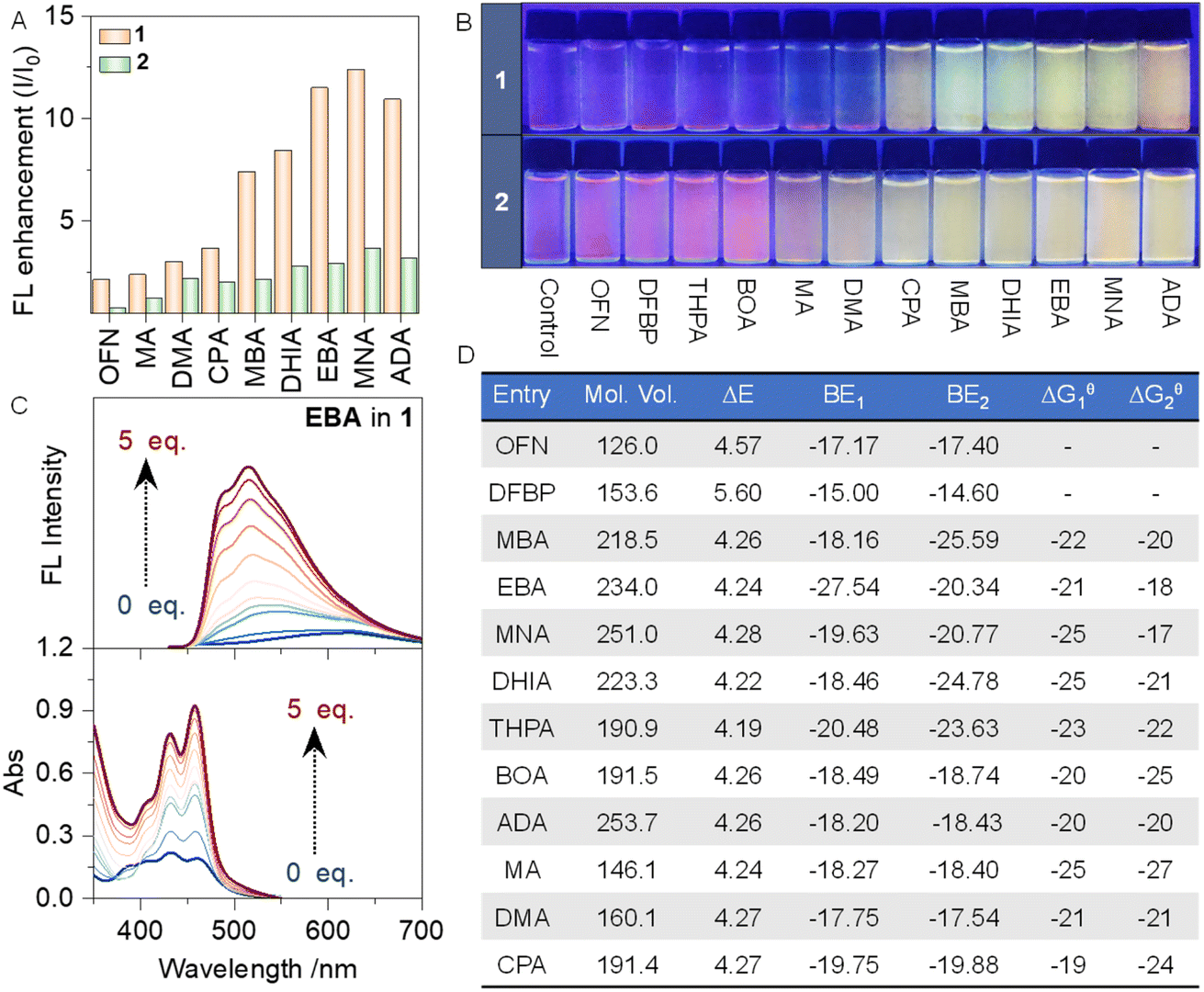 | ||
| Fig. 1 (A) Bar chart showing the fluorescence enhancement multiples of different systems. (B) Photographs of different system solutions under UV irradiation (0.05 mmol L−1 in MeCN:H2O = 2:8, v/v). (C) Fluorescence emission spectra and UV-vis absorption spectra of 1-EBA (0.05 mmol L−1 in MeCN:H2O = 2:8). (D) Summary table of data for each system (Mol. Vol. = molecule volume/cm3 mol−1, ΔE = energy gap = ELUMO − EHOMO/eV mol−1, BE = binding energy/kcal mol−1, ΔGθ = change in Gibbs free energy/kJ mol−1). | ||
In addition to the theoretical validation, experimental studies were carried out simultaneously. The fluorescence emission spectrum of the host molecule was measured with a fixed concentration of 0.05 mmol L−1 and an excitation wavelength of 400 nm. Subsequently, 5 molar eq. of the guest molecules DFBP and OFN were added to the host solution, and fluorescence tests were conducted again under the same conditions (Fig. S5 and S6†). A broad and strong fluorescence emission peak appeared beyond 450 nm, which can be readily identified as the characteristic emission peak of the perylene moiety. From the normalized fluorescence spectra, we observed a certain degree of blue shift in the system upon the addition of OFN compared to the host alone, while no significant changes were observed with DFBP. The blue shift is a typical characteristic of AP complexation. In contrast, the lack of significant interaction with DFBP is attributed primarily to the mismatch between the face-to-face interaction mode of π-hole/π interactions and the twisted conformation of DFBP. This highlights the high sensitivity of AP interactions to conformational matching, a key feature of AP interactions. Based on this, a series of OFN derivatives were designed (Scheme 1), and the fluorescence emission spectra were collected (Fig. S4–S15†) to statistically analyze the fluorescence intensity enhancement factors for each system (Fig. 1A). Under UV lamp irradiation at 365 nm (Fig. 1B), they clearly illustrate the fluorescence changes of the various systems. The corresponding UV-vis spectra of the systems were also measured simultaneously (Fig. S16–S27†), with the titration curve of the 1-EBA system presented in Fig. 1C. As the guest molecules were introduced, a significant enhancement in both UV absorption and fluorescence was observed, accompanied by a blue shift in the fluorescence spectrum. This demonstrates the successful use of the modified OFN as a barrier molecule. Interestingly, computational results indicate that, from an energetic perspective, the energy gap of the amination-modified guest molecules is less favorable for molecular barrier effects than OFN (Fig. S28†). However, these modified guest molecules exhibit superior performance that OFN cannot match. Although our strategy slightly reduces the energy gap of the OFN fragment, it remains greater than the energy gaps of the two host molecules, thereby allowing it still to function as a molecular barrier from an energetic standpoint. Additionally, the molecular engineering strategy of amination introduces another influencing factor: steric hindrance. This spatial hindrance can force the two phenanthrene segments to move apart, thereby facilitating the molecular barrier function from another angle. Subsequently, the influence of the structure–activity relationship was analyzed.
