
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the nature of the second branch point in the catalytic mechanism of the Fe(II)/2OG-dependent ethylene forming enzyme†
Simahudeen Bathir
Jaber Sathik Rifayee
 ,
Midhun George
Thomas
and
Christo Z.
Christov
*
,
Midhun George
Thomas
and
Christo Z.
Christov
*
Department of Chemistry, Michigan Technological University, Houghton, MI-49931, USA. E-mail: christov@mtu.edu
First published on 13th March 2025
Abstract
Ethylene-forming enzyme (EFE) has economic importance due to its ability to catalyze the formation of ethylene and 3-hydroxypropionate (3HP). Understanding the catalytic mechanism of EFE is essential for optimizing the biological production of these important industrial chemicals. In this study, we implemented molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) to elucidate the pathways leading to ethylene and 3HP formation. Our results suggest that ethylene formation occurs from the propion-3-yl radical intermediate rather than the (2-carboxyethyl)carbonato-Fe(II) (EFIV) intermediate, which conclusively acts as a precursor for 3HP formation. The results also explain the role of the hydrophobic environment surrounding the 2OG binding site in stabilizing the propion-3-yl radical, which defines their conversion to either ethylene or 3HP. Our simulations on the A198L EFE variant, which produces more 3HP than wild-type (WT) EFE based on experimental observations, predict that the formation of the EFIV intermediate was more favored than WT. Also, MD simulations on the EFIV intermediate in both WT and A198L EFE predicted that the water molecules approach the Fe center, which suggests the role of water molecules in the breakdown of the EFIV intermediate. QM/MM simulations on the EFIV intermediate of WT and A198L EFE predicted that the Fe-bound water molecule could provide a proton for the 3HP formation from EFIV. The study underscores the critical influence of the enzyme's hydrophobic environment and second coordination sphere residues in determining product distribution between ethylene and 3HP. These mechanistic insights lay a foundation for targeted enzyme engineering, aiming to improve the selectivity and catalytic efficiency of EFE in biological ethylene and 3HP production.
1. Introduction
Ethylene is an important organic compound due to its abundant use as an industrial chemical and a precursor in synthesizing several other organic molecules.1,2 Ethylene biosynthesis has gained focus recently due to the depletion of ethylene sources such as fossil fuels and the increase in global energy demand.3 The primary source of ethylene production involves burning carbon feedstocks from the fossil fuel industry.4 Biological sources have gained traction recently for a greener way to synthesize ethylene.5 Ethylene is produced through different biological processes by plants and microbes such as bacteria and fungi.5 It is made from various starting materials due to the action of certain enzymes. For example, in plants, ethylene is formed from methionine through oxidation of 1-aminocyclopropane-1-carboxylic acid (ACC) by an ACC oxidase enzyme.6–9 However, the toxic byproduct of this process (cyanide) has limited its use in large-scale ethylene production.10 Some microorganisms produce ethylene in trace quantities, which is not appealing to be considered for widespread exploration.11,12 However, in the case of bacteria such as Pseudomonas syringae and Ralstonia solanacearum and fungus Penicillium digitatum, ethylene is produced in large amounts through the catalysis of Ethylene-Forming Enzyme (EFE).13–16EFE is an iron(II)(Fe(II)) and 2-oxoglutarate (2OG)-dependent (Fe(II)/2OG) oxygenase containing a characteristic double sheet beta-helix (DSBH) fold with distinct reactivities (Fig. 1).17–20 The canonical reactions catalyzed by Fe(II)/2OG-dependent oxygenases involve hydroxylation, epimerization, desaturation, cyclization, and ring-expansion reactions of the substrate through a reactive ferryl intermediate.21,22
 | ||
| Fig. 1 (a) Overall fold of EFE (PDB ID: 5v2y). Violet-colored regions represent the β-sheets, and teal-colored regions represent the α-helices. (b) The skeletal and 3D representation of the computationally modeled EFE active site depicting Fe(III) bonded with the facial triad, 2OG, superoxide, and substrate L-Arg. Non-polar hydrogens are hidden for clarity. | ||
EFE conforms to these functionalities only in its minor reaction involving the two-electron oxidative decarboxylation of co-substrate 2-OG to form succinate and Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ferryl) that hydroxylates the native substrate L-arginine (L-Arg) at C5 carbon (RO), which further undergoes oxidative fragmentation to form pyrroline 5-carboxylate (P5C) and guanidine.23 The major pathway of EFE, however, involves the atypical breakdown of prime substrate 2OG through four-electron oxidation to form ethylene (EF) (Fig. 2a). Although L-Arg is not involved in the EF pathway, it has been reported that the binding of L-Arg is essential for the reactions catalyzed by EFE.17 Also, very recently, a third reaction catalyzed by EFE was experimentally determined by Bollinger et al., which involved the breakdown of 2OG to 3-hydroxypropionate (3HP),24,25 an important feedstock used to synthesize a wide range of chemicals, including acrylic acid, malonic acid, acrylamide, acrylonitrile, and poly-3HP based polymers.26 The reaction mechanism of EFE initiates through the binding of dioxygen (O2) to Fe, which was proposed to give an Fe(III)–OO˙− intermediate (Fig. 1b);27 however, it is yet to be experimentally determined. The branch point between the EF:RO pathway was postulated to be at the O2 activation step of the mechanism where the attack of distal oxygen (Od) of Fe(III)–OO˙− on the C2 carbon of 2OG leads to the formation of peroxy-succinate (RO-I) through C1 decarboxylation which subsequently proceeds to the RO pathway while the succinyl-peroxo-carbonato-Fe(II) (EFI) formation through O2 insertion between C1–C2 proceeds to the EF pathway (Fig. 2b).24,28,29 The RO-I intermediate undergoes cleavage of the Op–Od bond to form ferryl and succinate (RO-II). The ferryl intermediate abstracts hydrogen from the C5-carbon of L-Arg to form the hydroxo-Fe(III) complex and C5-carbon radical (RO-III). The subsequent rebound of the hydroxo group to the C5-carbon results in C-5 hydroxylated L-Arg (RO-IV).23 In contrast, the EFI intermediate was computationally proposed to undergo Op–Od bond homolytic cleavage to form a succinyl radical and C1-derived carbonato-Fe(III) (EFII) intermediate.20,24,29,30 The succinyl radical was proposed to initiate radical decarboxylation to give a propion-3-yl radical (EFIII). The radical coupling of C3 of the propion-3-yl radical to carbonato-Fe(III) leads to the formation of the (2-carboxyethyl)carbonato-Fe(II) (EFIV) intermediate. The second branch point that leads to ethylene and 3HP production is along the EF pathway; however, the intermediate state at which this branching occurs is still under discussion. Bollinger et al. proposed that the branching occurs at the EFIV state.24,28 The EFIV intermediate was proposed to undergo a Grob-like polar concerted fragmentation to give CO2, ethylene, and carbonate or partial fragmentation to give 3HP and C2-derived CO2.24,28 However, the suggested pathway was based on isotope labeling and product analysis with different 2OG modifications, considering the steric effects and stringent requirements for the anti-periplanar disposition of the electrofuge (carboxylate of EFIV) and nucleofuge (carbonate of EFIV).24,28 In addition to the steric effects, there would be electronic factors upon which the fate of the ethylene and 3HP formation depends. A recent computational study on the catalytic mechanism of EFE suggested ethylene formation along with forming Fe(II)-pyro-carbonates, claiming an agreement with experimental Mössbauer data.31 However, the study did not explore the formation of 3HP and the factors responsible for the second branching.31 Hence, it is important to understand this reaction mechanism and define the second branch point of the EF pathway that diverges to ethylene and 3HP production.
