
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchable colossal anisotropic thermal expansion in a spin crossover framework†
Si-Guo
Wu
 ,
Wen
Cui
,
Ze-Yu
Ruan
,
Zhao-Ping
Ni
* and
Ming-Liang
Tong
*
,
Wen
Cui
,
Ze-Yu
Ruan
,
Zhao-Ping
Ni
* and
Ming-Liang
Tong
*
Key Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, School of Chemistry, Institute of Green Chemistry and Molecular Engineering, Guangdong Basic Research Center of Excellence for Functional Molecular Engineering, Sun Yat-Sen University, Guangzhou 510006, P. R. China. E-mail: nizhp@mail.sysu.edu.cn; tongml@mail.sysu.edu.cn
First published on 8th April 2025
Abstract
Advanced materials with tunable thermal expansion properties have garnered significant attention due to their potential applications in thermomechanical sensing and resistance to thermal stress. Here, switchable colossal anisotropic thermal expansion (ATE) behaviors are realized in a Hofmann-type framework [Fe(bpy-NH2){Au(CN)2}2]·iPrOH (Fe·iPrOH, bpy-NH2 = [4,4′-bipyridin]-3-amine) through a three-in-one strategy: a vibrational mechanism, an electronic mechanism and molecular motion. Spin crossover (SCO) centers coordinate with dicyanoaurate linkers to form flexible wine-rack frameworks, which exhibit structural deformations driven by host–guest interactions with iPrOH molecules. By means of the vibrational mechanism, a scissor-like motion driven by the rotation of dicyanoaurate is observed within the rhombic grids, resulting in the emergence of colossal ATE in the high temperature region. When the spin transition comes into play, the electronic mechanism is predominant to form reverse ATE behavior, which is associated with host–guest cooperation involving significant molecular motion of the iPrOH guest and adaptive deformation of the host clathrate. A remarkably high negative thermal expansion coefficient up to −7.49 × 105 M K−1 accompanied by abrupt SCO behavior is observed. As a proof of concept, this study provides a novel perspective for designing dynamic crystal materials with tunable thermomechanical properties by integrating various ATE-related elements into a unified platform.
Introduction
Thermal expansion, referring to the changes in size with response to temperature fluctuations, is an inherent characteristic of matter. Due to the increasing atomic vibrations upon heating, crystalline materials commonly experience modest positive thermal expansions (PTEs) with thermal expansion coefficients (α) less than 20 M K−1 along all principal axes.1 In rare cases, materials can demonstrate anisotropic thermal expansion (ATE) in the lattice where expansion occurs in one direction while contraction takes place in another.2–6 Unusual thermal expansions, such as colossal ATE (α > 100 M K−1), have emerged as a hot topic in solid-state chemistry and functional materials for their potential applications in thermomechanical sensing and resistance against thermal stress.4,7–11Chemical diversity enables versatile crystal engineering strategies for the design of materials exhibiting colossal ATE. As shown in Fig. 1a, flexible structures (cyanides,2,12–14 metal oxides/fluorides,15–17 metal–organic frameworks18–20etc.) featuring wine-rack or hinge-like scaffolds can facilitate ATE via a vibrational mechanism involving transverse vibrational displacement of bridging atoms.21 In addition, the couplings of geometric deformations in lattices and phase transitions (such as spin crossover (SCO),22–24 charge transfer25,26 and electric polarization27,28) can significantly enhance ATE performance within a narrow temperature window through the electronic mechanism (Fig. 1b). Besides, considerable ATE behavior can also be induced by collective rotation/reorientation in dynamic molecular crystals (Fig. 1c).11,29–31 However, the efficacy of this strategy is limited since molecular motions are always constrained by intermolecular interactions and steric hindrance in crystalline materials.
 | ||
| Fig. 1 Schematics of ATE regimes. (a) ATE via the vibrational mechanism in a wine-rack framework; (b) ATE via the electronic mechanism in an SCO complex; (c) ATE via collective motion in a dynamic molecular crystal; (d) three-in-one strategy for switchable ATE in this work. | ||
Advanced materials with tunable thermal expansion capabilities are of great interest for high-tech scenarios. However, delicate design of ATE structures and efficient control of lattice deformations are still challenging.32–36 By integrating multiple potential ATE-involving elements (e.g., flexible structures, phase transitions, molecular motion, etc.) into a unified crystalline material, switchable thermomechanical properties can potentially be manipulated via both vibrational and electronic mechanisms. Herein, a Hofmann-type framework [Fe(bpy-NH2){Au(CN)2}2]·iPrOH (Fe·iPrOH, bpy-NH2 = [4,4′-bipyridin]-3-amine) was constructed by a three-in-one strategy: a vibrational mechanism, an electronic mechanism and molecular motion (Fig. 1d). Specifically, spin crossover centers are assembled with cyanometallate linkers to form the flexible wine-rack framework, while iPrOH molecules are incorporated into the lattice and exhibit deformation driven by host–guest synergy. In the high temperature region, flexing motions within rhombic grids are dominated by the vibrational mechanism, leading to a linear ATE phenomenon. During SCO at lower temperature, reverse ATE is induced by the electronic mechanism, where the host framework and iPrOH guest undergo adaptive deformations in response to variations in internal stress. Note that the reorientation of iPrOH molecules plays an important role in modulating the SCO/ATE kinetics; in turn, the structural transformations caused by spin transition facilitate molecular motions in the lattice.