Based on the type of substituents, the guest molecules can be classified into three categories: aliphatic type, which include MA, DMA, CPA, and ADA; aromatic type, including MBA, EBA, MNA, and DHIA; and acidic hydrogen type, which include BOA and THPA. The third type, due to the presence of acidic hydrogen in the side chains, leads to increased water solubility, which hinders effective aggregation with the host. Therefore, it is not surprising that this category exhibits inferior performance. In the first type, it is evident that the main difference between the four molecules lies in the size of their steric hindrance, which increases in the following order: MA, DMA, CPA, and ADA. Correspondingly, the fluorescence enhancement factors also follow this order, demonstrating that greater steric hindrance is beneficial for fluorescence enhancement. Introducing aryl groups into the second type of guest molecule resulted in a substantial increase in fluorescence enhancement. This can be attributed to the positive influence of π–π stacking or π–hydrogen bond interactions. However, expanding the benzene ring to a naphthalene ring did not yield better results compared to the benzene ring. This indicates that, while the presence of the aryl structure is crucial, the extent of the conjugated system has only a subtle impact. Subsequently, the binding constant of the host–guest interaction was determined by fitting the UV-vis titration, and the corresponding ΔGθ of the interaction was calculated based on the binding constant (Fig. 1D; the fitting method is detailed in the ESI†). It can be observed that, regardless of the steric hindrance of the guest molecules in the system, ΔGθ remains stable at approximately 17–27 kJ mol−1. Simultaneously, fluorescence titration was also utilized to perform fitting and calculate the corresponding Gibbs free energy. However, due to the numerous factors affecting fluorescence, this data is provided for reference only (Fig. S4–S15†).
Additionally, the molecular volumes (MV) of the guest molecules were calculated to provide a characterization of steric hindrance, and their relationships with both fluorescence enhancement factors and binding energies were evaluated (Fig. 2A). As the molecular volume increases, the degree of fluorescence enhancement also rises. For 2, the interaction with the guest molecules exhibits a linear correlation, whereas for 1, the slope suddenly increases around a molecular volume of 190 cm3 mol−1. For guest molecules with volumes smaller than 190 cm3 mol−1, the substituents are alkyl groups, while aryl-substituted derivatives appear after 190 cm3 mol−1. This suggests the presence of other non-covalent interactions of aryl groups, which enhance the AP interaction, leading to increased fluorescence. Compound 2 does not show sensitivity to aryl groups compared to 1, due to 1 possessing a fully rigid structure: starting from the pyridine fragment, the nitrogen atom forms hydrogen bonds with the acidic hydrogen on the amide at the α-position, which locks the two side chains. The amide group is further stabilized by conjugation through the benzene ring. In contrast, 2 exhibits weaker intermolecular interactions and is significantly smaller and less rigid than 1. It is precisely this rigid nature of 1 that allows it to establish stronger weak interactions with guest molecules, making it more responsive to aryl groups. In the plot of the relationship between binding energy and molecular volume (Fig. 2B and C), the correlation is moderate due to the presence of multiple influencing factors. However, a general trend of increasing binding energy with larger molecular volume can be observed, revealing the prominent steric effects.
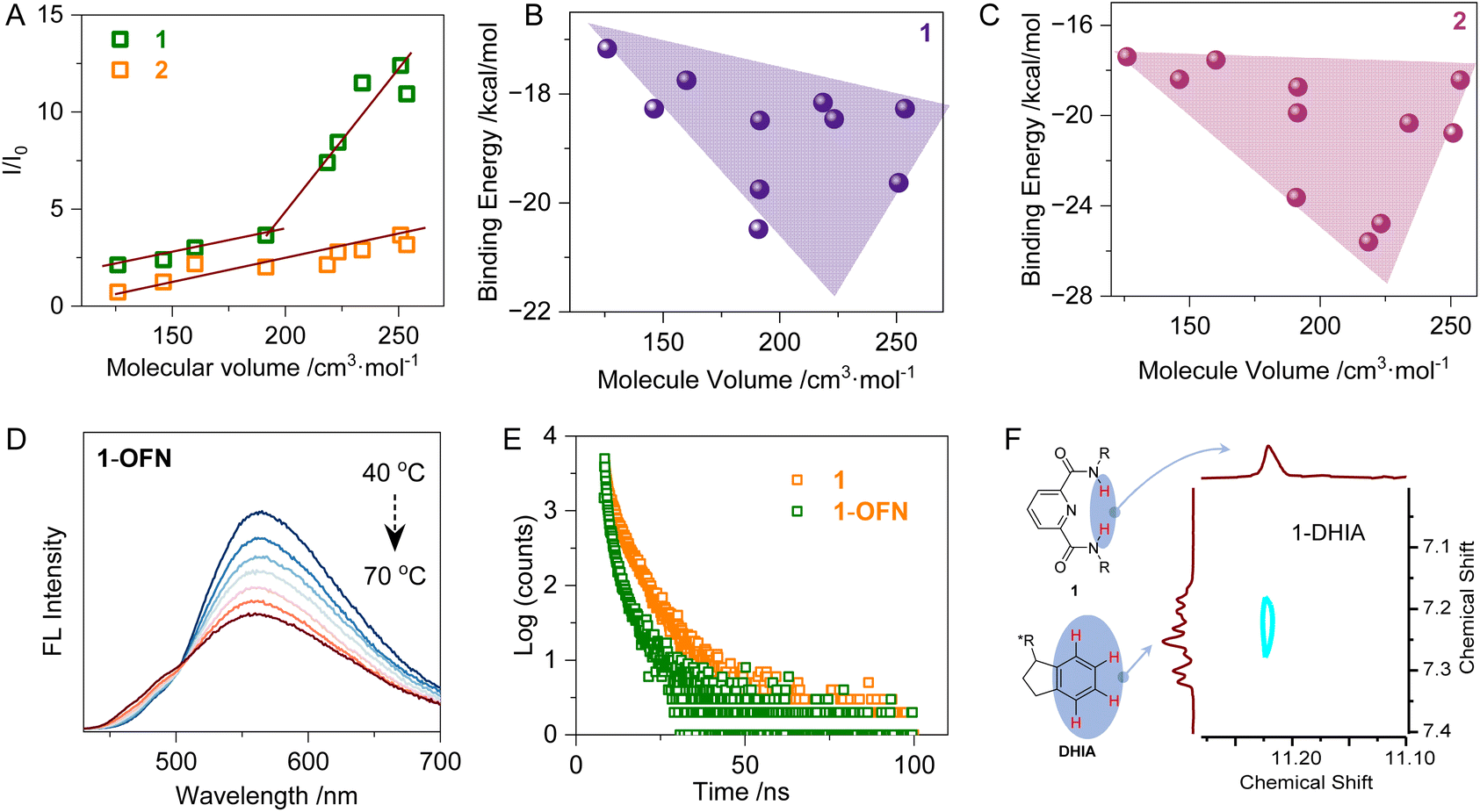 | ||
| Fig. 2 (A) Plot of the relationship between molecular volume and fluorescence enhancement factor. (B) Plot illustrating the correlation between molecular volume and fluorescence enhancement for Host 1. (C) Plot depicting the correlation between molecular volume and fluorescence enhancement for Host 2. (D) Temperature-dependent fluorescence spectra for 1-OFN (0.05 mmol L−1 in MeCN:H2O = 2:8, and 5 eq. OFN as 1, excitation wavelength 400 nm). (E) Fluorescence decay curve of 1 and 1-OFN (0.05 mmol L−1 in MeCN:H2O = 2:8, and 5 eq. OFN as 1, excitation wavelength 400 nm, and probe wavelength 550 nm). (F) NOESY spectra of DHIA with Host 1 (1.0 mmol L−11 in DMSO-d6:CDCl3 = 1:1, and 15 eq. DHIA as 1). | ||
To further investigate the luminescent properties, emission lifetimes and temperature-dependent fluorescence emission spectra were obtained (Fig. 2D and E). Most systems exhibited a trend of thermal quenching in fluorescence emission (Fig. S29†). As the temperature increased, the enhanced thermal motion of the molecules led to more frequent collisions between molecules and with the solvent, which in turn increased non-radiative transitions. Consequently, fluorescence emission was reduced. Interestingly, in the system of 2 and DHIA, the fluorescence exhibited thermal enhancement. During the heating process, two competing effects are at play: one is the enhancement of non-radiative transitions, which causes fluorescence quenching, and the other is the disaggregation of the molecular assembly due to enhanced supramolecular interactions and solvation. In most systems, the result of this competition is fluorescence thermal