
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinker driven site-specific catalysis in atomically precise silver cluster assemblies†
Priyanka
Chandrashekar
a,
Arun
Karmakar
 b,
Ravari Kandy
Aparna
a,
Laddi
Singh
c,
Pradip Kumar
Mondal
d,
Subrata
Kundu
*b,
Kalishankar
Bhattacharyya
*c and
Sukhendu
Mandal
*a
b,
Ravari Kandy
Aparna
a,
Laddi
Singh
c,
Pradip Kumar
Mondal
d,
Subrata
Kundu
*b,
Kalishankar
Bhattacharyya
*c and
Sukhendu
Mandal
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala 69551, India. E-mail: sukhendu@iisertvm.ac.in
bElectrochemical Process Engineering (EPE) Division, CSIR-Central Electrochemical Research Institute (CECRI), Karaikudi, Tamil Nadu 630006, India. E-mail: kundu.subrata@gmail.com
cDepartment of Chemistry, IIT Guwahati, Assam 781039, India. E-mail: ksb@iitg.ac.in
dElettra-Sincrotrone Trieste, S.S. 14 Km 163.5 in Area Science Park, Basovizza, Trieste, 34149, Italy
First published on 26th February 2025
Abstract
Metal nanoclusters (NCs) exhibit potential as catalysts for electrochemical studies, providing atomic-level insights into mechanisms. However, it remains elusive to construct an integrated catalyst with a molecular-level understanding of its mechanism, especially in silver cluster assemblies. In this study, we have shown that atomically precise Ag12 cluster assemblies Ag12-py, Ag12-pyz, Ag12-bpy, Ag12-bpa, Ag12-azopy, (where Ag12 = secondary building unit, Py = pyridine, pyz = pyrazine, bpy = 4,4′-bipyridine, bpa = 1,2-bis(4-pyridyl)ethane, and azopy = 4,4′-azopyridine) serve as paradigms for demonstrating the hydrogen evolution reaction (HER), where the catalytic activity is fine-tuned using two functional units: the cluster core and the linkers. The atomic resolution of such catalysts allows tracing the reaction process via experiments coupled with theory and structural analysis. Site-specific catalysis for Ag12-pyz induced by metal cluster assembly and linker synergy can be accurately elucidated to dominate in the series. Taking advantage of the pyrazine linker due to its lower basicity and the isotropic nature of inter-cluster interactions in Ag12-pyz, it shows enhanced catalytic activity and selective hydrogen adsorption at the sulfur site, different from others in the series with nearly five times higher efficiency. This work on a series of silver cluster assemblies provides a substantial structural model to understand the catalyst's active site and activity, further driving advancements in functional cluster-based assemblies.
Introduction
Atomically precise noble metal nanoclusters (NCs), which bridge molecular organometallic complexes and plasmonic metal nanoparticles (NPs),1,2 comprising only a few atoms, allow excellent control over the total structure, exhibiting unique electronic and optical properties, including molecular-like transitions, tunable photoluminescence, and high catalytic activity.3–5 The manipulation of these properties is primarily achieved through control over the cluster core and protective ligands. The distinctive geometry and electronic structure make them particularly attractive in catalysis, due to their high surface-to-volume ratio, atomic precision structure, rich surface chemistry, and low coordination number.5–8 These clusters can indeed be used as ideal model catalysts for correlating catalytic properties with their atom packing structures.8–12 However, the major challenge is the insufficient stability leading to aggregation. Therefore, it requires strategies to stabilize NC structures while preserving cluster integrity.13–16To address these challenges, recent progress has been made in utilizing NCs as nanoscale building blocks for superstructures17–19 through assembly processes by employing bridging ligands, known as cluster-assembled materials (CAMs).20–22 These CAMs not only enhance the stabilization of NCs but also merge the functionalities of NCs and linkers.23 They exhibit superior properties and multiple tunable components, compared to their cluster building blocks.24–26 This approach provides a potential solution for stabilizing NCs while simultaneously enhancing catalytic performance by facilitating improved electron transport and chemical interactions between clusters.