Results and discussion
Synthesis and structural characterization
Fe·iPrOH was prepared stoichiometrically via slow diffusion of KAu(CN)2 and bpy-NH2 ligands into Fe(ClO4)2·6H2O in a iPrOH/H2O mixture. Single crystal X-ray diffraction (SC-XRD) measurements indicated that Fe·iPrOH crystalizes in an orthorhombic Ibca space group (Table S1†). The asymmetric unit contains only one half of the formula. A C2 rotation axis along the c-axis is present within the bpy-NH2 ligand, where the amino group displays positional disorder with an occupation ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Additionally, the iPrOH guest molecule is associated with a C2 rotation axis along the b-axis and exhibits two-fold disorder with an occupation ratio of 1:1 at all measured temperatures (Fig. S1†). As shown in Fig. 2a, each FeII ion equatorially coordinates to four [Au(CN)2]− linkers in the ab-plane to form an [Fe{Au(CN)2}2]n layer. The almost-linear Fe–NC–Au–CN–Fe linkages lie parallel to the crystallographic 〈
:1. Additionally, the iPrOH guest molecule is associated with a C2 rotation axis along the b-axis and exhibits two-fold disorder with an occupation ratio of 1:1 at all measured temperatures (Fig. S1†). As shown in Fig. 2a, each FeII ion equatorially coordinates to four [Au(CN)2]− linkers in the ab-plane to form an [Fe{Au(CN)2}2]n layer. The almost-linear Fe–NC–Au–CN–Fe linkages lie parallel to the crystallographic 〈![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10〉 and 〈0〉 directions. The adjacent heterometallic cyanido-bridged layers are connected by bpy-NH2 ligands along the c-axis, giving rise to a sub-lattice framework with pcu topology. The potential pores of this framework enable double interpenetration of identical frameworks which are related to each other by weak aurophilic interactions (Fig. 2b). The pore space for each formula unit is occupied by one two-fold disordered iPrOH molecule which interacts with the interpenetrating skeletons with H-bonding.
10〉 and 〈0〉 directions. The adjacent heterometallic cyanido-bridged layers are connected by bpy-NH2 ligands along the c-axis, giving rise to a sub-lattice framework with pcu topology. The potential pores of this framework enable double interpenetration of identical frameworks which are related to each other by weak aurophilic interactions (Fig. 2b). The pore space for each formula unit is occupied by one two-fold disordered iPrOH molecule which interacts with the interpenetrating skeletons with H-bonding.
 | ||
| Fig. 2 The perspective view (a) and double interpenetration (b) of the Fe·iPrOH framework. The lattice conformation of Fe·iPrOH at 100 K (c) and 298 K (d) viewed along the c-axis. The two-fold disordered iPrOH molecules are marked in distinct shades of color and the green dashed lines stand for the host–guest H-bonding interactions. The black boxes indicate the unit cell dimensions. Hydrogen atoms are omitted for clarity. | ||
Variable temperature SC-XRD measurements reveal structural evolution of Fe·iPrOH. Typically, the average Fe–N bond length alters from 2.167(4) Å at 298 K to 1.948(5) Å at 100 K, hinting at the spin transition from HS to LS states (Table S4†). Accordingly, the unit cell volume decreases by 6.5% with colossal anisotropic thermal expansion (Δa = −0.834 Å, Δb = 1.637 Å, and Δc = 0.776 Å). The decline in the c axis can be attributed to the shrinkage of Fe–Npy bonds mediated by rigid bpy-NH2 pillars. By contrast, the thermal variations in the ab-plane are due to scissor-like motion of the flexible [Fe{Au(CN)2}2]n layer (Fig. 2c and d). Amid the spin transition from HS to LS, massive strain accumulated due to the significant contraction in the b direction must be compensated for by negative thermal expansion along the a-axis to ensure the structural integrity. This situation is also accompanied by the reorientations of [Au(CN)2]− linkers and iPrOH molecules. The Fe1–N1![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C1/Fe1–C2N2 angles change from 171.0°/160.8° at 298 K to 177.4°/169.4° at 100 K (Table S4†). On the other hand, iPrOH undergoes an adaptive molecular rotation and deformation to match the more condensed packing with enhanced H-bonding interactions.