O (ferryl) that hydroxylates the native substrate L-arginine (L-Arg) at C5 carbon (RO), which further undergoes oxidative fragmentation to form pyrroline 5-carboxylate (P5C) and guanidine.23 The major pathway of EFE, however, involves the atypical breakdown of prime substrate 2OG through four-electron oxidation to form ethylene (EF) (Fig. 2a). Although L-Arg is not involved in the EF pathway, it has been reported that the binding of L-Arg is essential for the reactions catalyzed by EFE.17 Also, very recently, a third reaction catalyzed by EFE was experimentally determined by Bollinger et al., which involved the breakdown of 2OG to 3-hydroxypropionate (3HP),24,25 an important feedstock used to synthesize a wide range of chemicals, including acrylic acid, malonic acid, acrylamide, acrylonitrile, and poly-3HP based polymers.26 The reaction mechanism of EFE initiates through the binding of dioxygen (O2) to Fe, which was proposed to give an Fe(III)–OO˙− intermediate (Fig. 1b);27 however, it is yet to be experimentally determined. The branch point between the EF:RO pathway was postulated to be at the O2 activation step of the mechanism where the attack of distal oxygen (Od) of Fe(III)–OO˙− on the C2 carbon of 2OG leads to the formation of peroxy-succinate (RO-I) through C1 decarboxylation which subsequently proceeds to the RO pathway while the succinyl-peroxo-carbonato-Fe(II) (EFI) formation through O2 insertion between C1–C2 proceeds to the EF pathway (Fig. 2b).24,28,29 The RO-I intermediate undergoes cleavage of the Op–Od bond to form ferryl and succinate (RO-II). The ferryl intermediate abstracts hydrogen from the C5-carbon of L-Arg to form the hydroxo-Fe(III) complex and C5-carbon radical (RO-III). The subsequent rebound of the hydroxo group to the C5-carbon results in C-5 hydroxylated L-Arg (RO-IV).23 In contrast, the EFI intermediate was computationally proposed to undergo Op–Od bond homolytic cleavage to form a succinyl radical and C1-derived carbonato-Fe(III) (EFII) intermediate.20,24,29,30 The succinyl radical was proposed to initiate radical decarboxylation to give a propion-3-yl radical (EFIII). The radical coupling of C3 of the propion-3-yl radical to carbonato-Fe(III) leads to the formation of the (2-carboxyethyl)carbonato-Fe(II) (EFIV) intermediate. The second branch point that leads to ethylene and 3HP production is along the EF pathway; however, the intermediate state at which this branching occurs is still under discussion. Bollinger et al. proposed that the branching occurs at the EFIV state.24,28 The EFIV intermediate was proposed to undergo a Grob-like polar concerted fragmentation to give CO2, ethylene, and carbonate or partial fragmentation to give 3HP and C2-derived CO2.24,28 However, the suggested pathway was based on isotope labeling and product analysis with different 2OG modifications, considering the steric effects and stringent requirements for the anti-periplanar disposition of the electrofuge (carboxylate of EFIV) and nucleofuge (carbonate of EFIV).24,28 In addition to the steric effects, there would be electronic factors upon which the fate of the ethylene and 3HP formation depends. A recent computational study on the catalytic mechanism of EFE suggested ethylene formation along with forming Fe(II)-pyro-carbonates, claiming an agreement with experimental Mössbauer data.31 However, the study did not explore the formation of 3HP and the factors responsible for the second branching.31 Hence, it is important to understand this reaction mechanism and define the second branch point of the EF pathway that diverges to ethylene and 3HP production.
 | ||
| Fig. 2 (a) Reactions catalyzed by the ethylene-forming enzyme (EFE). (b) Mechanistic hypothesis proposed by this study for the three reactions EFE catalyzes. Green dots denote the key branch points. R–H represents the L-Arg substrate. | ||
Although several proposals have postulated the structural determinants responsible for this unique reactivity, such as the hydrophobic environment lining the 2OG, the effect of L-Arg substrate orientation, and the role of 2OG conformation,17,19,24,28,29 the mechanism of the unique EFE reactions in ethylene and 3HP production still remains unclear. It is important to understand the nature of the second branch point along the EF pathway and to delineate the factors responsible for diverging reactivities and the reasons for the lower output of 3HP formation in WT EFE compared to ethylene. Recent experimental studies on EFE variant A198L showed improved 3HP formation compared to WT.24 The effect of this substitution on the conformational dynamics, the second coordination sphere (SCS), and long-range (LR) interactions needs to be understood to define the factors causing improved 3HP production in the A198L EFE variant. Computational simulations have been widely implemented to study the reaction mechanism of non-heme Fe(II)/2OG enzymes, providing novel insights into the unique reaction mechanism of each enzyme based on molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) calculations.32–41 The studies have shown the complex effects of the SCS and LR interactions on the reaction energetics, intermediates, and the overall reaction path.29,32,42–46 Hence, to determine the second branchpoint of the EFE reaction mechanism and further elucidate the role of SCS and LR residues, we implemented combined MD and QM/MM methods.
2. Methods
2.1 System preparation
The initial structure of EFE was obtained from the crystal structure (PDB ID: 5v2y)17 with L-Arg, 2OG in an offline fashion (C1 carboxylate of 2OG bound trans to H268), and an Fe(II)-analog Mn(II) from the RCSB protein data bank. A high-quality structure of the actual reactant complex (Fe(II)-complex) has recently become available (PDB: 6vp4);23 however, overlaying it with the Mn(II) complex structure (PDB ID: 5v2y)17 showed that the two structures are highly superimposable (RMSD = 0.17 Å); therefore, to maintain consistency with our previous calculations,29,46,47 we used the 5v2y crystal structure in this study. The obtained structure was remodeled by replacing Mn with Fe and modifying the water molecule attached trans to the H189 to dioxygen (O2) to represent the offline Fe(III)–OO˙− intermediate. The system WT-EFE-Fe(III)–OO˙−·L-Arg (WT-EFE) thus obtained has Fe with bidentate coordination to 2OG and monodentate coordination to two histidines H189 and H268 and an aspartate D191. The parameters for L-Arg and 2OG were obtained using the generalized Amber force field (GAFF)48 implemented in the antechamber suite in AmberTools20.49 The metal center parameters at a high spin (HS) Fe(III)–OO˙− intermediate level, including bond and angle force constants, were obtained using the Metal Center Parameter Builder (MCPB.py v3.0)50 of AmberTools20. The remaining protein residues were modeled using an Amber ff14SB force field.51 The Na+ counter ions were added to the system for neutralization using a leap module in Amber20.49 Finally, the protein system was solvated with TIP3P52 water molecules, extending up to 10 Å from the farthest point of the protein surface. For the A198L variant, the alanine residue at the 198th position was replaced by a leucine residue using the Chimera tool.53 The same procedure was repeated for the variant to obtain the A198L-EFE-Fe(III)–OO˙−·L-Arg (A198L-EFE) system. Similarly, the system was modified accordingly for the EFIV intermediate, and the parameters were obtained using the MCPB techniques mentioned above. The EFIV intermediate for WT EFE (WT-EFE-EFIV) and A198L variant (A198L-EFE-EFIV) systems were obtained using the same procedure for further calculations.2.2 Molecular dynamics simulations
Initially, the solvated systems were minimized in two steps. First, the solvent molecules were minimized for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps with a restraint of 100 kcal mol−1 on the solute molecules. Following this, the whole system was minimized for 10000 steps. The minimizations were first carried out using the steepest descent (5000 steps) and then the conjugate gradient (5000 steps) method. The systems were then subjected to controlled heating under NVT ensemble from 0 to 300 K using a Langevin thermostat54,55 with a collision frequency of 1 ps−1 for 250 ps with mild restraint using a harmonic potential of 50 kcal mol−1 Å2 on the solute molecules. The periodic boundary conditions were implemented in all simulations. Long-range electrostatic interactions were calculated with the particle mesh Ewald method56,57 with a direct space and van der Waals cutoff of 10 Å. The SHAKE58 algorithm was used to constrain the bonds involving hydrogens. Following heating, the system undergoes MD simulation for 1 ns to achieve a uniform density with a weak restraint on the solute molecules using periodic boundary conditions. Subsequently, the systems were equilibrated for 3 ns at 300 K in an NPT ensemble without restraint. During the simulations, the pressure was maintained at 1 bar using the Berendsen barostat.59 The production simulations were performed for 1 μs each with a timestep of 2 fs in an NPT ensemble with pressure set at 1 bar and a constant pressure coupling of 2 ps. The GPU version of Amber20 was used for the production simulations.49 The hydrogen bond analysis was carried out using the CPPTRAJ60 module of Amber20. The principal component analysis (PCA) and dynamic cross-correlation analysis (DCCA) were performed on the backbone atoms on the equilibrated portion of the production simulation using the Bio3D61 module in the R programming language.