The electrocatalytic hydrogen evolution reaction (HER) is a fascinating and sustainable strategy to produce molecular hydrogen, which is considered an excellent fuel for the future. Hydrogen has high energy density and produces water as the sole byproduct upon combustion, making it an ideal candidate to replace fossil fuels, which cause many detrimental environmental issues.27–29 Despite the inherent inertness or low HER activity of coinage metals such as gold, silver, and copper, the nanoscale regime introduces distinctive material properties, deviating from their bulk counterparts and offering potential avenues for enhanced catalytic activity.30–34
Ligand-protected metal NCs, such as Ag, Au, and Pt, have been minimally explored as catalysts,13,35–39 and their assembled materials remain largely unstudied.40–43 A more in-depth investigation is needed to understand the relationship between the structure of the electrocatalyst and catalytic performance at the atomic level.44–50 Clusters with identical surface ligands and core atoms, but different linkers, offer a unique opportunity to isolate and study the factors influencing overall performance. In this context, we focus on the synthesis and catalytic application of five silver cluster assemblies such as Ag12-py {[Ag12(C6H11S)6(CF3COO)6(C5H5N)6]·4H2O}n,25 Ag12-pyz [Ag12(C6H11S)6(CF3COO)6(C4H4N2)3]n, Ag12-bpy [Ag12(C6H11S)6(CF3COO)6(C10H8N2)3]n, Ag12-bpa [Ag12(C6H11S)6(CF3COO)6(C12H12N2)3(DMF)2]n, and Ag12-azopy [Ag12(C6H11S)6(CF3COO)6(C10H8N4)3(DMF)2]n. By working with precise Ag12 structured systems, we obviated the problem of accurately analyzing the origin of the difference in the catalytic activities, which plagues the study in structure-catalysis analysis. These assemblies exhibit varying activity levels, where the gradual lengthening of ligand systems from pyridine to azopyridine alters cluster arrangement. The Ag12-pyz CAM displayed very good catalytic performance for the HER even at lower over-potentials. The low basicity of the pyrazine linker increases the electropositivity of the silver atom, shifting the catalytic active site from silver to sulfur. We propose that the linker basicity and assembly-induced cluster arrangements in these five assemblies define the active center and activity. This structure–property correlation, combined with experimental and theoretical insights, offers a promising model for cluster chemistry and electrocatalysis research.
Results and discussion