C1/Fe1–C2N2 angles change from 171.0°/160.8° at 298 K to 177.4°/169.4° at 100 K (Table S4†). On the other hand, iPrOH undergoes an adaptive molecular rotation and deformation to match the more condensed packing with enhanced H-bonding interactions.
Anisotropic thermal expansion and SCO behavior
To investigate the ATE behavior in Fe·iPrOH, temperature-dependent unit cell parameters are monitored at 100–300 K (Fig. 3). Within the temperature range of 225–300 K, the a-axis and b-axis of Fe·iPrOH exhibit nearly linear PTE and NTE, respectively, with coefficients of thermal expansion (CTEs) calculated to be αa = 463(14) M K−1 and αb = −166(14) M K−1 using the PASCal web tool37,38 (Fig. S32 and Table S8†). This ATE can be well described by the vibrational mechanism commonly reported in wine-rack framework materials. Further proof comes from the Zn-analogue (Zn·iPrOH) wherein the a-axis and b-axis undergo similar PTE and NTE in the same temperature range. The CTEs for Zn·iPrOH are αa = 337(8) M K−1 and αb = −157(9) M K−1 (Table S11†), comparable to those observed in Ag3[Co(CN)6]2 and KMn[Ag(CN)2]3.13 At around 225 K, the ATE of the a-axis and b-axis switches to NTE and PTE owing to the abrupt spin transition. Below 220 K, thermal expansion behavior is imperceptible in Fe·iPrOH, which is due to the closer packing and the void-free structure in the LS state (Fig. S37 and Table S10†). In contrast, the linear ATE is maintained over the whole temperature range in Zn·iPrOH due to the absence of SCO capability.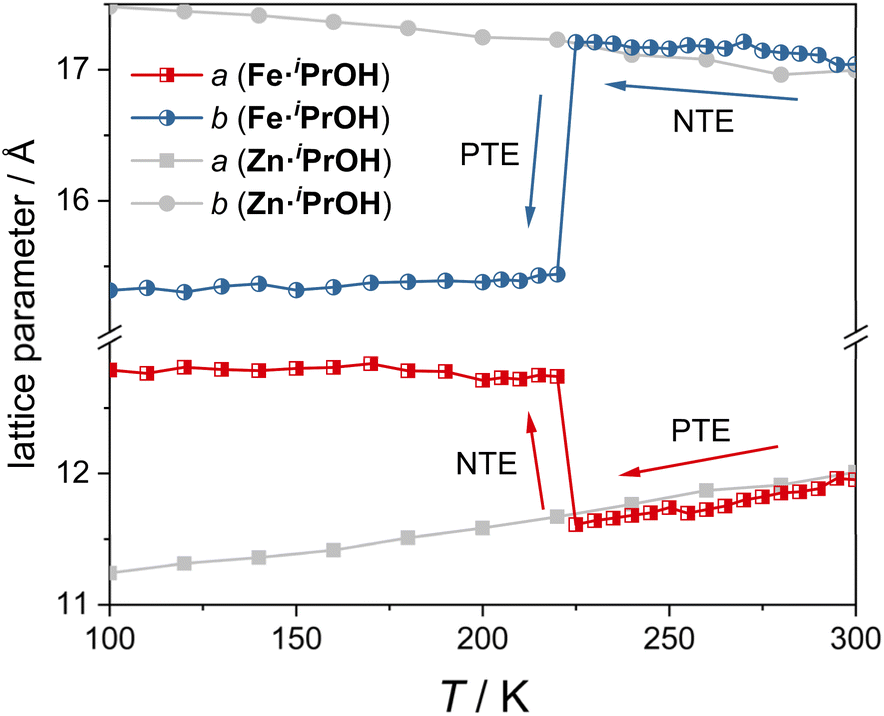 | ||
| Fig. 3 Temperature-dependent unit cell vectors (a-axis and b-axis) in Fe·iPrOH and its Zn-analogue. | ||
To get further insight into the ATE originated from the spin transition, detailed unit cell parameters were recorded for the single crystal of Fe·iPrOH with an interval of 1 K upon heating and cooling modes (210–240 K). As depicted in Fig. 4a, an abrupt and hysteretic event was detected for crystallographic axes with critical temperatures (Tc) of Tcoolc = 220 K and Theatc = 231 K. The related principal axis parameters were modelled using the sigmoidal function,23,39 and the best-fit results indicate that the spin transitions take place within extremely narrow temperature regions (<0.1 K, Table S9†). Significant variations of the lattice parameters imply evident deformation of crystal shape in response to the rearrangement of electronic configuration. Fig. 4b reveals the crystal images recorded at 210 K and 250 K. The obvious volume contraction and color change from yellow to red indicate the spin transition from HS to LS states. The anisotropic transformation can be checked from the crystallographic (001) plane with the angle altering from 69.6° to 79.0°, in line with the scissor-like motion of the rhombic Hofmann-type clathrate (vide infra). The CTEs are extracted from the sigmoidal function with maxima αcoola = −7.49 × 105 M K−1, αcoolb = 3.79 × 105 M K−1, and αcoolc = 9.36 × 104 M K−1 and αheata = −3.89 × 105 M K−1, αheatb = 3.67 × 105 M K−1, and αheatc = 9.54 × 104 M K−1 (Fig. 4c). The uniaxial NTE is four orders of magnitude higher than that in prototypical NTE materials (such as Cd(CN)2 (ref. 40) and ZrW2O8 (ref. 15)) and two orders of magnitude higher than the record in SCO materials.24 Note that the huge CTEs in Fe·iPrOH are caused by the abrupt SCO behavior within the extremely narrow temperature range. The crystals tend to crack after undergoing three cycles of the cooling and heating process, which is attributed to the significant structural changes associated with the abrupt SCO behavior.