000 steps with a restraint of 100 kcal mol−1 on the solute molecules. Following this, the whole system was minimized for 10000 steps. The minimizations were first carried out using the steepest descent (5000 steps) and then the conjugate gradient (5000 steps) method. The systems were then subjected to controlled heating under NVT ensemble from 0 to 300 K using a Langevin thermostat54,55 with a collision frequency of 1 ps−1 for 250 ps with mild restraint using a harmonic potential of 50 kcal mol−1 Å2 on the solute molecules. The periodic boundary conditions were implemented in all simulations. Long-range electrostatic interactions were calculated with the particle mesh Ewald method56,57 with a direct space and van der Waals cutoff of 10 Å. The SHAKE58 algorithm was used to constrain the bonds involving hydrogens. Following heating, the system undergoes MD simulation for 1 ns to achieve a uniform density with a weak restraint on the solute molecules using periodic boundary conditions. Subsequently, the systems were equilibrated for 3 ns at 300 K in an NPT ensemble without restraint. During the simulations, the pressure was maintained at 1 bar using the Berendsen barostat.59 The production simulations were performed for 1 μs each with a timestep of 2 fs in an NPT ensemble with pressure set at 1 bar and a constant pressure coupling of 2 ps. The GPU version of Amber20 was used for the production simulations.49 The hydrogen bond analysis was carried out using the CPPTRAJ60 module of Amber20. The principal component analysis (PCA) and dynamic cross-correlation analysis (DCCA) were performed on the backbone atoms on the equilibrated portion of the production simulation using the Bio3D61 module in the R programming language.
2.3 QM/MM simulations
Representative snapshots from the equilibrated regions of the 1 μs MD trajectory were obtained for QM/MM calculations. Water molecules beyond 12 Å from each atom on the protein surface were truncated. QM/MM simulations were conducted using the ChemShell program:62 DL_POLY63 for the MM implementation and Turbomole64 for the QM region. The polarizing effect of the protein environment on the QM region was accounted for using an electrostatic embedding scheme.65 Hydrogen link atoms were used to cap the QM/MM boundaries using a charge shift model.65 The facial triad residues (H189, D191, and H268) with Fe, 2OG, and L-Arg substrates defined the QM regions for WT and A198L EFE systems. In the case of EFIV systems, the EFIV intermediate with the facial triad residues, 2OG, L-Arg, and a water molecule near Fe was included in the QM region. The protein region within 8 Å from the QM region was defined as the flexible MM region, and the rest of the system beyond 8 Å was fixed. The Amber ff14SB forcefield was used for the MM region.51 Experimental studies demonstrated a quintet spin state as the ground state for non-heme Fe enzymes.66 Multiple computational studies on the O2 activation reaction in non-heme Fe(II)/2OG-dependent enzymes have shown that although triplet, quintet, and septet spin states are possible, the reaction proceeds through the HS quintet state.20,41,67–70 Hence, we used the HS quintet spin state of EFE-Fe(III)–OO˙−·L-Arg for our reaction path calculations. The geometry optimization and frequency calculations used the def2-SVP71 basis set with the DFT-B3LYP (QM(B1)/MM) method. The potential energy surface (PES) scans with specific reaction coordinates for the reaction under study were implemented on the optimized reaction complexes (RCs) with a step size of 0.1 Å to obtain transition states (TSs), intermediate (IM), and product complexes (PCs). Without any constraints, the highest energy points along the PES are optimized using the dimer method72 implemented in the DL-FIND73 optimizer. Frequency calculations were performed to confirm the minima and transition states. To further refine the energies, single point (SP) energy calculations were performed on the optimized geometries using a large all-electron def2-TZVP71 basis set (QM(B2)/MM). The zero-point energies from frequency calculations were added to B2 energy to obtain zero-point corrected QM(B3)/MM energies. The results are discussed at the QM(B3)/MM level of energies. Additionally, we calculated SP energy on the QM/MM geometries of the stationary points with different functionals, including GGA functionals such as PBE and B97-D and double hybrid functionals such as B2-PLYP. The calculations revealed that although the performance of the different functionals varies, the energies obtained at B97-D are closer to those at B3LYP (Tables S1–S3†). The QM/MM method with the B3LYP functional implemented in this study has been widely used29,42,74–76 and proven efficient in identifying important intermediates along the EFE reaction pathway,29 including succinyl-peroxo-carbonato-Fe(II) (EFI), which was later proven by experimental studies.28 Natural Orbital (NO) and Spin Natural Orbital (SNO) analyses were carried out using the Gaussian 16 package.77 The coordinates of the QM/MM optimized structures with the absolute energies obtained during the simulations are given in the ESI.†3. Results and discussion
3.1 How is ethylene formed? From the propion-3-yl radical or EFIV intermediate?
Despite several experimental and computational efforts,17–20,24,29,78 how ethylene is formed from 2OG is still unknown. Bollinger et al. proposed the formation of an unusual EFIV intermediate (Fig. 2b), which undergoes a Grob-like fragmentation to form ethylene or partial fragmentation to form 3-hydoxypropionate.24,28 To understand the feasibility of this mechanism, we carried out QM/MM calculations on two snapshots obtained from MD simulations of the WT-EFE system.29 MD simulations of the WT-EFE system were already discussed in our previous study.29 We chose the two snapshots considering the closer orientation of SCS residues surrounding the 2OG co-substrate, which was experimentally shown to influence reactivity.24 | ||
| Fig. 3 (a) Reaction profile of the O2 activation mechanism from the WT1 snapshot. Relative energies are given in kcal mol−1 at QM(B2)/MM (black) and QM(B3)/MM (red) levels. The profile was plotted at QM(B3)/MM levels. (b) Representations of QM/MM optimized structures obtained during the O2 activation mechanism from the EFE WT1 snapshot. Non-polar hydrogens are hidden for clarity. Distances are mentioned in Å. | ||
We could not optimize a stable propion-3-yl radical derived from the WT1 snapshot. Subsequent breakdown of the propion-3-yl radical to ethylene could be correlated with the reduced hydrophobic interactions surrounding the radical in WT1-EFII and WT1-TS3. For example, L173 has positioned one of its methyl groups away from the C3–C4 bond (Fig. 4). The results suggested that the EFIV intermediate might not be required for ethylene formation. All intermediate states identified in the C2–C3 bond breakage PES after WT1-TS3 converge to ethylene upon free optimization (leading to spontaneous breakage of the C4–C5 bond), reinforcing that ethylene formation happens through sequential breakage of the C2–C3 and C4–C5 bonds.