Ag12-py, Ag12-pyz, Ag12-bpy, Ag12-bpa, and Ag12-azopy were prepared by a “one pot method”. The (Ag-SCy)n (SCy-cyclohexanethiol) and AgCF3COO precursors were dissolved in solvent, followed by a dropwise addition of different linkers to yield five distinct assembly patterns: 2 three-dimensional extended Ag12-pyz and Ag12-bpa CAMs and 2 two-dimensional extended Ag12-bpy and Ag12-azopy CAMs, created by covalently linking the clusters with bidentate linkers (pyz = pyrazine, bpy = 4,4′-bipyridine, bpa = 1,2-bis(4-pyridyl)ethane, and azopy = 4,4′-azopyridine). The inter-cluster distance and dimensionality depend on the linker length and flexibility. Long rigid linkers, such as bpy and azopy, tend to facilitate two-dimensional assemblies (Fig. 1 and 2). In contrast, more flexible linkers like bpa and the shorter rigid pyz linker promote the formation of three-dimensional structures. Non-covalent interactions extended the clusters with monodentate pyridine in Ag12-py (py = pyridine) (Fig. 1).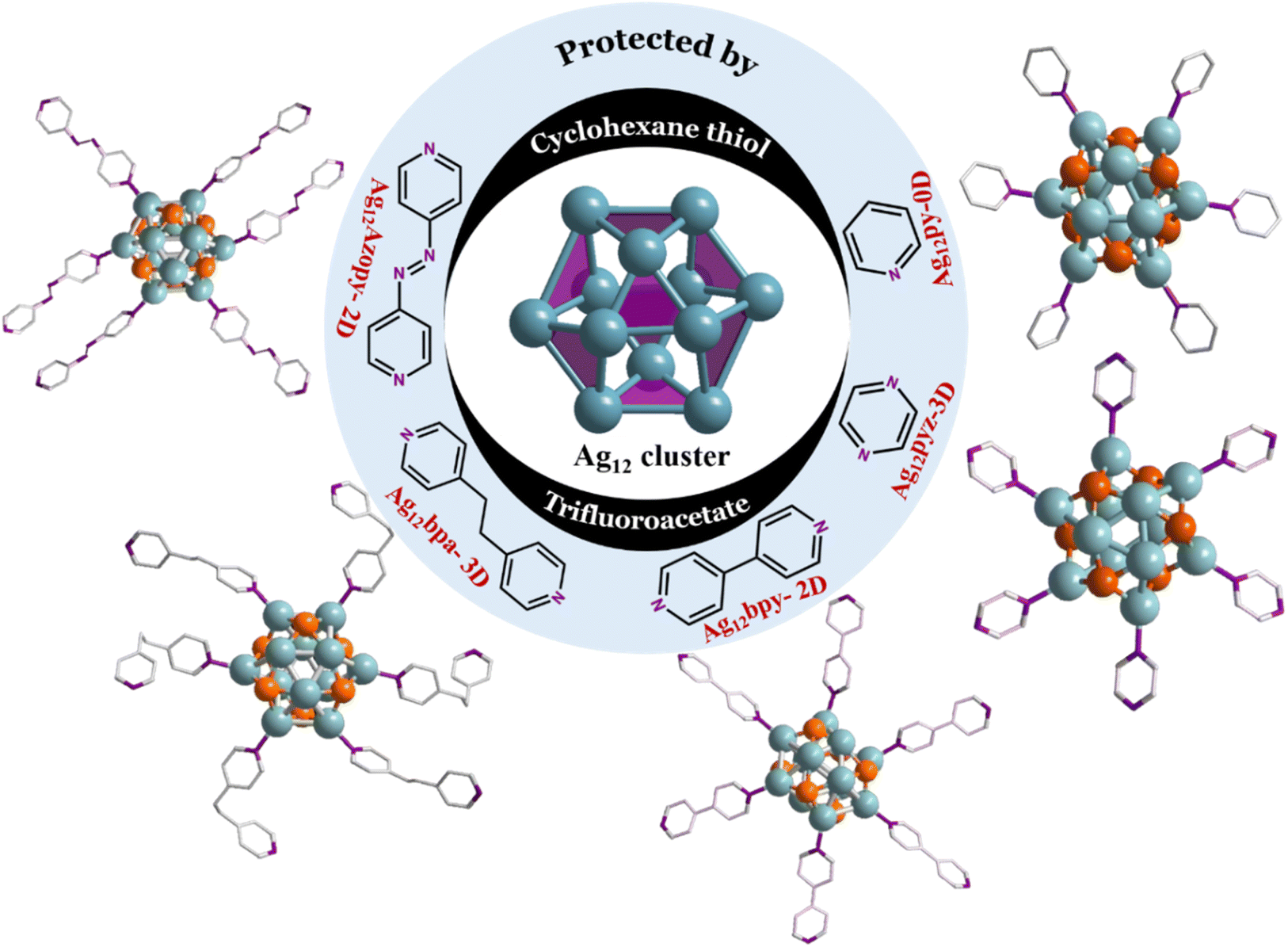 | ||
| Fig. 1 (a) Schematic representation (cluster core with the linker) of the five silver nanocluster assemblies. The core constituting Ag12 (center) is protected by cyclohexane thiol and trifluoroacetate ligands. The assemblies (right to left) are Ag12-py (0D NC), Ag12-pyz (3D CAM), Ag12-bpy (2D), Ag12-bpa (3D), and Ag12-azopy (2D). The color code: Ag, blue; S, orange; N, violet; C, grey. All O, F, and H atoms are omitted for clarity. | ||
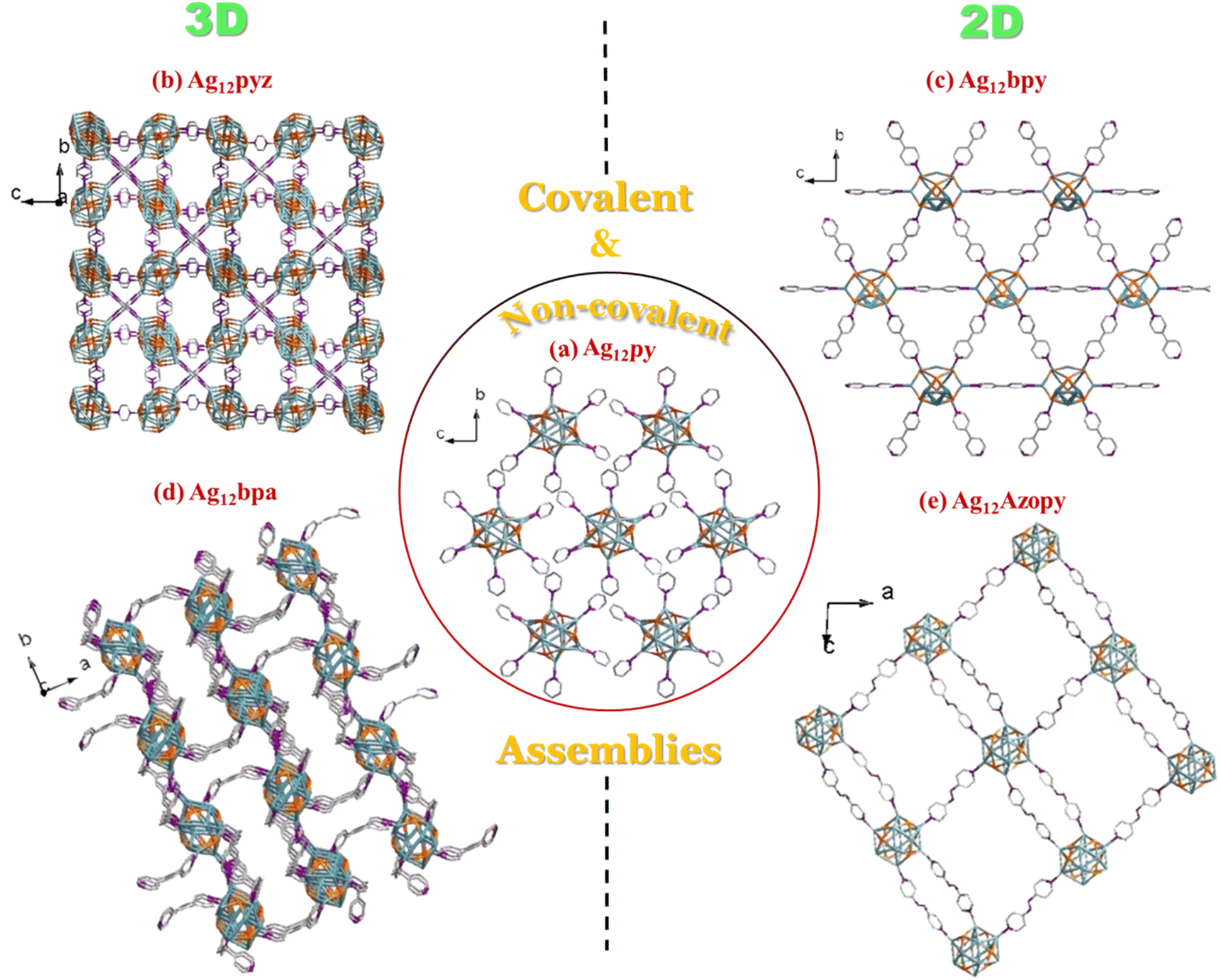 | ||
| Fig. 2 Schematic representation of the four covalent assemblies (covalent bonding between the Ag of the cluster and the N of the different N-based linkers) and a non-covalent assembly. (a) Ag12-py NC extends via non-covalent interactions with the pyridine ligand, viewed along the a direction. (b) The Ag12 clusters in Ag12-pyz extend in three dimensions, and the figure is viewed along the a direction and slightly tilted to see the three-dimensional representation. (c) The bpy linker in Ag12-bpy extends the cluster in two-dimensional hexagonal-like representation viewed along the a direction. (d) The flexibility of the bpa linker in Ag12-bpa extends the cluster three-dimensionally, which is viewed along the c direction and slightly tilted to see the assembly extended in the c direction. (e) The azopy linker in Ag12-azopy extends the cluster two-dimensionally, which is viewed along the b direction. The color code: Ag, blue; S, orange; N, violet; C, grey. All O, F, and H atoms are omitted for clarity. | ||