 | ||
| Fig. 4 (a) Crystallographic axis parameters (circle) for Fe·iPrOH in the temperature range of 200–250 K. Solid lines indicate the best-fitting results using the sigmoidal function. (b) The crystal images of Fe·iPrOH recorded at 210 and 250 K. (c) The calculated thermal expansion coefficients according to unit cell parameters. (d) Variable-temperature molar magnetic susceptibility measured in the settle mode for Fe·iPrOH. (e) The DSC curves of Fe·iPrOH at 10 K min−1. | ||
Variable-temperature molar magnetic susceptibility for Fe·iPrOH was measured in settle mode (Fig. 4d). Upon cooling, the χMT value changes from 3.99 cm3 mol−1 K to 0.26 cm3 mol−1 K, hinting at a HS-to-LS transition. Upon warming, the steep SCO profile shifts toward a higher temperature region. The 1st derivative curves of magnetic data reveal a thermal hysteresis with Tcoolc = 222 K and Theatc = 232 K (Fig. 4d, inset). The entropy-driven SCO behavior was further verified by differential scanning calorimetry (DSC) measurement with a pair of exothermic/endothermic peaks lying at 219/233 K (Fig. 4e). The variations of enthalpy are −12.20/11.54 kJ mol−1 in the cooling/heating modes, in accordance with the spin transition of Fe(II) complexes.41 The hysteretic loops of in-phase dielectric constant ε′ also prove the SCO behavior in Fe·iPrOH (Fig. S27†). The kinetic phenomenon of SCO profiles is negligible at 1–10 K min−1 scanning rates, confirming the significant cooperative interactions in this system (Fig. S28†).
Mechanisms for ATE
To understand the switching regimes of the linear ATE and sigmoidal ATE in Fe·iPrOH, the flexing motions within the rhombic grid [M{Au(CN)2}2]n are studied in this system and clues are drawn from the rotations of [Au(CN)2]− motifs and/or the host–guest cooperation.For the linear ATE, the vibrational mechanism can be explored by comparing the variable-temperature crystal structures of the diamagnetic Zn-analogue (Fig. 5a). First, the Zn⋯Zn distance (10.4 Å) linked by one [Au(CN)2]− remains almost unchanged at both 103 and 300 K in Zn·iPrOH, revealing that the scissor-like motion within the rhombic grid is merely derived from atomic displacements and reorientations of the [Au(CN)2]− bridging motifs since the [Au(CN)2]− motifs prefer transverse vibrational displacement rather than the stretching one within the ab-plane. Upon heating from 103 to 300 K, [Au(CN)2]− linkers undergo rotations away from the b-axis with the related angle increasing from 24.9° to 28.4° (more details in Fig. S18 and Table S7†). The Zn1–N1C1/Zn1–N2C2 angles alter from 168.0°/158.1° to 171.7°/159.3° because of the more linear linkages of Zn–NC–Au–CN–Zn (Table S5†). Accordingly, the octahedral distortion parameter (∑) defined as the sum of the deviations from 90° for twelve cis-bond angles alters from 16.76° to 8.8°. These conformational changes lead to a “close” scissor-like motion with expansion along the a-axis and contraction along the b-axis. As a result, the Zn⋯Zn⋯Zn angle of the rhombic grid increases from 65.9° to 70.5° (Fig. 5a inset). Similarly, the Fe⋯Fe⋯Fe angle in HS Fe·iPrOH increases from 67.7° at 225 K to 69.7° at 298 K (Table S4†).