 | ||
| Fig. 4 SCS interactions around the succinyl radical EFII in WT1-EFII (left) and WT2-EFII (right). Distances are denoted by the yellow dashed line and labeled in Å, and salt bridge interactions are denoted by the green dashed line. Active site residues are labeled in black, and SCS residues in red. Non-polar hydrogens are hidden for clarity. | ||
 | ||
| Fig. 5 (a) Reaction profile of the O2 activation mechanism from the WT2 snapshot. Relative energies are given in kcal mol−1 at QM(B2)/MM (black) and QM(B3)/MM (red) levels. The profile was plotted at QM(B3)/MM levels. (b) Representations of QM/MM optimized structures obtained during the O2 activation mechanism from the EFE WT2 snapshot. Non-polar hydrogens are hidden for clarity. Distances are mentioned in Å. | ||
The WT2-EFII intermediate undergoes a C2–C3 bond cleavage, resulting in a stable propion-3-yl radical (WT2-EFIII) with an associated 20.3 kcal mol−1 barrier, which is higher compared to WT1-EFII C2–C3 bond breakage (8.6 kcal mol−1). At WT2-EFIII, the spin density of C3 carbon is −0.94, representing the presence of a radical (Fig. S2†). The stability of the propion-3-yl radical WT3-EFIII could be attributed to the hydrogen bonding and hydrophobic interactions with the SCS residues. At the WT2-EFII and WT2-TS3 states, we could see strong salt bridge interactions between the C5 carboxylate and R277 guanidino group, and there are increased hydrophobic interactions from L173 along the C3–C4 bond (Fig. 4). Contrary to WT1-EFII, both the δ1 and δ2 methyl groups of L173 face toward the C3–C4 bond in WT2-EFII and WT2-TS3. To explore the reactivity of the propion-3-yl radical, we performed QM/MM reaction path calculations using WT2-EFIII. Upon reducing the distance between the C3 carbon of the propion-3-yl radical and the Op atom of Fe(II)-carbonate, the propion-3-yl radical led to barrierless formation of ethylene (WT2-ethylene), which later rebinds to form the (2-carboxyethyl)carbonato-Fe(II) intermediate (WT2-EFIV′) with a barrier of 22.6 kcal mol−1 (calculated from WT2-ethylene) (Fig. S3a†).
To obtain WT2-EFIV directly from WT2-EFIII, we performed reaction path calculations with a modified reaction coordinate that included a slight restraint on the C4–C5 bond to prevent bond breakage that would lead to ethylene. This approach eventually formed WT2-EFIV with a 1.7 kcal mol−1 barrier (Fig. 5a). We, therefore, propose that the competition between C4–C5 bond breakage and C3–Op bond creation (coupling) determines the product distribution between ethylene and 3HP. Fig. 5b shows the optimized structures obtained during the O2 activation mechanism in WT2. We predict that WT2-EFIV may serve as a precursor for 3HP, and the formation of the EFIV intermediate determines the yield of 3HP. For comparison, the overlaid reaction profiles of WT1 and WT2 are given in the ESI (Fig. S4†).
However, our attempts to obtain 3HP from WT2-EFIV through direct C1–Op bond cleavage did not yield a product, indicating that a different mechanism is involved in the breakdown of EFIV to 3HP. Although our analysis suggests that ethylene is generated in competition with EFIV, we considered that it is important to assess whether ethylene formation might also be energetically feasible directly from EFIV, as this was proposed by Bollinger et al.24 Consequently, exploring ethylene formation from WT2-EFIV indicated that, although ethylene can be produced (WT2-ethylene′), the process could be unfeasible due to the high barrier of approximately 57.2 kcal mol−1 (Fig. S3b†).
Hence, based on our QM/MM calculations on two snapshots of WT EFE, we hypothesize that ethylene is directly formed from the breakage of the C2–C3 bond and C4–C5 bond sequentially. In contrast, a stabilized propion-3-yl radical might bind with carbonato-Fe(III) to form an EFIV intermediate, which, upon further reaction, leads to a 3HP product in agreement with the experimental reports.24,28 From the present calculations, the formation of ethylene from EFIV looks unlikely. However, the mechanism of 3HP formation from EFIV still needs to be explored.
 in which the superoxo anion radical is present in the
in which the superoxo anion radical is present in the  orbital (Fig. S7†), whereas, in WT2-RC, the electronic configuration of
orbital (Fig. S7†), whereas, in WT2-RC, the electronic configuration of  shows that the radical is present in the
shows that the radical is present in the  orbital (Fig. S8†). Previous computational studies suggested that the EF pathway is favored irrespective of the presence of the superoxo anion radical in either
orbital (Fig. S8†). Previous computational studies suggested that the EF pathway is favored irrespective of the presence of the superoxo anion radical in either  or
or  orbitals.29,47 The current calculations also agree with the earlier findings, as WT1-RC and WT2-RC snapshots followed the EF pathway. The NO and SNO analyses show that the Fe–Op bond is formed by the orbital overlap of the Fe dx2−y2 and the
orbitals.29,47 The current calculations also agree with the earlier findings, as WT1-RC and WT2-RC snapshots followed the EF pathway. The NO and SNO analyses show that the Fe–Op bond is formed by the orbital overlap of the Fe dx2−y2 and the  orbitals of O2 in WT1-RC. Conversely, the bonding is achieved by the overlap of Fe dxz and
orbitals of O2 in WT1-RC. Conversely, the bonding is achieved by the overlap of Fe dxz and  orbitals in WT2-RC. The transfer of two electrons from the σC1–C2 bond occurs when O2 attacks C2 of 2OG: one electron to the
orbitals in WT2-RC. The transfer of two electrons from the σC1–C2 bond occurs when O2 attacks C2 of 2OG: one electron to the  orbital and the other to the Fe dz2 orbital in WT1, whereas one electron is transferred to the
orbital and the other to the Fe dz2 orbital in WT1, whereas one electron is transferred to the 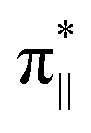 orbital and the other to the Fe dx2−y2 orbital in WT2. Thus, at WT1-EFI and WT2-EFI, the electronic configurations are
orbital and the other to the Fe dx2−y2 orbital in WT2. Thus, at WT1-EFI and WT2-EFI, the electronic configurations are  and
and  respectively. Subsequently, the Op–Od homolytic cleavage happens with the transfer of a β electron from the Fe dz2 orbital to the σOp–Od orbital of O2 in WT1 and from the Fe dx2−y2 orbital to the σOp–Od orbital of O2 in WT2 leading to a succinyl radical and carbonato-Fe(III) in WT1-EFII and WT2-EFII respectively. The succinyl radical leads to C2–C3 bond breakage by transferring an α electron from σC2–C3 to the C2 carboxylate, releasing C2-based CO2 and leaving the radical on the propion-3-yl group thus formed. This process is consistent in both WT1-EFIII and WT2-EFIII. However, upon free optimization of WT1-EFIII, an α electron from the σC4–C5 combines with the radical present on C3 carbon to form C3–C4-based ethylene. Meanwhile, the β electron from C5 carboxylate moves to the Fe center to form Fe(II)-carbonate. Conversely, the propion-3-yl radical was optimized with a single electron on C3 carbon in WT2-EFIII. Upon the close approach of the propion-3-yl radical to the carbonato-Fe(III) in WT2-TS4, the radical β electron is transferred to carbonato-Fe(III) to form the (2-carboxyethyl)carbonato-Fe(II) intermediate (WT2-EFIV). The electronic structure analysis of WT1 and WT2 suggests that the electronic rearrangement of the propion-3-yl radical determines the formation of either ethylene or the (2-carboxyethyl)carbonato-Fe(II) intermediate.
respectively. Subsequently, the Op–Od homolytic cleavage happens with the transfer of a β electron from the Fe dz2 orbital to the σOp–Od orbital of O2 in WT1 and from the Fe dx2−y2 orbital to the σOp–Od orbital of O2 in WT2 leading to a succinyl radical and carbonato-Fe(III) in WT1-EFII and WT2-EFII respectively. The succinyl radical leads to C2–C3 bond breakage by transferring an α electron from σC2–C3 to the C2 carboxylate, releasing C2-based CO2 and leaving the radical on the propion-3-yl group thus formed. This process is consistent in both WT1-EFIII and WT2-EFIII. However, upon free optimization of WT1-EFIII, an α electron from the σC4–C5 combines with the radical present on C3 carbon to form C3–C4-based ethylene. Meanwhile, the β electron from C5 carboxylate moves to the Fe center to form Fe(II)-carbonate. Conversely, the propion-3-yl radical was optimized with a single electron on C3 carbon in WT2-EFIII. Upon the close approach of the propion-3-yl radical to the carbonato-Fe(III) in WT2-TS4, the radical β electron is transferred to carbonato-Fe(III) to form the (2-carboxyethyl)carbonato-Fe(II) intermediate (WT2-EFIV). The electronic structure analysis of WT1 and WT2 suggests that the electronic rearrangement of the propion-3-yl radical determines the formation of either ethylene or the (2-carboxyethyl)carbonato-Fe(II) intermediate.