quenching. However, in the case of DHIA, the presence of π–π stacking leads to a more pronounced disaggregation effect. This favors the latter process, resulting in weaker thermal quenching for the 1-DHIA system compared to 1-OFN. In the 2 system, due to its flexibility, disaggregation is relatively weak at room temperature, but upon heating, there is more space for disaggregation to occur. The synergy between these factors leads to the observed fluorescence thermal enhancement in the 2-DHIA system. To further investigate this, nuclear overhauser effect spectroscopy (NOESY) was conducted, providing insights into the H–H correlations between DHIA and each of the 1 and 2 (Fig. 2F and S30†). A peak was observed in the NOESY spectrum between the hydrogens on the amide of the host and the aromatic hydrogens of DHIA, confirming the existence of N–H⋯π interactions between the two molecules. Additionally, through variable-temperature NMR analysis (Fig. S30†), we observed a high-field shift in both the amide hydrogen and aromatic hydrogen with increasing temperature. This phenomenon can be attributed to the disruption of hydrogen bonding as the temperature rises, a behavior consistent with the characteristics of N–H⋯π interactions, further supporting their presence. The fluorescence lifetimes of all the systems are in the nanosecond range (Fig. 2E and S31†). Most systems exhibit a reduction in fluorescence lifetime after the guest molecules are incorporated into the assemblies. This could be attributed to the presence of AP interactions, which accelerate the internal conversion of the host molecules. Additionally, the guest molecules may play a role in stabilizing the conformation of the host, reducing non-radiative transitions, and consequently leading to a shortened fluorescence lifetime. Interestingly, in the 2-OFN system, due to the flexibility of 2, the addition of OFN does not lead to a more rigid conformation. Instead, it increases the opportunities for non-radiative transitions, resulting in an extended fluorescence lifetime. In addition, the 1H NMR spectra of each system were measured in a deuterated DMSO:CHCl3 (1:1 v/v) solvent mixture. The aromatic protons of the two host molecules exhibit distinct degrees of high-field shifts. This phenomenon is further corroborated by observations in solid-state NMR, which provide strong support for the presence of AP interactions (Fig. S32†).
To further investigate how AP interactions affect molecular aggregation in the assembled state, molecular dynamics (MD) simulations were performed (Fig. 3 and S33†). Building units were inserted into the box at a low density to reflect its relatively low solubility. Water was then fully added to the box as the solvent. The particle mesh ewald (PME) method and the Verlet list scheme were selected to handle electrostatic and non-bonded interactions, respectively. Energy minimization was performed using the steepest descent method. For temperature control, the improved Berendsen thermostat, V-rescale, was chosen with the reference temperature set to 298.15 K. For pressure control, the Parrinello–Rahman scheme was applied, maintaining a uniform pressure of 1 bar in all directions. Under these conditions, MD simulations were conducted for the two hosts and their assembly systems with OFN and ADA, respectively. The time step was set to 0.001 ps, with a total of 15,000,000 steps, corresponding to a total simulation time of 15 ns. It can be observed that the potential energy stabilized within several hundred nanoseconds of the simulation (Fig. 3D). Overview snapshots were taken at 0 ns and 15 ns, and a close-up view of the 15 ns results is provided (Fig. 3A–C). In 1, distinct π–π stacking is observed between perylenes and picolinamide cores. When OFN is introduced, it binds to 1 through AP interactions and inserts itself between the two perylene units, thereby disrupting the aggregation of 1. When ADA is introduced, AP interactions also occur. However, due to the spatial hindrance caused by the side chains of the guest molecules, continuous aggregated structures do not form; instead, they disperse into individual AP-bound complexes. This also results in the 1-ADA system having a slightly larger solvent accessible area compared to the 1-OFN system (Fig. 3F). With the insertion of the guest molecules, the reduction in solvent accessible area at equilibrium is smaller than that of the system containing 1 alone, indicating that the degree of aggregation in the system is reduced. The radial distribution function (RDF) was also investigated (Fig. 3E). The carbon atom closest to the negatively charged center of the perylene fragment was designated (Fig. S34†), and its RDF results were derived to represent the distribution between the perylene units of different hosts. The first peak was identified as the shortest distance between the two perylene units, serving as an indicator of the degree of π–π stacking. In the systems with added OFN and ADA, the peak shifts to the right, indicating that the presence of the guest molecules weakens the aggregation of the host molecules. In the 1-OFN system, an interaction mode with distance 4.56 Å was observed, representing the distance between two perylene units following insertion of OFN. However, in the 1-ADA system, this peak is not prominent due to the lack of continuous aggregation. A similar pattern is observed in 2.