 | ||
| Fig. 5 The lattice deformations within heterometallic cyanido-bridged rhombic grids for Zn·iPrOH (a) and Fe·iPrOH (b). Inset: schematic representations of the ab-plane geometric deformations caused by vibrational/electronic mechanisms. | ||
For the sigmoidal ATE, the electronic mechanism is revealed by checking the structural transformation of Fe·iPrOH at the same temperature but with distinct spin states (Fig. 5b). Amid the spin transition from LS to HS states, the radius of Fe(II) significantly increases from 75 pm to 92 pm due to the electron population at the higher eg orbital.42,43 Concomitantly, the expansion of each Fe–N length by ∼0.2 Å gives rise to an increase in the Fe⋯Fe distance from 10.0 to 10.4 Å. Driven by the internal stress caused by the thermal expansion of spin centers, the [Au(CN)2]− motifs reorientate to approach the b-axis (opposite to Zn·iPrOH) with the angle changing from 26.4° to 36.3° to ensures minimize lattice energy under constraint. Upon the LS-to-HS transition, the Fe1–N1C1/Fe1–N2C2 angles decrease from 177.8°/168.5° to 169.3°/160.0°, and Fe(II) adopts a more distorted octahedral geometry (∑Fe = 9.2°/16.48° in the LS/HS states). These conformational changes result in an “open” scissor-like motion with contraction along the a-axis and expansion along the b-axis. And the Fe⋯Fe⋯Fe angle of the rhombic grid significantly decreases from 79.3° to 67.7° (Fig. 5b inset).
The geometric deformation in the SCO lattice is also involved with the host–guest synergistic effect44,45 and commonly observed in amino-functionalized frameworks with asymmetric strong H-bonds.46,47 The conformations of guest molecules and related H-bonding interactions in this system are described in Fig. 6a–d. For Zn·iPrOH, H-bonds are formed between two-fold disordered iPrOH molecules and the double interpenetrating frameworks. The pyridyl moieties of bpy-NH2 ligands retain two-fold disorder at both 103 and 300 K, while the iPrOH molecules undergo a slight slippage and deformation with the angle of isopropyl altering from 99.5° to 111.7° in response to the ATE translation. For Fe·iPrOH, the HS structure (including the conformations of isopropyl from iPrOH and pyridyl from bpy-NH2) at 225 K is similar to those in Zn·iPrOH. Upon the HS-to-LS transition, significant molecular rotation and deformation take place in the iPrOH molecule with the angle of isopropyl altering from 97.6° to 123.4° (Fig. 6g). This flattening distortion of the guest operates in favor of the contraction of the host framework along the b-axis, and in turn, the pyridyl moieties in the host framework become ordered on account of the closer packing in the LS state. Variable temperature Raman spectra show strong correlation with SCO behavior and give a clue for the related conformational changes. Switchable stretching vibrations of the cyano group were detected near spin transition temperature (Fig. S25†). On the other hand, the vibrations of iPrOH, such as –CH3 bending (1431 cm−1)48 and C–C stretching (872/813 cm−1)49 modes, are nearly smeared out in the LS state, possibly due to the vibrational restraint in the shrunken lattice (Fig. 6h). When converting to the HS state, the corresponding vibrations of iPrOH recovered as expected (Fig. S26†). Therefore, the geometric deformation in the lattice is mediated by host–guest cooperation.
 | ||
| Fig. 6 The selected structures in the bc-plane of Fe·iPrOH (a) and (b), Zn·iPrOH (c) and (d) and Fe·tBuOH (e) and (f). The green/orange dashed lines stand for the host–guest H-bonding interactions and aurophilic interactions between pcu networks. Color code: cyaneous/carmine, Fe(LS/HS state); orange, Au; purple, I; red, Br; grey, C; blue, N; red, O. The H atoms are omitted for clarity. (g) The iPrOH and tBuOH viewed along the C–O bond. The numbers demonstrate the angles of isopropyl and tert-butyl moieties. (h) The selected Raman spectra of Fe·iPrOH recorded at 210 and 240 K. | ||
Generally, the interplay between host–guest chemistry and spin-state change has an impact on the SCO behavior.50–52 To evaluate the utility of the deformation in iPrOH toward collective SCO properties, a control experiment was conducted by introducing the guest molecule with a larger substituent to hamper the conformational deformation in the lattice. An analogue Fe·tBuOH was constructed with a similar framework except that iPrOH is replaced by a tBuOH molecule. As expected, no remarkable molecular deformation was observed in tBuOH during the spin transition taking advantage of the steric hindrance of the tert-butyl group (Fig. 6g). Meanwhile, the pyridyl moieties are crystallographically ordered in the HS and LS states (Fig. 6e and f). Magnetic measurement indicates that Fe·tBuOH displays a gradual and stepwise SCO behavior (Fig. S29†). Variable temperature SC-XRD experiments reveal ATE in unit cell parameters with maxima αcoola = −671 M K−1, αcoolb = 1267 M K−1, and αcoolc = 543 M K−1 and αheata = −752 M K−1, αheatb = 1494 M K−1, and αheatc = 615 M K−1 within the temperature range of 100–250 K (Fig. S41†). The values of ATE coefficients in Fe·tBuOH are almost three orders of magnitude smaller than those in Fe·iPrOH, confirming the vital role of molecular motion of the guest in accelerating the spin transition and lattice deformation.