3.2 Why SCS variant A198L changes the ratio between ethylene and 3HP?
Experimental studies on WT EFE indicate that 3HP was produced in a yield of less than 1%, significantly lower than the yields of ethylene and P5C.24,28 However, the SCS variant A198L was reported to produce high yields of 3HP (about 58–66%) compared to WT due to the steric bulk provided by the leucine residue, which perturbs the active site to favor 3HP formation over ethylene.24 To give an atomistic insight into how the A198L variant of EFE increases the production of 3HP, we carried out MD and QM/MM studies. | ||
| Fig. 6 The overall protein dynamics of A198L-EFE. (a) Dynamic cross-correlation matrix shows the regions of correlated and anticorrelated motions in the A198L variant. (b) Principal component analysis shows the flexible regions of the A198L variant. The circled regions show loss of correlated/anticorrelated motion in (a) and flexibility in (b) compared to the WT-EFE system. | ||
 | ||
| Fig. 7 (a) Reaction profile of the O2 activation mechanism in the A198L-EFE variant. Relative energies are given in kcal mol−1 at QM(B2)/MM (black) and QM(B3)/MM (red) levels. The profile was plotted at the QM(B3)/MM level. (b) Optimized structures obtained during the QM/MM simulations. Distances are mentioned in Å. | ||
The reaction path calculation with the same reaction coordinate as for WT1 led to the energetically favorable EF pathway, forming an Fe(II)-succinyl-peroxy carbonate intermediate (A198L-EFI) with a 9.8 kcal mol−1 activation barrier (Fig. 7). A198L-EFI then undergoes homolytic cleavage of the Op–Od bond, forming carbonato-Fe(III) and a succinyl radical (A198L-EFII). This succinyl radical intermediate subsequently undergoes C2–C3 bond cleavage, forming a propion-3-yl radical (A198L-EFIII) through a barrier of 15.0 kcal mol−1, which then led to the formation of the EFIV intermediate (A198L-EFIV) with an activation barrier of 1.3 kcal mol−1. The presence of succinyl and propion-3-yl radicals at A198L-EFII and A198L-EFIII, respectively, is confirmed by the spin density analysis shown in Fig. S11.†
The analysis of the SCS interactions shows that the two methyl groups of the substituted residue L198 form hydrophobic interactions with the C4 carbon of the succinyl radical (3.76 Å and 3.72 Å) in A198L-EFII. Similarly, the residues L173 and I186 also form hydrophobic interactions with the C3 carbon of the succinyl radical. Like WT2-EFII, these interactions help stabilize propion-3-yl radical formation and the subsequent approach of this radical to carbonato-Fe(III) to form A198L-EFIV. However, the A198L variant does not require additional restraints for this conversion as in WT2-EFIII because the additional methyl groups of L198 provide the necessary steric effects to form A198L-EFIV (Fig. 8). Based on EDA analysis, the residues E84, D253, and E285 give energetically stabilizing contributions to A198L-TS3 for C2–C3 bond breakage. Contrary to WT snapshots, the residues D91, R171, and D201 are involved in destabilizing A198L-TS3 (Fig. S12†). R171 is involved in hydrogen bonding interactions with C1 carboxylate (of carbonato-Fe(III)) and L-Arg, while E84 has hydrogen bonding with the guanidino group of L-Arg.
 | ||
| Fig. 8 Hydrophobic and salt bridge interactions surrounding the succinyl and propion-3-yl radicals during the formation of EFIV in A198L-EFE. Distances (yellow dashed lines) were tabulated and mentioned in Å. Green dashed lines denote the salt bridge interactions. Non-polar hydrogens were hidden for clarity. | ||
To explore if ethylene formation from the propion-3-yl radical is as favored in the A198L-EFE variant as in the WT, we performed reaction path calculation starting from A198L-EFIII by increasing the distance between C4 and C5 to facilitate ethylene formation. However, the simulation resulted in an EFIV intermediate rather than ethylene, aligning with experimental observations that in the A198L variant, 58–66% of the EF pathway flux is redirected toward the 3HP product. In agreement with the experimental studies,24 we elucidated that 3HP is formed from the EFIV intermediate. The ethylene product is formed with a barrier of 5 kcal mol−1 by applying a constraint on the distance between Op and C3 to prevent bond formation. Hence, the results suggest that ethylene formation is feasible in the A198L mutant, although not as favored as in the WT, and might require further structural rearrangements in the SCS interactions that keep the C3 carbon away from the Op. The calculations imply that in the A198L-EFE variant, the formation of the EFIV intermediate is facilitated compared to WT, thereby increasing the output of 3HP compared to WT EFE. The electronic configuration of A198L-RC is  in which the superoxo anion radical is present in the
in which the superoxo anion radical is present in the  orbital (Fig. S13†). The subsequent electronic rearrangement through the successive steps follows the same pathway as the WT2-RC pathway.
orbital (Fig. S13†). The subsequent electronic rearrangement through the successive steps follows the same pathway as the WT2-RC pathway.
3.3 Conformational dynamics of the (2-carboxyethyl)carbonato-Fe(II) intermediate in the WT and A198L EFE
The (2-carboxyethyl)carbonato-Fe(II) intermediate is unique for EFE24,28 as it was not indicated as part of the catalytic mechanism of Fe(II)/2OG-dependent oxygenases. To understand its conformational properties, we performed MD simulations on the EFIV intermediate in both WT and A198L variants.The simulation of the EFIV intermediate in WT EFE (WT-EFE-EFIV) shows a stable trajectory with an average RMSD of 1.52 Å (Fig. S14†). Compared to the Fe(III)–OO˙− dynamics, the L-Arg substrate has moved slightly away from the Fe(II)-center in WT-EFE-EFIV simulations (Fig. S15†). Furthermore, the hydrogen bonding analysis of WT-EFE-EFIV simulation revealed differences in the hydrogen bonding interactions of the L-Arg substrate. Notable changes include the lower stability (22%) of hydrogen bonding interactions between Nε of L-Arg and D191 carboxylate compared to WT-Fe(III)–OO˙− dynamics (64%). This loss in hydrogen bonding interaction leads to the slight movement of L-Arg away from the Fe(II) center. This observation could be attributed to the lack of the role of L-Arg in the EF pathway. Also, the carbonate group of the EFIV intermediate forms stable hydrogen bonding interactions with the guanidino group of Arg171 (28%). Also, we analyzed the 1 μs trajectory and observed that the water molecules often approach the Fe(II) center. Based on histogram analysis, more than 60% of the snapshots in the trajectory have at least one water molecule within 5 Å from the Fe(II) center (Fig. S16†). This is brought about by the slight displacement of the L-Arg substrate, allowing water molecules to access the Fe center. Based on this intriguing observation, we hypothesize that the further breakdown of the EFIV intermediate might involve the role of water molecules, possibly as proton donors.
We further implemented DCCA analysis to understand the LR correlated/anticorrelated interactions at the EFIV intermediate state. EFIV dynamics revealed loss of correlated and anticorrelated motions predominant in Fe(III)–OO˙−. The EFIV intermediate attached to the Fe center exhibited highly correlated motion with the other Fe-coordinated residues. The regions, including residues 80–93, 210–240, and 290–305, have limited correlated and anticorrelated motion with each other (Fig. S17†). The PCA analysis also showed a loss in the flexibility of these regions compared to WT-Fe(III)–OO˙− (Fig. S17†). These regions influence the substrate binding and are involved in the hydrophobic region surrounding the EFIV intermediate. This apparent change in the flexibility and the correlated motions could be related to the active site change and corresponding LR conformational changes, allowing the solvent molecules to access the Fe center. Overall conformational dynamics analysis of the EFIV intermediate in WT intriguingly revealed that more water molecules have access to the Fe center.