Single Crystal X-Ray Diffraction (SCXRD) data of Ag12-pyz crystals (Table S1 and Fig. S1a†) show that they crystallize in the Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (201) space group with the unit formula [Ag12(C6H11S)6(CF3COO)6(C4H4N2)3]n. The cluster core constitutes 12 Ag atoms held together by twelve argentophilic interactions with an ∼Ag⋯Ag distance of 2.93 Å (Table S5†), which is protected by six primary cyclohexane thiol groups in a μ4 fashion with an Ag–S distance of ∼2.51 Å and six auxiliary CF3COO− ligands via oxygen in μ2–η2 mode with an Ag–O distance of ∼2.40–2.51 Å. This Ag12(C6H11S)6(CF3COO)6 node connects with 6 pyrazine linkers via Ag–N bonds (∼2.30 Å) (Fig. S2†) to further connect 6 other clusters (Fig. S3†). A simple topology of this can be viewed in Fig. S4,† which repeats in three dimensions (Fig. 2b). The shortest Ag⋯Ag distance between two clusters in all three crystallographic directions is 7.40 Å (Fig. S5†).
(201) space group with the unit formula [Ag12(C6H11S)6(CF3COO)6(C4H4N2)3]n. The cluster core constitutes 12 Ag atoms held together by twelve argentophilic interactions with an ∼Ag⋯Ag distance of 2.93 Å (Table S5†), which is protected by six primary cyclohexane thiol groups in a μ4 fashion with an Ag–S distance of ∼2.51 Å and six auxiliary CF3COO− ligands via oxygen in μ2–η2 mode with an Ag–O distance of ∼2.40–2.51 Å. This Ag12(C6H11S)6(CF3COO)6 node connects with 6 pyrazine linkers via Ag–N bonds (∼2.30 Å) (Fig. S2†) to further connect 6 other clusters (Fig. S3†). A simple topology of this can be viewed in Fig. S4,† which repeats in three dimensions (Fig. 2b). The shortest Ag⋯Ag distance between two clusters in all three crystallographic directions is 7.40 Å (Fig. S5†).
Ag12-bpy crystallized in the Pnnm (no. 58) space group (Table S2 and Fig. S1b†), with the cluster unit formula [Ag12(C6H11S)6(CF3COO)6(C10H8N2)3]n. The 12 Ag atoms that constitute the core are held by 20 argentophilic interactions (∼Ag⋯Ag distance of 3.15 Å) (Table S5†). The silver core is protected by 6 cyclohexane thiol (Ag–S distance ∼2.47 Å) and six CF3COO− ligands (Ag–O distance ∼2.20–2.67 Å) in a similar fashion and further connected by six 4,4′-dipyridyl linkers (Fig. S6†) (Ag–N bonds ∼2.24–2.30 Å) to form a two-dimensionally extended solid (Fig. 2c and S7†), with the shortest Ag⋯Ag distances between two clusters in the bc plane being 11.57 and 11.38 Å (Fig. S8†).
Ag12-bpa [Ag12(C6H11S)6(CF3COO)6(C12H12N2)3(DMF)2]n crystallized in the C2/c (no. 15) space group (Table S3 and Fig. S1c†). The core which is composed of 12 silver atoms is held together by 18 argentophilic interactions (∼Ag⋯Ag distance of 3.10 Å) (Table S5†). The 6 cyclohexane thiol and 6 trifluoroacetate ligands protect the cluster in a similar fashion (Ag–S distance ∼2.49 Å and Ag–O distance ∼2.43–2.75 Å). The bpa linker via Ag–N bonds (∼2.27–2.29 Å) connects each cluster to six others (Fig. S9 and S10†) to form a three-dimensionally extended solid (Fig. 2d), with the shortest Ag⋯Ag distances between the neighboring clusters in the bc plane being 9.89 and 12.02 Å (Fig. S11†).