Conclusions
In summary, a Hofmann-type framework was constructed by the assembly of SCO centers, flexible cyanometallate linkers and deformable isopropanol guests. Notably, switchable colossal ATE behaviors were observed with a record-high negative thermal expansion coefficient up to −7.49 × 105 M K−1. Geometric transformations were found to be influenced either by vibrational or electronic mechanisms during structural deciphering. Moreover, it was discovered that the deformation of guest molecules in the lattice significantly impacted the collective magnetic and thermomechanical properties. These findings highlight that the multi-coupled relations between the ATE lattice and physicochemical properties (such as the spin transition and host–guest synergy) provide novel insights for functionalizing dynamic crystal materials and exploring ATE mechanisms.Experimental
Materials and methods
All the reagents and solvents were commercially available and used without further purification. The elemental analyses were performed on an Elementar Vario-ELCHNS elemental analyzer with fresh samples. The Fourier transform infrared (FT-IR) spectra were recorded using KBr tablets in the range of 4000–400 cm−1 on a Thermo Scientific Nicolet 6700. The powder X-ray diffraction (PXRD) patterns were recorded on a Rigaku D-MAX 2200 VPC X-ray diffractometer using Cu-Kα (λ = 1.54178 Å) radiation. The thermogravimetric analyses (TGA) were performed on a Netzsch TG209 F1 Libra under a N2 flow with a heating rate of 10 K min−1 from ambient temperature to 900 °C. Differential Scanning Calorimetry (DSC) was carried out on fresh samples soaked in a little mother liquor and performed in an aluminium crucible using a NETZSCH5 with a sweeping rate of 10 K min−1 under a N2 atmosphere.When the anisotropic thermal expansion is dominated by the electronic mechanism, the coefficients of thermal expansion of crystallographic parameters were calculated using the sigmoidal function:39
 | (1) |
The sigmoidal function is usually used for empirical fitting of the spin crossover trasnition.23,24 The equation parameters represent A, the change of lattice parameters amid spin transitions (negative for NTE and positive for PTE); B, the sigmoidal center, equal to the critical temperature of the spin transition; C, the sigmoidal width, equal to the abruptness of SCO behavior; D, the momentum transfer value of the low-spin state.
The thermal expansion coefficients α are proportional to the 1st derivative of L(T) and can be calculated using eqn (2):
 | (2) |
:2) was added carefully. 5 mL iPrOH containing 0.1 mmol Fe(ClO4)2·6H2O was laid on the top. Yellow single crystals suitable for X-ray diffraction were isolated after two weeks. Yield: ∼20%. Elemental analysis: Calcd for C17H17Au2FeN7O: C 26.01, H 2.18, N 12.49; found: C 25.92, H 2.18, N 12.56.
:2) and then transferred into a test tube. 5 mL solution of tBuOH and H2O (v:v = 4:1) was added as the buffer layer. 2 mL tBuOH containing 0.1 mmol Fe(ClO4)2·6H2O was laid on the top. Yield: ∼60%. Elemental analysis: calcd for C18H19Au2FeN7O: C 27.05, H 2.40, N 12.27; found: C 27.19, H 2.46, N 12.26.
Author contributions
S. G. W., Z. P. N. and M. L. T. designed the study and wrote the manuscript. S. G. W. and W. C. synthesized the materials and performed the experimental measurements. Z. Y. R. performed the magnetic measurements. M. L. T. supervised the research. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† The crystallography data have been deposited at the Cambridge Crystallographic Data Centre (CCDC). CCDC 2386640–2386647 contain the supplementary crystallographic data for this paper. These data can be obtained free from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures/.Acknowledgements
This work was supported by the NSFC (grant no. 22488101, 22075323 and 21773316), the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (23ptpy73 and 24xkjc003), and the Science and Technology Program of Guangzhou, China (2024A04J4433).Notes and references
- R. S. Krishnan, R. Srinivasan and S. Devanarayanan, Thermal Expansion of Crystals, Pergamon, New York, 1979 Search PubMed.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS PubMed.