To provide some structural background about the catalytic differences between the WT and A198L EFE forms, we explored the conformational dynamics of the EFIV intermediate in the A198L variant (A198L-EFE-EFIV) (Fig. S18†). Similar to WT-EFE-EFIV, the hydrogen bonding interactions between Nε of L-Arg and D191 carboxylate are less prominent in A198L-EFE-EFIV. The C5 carboxylate of the EFIV intermediate forms stable hydrogen bonding interactions with the guanidino group of R277, similar to the WT-EFE-EFIV system and WT Fe(III)–OO˙− system. Interestingly, the hydrogen bonding between R171 and the carbonate group of EFIV is relatively more stable (33%) than that in WT-EFE-EFIV. We also observe the approach of solvent molecules to the Fe center (within 5 Å), which is observed in more than 60% of the trajectory, similar to the WT-EFE-EFIV system (Fig. S19†). This observation reiterates the intriguing nature of the EFIV intermediate, which allows water molecules to access the Fe(II) center.
DCCA analysis shows that the correlated motions observed in WT-EFE-EFIV and WT Fe(III)–OO˙− forms have been lost in the A198L-EFE-EFIV form (Fig. S20†). However, there is an increased anticorrelated motion between the region forming β4 and β5 sheets (residues 87–92) and the region forming α8 and α9 (residues 186–202) helices. These regions have the residues involved in substrate stabilization. This observation could be related to the change in the active site with increased solvent access to the active site. The conformational dynamics of A198L-EFE-EFIV revealed changes in the SCS interactions and correlated motions, which could help in the reaction involving the breakdown of the EFIV intermediate.
3.4 Mechanism of 3HP formation
The breakdown of the EFIV intermediate has been proposed to form either ethylene or 3HP. While the conversion of EFIV to ethylene is energetically unfavorable, the formation of 3HP from its partial breakdown should be explored further. Direct breakdown of EFIV through decarboxylation of C1 carboxylate to 3HP is also not feasible. However, MD simulations of the EFIV intermediate in WT and A198L variants show that water molecules can access the Fe(II) center, suggesting the potential role of water in the breakdown of EFIV. Previous computational studies have explored the role of water molecules in the formation of ethylene.20 Participation of water in the catalytic mechanism by binding to the metal center and serving as proton donors was proposed in several enzymes, including isopenicillin N synthase82 and pterin-dependent hydroxylases.83 To explore this hypothesis in the case of 3HP formation, we conducted QM/MM simulations to define the reaction mechanism of 3HP formation in WT and A198L EFE variants, with water acting as a proton donor. | ||
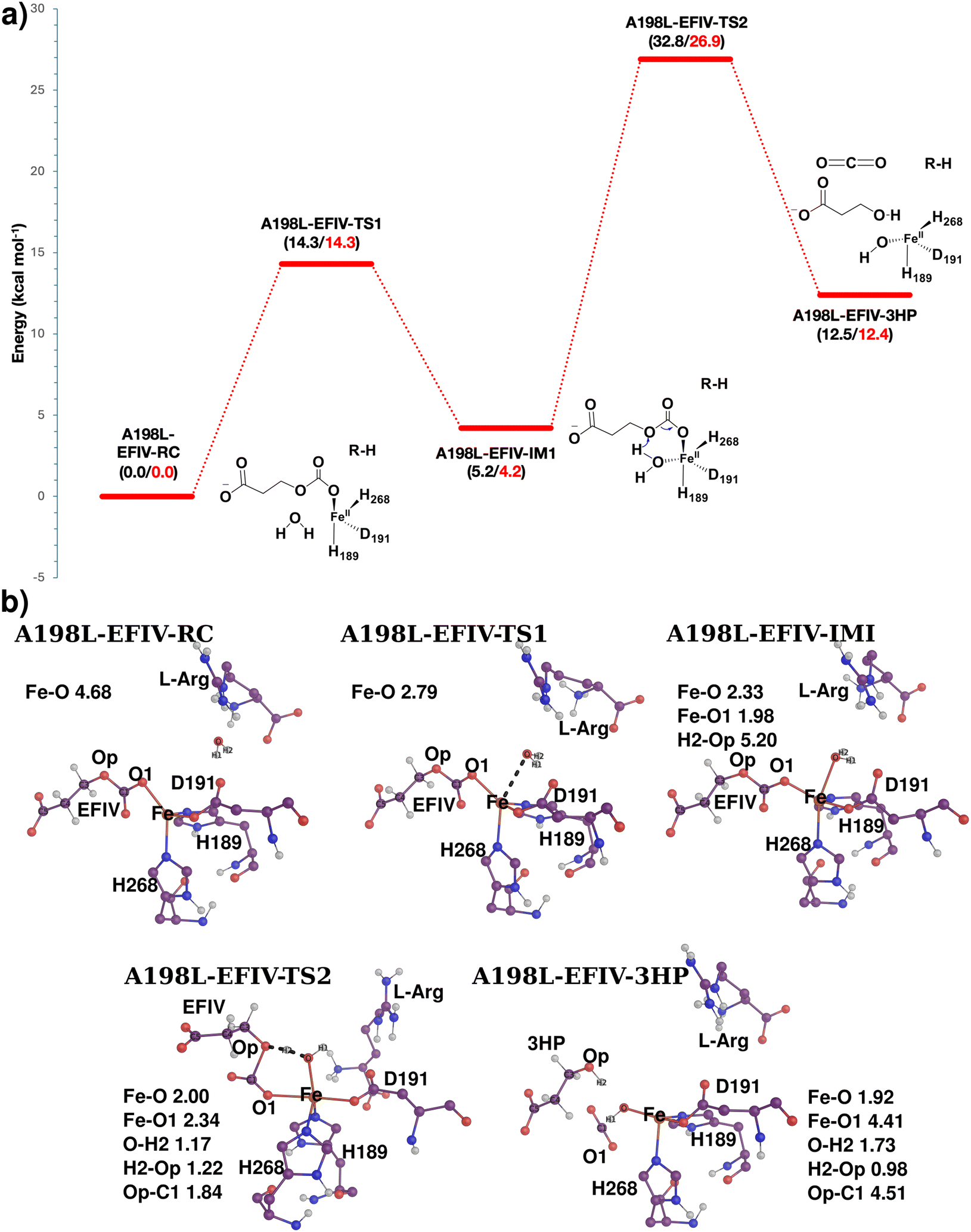
| Fig. 9 (a) Reaction profile of 3HP formation in the WT-EFE-EFIV system. Relative energies are given in kcal mol−1 at QM(B2)/MM (black) and QM(B3)/MM (red) levels. The profile was plotted at QM(B3)/MM levels. The pink arrow indicates electron movement in ethylene formation, and the blue indicates electron movement in 3HP formation. (b) The optimized geometries of the intermediates and transition states obtained during the QM/MM simulations. | ||
 | ||
| Fig. 10 (a) Reaction profile of the 3HP formation mechanism in A198L EFE. Relative energies are given in kcal mol−1 at QM(B2)/MM (black) and QM(B3)/MM (red) levels. The profile was plotted at QM(B3)/MM levels. (b) The optimized geometries of the intermediates and transition states obtained during the QM/MM simulations. | ||
3.5 Alternate sources of hydrogen for EFIV breakdown
In addition to the Fe(II)-coordinated water molecule as a proton source, we explored a potential proton transfer from the protonated SCS residues in EFIV (Fig. S23†). In particular, we attempted proton transfer from R171 – one of the SCS residues involved in C1 stabilization of 2OG and carbonate stabilization in EFIV, as studies suggest that arginine can be involved in proton transfer reactions.84 Despite several attempts using different combinations of reaction coordinates, the formation of 3HP or ethylene is either not achieved or energetically unfavorable. We also explored proton transfer from a water molecule through the E84 residue, which did not lead to stable products. Additionally, we explored proton transfer from non-coordinated water molecules, but it did not lead to 3HP or ethylene products. Based on our calculations, Fe(II)-coordinated water can be the viable proton source for forming 3HP.4. Conclusions
The current study provides a comprehensive account of the mechanism of EFE, particularly the second branch point along the EF pathway, which determines the formation of ethylene and 3HP. We employed combined MD and QM/MM simulations to understand the mechanism. Our primary goal was to describe the EF pathway through conformational analysis of intermediates obtained during the reaction, including the succinyl radical (EFII), the propion-3-yl radical (EFIII), and the (2-carboxyethyl)carbonato-Fe(II) (EFIV) intermediate. We presented the critical conformational and electronic factors related to these intermediates and their outcomes, particularly the hydrophobic environment around 2OG, SCS, and LR residues that govern the stability and fate of these intermediates. Contrary to the previous proposal, we propose a pathway in the EFE reaction mechanism where the second branch point of the EF pathway occurs at the EFIII intermediate state instead of EFIV.24,28Our simulations suggested that ethylene formation happens through a pathway involving sequential bond breakages of the C2–C3 bond followed by the C4–C5 bond of the succinyl radical through a branch point propion-3-yl radical (EFIII) intermediate. QM/MM simulations on two snapshots of the WT EFE system predicted that the competition between C4–C5 bond breakage and C3–Op bond coupling determines the fate of the EFIII intermediate to form ethylene as in WT1, or EFIII approaches and binds to carbonato-Fe(III) to form the 2-carboxyethylcarbonato-Fe(II) (EFIV) intermediate as in WT2, respectively. The results predicted that the fine-tuning of the hydrophobic SCS residues surrounding the propion-3-yl radical (2OG binding site) plays a key role in stabilizing the propion-3-yl radical. More relaxed hydrophobic SCS interactions surrounding the EFIII intermediate lead to its rapid decomposition to ethylene, whereas stable EFIII with tighter hydrophobic interactions surrounding it eventually led to the formation of the EFIV intermediate. Our efforts to simulate ethylene formation from EFIV have not provided any feasible pathway.