Ag12-azopy [Ag12(C6H11S)6(CF3COO)6(C10H8N4)3(DMF)2]n crystallized in the C2/c (no. 15) space group (Table S4 and Fig. S1d†). The 12 Ag atoms constituting the cluster core are held by 20 argentophilic interactions (∼Ag⋯Ag distance of 3.15 Å) (Table S5†). The cyclohexane thiol and trifluoroacetate ligands protect the cluster, and this is further covalently linked by 6 azopy linkers (Fig. S12†) to form a two-dimensionally extended solid (Fig. 2e and S13†). The shortest Ag⋯Ag distances with the neighbouring clusters in the ac plane are 13.36 and 13.46 Å (Fig. S14†).
The Ag12-NC, {[Ag12(C6H11S)6(CF3COO)6(C5H5N)6]·4H2O}, a non-covalently extended solid that is protected by pyridine (Fig. 2a, S15 and S16†), was earlier synthesized by us.25 This aids in correlating with the four new, two- and three-dimensionally extended solids (Fig. 1 and 2). Overall, five assemblies having a similar metal core and protecting ligands, differing in their assembly pattern, have been studied, with the shortest intercluster distance in Ag12-pyz and the longest in Ag12-azopy, based on the linker connecting them. More compact assemblies or higher dimensional structures tend to show shorter Ag⋯Ag inter- and intra-cluster distances. The shortest linker pyz connecting the clusters three-dimensionally (Ag12-pyz) not only has reduced inter-cluster Ag⋯Ag distances but also has reduced intra-cluster Ag⋯Ag distances and the series can be found in Table S6.† Covalent assemblies offer a tunable approach to achieving desired structural compactness, which is less controllable in non-covalent assembly.
The Powder X-Ray Diffraction (PXRD) pattern verified the crystallinity and phase purity of all the bulk samples, and the resulting unaltered pattern indicates its structural stability (Fig. S17–S21†). The Scanning Electron Microscopy (SEM) images of Ag12-pyz show an octahedral morphology (Fig. S22†), Ag12-bpy and Ag12-azopy show a hexagonal morphology (Fig. S23 and S25†) and Ag12-bpa has a block-like morphology (Fig. S24†), all of which are similar to their optical microscope images (Fig. S1†). The chemical composition of these samples was validated by SEM Energy-Dispersive X-Ray Spectroscopy (EDS) analysis, confirming the presence of all the desired elements (Fig. S22–S25†). X-Ray Photoelectron Spectroscopy (XPS) analysis further elucidated the chemical identity and oxidation state of the respective elements in all the samples (Fig. S26–S29†). The survey spectra confirmed the presence of all the desired elements, namely Ag, S, N, O, and F. The binding energies of Ag 3d5/2 and Ag 3d3/2 were 368.85 eV and 374.85 eV for Ag12-pyz, 368.57 eV and 374.57 eV for Ag12-bpy, 368.37 eV and 374.37 eV for Ag12-bpa, and 368.59 eV and 374.59 eV for Ag12-azopy, respectively, which confirms the presence of silver in the +1 oxidation state. Furthermore, the binding energy of S, approximately at 163 eV, indicates the presence of bridging sulfur. The four CAM structures (Ag12-pyz, Ag12-bpy, Ag12-bpa and Ag12-azopy) exhibit enhanced thermal stability up to ∼130 °C as determined by thermogravimetric analysis (TGA) (Fig. S30a†). However, they display varying degrees of thermal structural integrity at elevated temperatures, as indicated by PXRD analysis (Fig. S30b†). Ag12-pyz assembly exhibits enhanced structural stability at higher temperatures.
The electrochemical studies were carried out in 0.5 M H2SO4 with pH = 0.3 as the electrolyte; all five assemblies were loaded on carbon cloth for fabricating the working electrode. Linear sweep voltammetric (LSV) analysis of the samples carried out at a scan rate of 5 mV s−1 revealed the overpotentials of the materials at a current density of 10 mA cm−2 (Fig. 3a). It was observed that the Ag12-pyz CAM delivers a much lower overpotential of 122 mV compared to Ag12-azopy, Ag12-py, Ag12-bpa, and Ag12-bpy, which show overpotentials of 205, 230, 401, and 575 mV, respectively, to drive the same current density of 10 mA cm−2. The studies regarding the current density-dependent activity of the materials were carried out by calculating the overpotential delivered by the material at different current densities. The obtained values are shown in the bar diagram (Fig. 3b).