- Q. Li, K. Lin, Z. Liu, L. Hu, Y. Cao, J. Chen and X. Xing, Chem. Rev., 2022, 122, 8438–8486 CrossRef CAS PubMed.
- D. Das, T. Jacobs and L. J. Barbour, Nat. Mater., 2010, 9, 36–39 CrossRef CAS PubMed.
- X. G. Zheng, H. Kubozono, H. Yamada, K. Kato, Y. Ishiwata and C. N. Xu, Nat. Nanotechnol., 2008, 3, 724–726 CrossRef CAS PubMed.
- A. D. Fortes, E. Suard and K. S. Knight, Science, 2011, 331, 742–746 CrossRef CAS PubMed.
- L. Zhang, J. B. Bailey, R. H. Subramanian, A. Groisman and F. A. Tezcan, Nature, 2018, 557, 86–91 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- C. Lind, Materials, 2012, 5, 1125–1154 CrossRef CAS PubMed.
- B. B. Rath, G. Gallo, R. E. Dinnebier and J. J. Vittal, J. Am. Chem. Soc., 2021, 143, 2088–2096 CrossRef CAS PubMed.
- Z.-S. Yao, H. Guan, Y. Shiota, C.-T. He, X.-L. Wang, S.-Q. Wu, X. Zheng, S.-Q. Su, K. Yoshizawa, X. Kong, O. Sato and J. Tao, Nat. Commun., 2019, 10, 4805 CrossRef PubMed.
- A. L. Goodwin, K. W. Chapman and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 17980–17981 CrossRef CAS PubMed.
- A. B. Cairns, A. L. Thompson, M. G. Tucker, J. Haines and A. L. Goodwin, J. Am. Chem. Soc., 2012, 134, 4454–4456 CrossRef CAS PubMed.
- Q. Gao, J. Wang, A. Sanson, Q. Sun, E. Liang, X. Xing and J. Chen, J. Am. Chem. Soc., 2020, 142, 6935–6939 CrossRef CAS PubMed.
- T. A. Mary, J. S. O. Evans, T. Vogt and A. W. Sleight, Science, 1996, 272, 90–92 CrossRef CAS.
- L. Hu, J. Chen, J. Xu, N. Wang, F. Han, Y. Ren, Z. Pan, Y. Rong, R. Huang, J. Deng, L. Li and X. Xing, J. Am. Chem. Soc., 2016, 138, 14530–14533 CrossRef CAS PubMed.
- D. Wendt, E. Bozin, J. Neuefeind, K. Page, W. Ku, L. Wang, B. Fultz, A. V. Tkachenko and I. A. Zaliznyak, Sci. Adv., 2019, 5, eaay2748 CrossRef CAS PubMed.
- S. Henke, A. Schneemann and R. A. Fischer, Adv. Funct. Mater., 2013, 23, 5990–5996 CrossRef CAS.
- C. Yang, X. Wang and M. A. Omary, Angew. Chem., Int. Ed., 2009, 48, 2500–2505 CrossRef CAS PubMed.
- S. J. Baxter, A. Schneemann, A. D. Ready, P. Wijeratne, A. P. Wilkinson and N. C. Burtch, J. Am. Chem. Soc., 2019, 141, 12849–12854 CrossRef CAS PubMed.
- A. L. Goodwin and C. J. Kepert, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 140301 CrossRef.
- H. J. Shepherd, T. Palamarciuc, P. Rosa, P. Guionneau, G. Molnár, J.-F. Létard and A. Bousseksou, Angew. Chem., Int. Ed., 2012, 51, 3910–3914 CrossRef CAS PubMed.
- B. R. Mullaney, L. Goux-Capes, D. J. Price, G. Chastanet, J.-F. Létard and C. J. Kepert, Nat. Commun., 2017, 8, 1053 CrossRef PubMed.
- H.-Y. Sun, Y.-S. Meng, L. Zhao, N.-T. Yao, P.-D. Mao, Q. Liu, F.-F. Yan, H. Oshio and T. Liu, Angew. Chem., Int. Ed., 2023, 62, e202302815 CrossRef CAS PubMed.
- M. Azuma, W.-t. Chen, H. Seki, M. Czapski, S. Olga, K. Oka, M. Mizumaki, T. Watanuki, N. Ishimatsu, N. Kawamura, S. Ishiwata, M. G. Tucker, Y. Shimakawa and J. P. Attfield, Nat. Commun., 2011, 2, 347 CrossRef PubMed.
- J.-X. Hu, Y. Xu, Y.-S. Meng, L. Zhao, S. Hayami, O. Sato and T. Liu, Angew. Chem., Int. Ed., 2017, 56, 13052–13055 CrossRef CAS PubMed.
