
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic surface reconstruction engineers interfacial water structure for efficient alkaline hydrogen oxidation†
Chaoyi
Yang
,
Zihao
Dai
,
Jianchao
Yue
,
Guangqin
Wang
and
Wei
Luo
 *
*
College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: wluo@whu.edu.cn
First published on 13th February 2025
Abstract
Investigating the dynamic evolution of the catalyst and regulating the structure of interfacial water molecules participating in the hydrogen oxidation reaction (HOR) are essential for developing highly efficient electrocatalysts toward the practical application of anion exchange membrane fuel cells. Herein, we report an efficient strategy to activate hexagonal close-packed PtSe catalyst through in situ reconstruction that undergoes dynamic Se leaching and phase transition during linear sweep voltammetry cycles. The obtained Pt–Se catalyst presents as a surface Se atom-modified face-centered-cubic Pt-based nanocatalyst, and it exhibited remarkable catalytic performance in the alkaline HOR, showing an intrinsic activity of 0.552 mA cm−2 (j0,s) and a mass activity of 1.084 mA μg−1 (jk,m @ 50 mV). The experimental results, including in situ surface-enhanced infrared absorption spectroscopy and density functional theory calculations suggest that the accumulated electrons on the surface-decorated Se of Pt–Se can induce the regulation of the interfacial water structure between the electrode and electrolyte surface in the electric double-layer region. Consequently, the migration of OH− species from the electrolyte to the catalyst surface can be apparently accelerated within this disordered water network, which together with the optimized intermediate thermodynamic binding energies, contribute to the enhanced alkaline HOR activity.
Introduction
For future environmentally friendly energy demands, the hydrogen fuel cell is one of the most promising candidates for the replacement of fossil fuels.1,2 The anion exchange membrane fuel cell (AEMFC) has been recognized as a competitive energy conversion device because of its merits of high energy density, zero carbon emission, and sustainability.3–5 Different from the well-developed cathodic oxygen reduction reaction (ORR) within precious metal-free catalysts under alkaline media, severe dependence on the high-loading expensive noble metal catalysts for the alkaline anode hydrogen oxidation reaction (HOR) has greatly hindered practical applications of the AEMFC.6–8Recently, the dynamic evolution of catalysts has been recognized during electrocatalysis, and the effects of pre-catalyst reconstruction in various electrochemical catalytic processes, including the oxygen evolution reaction,9–11 carbon dioxide reduction reaction,12–14 nitrate reduction reaction,15,16 and hydrogen evolution reaction,17,18 have been widely investigated. The reconstructed structure and the redistribution of elements during the dynamic evolution can lead to the boosting of the catalytic activity, which is beneficial for the design of advanced electrocatalysts.19,20 Identifying the dynamic evolution process and activating the catalytic performance through reconstruction are essential for the rational design of advanced HOR electrocatalysts under alkaline electrolyte, but unfortunately, they have been rarely investigated.
During the alkaline HOR process, the OH species have received considerable attention owing to their engagements in Volmer step, which is known as the rate-determining step.21,22 Through advanced spectroscopy technology, the adsorbed OH species has been detected during the alkaline HOR process by Li et al., directly demonstrating the involvement of OHad.23 Markovic et al. proved that metals with improved hydroxyl adsorption may present much higher alkaline HOR activities,24 guiding the direction for the rational design of highly efficient alkaline HOR electrocatalysts. Consequently, many studies have mainly focused on the thermodynamic energetics of OHad, which can dictate the catalytic activity derived from the charge redistribution and the electronic adjustment-induced adsorption behavior modulation.25–27
Besides, there has been growing recognition that the catalytic performance does not solely depend on the inherent structures of catalysts but is also shaped by the electrochemical interface and the electric-double-layer (EDL) structure formed at the interface between the electrode and electrolyte.28 However, due to the decreased flexibility of the interfacial water network in alkaline media, additional energy is needed for the migration of OH species from electrolyte to the catalyst surface, thereby leading to the sluggish HOR kinetics under alkaline electrolytes.29–31 Therefore, rational regulation of the interfacial water structure and decreased rigidity of water networks to facilitate OH species across the EDL can be an efficient method to accelerate the alkaline HOR kinetic, but it remains challenging.
Herein, we propose an efficient strategy through in situ reconstruction to boost the HOR performance of hexagonal close-packed (hcp) PtSe alloys under alkaline electrolytes. Compared with hcp-PtSe alloys with negligible catalytic performance, face-centered-cubic (fcc) Pt–Se formed through the dynamic leaching of Se atoms and the follow-up phase transition during the electrocatalysis process displays significantly boosted HOR performance under an alkaline electrolyte (Fig. 1a), with an intrinsic activity of 0.552 mA cm−2 (j0,s) and mass activity of 1.084 mA μg−1 (jk,m @ 50 mV).
 | ||
| Fig. 1 (a) Schematic illustration for the activation process of the leaching of Se and phase transition on the PtSe alloy during the HOR process. The green balls denote the Se atoms, and the silver balls denote the Pt atoms. (b) The activation process for PtSe2 under the HOR polarization scans. (c) XRD patterns for PtSe2 during the activation process. (d) Typical TEM image and the corresponding size distribution for Pt–Se-2. (e) HAADF-STEM image and (f) atomic-resolution HAADF-STEM image of Pt–Se-2. (g) HAADF-STEM image and corresponding EDX mappings of Pt–Se-2. | ||
By the analyses of in situ surface-enhanced infrared absorption spectroscopy (SEIRAS) and the theoretical calculations, it is clear that the rigid water network on the interface of Pt–Se has been largely impaired. Regulation of the water structure by inducing surface-decorated Se atoms leads to facilitated migration of OH species to the catalyst surface, which together with the optimized intermediate adsorption, contributes to the enhanced alkaline HOR performance.