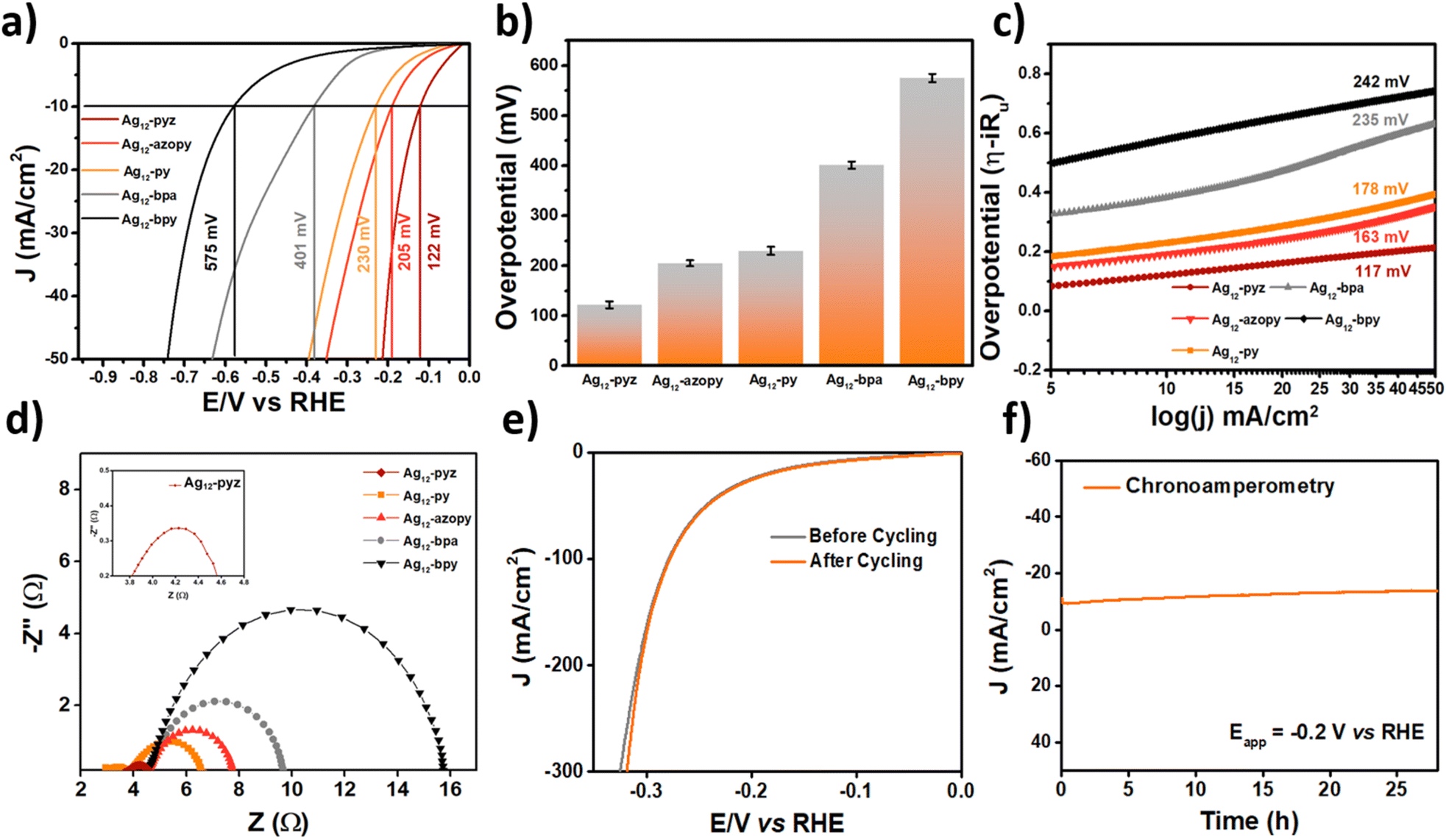 | ||
| Fig. 3 HER performance of the catalysts. (a) LSV curves of Ag12-pyz, Ag12-azopy, Ag12-py, Ag12-bpa, and Ag12-bpy, (b) bar diagram representing the current density at different overpotentials of Ag12-pyz, Ag12-azopy, Ag12-py, Ag12-bpa, and Ag12-bpy, (c) Tafel slopes of all five assemblies, (d) Nyquist plots of all the structures (inset: Ag12-pyz), (e) polarization curves of Ag12-pyz before and after 2000 cycles, and (f) chronoamperometry measurements for stability. | ||
The charge transfer kinetics at the electrode–electrolyte interface were analyzed using the Tafel slopes of the materials obtained from the iR-corrected LSV plots, which were found to be 117, 163, 178, 235, and 242 mV dec−1 for Ag12-pyz, Ag12-azopy, Ag12-py, Ag12-bpa, and Ag12-bpy, respectively (Fig. 3c). The values of the Tafel slopes suggest that Ag12-pyz shows the fastest charge-transfer kinetics among all the samples and follows the Volmer–Heyrovsky mechanism with the Heyrovsky step as the rate-determining step.51 Electrochemical impedance spectroscopic analysis revealed the charge transfer resistance (Rct) at the electrode–electrolyte interface. Ag12-pyz showed an Rct as low as 0.68 Ω, whereas for Ag12-azopy, Ag12-py, Ag12-bpa, and Ag12-bpy, the Rct values were found to be 3.1, 3.52, 5.1, and 11.1 Ω, respectively (Fig. 3d).