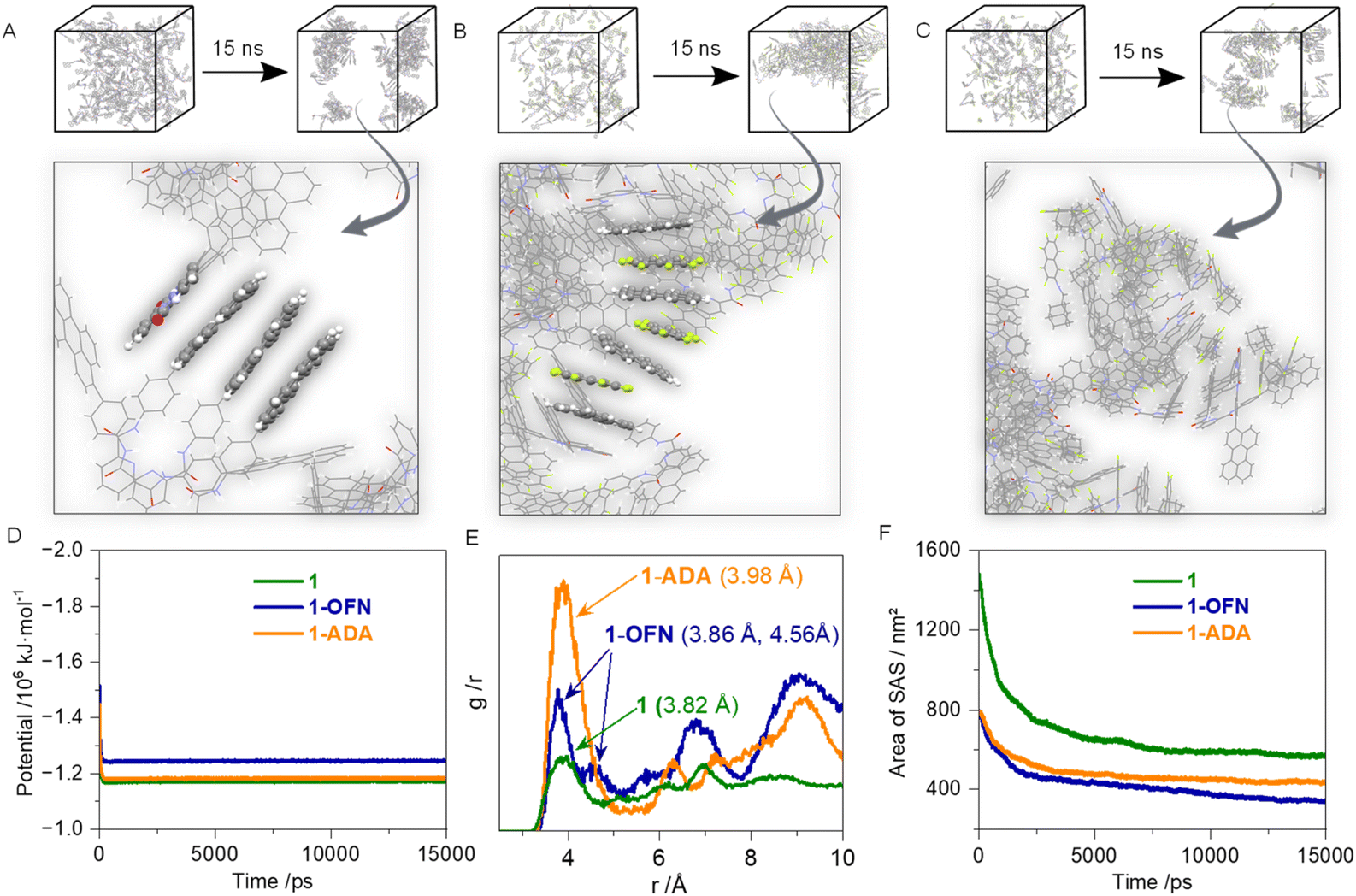 | ||
| Fig. 3 MD simulation. (A) System of host 1. (B) System of host 1 and OFN. (C) System of host 1 and ADA. (D) Potential energy of the process. (E) Radial distribution function (RDF) profiles. (F) Solvent accessible area. | ||
To gain a deeper understanding of the components of the AP interactions, energy decomposition was performed using symmetry-adapted perturbation theory (SAPT) at the SAPT2+/aug-cc-pVDZ basis set level (Fig. 4A, B and S35†). The AP interaction is decomposed into four components: dispersion, electrostatics, induction, and exchange, with the first three contributing to stabilization. In the 1-DHIA coasssembly, the dispersion contribution to the AP interaction is significantly increased compared to OFN, while the electrostatic contribution is reduced. This suggests a tighter arrangement in the 1-OFN system, whereas in 1-DHIA, the steric hindrance from the side chains leads to a looser arrangement, which more effectively disrupts the aggregation of the system. The transient absorption (TA) spectrum of the system was also tested to explore the interaction mechanism of the guest molecules (Fig. 4C–H and S36†). At 0 ns, representing the ground state, no absorption peak is observed at 700 nm. Upon excitation of the system with 350 nm pump light around 1 ps, the 0.1 ns mark represents the absorption of the excited state, and this peak is attributed to the S1–Sn transition of the local excited state. The negative absorption observed before 600 nm is caused by the fluorescence emission of the molecule itself. As time progresses, the absorption peak gradually weakens, and by around 7 ns, it has almost completely decayed and disappeared. It should be noted that during the decay process, the absorption peak also undergoes a slight red shift. This occurs because, after reaching the excited state, the changes in the permanent dipole moment alter the interactions between the host and guest molecules. As a result, the most stable binding geometry in the ground state no longer corresponds to the equilibrium configuration in the excited state, leading to disruption in the molecular alignment. Then, the system relaxes towards a new stable configuration, leading to the slight red shift in the absorption peak. Subsequently, the decay process was subjected to kinetic fitting, yielding the time constant for this process, which represents the average lifetime of the excited state. It can be observed that with the addition of OFN and DHIA, the excited-state lifetime of 1 is significantly reduced. This is because the rate at which excited-state 1 returns to the ground state increases after binding with the guest molecules, with the effect being particularly pronounced in the case of DHIA.