Results and discussion
PtSex alloys (x = 1.5, 2, 3) with different Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Se ratios were successfully synthesized through a colloid method (the detailed experimental process is shown in the ESI†), and the corresponding X-ray diffraction (XRD) in Fig. S1† is depicted with the broad peak at approximately 30° and 50°, which was assigned to the hexagonal close-packed structure.32,33 The HOR performance of the PtSe alloys was tested by rotating disk electrode (RDE) with a 3-electrode system using H2-saturated 0.1 M KOH electrolyte. As shown in Fig. 1b and S2,† PtSex presents gradually increased current densities during the linear sweep voltammetry (LSV) cycles, and the HOR performance finally reaches the steady state after several LSV sweeps, indicating the dynamic evolution of PtSex during the HOR activation process. The activation processes for PtSex are also prolonged with increasing content of Se. The stabilized catalysts after in situ reconstruction are noted as Pt–Se-x (x = 1.5, 2, 3), and Pt–Se-2 presents the highest alkaline HOR performance among the three activated catalysts (vide infra).
:Se ratios were successfully synthesized through a colloid method (the detailed experimental process is shown in the ESI†), and the corresponding X-ray diffraction (XRD) in Fig. S1† is depicted with the broad peak at approximately 30° and 50°, which was assigned to the hexagonal close-packed structure.32,33 The HOR performance of the PtSe alloys was tested by rotating disk electrode (RDE) with a 3-electrode system using H2-saturated 0.1 M KOH electrolyte. As shown in Fig. 1b and S2,† PtSex presents gradually increased current densities during the linear sweep voltammetry (LSV) cycles, and the HOR performance finally reaches the steady state after several LSV sweeps, indicating the dynamic evolution of PtSex during the HOR activation process. The activation processes for PtSex are also prolonged with increasing content of Se. The stabilized catalysts after in situ reconstruction are noted as Pt–Se-x (x = 1.5, 2, 3), and Pt–Se-2 presents the highest alkaline HOR performance among the three activated catalysts (vide infra).
As the activation process proceeds, the cyclic voltammetry (CV) curves (Fig. S3†) display a flat current in the hydrogen underpotential desorption region at the beginning, and then gradually show the underpotential desorption peaks of H (Hupd) on Pt, implying the exposure of Pt atoms and the potential phase transformation during the activation.34,35 The XRD patterns (Fig. 1c) during the activation process clearly indicate that the crystalline phase of PtSe2 gradually changes from the initial hcp phase to the fcc phase of Pt–Se-2, which is well matched with the Pt standard (PDF# 89-7382). A different ratio of Pt–Se-x also shows the same XRD patterns after the activation (Fig. S4†). Furthermore, the precise composition of PtSe2 alloy and activated Pt–Se-2 was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES), as shown in Table S1.†
It is clear that the content of Se is significantly decreased after the activation, indicating that the majority of Se atoms are leached during the activation, and only a portion of them can be stabilized in the final catalyst. As the amount of Se feed grows, the ratio of stabilized Se on catalysts is enhanced as well. Through transmission electron microscopy (TEM), as shown in Fig. 1d, the morphology of Pt–Se-2 consists of small nanoparticles uniformly distributed on the XC-72R carbon with an average size of 1.89 nm. The TEM image of PtSe2 also shows evenly distributed small nanoparticles on the substitute, implying that there is little influence of activation on morphology (Fig. S5†).
To further investigate the structure of the activated catalyst and identify the existing form of Se after activation, high-angle annular dark-field scanning TEM (HAADF-STEM) was employed. In Fig. S6,† hexagonally packed atoms are shown in the HAADF-STEM image of PtSe2 alloy, which is in accordance with the XRD results. Pt and Se elements are evenly spread on the nanoparticles, as revealed by energy-dispersive X-ray spectroscopy (EDX, Fig. S7†). The HAADF-STEM image of Pt–Se-2 demonstrates the apparent crystallinity of the inner part of the nanoparticles, while the outside shows obvious amorphous atomic layers (Fig. 1e). As shown in Fig. 1f, the atomic-resolved lattice spacings of the inner part of Pt–Se-2 are measured to be 0.195 nm and 0.225 nm, which are attributed to the (200) facet and (111) facet of fcc Pt, respectively.
The angle between the (200) and (111) planes is 55°, which further indicates the face-centered-cubic phase. The corresponding fast Fourier transform (FFT) of the STEM image in Fig. S8† also shows that the crystal lattice orientation is based on the [0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1] crystallographic direction of fcc Pt, which is in accordance with the XRD pattern. The EDX mappings (Fig. 1g) and the line scan signals (Fig. S9†) indicate that the remaining Se atoms prefer to stabilize on the surface around the crystalline phase. In addition, the surface elements of Pt–Se-2 were also determined by X-ray photoelectron spectroscopy (XPS), as shown in Table S2.† Compared with the Se/Pt ratio in the bulk of Pt–Se-2 obtained by ICP-AES, the higher atomic ratio of Se/Pt determined by XPS further confirms the surface decoration of Se atoms in Pt–Se-2.
1] crystallographic direction of fcc Pt, which is in accordance with the XRD pattern. The EDX mappings (Fig. 1g) and the line scan signals (Fig. S9†) indicate that the remaining Se atoms prefer to stabilize on the surface around the crystalline phase. In addition, the surface elements of Pt–Se-2 were also determined by X-ray photoelectron spectroscopy (XPS), as shown in Table S2.† Compared with the Se/Pt ratio in the bulk of Pt–Se-2 obtained by ICP-AES, the higher atomic ratio of Se/Pt determined by XPS further confirms the surface decoration of Se atoms in Pt–Se-2.