From the LSV, Tafel slope and Rct analysis, it was observed that, among the five catalysts, Ag12-pyz exhibits the best HER activity. We also conducted some experiments to analyze the stability of the Ag12-pyz catalyst. The dynamic stability of Ag12-pyz was studied using accelerated degradation (AD) studies for 1000 cycles at a 200 mV s−1 scan rate. The material maintained its stability after the AD process (Fig. 3e). Chronoamperometric analysis carried out at a constant potential of −0.2 V also proved the high static stability of the material for 28 hours (Fig. 3f). The amount of silver in the electrolyte of chronoamperometric experiments was found to be 0.626 ppm using ICP-MS analysis, which further corroborates the stability of the catalyst. The stability of Ag12-pyz in water is shown in Fig. S31.† The post-catalytic PXRD of Ag12-pyz coated on carbon tape confirms that the cluster is intact after the catalysis cycle (Fig. S32†). The SEM-EDS analysis and XPS survey spectrum also confirm the presence of all the necessary elements: Ag, S, O, N, and F after the catalysis cycle (Fig. S33 and S34†). The binding energy values of silver at 368.9 and 374.9 eV are similar to that of the pre-catalytic Ag12-pyz CAM, which further confirms the silver oxidation state of +1 and also the catalyst stability.
To gain further insight into the observed catalytic activity, computationally we measured the hydrogen adsorption free energy (ΔGH) as a descriptor for understanding HER reactivity.52 Initially, we performed the adsorption of a hydrogen atom at various accessible sites among the Ag and S core atoms within the optimized clusters. The ΔGH energies at different conformers of the clusters are depicted in Fig. 4a and b. Based on the optimized structures after hydrogen adsorption, it is shown that except for the Ag12-pyz, hydrogen adsorption at the Ag site is energetically favourable compared to that at the S site. In the Ag12-azopy, Ag12-py, Ag12-bpa, and Ag12-bpy clusters, hydrogen adsorption is more favourable by over 5.09, 6.8, 7.55, and 0.57 kcal mol−1, respectively, at the bridge Ag–Ag sites compared to the S sites (Table 1). It is important to note that, according to the Sabatier principle, both highly exothermic and highly endothermic hydrogen adsorption are detrimental to the HER. In contrast, Ag12-pyz shows hydrogen adsorption at the apical S sites, which is energetically favourable by 5.8 kcal mol−1 compared to adsorption at the bridge Ag–Ag site. The calculated ΔGH values from the optimized structures (Fig. S35†) are shown in Fig. 4a and b, indicating that Ag12-pyz could exhibit more effective HER activity compared to the others, aligning well with the experimentally measured overpotential values.
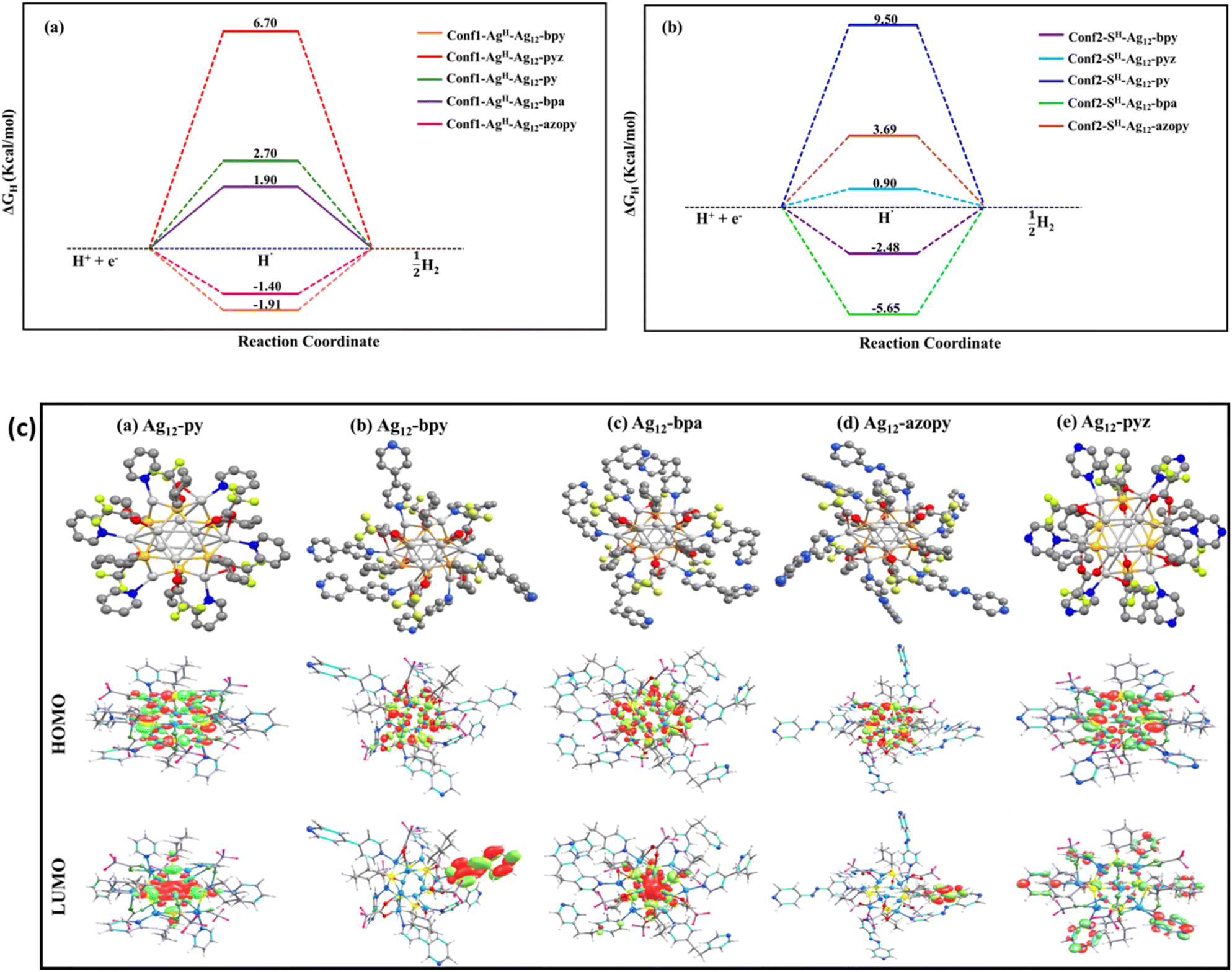 | ||
| Fig. 4 (a) Calculated free energy profile of the HER at equilibrium potential (U = URHE): hydrogen adsorption at the Ag site, (b) hydrogen adsorption at the S site. (c) Optimized structure and occupied and unoccupied molecular orbitals, (a) Ag12-py, (b) Ag12-bpy, (c) Ag12-bpa, (d) Ag12-azopy, and (e) Ag12-pyz clusters. Color code: Ag, light grey; S, yellow; O, red; N, blue; C, grey; F, greenish yellow. | ||