- J. Chen, L. Hu, J. Deng and X. Xing, Chem. Soc. Rev., 2015, 44, 3522–3567 RSC.
- P. Hu, J. Chen, J. Deng and X. Xing, J. Am. Chem. Soc., 2010, 132, 1925–1928 CrossRef CAS PubMed.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- Z.-S. Yao, M. Mito, T. Kamachi, Y. Shiota, K. Yoshizawa, N. Azuma, Y. Miyazaki, K. Takahashi, K. Zhang, T. Nakanishi, S. Kang, S. Kanegawa and O. Sato, Nat. Chem., 2014, 6, 1079 CrossRef CAS PubMed.
- X. Ding, D. K. Unruh, L. Ma, E. J. van Aalst, E. W. Reinheimer, B. J. Wylie and K. M. Hutchins, Angew. Chem., Int. Ed., 2023, 62, e202306198 CrossRef CAS PubMed.
- I. Grobler, V. J. Smith, P. M. Bhatt, S. A. Herbert and L. J. Barbour, J. Am. Chem. Soc., 2013, 135, 6411–6414 CrossRef CAS PubMed.
- Z. Chen, G. D. Stroscio, J. Liu, Z. Lu, J. T. Hupp, L. Gagliardi and K. W. Chapman, J. Am. Chem. Soc., 2023, 145, 268–276 CrossRef CAS PubMed.
- H.-L. Zhou, Y.-B. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 6917 CrossRef CAS PubMed.
- Q. Gao, J. Chen, Q. Sun, D. Chang, Q. Huang, H. Wu, A. Sanson, R. Milazzo, H. Zhu, Q. Li, Z. Liu, J. Deng and X. Xing, Angew. Chem., Int. Ed., 2017, 56, 9023–9028 CrossRef CAS PubMed.
- J. Chen, Q. Gao, A. Sanson, X. Jiang, Q. Huang, A. Carnera, C. G. Rodriguez, L. Olivi, L. Wang, L. Hu, K. Lin, Y. Ren, Z. Lin, C. Wang, L. Gu, J. Deng, J. P. Attfield and X. Xing, Nat. Commun., 2017, 8, 14441 CrossRef CAS PubMed.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- M. Lertkiattrakul, M. L. Evans and M. J. Cliffe, J. Open Source Softw., 2023, 8, 5556 CrossRef.
- R. M. van der Veen, O.-H. Kwon, A. Tissot, A. Hauser and A. H. Zewail, Nat. Chem., 2013, 5, 395–402 CrossRef CAS PubMed.
- K. W. Chapman, P. J. Chupas and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 15630–15636 CrossRef CAS PubMed.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed., 1994, 33, 2024–2054 CrossRef.
- R. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- X. Li, D. Zhang, Y. Qian, W. Liu, C. Mathonière, R. Clérac and X. Bao, J. Am. Chem. Soc., 2023, 145, 9564–9570 CrossRef CAS PubMed.
- J.-P. Xue, Y. Hu, B. Zhao, Z.-K. Liu, J. Xie, Z.-S. Yao and J. Tao, Nat. Commun., 2022, 13, 3510 CrossRef CAS PubMed.
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS PubMed.
- W. Liu, Y.-Y. Peng, S.-G. Wu, Y.-C. Chen, M. N. Hoque, Z.-P. Ni, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2017, 56, 14982–14986 CrossRef CAS PubMed.
- R. Turo-Cortés, C. Bartual-Murgui, J. Castells-Gil, M. C. Muñoz, C. Martí-Gastaldo and J. A. Real, Chem. Sci., 2020, 11, 11224–11234 RSC.
- Y. Yu, Y. Wang, N. Hu, K. Lin, X. Zhou and S. Liu, J. Raman Spectrosc., 2014, 45, 259–265 CrossRef CAS.
- B. Yang, X. Cao, S. Wang and C. Sun, Optik, 2020, 204, 163544 CrossRef CAS.
- Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767–4771 CrossRef CAS PubMed.
- M. J. Murphy, K. A. Zenere, F. Ragon, P. D. Southon, C. J. Kepert and S. M. Neville, J. Am. Chem. Soc., 2017, 139, 1330–1335 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystal data (Fig. S1–S18 and Tables S1–S7), powder X-ray diffraction patterns (Fig. S19 and S21), thermogravimetric analysis (Fig. S22 and S23), infrared and Raman spectra (Fig. S24–S26), dielectric properties (Fig. S27), magnetic properties (Fig. S28 and S29) and thermal expansion properties (Fig. S30–S47 and Tables S8–S13). CCDC 2386640–2386647. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08032g |
| This journal is © The Royal Society of Chemistry 2025 |