XPS was applied to probe the electronic states of Pt and Se in Pt–Se-2. The high-resolution XPS spectra of Pt 4f (Fig. 2a) can be deconvolved into four peaks, corresponding to Pt0 4f7/2 and Pt0 4f5/2 electrons at 71.3 eV and 74.6 eV, and Pt2+ 4f7/2 and Pt2+ 4f5/2 electrons at 72.4 eV and 75.7 eV, respectively.36 The valence state of Pt in Pt–Se-2 is slightly higher than that of Pt (Fig. S10†), indicating the electron transfer from Pt to Se atoms. Compared with the PtSe2 alloy (Fig. S11†), the peak position of Pt for Pt–Se-2 is slightly shifted to the lower binding energies, which may contribute to the decreasing content of Se. In addition, the Se 3d spectrum (Fig. 2b) shows the peaks located at 52.5 eV and 53.4 eV, which can be assigned to 3d5/2 and 3d3/2 of selenide, respectively.
 | ||
| Fig. 2 XPS spectra of (a) Pt 4f and (b) Se 3d for Pt–Se-2. (c) Pt L3-edge XANES spectra and (d) corresponding FT-EXAFS spectra for Pt–Se-2 in reference to Pt foil and PtO2. (e) WT for the k3-weighted EXAFS of the Pt L3-edge for Pt–Se-2, Pt foil, and PtO2. | ||
Compared with the Se powder (Fig. S12†), the downshift of the valence state indicates the electron acception in Se of Pt–Se-2, further proving the electron transfer process in Pt–Se-2. Specifically, no obvious peak appears at approximately 59 eV, implying that no selenates existed on the surface.37,38 Considering the relatively low potential of the HOR process, the Se atoms were merely leaching and did not oxidize on the surface during the LSV scans, leading to the Se atoms in Pt–Se-2 maintaining the zero-valence state after the activation, and the same with that before activation (Fig. S11†). Additionally, X-ray adsorption spectroscopy (XAS) in Pt L3-edge was performed to further determine the electronic state on Pt–Se-2.
As shown in Fig. 2c, the Pt L3-edge X-ray adsorption near edge structure (XANES) of Pt–Se-2 shows the relatively low intensity of a white line close to that of Pt foil, revealing the metallic state of Pt in Pt–Se-2 and the slight electron loss in Pt, in accordance with the XPS results. The Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra (Fig. 2d) and the wavelet transforms (WT) for k3-weighted EXAFS signals (Fig. 2e) of Pt L3-edge confirm the existence of Pt–Se and Pt–Pt bonds and the lack of Pt–O bonds, further revealing that Pt atoms are not oxidized during the activation process.
The HOR activities of Pt–Se-x samples were evaluated by LSV in fresh 0.1 M KOH electrolyte, as shown in Fig. S13.† Calculating the linear fitting of micropolarization regions from −5 to 5 mV through the simplified Butler–Volmer equation (Fig. S14†), the exchange current density (j0) normalized by the geometric area of the glassy carbon electrode (GCE) was obtained.39,40 With increasing Se, the exchange current density (j0) of Pt–Se-x initially increases and then decreases, indicating the existence of the optimized Se content for the HOR performance. Fig. S15† shows the LSV curves of Pt–Se-x at rotation rates from 2500 to 400 rpm and the corresponding fitting of the Koutecky–Levich plots.41
Calculation was performed using the Butler–Volmer equation, and Tafel plots of kinetic current densities (jk) were obtained in Fig. S16,† still presenting the same tendency as j0. The mass activities (jk,m) were normalized by the mass of the Pt content obtained from the ICP-AES results (Fig. S17†), and show the same volcano correlation, confirming that Pt–Se-2 possesses the highest mass activity at 50 mV among the three catalysts. Through Cu underpotential deposition (Cu-UPD) stripping, electrochemical surface areas (ECSAs) were procured to further evaluate the intrinsic activities (j0,s) of Pt–Se-x (Fig. S18†). After normalization of the exchange current density (j0) by the ECSA, Pt–Se-2 still showed much higher intrinsic activity (j0,s) compared with Pt–Se-1 and Pt–Se-3 (Fig. S17†).
To further estimate the HOR performance of Pt–Se-2, commercial Pt was considered for comparison. Using the LSV curves of Pt and Pt–Se-2 in Fig. 3a, the exchange current density (j0) was calculated by the micropolarization regions (Fig. 3b) of Pt–Se-2, and was apparently higher than that of commercial Pt. The different rotating rates of LSV curves of Pt–Se-2 and Pt appear in Fig. 3c and S19.† The kinetic current density (jk) of Pt–Se-2 reached 7.13 mA cm−2 at 50 mV, exceeding 4.24 mA cm−2 for Pt (Fig. 3d). The calculated mass activity (jk,m) of Pt–Se-2 was 1.084 mA μg−1 at 50 mV (Fig. 3e), which is over two times higher than that of commercial Pt (0.416 mA μg−1).