We also carried out the QM/MM simulations on the A198L EFE variant, which was experimentally shown to have increased 3HP formation, to verify that 3HP formation comes from EFIV. Simulations on the A198L EFE predicted that the variant showed tighter SCS interactions around the propion-3-yl radical, which facilitates the formation of the EFIV intermediate. However, the ethylene formation from the propion-3-yl radical in A198L EFE required additional restraints to prevent its bonding with carbonato-Fe(III), suggesting that additional conformational/SCS rearrangement is required in A198L EFE to form ethylene. Furthermore, we propose that the fate of the EFIII intermediate dictates the product distribution of ethylene and 3HP.
Moreover, our study explored the EFIV intermediate in WT and A198L EFE using MD simulation, which predicted that water molecules, facilitated by enzyme flexibility, can access the Fe(II) center. This provides a rationale for a mechanism where a water molecule bound to the Fe(II) center can act as a proton donor, aiding in 3HP formation. Our QM/MM simulations revealed that the Fe(II)-coordinated water molecules transfer a proton to the EFIV intermediate with simultaneous decarboxylation, giving 3HP in both WT and A198L EFE with comparable energies. However, the difference in the product distribution of ethylene and 3HP between WT (approx. 99:1) and A198L (approx. 60:30) could be attributed to the enhanced facilitation of the EFIV intermediate formation in A198L EFE compared to WT.
Overall, our computational study provides important insight into the recent experimental studies24,28 about the reaction mechanism of EFE, emphasizing the role of the SCS in tuning the reaction product distribution. Thus, the results provided in this study in the longer term can guide future enzyme redesign efforts of EFE for enhanced production of ethylene or 3HP.
Data availability
The data supporting this article have been included in the ESI.†Author contributions
Conceptualization, C. Z. C.; methodology, C. Z. C. and S. B. J. S. R.; investigation and formal analysis, S. B. J. S. R.; discussion of results S. B. J. S. R., M. G. T., C. Z. C. writing – original draft, S. B. J. S. R. and C. Z. C.; writing – review and editing, S. B. J. S. R., M. G. T., C. Z. C.; visualization, S. B. J. S. R.; funding acquisition, C. Z. C.; supervision, C. Z. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank our collaborators, Prof. Robert P. Hausinger and Prof. Jian Hu from Michigan State University, for the valuable and helpful discussions. This research was supported by the National Science Foundation grant 2203630 to CZC.References
-
F. B. Abeles, P. W. Morgan and M. E. Saltveit, Ethylene in Plant Biology, Elsevier Science, 2012 Search PubMed
.
- A. B. Bleecker and H. Kende, Annu. Rev. Cell Dev. Biol., 2000, 16, 1–18 CrossRef CAS PubMed
- M. Ghanta, D. Fahey and B. Subramaniam, Appl. Petrochem. Res., 2014, 4, 167–179 CrossRef CAS
-
K. M. Sundaram, M. M. Shreehan and E. F. Olszewski, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Ltd, 2010, pp. 1–39 Search PubMed
- R. Fluhr, A. K. Mattoo and D. R. Dilley, Crit. Rev. Plant Sci., 1996, 15, 479–523 CAS
- D. G. McRae, J. A. Coker, R. L. Legge and J. E. Thompson, Plant Physiol., 1983, 73, 784–790 CrossRef CAS PubMed
- J. G. Dong, J. C. Fernández-Maculet and S. F. Yang, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9789–9793 CrossRef CAS PubMed
- Z. Zhang, J.-S. Ren, I. J. Clifton and C. J. Schofield, Chem. Biol., 2004, 11, 1383–1394 CrossRef CAS PubMed
- R. P. Hausinger, S. B. J. S. Rifayee, M. G. Thomas, S. Chatterjee, J. Hu and C. Z. Christov, RSC Chem. Biol., 2023, 4, 635–646 RSC
- G. D. Peiser, T.-T. Wang, N. E. Hoffman, S. F. Yang, H. Liu and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3059–3063 CrossRef CAS PubMed
- T. Ogawa, M. Takahashi, T. Fujii, M. Tazaki, H. Fukuda and J. Ferment, Bioengineering, 1990, 69, 287–291 CAS
- S. B. Primrose, J. Gen. Microbiol., 1977, 98, 519–528 CrossRef CAS PubMed
- K. Nagahama, T. Ogawa, T. Fujii, M. Tazaki, S. Tanase, Y. Morino and H. Fukuda, Microbiology, 1991, 137, 2281–2286 CrossRef CAS PubMed
- H. Fukuda, T. Ogawa, M. Tazaki, K. Nagahama, T. Fujiil, S. Tanase and Y. Morino, Biochem. Biophys. Res. Commun., 1992, 188, 483–489 CrossRef CAS PubMed
- M. Goto, Y. Ishida, Y. Takikawa and H. Hyodo, Plant Cell Physiol., 1985, 26, 141–150 CAS
- M. Goto and H. Hyodo, Plant Cell Physiol., 1987, 28, 405–414 CAS
- S. Martinez, M. Fellner, C. Q. Herr, A. Ritchie, J. Hu and R. P. Hausinger, J. Am. Chem. Soc., 2017, 139, 11980–11988 CrossRef CAS PubMed
- Z. Zhang, T. J. Smart, H. Choi, F. Hardy, C. T. Lohans, M. I. Abboud, M. S. W. Richardson, R. S. Paton, M. A. McDonough and C. J. Schofield, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4667–4672 CrossRef CAS PubMed
- S. Martinez and R. P. Hausinger, Biochemistry, 2016, 55, 5989–5999 CrossRef CAS PubMed
- J. Xue, J. Lu and W. Lai, Phys. Chem. Chem. Phys., 2019, 21, 9957–9968 RSC
- Md. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS PubMed
- R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2008, 39, 21–68 CrossRef PubMed
- R. A. Copeland, K. M. Davis, T. K. C. Shoda, E. J. Blaesi, A. K. Boal, C. Krebs and J. M. Bollinger, J. Am. Chem. Soc., 2021, 143, 2293–2303 CrossRef CAS PubMed
- E. J. Burke, R. A. Copeland, Y. Dixit, C. Krebs and J. M. Bollinger, J. Am. Chem. Soc., 2024, 146, 1977–1983 CrossRef CAS PubMed
- S. Dhingra, Z. Zhang, C. T. Lohans, L. Brewitz and C. J. Schofield, J. Biol. Chem., 2024, 300, 107546 CrossRef CAS PubMed
- X.-R. Jiang, X. Yan, L.-P. Yu, X.-Y. Liu and G.-Q. Chen, Nat. Commun., 2021, 12, 1513 CrossRef CAS PubMed