 | ||
| Fig. 4 (A) Energy decomposition of the AP interaction between 1 and OFN. (B) Energy decomposition of the AP interaction between 1 and OFN. (C and F) Transient absorption spectrum and kinetic fitting of 1, following photoexcitation with ∼70 fs ultraviolet pulses at 350 nm. (D and G) Transient absorption spectrum and kinetic fitting of 1-OFN, following photoexcitation with ∼70 fs ultraviolet pulses at 350 nm. (E and H) Transient absorption spectrum and kinetic fitting of 1-DHIA. | ||
After establishing the structure–property relationship, the microscopic morphology and structure of the assemblies were further investigated using a scanning electron microscope (SEM) and small-angle X-ray scattering (SAXS) (Fig. 5). The system was assembled at a concentration of 0.05 mmol L−1 in an MeCN:H2O = 2:8 (v/v) solvent mixture. The morphology of individual 1 after assembly was observed to be a rod-like structure (Fig. 5A). In its SAXS pattern (Fig. S37†), three peaks with a ratio of 1q:2q:3q were identified, confirming a layered stacking arrangement. Thus, it can be reasonably inferred that 1 forms the rod-like structure through this layered stacking mode. Upon the addition of OFN and chiral DHIA, no significant changes in the microscopic morphology were observed. However, SAXS also confirmed the presence of a hexagonal packing mode. When ADA, with a larger steric hindrance, was added, the morphology similarly showed no significant changes. Systems with other guest molecules were also tested and found to exhibit rod-like structures (Fig. S38†). The likely reason, as frequently mentioned earlier, is the relatively rigid structure, which leads to a more rigid stacking arrangement. This characteristic is further demonstrated in the subsequent discussion on chiral transfer. In contrast, the incorporation of guest molecules into the more flexible conformation of 2 showed a significant response. 2 itself forms long, sheet-like structures. Upon the addition of OFN, the sheets transformed into circular shapes. When chiral DHIA was added, elliptical sheets were formed, intertwining in a chiral manner. RDHIA induced a right-handed helix, while SDHIA induced a left-handed helix. Other chiral guest molecules also successfully yielded chiral sheet-like dimeric microstructures, indicating an evolution of chirality in the guest molecules, transitioning from the inherent point chirality of the guest molecules to a higher-order supramolecular chirality (Fig. S39†). Simultaneously, another intriguing phenomenon was observed: as the steric hindrance of the added guest molecules increased, the aspect ratio of the assembled structures gradually increased (Fig. S40†). It is reasonable to infer that greater steric hindrance from the guest molecules hinders lateral aggregation, leading to geometries with larger length-to-width ratios.
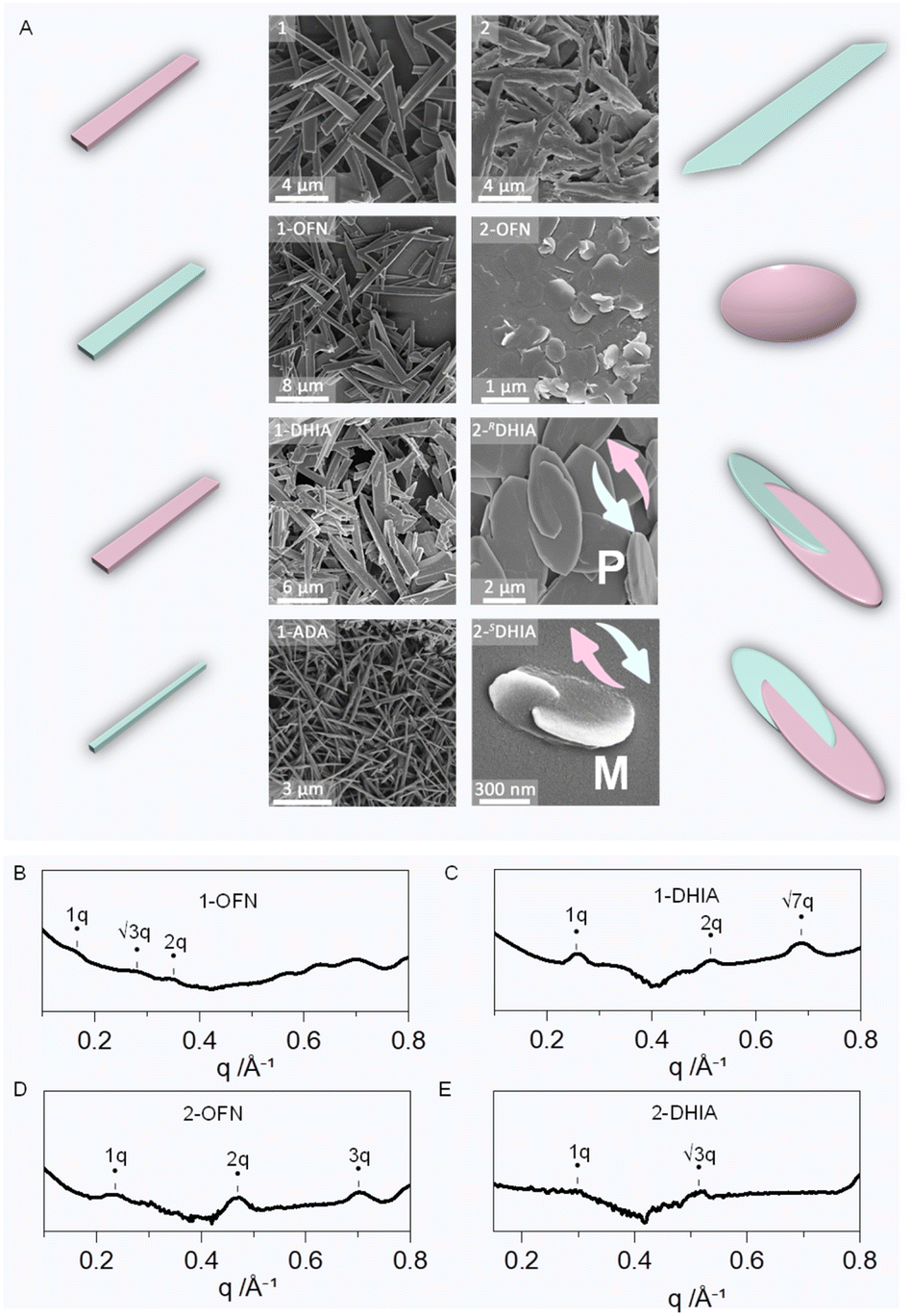 | ||
| Fig. 5 (A) Scanning electron microscope (SEM) images of the assemblies of different systems (0.1 mmol L−1 in MeCN:H2O = 2:8, and 5 eq. guest as the host). (B) Small-angle X-ray scattering (SAXS) pattern of 1-OFN. (C) SAXS spectra of 1-DHIA. (D) SAXS spectra of 2-OFN. (E) SAXS spectra of 2-DHIA. | ||
The chiroptical properties of the systems were evaluated. A multi-peaked exciton band around 450 nm can be observed, which corresponds to the characteristic absorption peaks of the perylene of 1. In the CD spectrum, the signal around 350 nm can be attributed to the Cotton effect of the chiral guest molecules. Additionally, in the 2-MBA system, CD signals appear around 450 nm, which fall within the absorption region of the perylene moiety. This indicates that chirality has been transferred from the guest to the host. The reason for this can be attributed to the spatial geometry during the assembly process, where the guest and host are brought closer together by the AP interaction. The chiral guest, lacking a second-order symmetry element, fails to provide a symmetrical spatial environment for the host. This asymmetry in the electronic distribution of the guest induces an asymmetric deformation of the host's phenanthrene segment, leading to a certain selectivity in chiral absorption. Consequently, a Cotton effect is observed in the absorption region of the phenanthrene rings, facilitating the transfer of chirality from