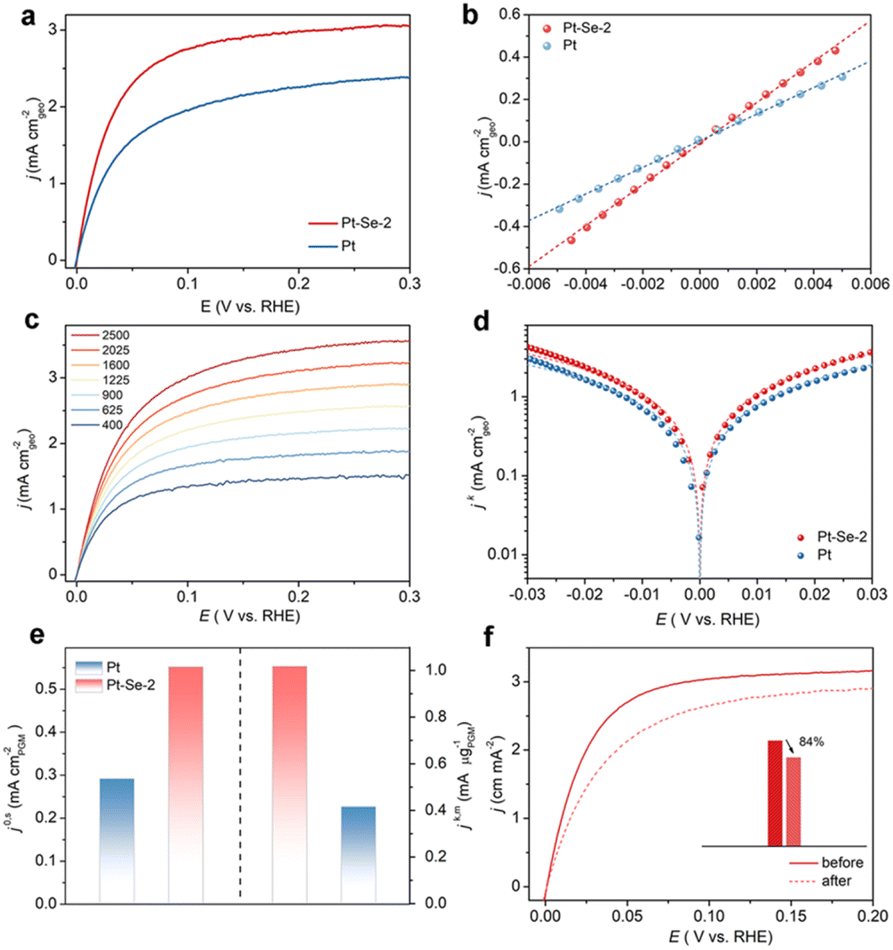 | ||
| Fig. 3 (a) The HOR polarization curves for Pt–Se-2 and commercial Pt at the rotating speed of 1600 rpm with a scan rate of 10 mV s−1 in H2-saturated 0.1 M KOH. (b) Linear fitting curves in the micro-polarization region of HOR polarization curves for Pt–Se-2 and commercial Pt. (c) Polarization curves for Pt–Se-2 in H2-saturated 0.1 M KOH electrolyte at rotating speeds varied from 2500 to 400 rpm. (d) Tafel plots with the Butler–Volmer fitting lines for Pt–Se-2 and commercial Pt. (e) Comparison of the specific activities (j0,s) and mass activities (jk,m) (@ 50 mV) of Pt–Se-2 and commercial Pt. (f) HOR polarization curves for Pt–Se-2 in H2-saturated 0.1 M KOH at a rotating speed of 1600 rpm before and after 1000 CV cycles. | ||
After normalization of the ECSA by Cu stripping, Pt–Se-2 displayed a remarkable intrinsic activity (j0,s) of 0.552 mA cm−2, showing a 2-fold increase compared with commercial Pt (0.292 mA cm−2) (Fig. 3e and S20†). Remarkably, the intrinsic activity (j0,s) and mass activity of Pt–Se-2 (jk,m) (@ 50 mV) exceed those of most noble metal-based HOR catalysts reported in previous studies (Table S3†). The stability of Pt–Se-2 was also evaluated through the accelerated degradation test (ADT).42 As depicted in Fig. 3f, Pt–Se-2 maintained more than 80% of the current density after 1000 cycles, while commercial Pt underwent almost 40% decay during the HOR activity (Fig. S21†), implying a significant increase in stability with the protection of surface-modified Se. After the stability test, Pt–Se-2 still maintained the previous crystalline phase (Fig. S22†), morphology (Fig. S23†), nanoparticle size (Fig. S24†), and chemical states (Fig. S25†).
Considering that natural gas reforming is still the most widely used method for the production of H2, the inevitable CO in H2 may poison the active sites during the HOR process and lead to the anti-CO property becoming the desirable characteristic for HOR catalysts. To evaluate the CO tolerance of the catalysts, long-term electrocatalysis in CO-containing (100 ppm) H2-saturated 0.1 M KOH was tested to poison the catalysts. As shown in Fig. S26,† commercial Pt only maintained a 64.9% HOR performance after CO poisoning, while Pt–Se-2 maintained 79.3% activity compared with that before the CO poisoning, suggesting the improved CO tolerance on Pt–Se-2.
In situ surface-enhanced infrared absorption spectroscopy (SEIRAS) within the potential range from 0 V to 0.2 V was applied to investigate the interfacial water structure of the catalysts. As shown in Fig. 4a and b, the bands from 3000 to 3600 cm−1 contributed to the O–H stretching mode of interfacial water, and were deconvoluted into three distinct components. Specifically, the bands located near 3200 cm−1 were assigned to tetrahedrally coordinated H-bond water (i.e., ice-like water); the higher bands at approximately 3400 cm−1 were assigned to trihedrally coordinated H-bond water (i.e., liquid-like water); the highest bands positioned at 3600 cm−1 were attributed to weak-bond water (i.e., free water).43–45 It is clear that the ice-like water nearly occupies half the proportion among the three components on Pt, implying that the interfacial water structure on the surface of Pt is rigid with strong hydrogen bonding (Fig. 4c).31,46
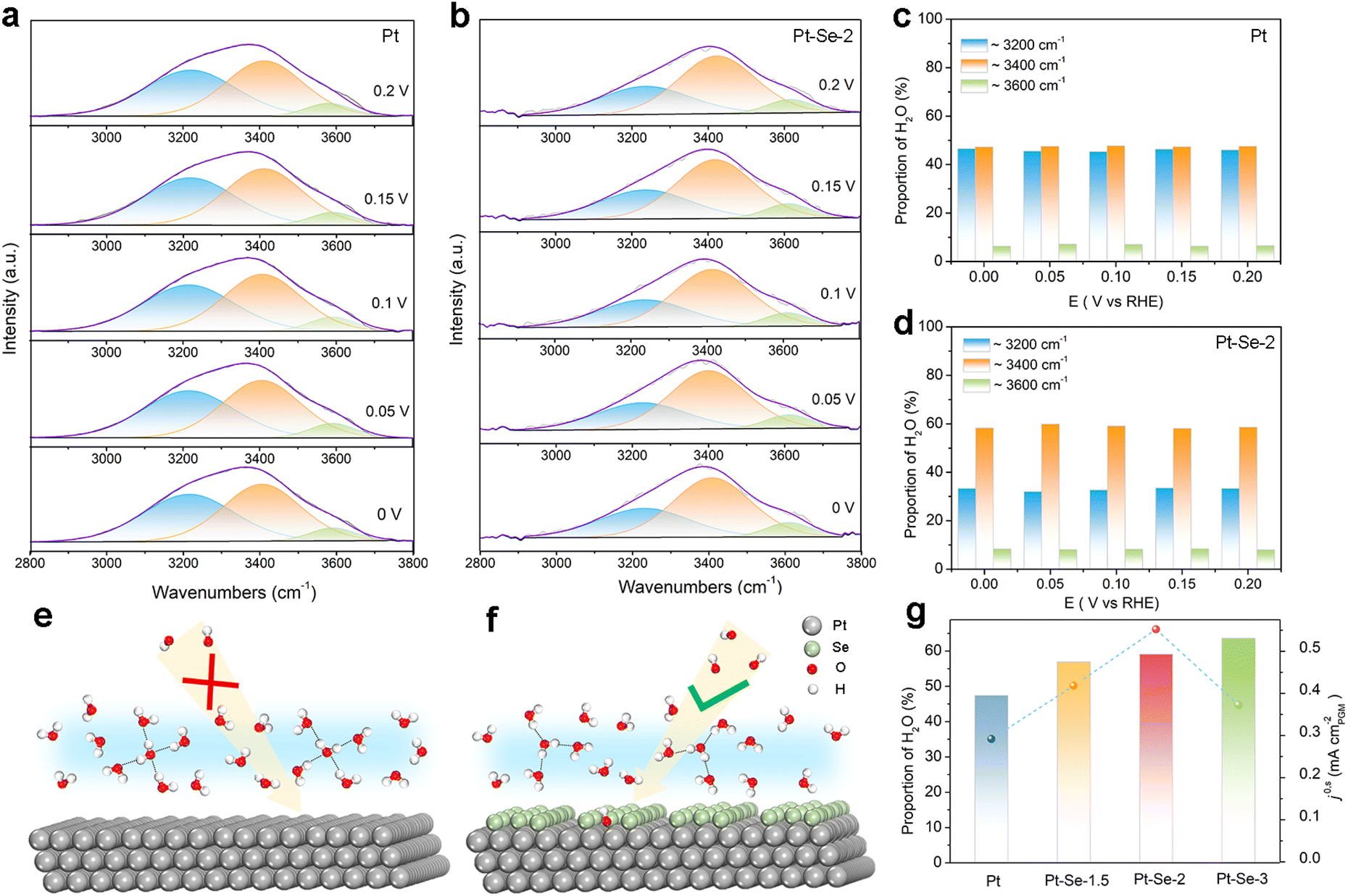 | ||
| Fig. 4 Deconvolution of the O–H stretching vibration features of in situ SEIRAS spectra recorded at potentials from 0 V to 0.2 V vs. RHE for (a) Pt and (b) Pt–Se-2 in 0.1 M KOH. The proportion of the three types of water molecules from the deconvolution of the O–H stretching vibration features of (c) Pt and (d) Pt–Se-2. The proposed mechanism for (e) Pt and (f) Pt–Se-2. The green balls denote the Se atoms, the silver balls denote the Pt atoms, the red balls denote the O atoms, and the white balls denote H atoms. (g) Plots of the intrinsic activities (j0,s) of the alkaline HOR vs. the proportion of the liquid-like water for Pt, Pt–Se-1.5, Pt–Se-2, and Pt–Se-3. | ||