- S. S. Chaturvedi, M. G. Thomas, S. B. J. S. Rifayee, W. White, J. Wildey, C. Warner, C. J. Schofield, J. Hu, R. P. Hausinger, T. G. Karabencheva-Christova and C. Z. Christov, Chem.–Eur. J., 2023, 29, e202300138 CrossRef CAS PubMed
- R. A. Copeland, S. Zhou, I. Schaperdoth, T. K. C. Shoda, J. M. Bollinger and C. Krebs, Science, 2021, 373, 1489–1493 CrossRef CAS PubMed
- S. S. Chaturvedi, R. Ramanan, J. Hu, R. P. Hausinger and C. Z. Christov, ACS Catal., 2021, 11, 1578–1592 CrossRef CAS
- C.-C. G. Yeh, S. Ghafoor, J. K. Satpathy, T. Mokkawes, C. V. Sastri and S. P. de Visser, ACS Catal., 2022, 12, 3923–3937 CrossRef CAS
- C. Wang and E. Sayfutyarova, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-pz39d
- S. P. de Visser, Chem.–Eur. J., 2020, 26, 5308–5327 CrossRef CAS PubMed
- H. S. Ali, J. Warwicker and S. P. de Visser, ACS Catal., 2023, 13, 10705–10721 CrossRef CAS
- D. W. Kastner, A. Nandy, R. Mehmood and H. J. Kulik, ACS Catal., 2023, 13, 2489–2501 CrossRef CAS
- R. Mehmood, V. Vennelakanti and H. J. Kulik, ACS Catal., 2021, 11, 12394–12408 CrossRef CAS
- H. Chen, K.-B. Cho, W. Lai, W. Nam and S. Shaik, J. Chem. Theory Comput., 2012, 8, 915–926 CrossRef CAS PubMed
- H. J. Kulik and C. L. Drennan, J. Biol. Chem., 2013, 288, 11233–11241 CrossRef CAS PubMed
- T. Borowski, H. Noack, M. Radoń, K. Zych and P. E. M. Siegbahn, J. Am. Chem. Soc., 2010, 132, 12887–12898 CrossRef CAS PubMed
- K. Dutta Dubey, B. Wang, M. Vajpai and S. Shaik, Chem. Sci., 2017, 8, 5335–5344 RSC
- B. Wang, Z. Cao, D. A. Sharon and S. Shaik, ACS Catal., 2015, 5, 7077–7090 CrossRef CAS
- S. Ye, C. Riplinger, A. Hansen, C. Krebs, J. M. Bollinger Jr and F. Neese, Chem.–Eur. J., 2012, 18, 6555–6567 CrossRef CAS PubMed
- S. O. Waheed, S. S. Chaturvedi, T. G. Karabencheva-Christova and C. Z. Christov, ACS Catal., 2021, 11, 3877–3890 CrossRef CAS
- R. Ramanan, S. S. Chaturvedi, N. Lehnert, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, Chem. Sci., 2020, 11, 9950–9961 RSC
- S. S. Chaturvedi, S. B. Jaber Sathik Rifayee, S. O. Waheed, J. Wildey, C. Warner, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, JACS Au, 2022, 2, 2169–2186 CrossRef CAS PubMed
- S. B. J. S. Rifayee, S. S. Chaturvedi, C. Warner, J. Wildey, W. White, M. Thompson, C. J. Schofield and C. Z. Christov, Chem.–Eur. J., 2023, 29, e202301305 CrossRef CAS PubMed
- M. G. Thomas, S. B. Jaber Sathik Rifayee and C. Z. Christov, ACS Catal., 2024, 14, 18550–18569 CrossRef CAS PubMed
- S. S. Chaturvedi, S. B. Jaber Sathik Rifayee, R. Ramanan, J. A. Rankin, J. Hu, R. P. Hausinger and C. Z. Christov, Phys. Chem. Chem. Phys., 2023, 25, 13772–13783 RSC
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed
-
D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, Amber 2020, University of California, San Francisco, 2020. Search PubMed
- P. Li and K. M. Merz, J. Chem. Inf. Model., 2016, 56, 599–604 CrossRef CAS PubMed
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed
- R. L. Davidchack, R. Handel and M. V. Tretyakov, J. Chem. Phys., 2009, 130, 234101 CrossRef PubMed
- J. Liu, D. Li and X. Liu, J. Chem. Phys., 2016, 145, 024103 CrossRef PubMed
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed
- B. J. Grant, A. P. C. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. D. Caves, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, WIREs Computational Molecular Science, 2014, 4, 101–110 CrossRef CAS
- W. Smith, C. W. Yong and P. M. Rodger, Mol. Simul., 2002, 28, 385–471 CrossRef CAS
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed
- M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700–733 CrossRef CAS
- S. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon and W. Nam, Nat. Commun., 2014, 5, 5440 CrossRef CAS PubMed
- A. R. Diebold, C. D. Brown-Marshall, M. L. Neidig, J. M. Brownlee, G. R. Moran and E. I. Solomon, J. Am. Chem. Soc., 2011, 133, 18148–18160 CrossRef CAS PubMed
- D. Fang, R. L. Lord and G. A. Cisneros, J. Phys. Chem. B, 2013, 117, 6410–6420 CrossRef CAS PubMed
- X. Song, J. Lu and W. Lai, Phys. Chem. Chem. Phys., 2017, 19, 20188–20197 RSC
- A. Wójcik, M. Radoń and T. Borowski, J. Phys. Chem. A, 2016, 120, 1261–1274 CrossRef PubMed
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed
- S. S. Chaturvedi, R. Ramanan, N. Lehnert, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, ACS Catal., 2020, 10, 1195–1209 CrossRef CAS PubMed
- R. Ramanan, S. O. Waheed, C. J. Schofield and C. Z. Christov, Chem.–Eur. J., 2021, 27, 11827–11836 CrossRef CAS PubMed
- J. Liu, Z. Wang, X. Sang, X. Zhang and B. Wang, ACS Catal., 2023, 5808–5818 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016. Search PubMed
- S. Chatterjee, M. Fellner, J. A. Rankin, M. G. Thomas, S. B. J S Rifayee, C. Z. Christov, J. Hu and R. P. Hausinger, Biochemistry, 2024, 63(8), 1038–1050 CrossRef CAS PubMed
- S. Bhunia, A. Ghatak and A. Dey, Chem. Rev., 2022, 122, 12370–12426 CrossRef CAS PubMed
- J. C. Lewis, Acc. Chem. Res., 2019, 52, 576–584 CrossRef CAS PubMed
- N. Lehnert, J. Inorg. Biochem., 2012, 110, 83–93 CrossRef CAS PubMed
- M. Lundberg, P. E. M. Siegbahn and K. Morokuma, Biochemistry, 2008, 47, 1031–1042 CrossRef CAS PubMed
- A. Bassan, M. R. A. Blomberg and P. E. M. Siegbahn, Chem.–Eur. J., 2003, 9, 106–115 CrossRef CAS PubMed
- B. L. Greene, G. E. Vansuch, C.-H. Wu, M. W. W. Adams and R. B. Dyer, J. Am. Chem. Soc., 2016, 138, 13013–13021 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Coordinates of the optimized structures and the absolute energies obtained during QM/MM simulations and additional images. See DOI: https://doi.org/10.1039/d4sc08378d |
| This journal is © The Royal Society of Chemistry 2025 |