the guest to the host through non-covalent interactions. In the assemblies of 2 with other guest molecules, only the system with BOA did not achieve chirality transfer. This is because BOA contains a hydroxyl group, which imparts greater hydrophilicity compared to the other guest molecules, leading to less effective assembly. Consequently, chirality transfer did not occur. Furthermore, no Cotton effect was observed in the absorption region of perylene for the systems involving 1, which aligns with the earlier observations from SEM regarding the relatively rigid structure. The chiroptical luminescent properties of the systems were investigated (Fig. S41†). In the systems where chirality transfer occurred, CPL was achieved. The fluorescence emission of 2-DHIA at 550 nm exhibited strong circular polarization, with a glum value reaching as high as 0.02 (Fig. 6I). Thus, the system has achieved chirality transfer and CPL, while also demonstrating the evolution of chirality in its micro-morphology.
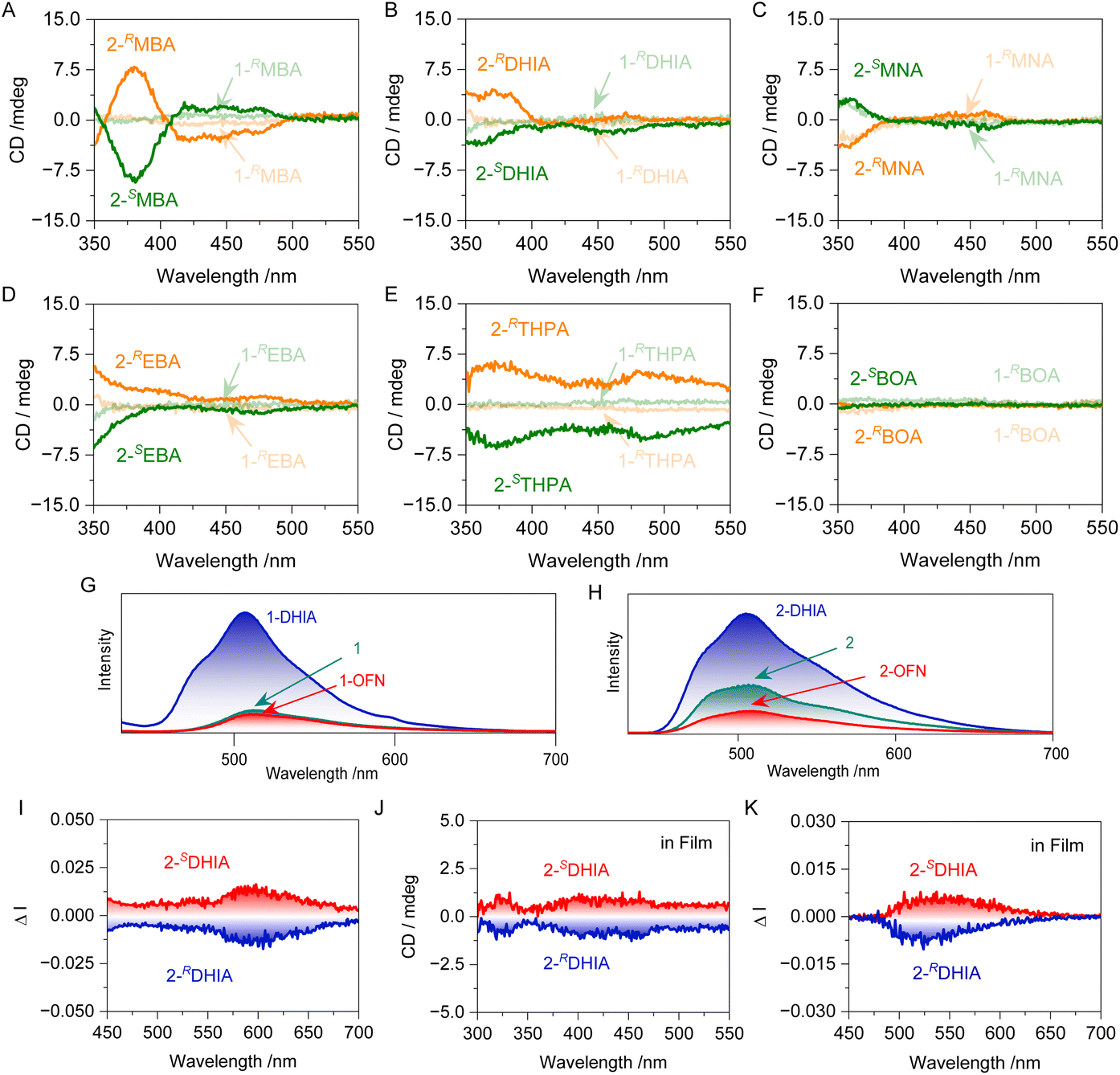 | ||
| Fig. 6 (A–F) Circular dichroism (C and D) spectra of different systems in an MeCN:H2O = 2:8 (v/v) solvent. (G) Fluorescence of system 1 in thin film. (H) Fluorescence of system 1 in thin film. (I) CPL spectra of 2-DHIA. (J) CD spectra of 2-DHIA in thin film. (K) CPL spectra of 2-DHIA in thin film (all the solutions mentioned above are 0.05 mmol L−1 MeCN:H2O = 2:8 (v/v) solutions, with a concentration of 5 × 10−7 mol g−1 in the film and the guest molecules present in a 5-fold excess in the assembly systems). | ||
To facilitate the practical application of the molecular barrier strategy, we undertook the following attempts. Given the excellent performance of films in various fields, particularly in luminescent materials, this strategy was initially tested for application in films. PMMA was dissolved in tetrahydrofuran (THF), and this solution was used as the stock solution to prepare solution 2. After placing the solution in a mold and allowing it to dry at room temperature, the film was obtained by demolding, with a concentration of 2 in the film of 5 × 10−7 mol g−1. The fluorescence emission of the film was tested to study the molecular barrier effect in polymer solution, yielding results consistent with those observed in the solution-based assemblies (Fig. 6G and H). Further testing of the CD and CPL (glum = 0.02) of 1-DHIA produced ideal chiral signals (Fig. 6J and K), suggesting that the molecular engineering strategy established in solution also holds true in polymer solution. In contrast, its performance as a pigment and ink was also investigated. The host molecules were dissolved in THF as a 0.05 mmol L−1 solution and used to impregnate filter paper, which was then dried. The process was repeated with a THF solution of other guests, resulting in filter paper that exhibited different colors (Fig. 7A). Under UV light irradiation at 365 nm, photographs clearly reveal the differences in color of the filter paper soaked in various systems. Selecting the well-performing DHIA as the primary test target allowed for the display (Fig. 7C). We prepared filter paper containing compound 1 as a display module, and then selectively applied DHIA as a fluorescent ink on the module, achieving a digital display transition from 888 to 666. Additionally, another intriguing phenomenon was observed: the incorporation of DHIA enhanced the adsorption capacity of 1 on the filter paper. After washing, 1-DHIA was significantly retained, while most of 1 was washed away, thereby improving the contrast of the display. This effect may be attributed to DHIA's exceptional hydrogen bonding potential as a derivative of perfluorinated aromatics, facilitating interactions with the hydroxyl groups on the filter paper fibers. Furthermore, the interaction with the host via the AP effect serves as a bridge, enhancing the adsorption capacity of the material on the filter paper. In contrast, we explored its potential as a fluorescent ink. Application tests using stamping and painting methods demonstrated its excellent potential for use, successfully achieving high-contrast stamp impressions and smooth, clear painting, while also exhibiting a degree of wash resistance (Fig. 7D and E). This paves the way for further exploration of the interactions between host–guest systems and their applications in various fields.