This rigid water structure may lead to a much higher energy barrier for the reorganization of interfacial water, hindering the H2O*/OH* transfer process (Fig. 4e).30 Functioning as electron acceptors, the surface-decorated Se on Pt–Se-2 can be used to reorient the interfacial water molecules.31,47–49 It is clear that on Pt–Se-2, the proportion of ice-like water apparently decreases, while the proportion of liquid-like water at approximately 3400 cm−1 is significantly enhanced (Fig. 4d). It has been reported that this type of asymmetric coordinate water is more flexible than ice-like water.44,50 Therefore, a relatively disordered interfacial water network can be generated on the surface of Pt–Se-2, which can efficiently accelerate the H2O*/OH* transport across the EDL, thereby leading to an enhanced number of available OH species on the surface, and subsequent accelerated HOR kinetics in alkaline electrolyte (Fig. 4f).31,50
It has also been reported that compared with the ice-like water, the water molecules located at higher frequencies are more sensitive to the local electric field and much closer to the surface,50,51 which may lead to stronger electron interactions between water molecules and the surface of catalysts, accelerating the charge transfer and promoting the reaction at the same time.43 The in situ SEIRAS of the catalysts with different amounts of Se appears in Fig. S27.† As the amount of Se increases, the proportion of the ice-like water decreases, indicating the strong water structure regulation of the surface-decorated Se. However, the volcano relationship between HOR activity and the content of Se indicates that the induced high Se content may lead to a decrease in H donors and acceptors, and unfavorable HOR kinetics (Fig. 4g).52,53
The synergistic employment of density functional theory (DFT) calculations provides a deeper comprehension for the mechanism of Pt–Se-x in the HOR process. The Se atoms binding on the hollow sites of the Pt (111) facet were used as a model for Pt–Se-2, and a model for Pt with the (111) facet exposed was also established for comparison (Fig. S28†). As shown in Fig. 5a, the Pt electrons on the surface tend to flow to the Se atoms, which is consistent with the increase in valence of Pt in the XPS results, leading to electron accumulation on the Se site. According to the optimal theoretical structures of the H2O adsorption on Pt and Pt–Se-2 (Fig. S29†), it is clear that the H2O molecule tends to transfer from O-down to parallel within the inducing of Se.31,47,53
 | ||
| Fig. 5 (a) Calculated differential charge density for Pt–Se-2. ELF evaluation for H2O adsorption on (b) Pt and (c) Pt–Se-2. (d) The adsorption energy of OH* on Pt and Pt–Se-2 with the corresponding optimal theoretical structure. (e) The p-DOS of Pt 4d orbitals and O 2p orbitals in Pt and Pt–Se-2. (f) CO stripping curves of Pt and Pt–Se-2 in CO-saturated 0.1 M KOH. (g) The reaction path of Pt and Pt–Se-2, and the corresponding optimized structure. | ||
Electron localization function (ELF) evaluations were also employed for the excess kinetic energy density.54 As shown in Fig. 5b, there was strong binding of Pt–O in this ‘O-down’ structure on the surface of Pt. Through the topological image in Fig. 5c, it is clear that the strong distribution of valence electrons in Se atoms leads to increased electron localization and formation of strong bond interactions with the O atoms of water molecules, confirming the structural transformation of interfacial water presented in the in situ SEIRAS.54–56 In addition to the transformation of water structures, the adsorption energy can also be regulated through electron redistribution, due to the introduction of Se. The OH binding energy of Pt–Se-2 was clearly enhanced compared with that of Pt (Fig. 5d), which can also contribute to the improvement in HOR performance.
According to the orbital coupling state diagram of Pt and Pt–Se-2 within the OH adsorption state (Fig. 5e), the electron cloud coincidence degree of Pt and O on Pt–Se-2 is obviously deeper than that of Pt, demonstrating increased filling of the bonding orbitals, and enhanced OH binding energy.57,58 Furthermore, the apparently lowered CO-stripping peak of Pt–Se-2 in Fig. 5f compared with Pt reveals the increasing OH adsorption within the introduction of Se, proving the conclusion of the calculation.59,60
To further examine the relationship between the Se concentration and HOR performance, modules with different amounts of Se were established to correspond to Pt–Se-1.5 and Pt–Se-3 (Fig. S30†). Similar to the water structure of Pt–Se-2, the water molecules also transfer to the parallel structure due to the inducement of the Se atoms on the surface (Fig. S31†). Through the theoretical calculation in Fig. S32† and the CO stripping curves in Fig. S33,† the OH binding energy of Pt–Se-x depicts the volcano correlation with the enhancement of Se, in accordance with the change in the HOR performance. In addition, the adsorption of hydrogen atoms is taken into consideration (Fig. S34 and S35†).
Compared with the Gibbs free energy of H (ΔGH) on Pt, the ΔGH of Pt–Se-x greatly decreased, and Pt–Se-2 demonstrated the ΔGH closest to zero, greatly promoting the HOR performance as well.61,62 Furthermore, the reaction paths for the HOR process of Pt and Pt–Se-2 are calculated in Fig. 5g. The steps of adsorption of H*/OH* and the formation of water are both spontaneous for Pt and Pt–Se-2, while the water desorption step and the adsorption of H* + OH* are endothermic. The adsorption of H* + OH* for Pt and Pt–Se-2 presents the highest energy barrier, which can be considered as the rate determining step (RDS). Because of the enhanced adsorption of OH, a decreased energy barrier is demonstrated on Pt–Se-2, and contributes to the promoted HOR process.
Conclusions
The HOR-inactive PtSe catalyst can be successfully activated though dynamic Se leaching and phase transition during LSV cycles. The obtained Pt–Se-2 sample, which is a surface Se atom-modified Pt-based nanocatalyst, presents elevated intrinsic activity of 0.552 mA cm−2 (j0,s) and mass activity of 1.084 mA μg−1 (jk,m @ 50 mV), outperforming most of the reported catalysts capable of participating in the alkaline HOR. Combining the experimental results, including in situ SEIRAS and DFT calculation, we reveal that the Se atoms on the surface can gather the electrons of the catalysts, and efficiently regulate the water structure, thereby leading to increased flexibility of the interfacial water network on the Pt–Se surface, and facilitated migration of OH− species across the EDL to the surface of the electrode, which together with the optimized thermodynamic adsorption, contributes to the enhanced alkaline HOR performance. Our work not only expands the horizon for the dynamic evolution of catalysts during the HOR process but also provides a new strategy for the rational design of advanced electrocatalysts through interfacial water structure engineering for the HOR and beyond.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
CY, ZD, JY, and GQ performed the material synthesis and electrochemical tests. CY performed the DFT calculations. WL supervised the work. CY and WL wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22272121). We thank the Core Facility of Wuhan University for performing the ICP-AES and XPS, and the Core Research Facilities of the College of Chemistry and Molecular Sciences for performing the TEM (JEM-2100 Plus) and XRD. The authors would like to acknowledge the Center for Electron Microscopy at Wuhan University for their substantial support to JEM-ARM200F, and the supercomputing system in the Supercomputing Center of Wuhan University for performing the DFT calculations. The authors also thank the BL11B in the Shanghai Synchrotron Radiation Facility (SSRF).Notes and references
- M. H. Tang, S. M. Zhang and S. L. Chen, Chem. Soc. Rev., 2022, 51, 1529–1546 RSC.
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed.
- Z. B. Cui, Z. H. Ren, C. Ma, B. W. Chen, G. Z. Chen, R. H. Lu, W. Zhu, T. Gan, Z. Y. Wang, Z. B. Zhuang and Y. H. Han, Angew. Chem., Int. Ed., 2024, 63, e202404761 CrossRef CAS PubMed.
- W. Y. Ni, T. Wang, F. Héroguel, A. Krammer, S. Lee, L. Yao, A. Schüler, J. S. Luterbacher, Y. S. Yan and X. L. Hu, Nat. Mater., 2022, 21, 804–810 CrossRef CAS PubMed.
- L. C. Wei, N. Fang, F. Xue, S. H. Liu, W. H. Huang, C. W. Pao, Z. W. Hu, Y. Xu, H. B. Geng and X. Q. Huang, Chem. Sci., 2024, 15, 3928–3935 RSC.
- W. Y. Ni, J. L. Meibom, N. Ul Hassan, M. Y. Chang, Y. C. Chu, A. Krammer, S. L. Sun, Y. W. Zheng, L. C. Bai, W. C. Ma, S. Lee, S. Jin, J. S. Luterbacher, A. Schüler, H. M. Chen, W. E. Mustain and X. L. Hu, Nat. Catal., 2023, 6, 773–783 CrossRef CAS.