 | ||
| Fig. 7 (A) Images of the impregnated filter paper under 365 nm UV lamp. (B) Images of the PMMA film containing 1 under sunlight and 365 nm UV light. (C) Fluorescent images of the filter paper contaminated with 1, displayed with DHIA and after washing with water. (D) Fluorescent images showing the effects of stamping and washing. (E) Fluorescent images of drawings made on filter paper 1 using DHIA ink. | ||
Conclusions
Perfluoroarenes, as a crucial subset of π-hole⋯π interactions, represent AP interactions that are widely applied in supramolecular and topochemical fields, with OFN serving as a typical example, which functions as an energy barrier molecule. However, research on their assembly in solution is limited, and there is a lack of attempts to diversify the modification of OFN. Therefore, the universality of OFN as an energy barrier molecule is also subject to verification. This study designed and synthesized a series of OFN derivatives, thoroughly investigating the relationship between their structures and their roles in AP interactions. Moreover, it has successfully transcended the limitations of OFN as a barrier molecule in solution. Additionally, the work considered the environment of the aromatic compounds interacting with OFN, designing both soft and hard guest molecules. A library of barrier molecules capable of adapting to diverse, complex environments in assembled solutions was successfully developed using an amination strategy through molecular engineering. It successfully achieved chirality transfer from guest to host, as well as the evolution of chirality from point chirality in the guests to the chiral micro-morphology and CPL. Finally, a display method was developed, utilizing different guests to create varying colors and brightness in the host, along with a highly luminous and water-resistant fluorescent ink, significantly expanding the application potential of the system in fluorescent displays. In summary, this study confirms the potential of the OFN fragment as a molecular barrier, demonstrating its effectiveness in assembled solutions and successfully overcoming limitations posed by complex environments. The findings provide valuable insights into the use of OFN-series compounds as energy barrier molecules and components within AP interactions, establishing their potential as reliable linking tools in supramolecular chemistry. Moreover, this research enhances the understanding of these compounds as barrier molecules capable of mitigating ACQ effects and improving quantum yield, thus envisioning promising applications in the development of advanced luminescent materials.Data availability
The data supporting this article have been included as part of ESI.†Author contributions
T. W.: conceptualization, investigation, formal analysis, methodology, resources, writing, supervision, funding acquisition, review & editing; Z. Z.: investigation; A. H.: conceptualization, funding acquisition, supervision, methodology, review & editing; P. X.: conceptualization, funding acquisition, resources, conceptualization, supervision, review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is also supported by the National Natural Science Foundation of China (No. 21901145 and 22171165). We also acknowledge the financial support from Youth cross-scientific innovation group of Shandong University (2020QNQT003) and the project of construction and management research of laboratory of Shandong University (sy20202202).Notes and references
- X. Kou, X. Xu, N. Gao, Y. Zhang, X. Huang, F. Chen, Q. Ke and Q. Meng, Food Hydrocolloids, 2024, 157, 110441 CrossRef CAS.
- Z. Liu, X. Dai, Y. Sun and Y. Liu, Aggregate, 2020, 1, 31 CrossRef.
- L. Yao, K. Fu, X. Wang, M. He, W. Zhang, P. Y. Liu, Y. P. He and G. Liu, ACS Nano, 2023, 17, 2159 CrossRef CAS PubMed.
- L. Xu, X. Miao, X. Ying and W. Deng, J. Phys. Chem., C, 2011, 116, 1061 CrossRef.
- N. Nerngchamnong, L. Yuan, D. C. Qi, J. Li, D. Thompson and C. A. Nijhuis, Nat. Nanotechnol., 2013, 8, 113 CrossRef CAS PubMed.
- X. Li, W. Ge, S. Guo, J. Bai and W. Hong, Angew. Chem., Int. Ed., 2023, 62, e202216819 CrossRef CAS PubMed.
- J. H. Mei, S. Lai, Y. N. Gong, W. J. Shi, J. H. Deng, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2024, e202413413 Search PubMed.