- J. Zheng, W. C. Sheng, Z. B. Zhuang, B. J. Xu and Y. S. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- S. L. Li, L. Shi, Y. J. Guo, J. Y. Wang, D. Liu and S. L. Zhao, Chem. Sci., 2024, 15, 11188–11228 RSC.
- Y. R. Xue, J. J. Fang, X. D. Wang, Z. Y. Xu, Y. F. Zhang, Q. Q. Lv, M. Y. Liu, W. Zhu and Z. B. Zhuang, Adv. Funct. Mater., 2021, 31, 2101405 CrossRef CAS.
- S. W. Zuo, Z. P. Wu, G. K. Zhang, C. L. Chen, Y. F. Ren, Q. Liu, L. R. Zheng, J. Zhang, Y. Han and H. B. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202316762 CrossRef CAS PubMed.
- S. L. Li, Z. C. Li, R. G. Ma, C. L. Gao, L. L. Liu, L. P. Hu, J. L. Zhu, T. M. Sun, Y. F. Tang, D. M. Liu and J. C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3773–3780 CrossRef CAS PubMed.
- J. Q. Sang, P. F. Wei, T. F. Liu, H. F. Lv, X. M. Ni, D. F. Gao, J. W. Zhang, H. F. Li, Y. P. Zang, F. Yang, Z. Liu, G. X. Wang and X. H. Bao, Angew. Chem., Int. Ed., 2022, 61, e202114238 CrossRef CAS PubMed.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. R. Qiu, A. Bergmann, B. R. Cuenya and O. M. Magnussen, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- J. Y. Duan, T. Y. Liu, Y. H. Zhao, R. O. Yang, Y. Zhao, W. B. Wang, Y. W. Liu, H. Q. Li, Y. F. Li and T. Y. Zhai, Nat. Commun., 2022, 13, 2039 CrossRef CAS PubMed.
- J. Yang, H. F. Qi, A. Q. Li, X. Y. Liu, X. F. Yang, S. X. Zhang, Q. Zhao, Q. K. Jiang, Y. Su, L. L. Zhang, J. F. Li, Z. Q. Tian, W. Liu, A. Q. Wang and T. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- F. Yang, P. Song, X. Ge, Y. Wang, T. Gunji, W. Zhang, X. Zhao and W. L. Xu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2301011120 CrossRef CAS PubMed.
- M. Li, H. Li, H. F. Fan, Q. F. Liu, Z. Yan, A. Q. Wang, B. Yang and E. R. Wang, Nat. Commun., 2024, 15, 6154 CrossRef CAS PubMed.
- Y. D. Pan, J. K. Gao, E. J. Lv, T. T. Li, H. Xu, L. Sun, A. Nairan and Q. C. Zhang, Adv. Funct. Mater., 2023, 33, 2303833 CrossRef CAS.
- J. X. Feng, X. S. Wang and H. Pan, Adv. Mater., 2024, 2411688 CrossRef CAS PubMed.
- C. Xie, W. Chen, Y. Y. Wang, Y. H. Yang and S. Y. Wang, Chem. Soc. Rev., 2024, 10852 RSC.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- Y. K. Feng, S. G. Lu, L. H. Fu, F. L. Yang and L. G. Feng, Chem. Sci., 2024, 15, 2123–2132 RSC.
- Y. H. Wang, X. T. Wang, H. J. Ze, X. G. Zhang, P. M. Radjenovic, Y. J. Zhang, J. C. Dong, Z. Q. Tian and J. F. Li, Angew. Chem., Int. Ed., 2021, 60, 5708–5711 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- Y. Yang, X. D. Sun, G. Q. Han, X. Liu, X. Y. Zhang, Y. F. Sun, M. Zhang, Z. Cao and Y. J. Sun, Angew. Chem., Int. Ed., 2019, 58, 10644–10649 CrossRef CAS PubMed.
- Y. Qiu, L. Xin, Y. W. Li, I. T. McCrum, F. M. Guo, T. Ma, Y. Ren, Q. Liu, L. Zhou, S. Gu, M. J. Janik and W. Z. Li, J. Am. Chem. Soc., 2018, 140, 16580–16588 CrossRef CAS PubMed.
- F. L. Yang, X. Bao, P. Li, X. W. Wang, G. Z. Cheng, S. L. Chen and W. Luo, Angew. Chem., Int. Ed., 2019, 58, 14179–14183 CrossRef CAS PubMed.
- P. Sebastián-Pascual, Y. Shao-Horn and M. Escudero-Escribano, Curr. Opin. Electrochem., 2022, 32, 100918 CrossRef.
- A. Montenegro, C. Dutta, M. Mammetkuliev, H. T. Shi, B. Y. Hou, D. Bhattacharyya, B. F. Zhao, S. B. Cronin and A. V. Benderskii, Nature, 2021, 594, 62–65 CrossRef CAS PubMed.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastian-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- K. A. Sun, X. Y. Wu, Z. W. Zhuang, L. Y. Liu, J. J. Fang, L. Y. Zeng, J. G. Ma, S. J. Liu, J. Z. Li, R. Y. Dai, X. Tan, K. Yu, D. Liu, W. C. Cheong, A. J. Huang, Y. Q. Liu, Y. Pan, H. Xiao and C. Chen, Nat. Commun., 2022, 13, 6260 CrossRef CAS PubMed.
- M. G. Kim, S. H. Kim, J. H. Jang, D. W. Lee, D. Choi, J. H. Park, K. S. Lee, N. J. Chen, C. Hu, Y. M. Lee and S. J. Yoo, Adv. Energy Mater., 2023, 13, 2203133 CrossRef CAS.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- M. F. Li, Z. P. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. H. Zhang, L. Gu, B. V. Merinov, Z. Y. Lin, E. B. Zhu, T. Yu, Q. Y. Jia, J. H. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. F. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- R. Samanta, B. K. Manna, R. Trivedi, B. Chakraborty and S. Barman, Chem. Sci., 2023, 15, 364–378 RSC.
- G. R. Xu, J. Bai, J. X. Jiang, J. M. Lee and Y. Chen, Chem. Sci., 2017, 8, 8411–8418 RSC.
- Q. L. Wen, Y. Lin, Y. Yang, R. J. Gao, N. Q. Ouyang, D. F. Ding, Y. W. Liu and T. Y. Zhai, ACS Nano, 2022, 16, 9572–9582 CrossRef CAS PubMed.
- Y. M. Shi, W. Du, W. Zhou, C. H. Wang, S. S. Lu, S. Y. Lu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 22470–22474 CrossRef CAS PubMed.
- Y. Y. Cong, B. L. Yi and Y. J. Song, Nano Energy, 2018, 44, 288–303 CrossRef CAS.
- C. Y. Yang, Y. B. Li, J. C. Yue, H. J. Cong and W. Luo, Chem. Sci., 2023, 14, 6289–6294 RSC.
- X. Y. Tian, P. C. Zhao and W. C. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- H. S. Wang and H. D. Abruña, J. Am. Chem. Soc., 2017, 139, 6807–6810 CrossRef CAS PubMed.
- S. Q. Zhu, X. P. Qin, Y. Yao and M. H. Shao, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef PubMed.
- M. Osawa, M. Tsushima, H. Mogami, G. Samjeské and A. Yamakata, J. Phys. Chem. C, 2008, 112, 4248–4256 CrossRef CAS.
- L. F. Shen, B. A. Lu, Y. Y. Li, J. Liu, Z. C. Huang-fu, H. Peng, J. Y. Ye, X. M. Qu, J. M. Zhang, G. Li, W. B. Cai, Y. X. Jiang and S. G. Sun, Angew. Chem., Int. Ed., 2020, 59, 22397–22402 CrossRef CAS PubMed.
- M. M. Wang, K. A. Sun, W. L. Mi, C. Feng, Z. K. Guan, Y. Q. Liu and Y. Pan, ACS Catal., 2022, 12, 10771–10780 CrossRef CAS.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef CAS PubMed.
- M. Cabán-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef PubMed.
- C. Y. Li, J. B. Le, Y. H. Wang, S. Chen, Z. L. Yang, J. F. Li, J. Cheng and Z. Q. Tian, Nat. Mater., 2019, 18, 697–701 CrossRef CAS PubMed.
- S. S. Li, L. Wu, Q. X. Liu, M. Y. Zhu, Z. H. Li, C. Wang, X. E. Jiang and J. H. Li, J. Am. Chem. Soc., 2023, 145, 26711–26719 CrossRef CAS PubMed.
- J. L. Bañuelos, E. Borguet, G. E. Brown, R. T. Cygan, J. J. DeYoreo, P. M. Dove, M. P. Gaigeot, F. M. Geiger, J. M. Gibbs, V. H. Grassian, A. G. Ilgen, Y. S. Jun, N. Kabengi, L. Katz, J. D. Kubicki, J. Lützenkirchen, C. V. Putnis, R. C. Remsing, K. M. Rosso, G. Rother, M. Sulpizi, M. Villalobos and H. C. Zhang, Chem. Rev., 2023, 123, 6413–6544 CrossRef PubMed.
- J. B. Le, A. Cuesta and J. Cheng, J. Electroanal. Chem., 2018, 819, 87–94 CrossRef CAS.
- L. Li, Z. D. Wei, X. Q. Qi, M. T. Li, X. Huang, M. M. Deng and S. K. Jiang, J. Phys. Chem. Lett., 2022, 13, 10550–10557 CrossRef PubMed.
- S. C. Zhu, R. O. Yang, H. J. W. Li, S. R. Huang, H. Z. Wang, Y. W. Liu, H. Q. Li and T. Y. Zhai, Angew. Chem., Int. Ed., 2024, 63, e202319462 CrossRef CAS PubMed.
- Z. Y. Luo, L. Zhang, L. Wu, L. Wang, Q. L. Zhang, X. Z. Ren and X. L. Sun, Nano Energy, 2022, 102, 107654 CrossRef CAS.
- D. Chen, R. H. Lu, R. H. Yu, Y. H. Dai, H. Y. Zhao, D. L. Wu, P. Y. Wang, J. W. Zhu, Z. H. Pu, L. Chen, J. Yu and S. C. Mu, Angew. Chem., Int. Ed., 2022, 61, e202208642 CrossRef CAS PubMed.
- M. Luo, J. Y. Cai, J. S. Zou, Z. Jiang, G. M. Wang and X. W. Kang, J. Mater. Chem. A, 2021, 9, 14941–14947 RSC.
- E. Nikolla, J. Schwank and S. Linic, J. Am. Chem. Soc., 2009, 131, 2747–2754 CrossRef CAS PubMed.
- L. F. Shen, B. A. Lu, X. M. Qu, J. Y. Ye, J. M. Zhang, S. H. Yin, Q. H. Wu, R. X. Wang, S. Y. Shen, T. Sheng, Y. X. Jiang and S. G. Sun, Nano Energy, 2019, 62, 601–609 CrossRef CAS.
- B. G. Mao, P. P. Sun, Y. Jiang, T. Meng, D. L. Guo, J. W. Qin and M. H. Cao, Angew. Chem., Int. Ed., 2020, 59, 15232–15237 CrossRef CAS PubMed.
- J. Deng, H. B. Li, S. H. Wang, D. Ding, M. S. Chen, C. Liu, Z. Q. Tian, K. S. Novoselov, C. Ma, D. H. Deng and X. H. Bao, Nat. Commun., 2017, 8, 14430 CrossRef CAS PubMed.
- F. L. Yang, P. Y. Han, N. Yao, G. Z. Cheng, S. L. Chen and W. Luo, Chem. Sci., 2020, 11, 12118–12123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08139k |
| This journal is © The Royal Society of Chemistry 2025 |