- G.-L. Li, Z. Zhuo, B. Wang, X.-L. Cao, H.-F. Su, W. Wang, Y.-G. Huang and M. Hong, J. Am. Chem. Soc., 2021, 143, 10920 CrossRef CAS.
- S. An, A. Hao and P. Xing, ACS Nano, 2021, 15, 15306 CrossRef CAS.
- Y. Xu, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2022, 61, e202113786 CrossRef CAS PubMed.
- A. G. Slater, L. M. A. Perdigão, P. H. Beton and N. R. Champness, Accounts Chem. Res., 2014, 47, 3417 CrossRef CAS PubMed.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072 CrossRef CAS.
- H. Xu, J. Cheng, X. Yang, Z. Liu, W. Li and Q. Li, ChemPhysChem, 2017, 18, 2442 CrossRef CAS PubMed.
- W. Wang, W. X. Wu, Y. Zhang and W. J. Jin, Chem. Phys. Rev., 2024, 5, 031303 CrossRef CAS.
- H. Ju, B. Wang, M. Li, J. Hao, W. Si, S. Song, K. Mei, A. C. Sue, J. Wang, C. Jia and X. Guo, J. Am. Chem. Soc., 2024, 146, 25290 CrossRef CAS PubMed.
- D. P. Sukumaran, K. Shoyama, R. K. Dubey and F. Wurthner, J. Am. Chem. Soc., 2024, 146, 22077 CrossRef.
- A. Hori, A. Shinohe, M. Yamasaki, E. Nishibori, S. Aoyagi and M. Sakata, Angew. Chem., Int. Ed., 2007, 46, 7617 CrossRef CAS.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352 CrossRef CAS PubMed.
- Y. Wang, H. Wu, W. Hu and J. F. Stoddart, Adv. Mater., 2021, 34, 2105405 CrossRef.
- Z. Huang and X. Ma, Cell Rep. Phys. Sci., 2020, 1, 100167 CrossRef CAS.
- H. Nie, Z. Wei, X. L. Ni and Y. Liu, Chem. Rev., 2022, 122, 9032 CrossRef CAS PubMed.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144 RSC.
- X. M. Chen, S. Zhang, X. Chen and Q. Li, ChemPhotoChem, 2022, 6, e202100256 CrossRef CAS.
- L. Guan, Z. Jiang, Y. Cui, Y. Yang, D. Yang and G. Qian, Adv. Opt. Mater., 2021, 9, 2002180 CrossRef CAS.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- M. Quan, X. Y. Pang and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201258 CrossRef CAS PubMed.
- L. Zhang, H.-X. Wang, S. Li and M. Liu, Chem. Soc. Rev., 2020, 49, 9095 RSC.
- J. Chen, Q. Huang, Q. Wang, Y. Ding, S. Lu, L.-H. Wang, S. Li and R. Wang, ACS Appl. Nano Mater., 2022, 5, 5993 CrossRef CAS.
- X. Duan, G. Q. Zhang, S. Ji, Y. Zhang, J. Li, H. Ou, Z. Gao, G. Feng and D. Ding, Angew. Chem., Int. Ed., 2022, 61, e202116174 CrossRef CAS.
- W. L. Zhou, W. Lin, Y. Chen and Y. Liu, Chem. Sci., 2022, 13, 7976 RSC.
- X. Cong, K. Ou, J. Ma, J. Xu, Y. Liao, Y. Yang and H. Wang, Chem. Eng. J., 2024, 481, 148355 CrossRef CAS.
- B. Liu, J. Gao, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2023, 62, e202305135 CrossRef CAS PubMed.
- M.-P. Zhuo, G.-P. He, Y. Yuan, Y.-C. Tao, G.-Q. Wei, X.-D. Wang, S.-T. Lee and L.-S. Liao, CCS Chem., 2021, 3, 413 CrossRef CAS.
- S. Bacchi, M. Benaglia, F. Cozzi, F. Demartin, G. Filippini and A. Gavezzotti, Chem.–Eur. J., 2006, 12, 3538 CrossRef CAS.
- X. Pang, H. Wang, X. R. Zhao and W. J. Jin, Crystengcomm, 2013, 151, 2722 RSC.
- G. Y. Lee, E. Hu, A. L. Rheingold, K. N. Houk and E. M. Sletten, J. Org. Chem., 2021, 86, 8425 CrossRef CAS PubMed.
- X. Pang, H. Wang, W. Wang and W. J. Jin, Cryst. Growth Des., 2015, 15, 4938 CrossRef CAS.
- H.-M. Tang, Z.-Y. Quan, B. Ding, X.-G. Wang, B. Tang, Z.-G. Huang and E.-C. Yang, Cryst. Growth Des., 2024, 24, 5211 CrossRef CAS.
- W. X. Wu, H. Wang and W. J. Jin, Cryst. Growth Des., 2018, 18, 6742 CrossRef CAS.
- A. Friedrich, I. E. Collings, K. F. Dziubek, S. Fanetti, K. Radacki, J. Ruiz-Fuertes, J. Pellicer-Porres, M. Hanfland, D. Sieh, R. Bini, S. J. Clark and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18907 CrossRef CAS.
- S. Li, Y. Lin and D. Yan, J. Mater. Chem. C, 2016, 4, 2527 RSC.
- H. Ye, G. Liu, S. Liu, D. Casanova, X. Ye, X. Tao, Q. Zhang and Q. Xiong, Angew. Chem., Int. Ed., 2018, 57, 1928 CrossRef CAS.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef.
- W. Wang, L. Luo, P. Sheng, J. Zhang and Q. Zhang, Chem.–Eur. J., 2020, 27, 464 CrossRef PubMed.
- S. An, M. Qiao, X. Jin, X. Chen, J. Su, L. Guo and Z. Zhang, Sci. China-Chem., 2023, 67, 512 CrossRef.
- H. Zhang, J. Han, X. Jin and P. Duan, Angew. Chem., Int. Ed., 2021, 60, 4575 CrossRef CAS.
- X. Deng, X. Yu, J. Xiao and Q. Zhang, Aggregate, 2021, 2, e35 CrossRef CAS.
- Y. Hu, K. Wang, Y. Wang and L. Ma, Dyes Pigment., 2023, 210, 110948 CrossRef CAS.
- Y. J. Wang, Z. Li, J. Tong, X. Y. Shen, A. Qin, J. Z. Sun and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 3559 RSC.
- W. Ma, X. Li, A. Hao and P. Xing, Sci. China-Chem., 2024, 67, 3482 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07859d |
| This journal is © The Royal Society of Chemistry 2025 |