| ΔGH (kcal mol−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ag12-pyz | Ag12-bpy | Ag12-bpa | Ag12-azopy | Ag12-py | |||||
| conf1-AgH | conf2-SH | conf1-AgH | conf2-SH | conf1-AgH | conf2-SH | conf1-AgH | conf2-SH | conf1-AgH | conf2-SH |
| 6.7 | 0.9 | −1.91 | −2.48 | 1.90 | −5.65 | −1.40 | 3.69 | 2.7 | 9.5 |
These energetic differences are further corroborated by the observation that the apical S sites in the Ag12-pyz cluster exhibit a higher negative charge, making them more prone to hydrogen adsorption compared to those in other analogues. After hydrogen adsorption at the S sites in Ag12-pyz, computed Bader charges indicate that it carries a negative charge (∼−0.155e), suggesting a greater electronic interaction of H with the sulphur active site than the others. For Ag12-py, Ag12-bpy, Ag12-bpa, and Ag12-azopy, the charges of hydrogen adsorbed on the S sites are −0.145e, −0.133e, −0.150e, and −0.133e, respectively. Changes in the linker within assemblies can influence basicity, which in turn affects catalytic activity. Specifically, linkers with higher basicity, such as py, bpy, bpa, and azopy, increase the electron density at Ag sites. This enhancement in electron density favors hydrogen binding at the Ag sites while reducing adsorption at the S sites. Overall, the trends in the binding energy at the S sites and the lower basicity of pyz result in superior performance in the HER compared to other linkers. It is important to note that the large negative charge density on the adsorbed S–H sites indicates the favourable charge transfer occurring from the catalyst surface to the adsorbed hydrogen. Hydrogen desorption would likely occur by the further adsorption of a proton from the electrolyte solution, followed by electron transfer, in a process known as the ‘Heyrovsky’ pathway. The adsorption of an additional hydrogen atom (Fig. S36†) in these clusters is energetically more favourable than the adsorption of a single hydrogen atom, further indicating that the HER follows the Volmer–Heyrovsky pathway as suggested by the Tafel slope analysis. Interestingly, the binding energy of the second hydrogen over the Ag12-pyz cluster is less stable compared to the other studied clusters, resulting in superior HER activity as observed experimentally.
The highest occupied molecular orbitals (HOMO) in these clusters are predominantly localized on the Ag and S atoms, with a minor contribution from the ligand. In contrast, the lowest unoccupied molecular orbitals (LUMO) exhibit a shift from the core of the cluster to the ligand. In Ag12-py and Ag12-bpa, the LUMO is primarily concentrated within the core, while in Ag12-bpy, Ag12-azopy, and Ag12-pyz clusters, the LUMO is dominated by ligand-centric contributions. This inversion in the distribution of the unoccupied orbital is attributed to the structural variation introduced by the different ligands (Fig. 4c). Fig. S37† presents the calculated total density of states (TDOS) and partial density of states (PDOS) for these clusters. From the PDOS spectra, it is evident that the valence band in these clusters is primarily occupied by contributions from the core Ag and S atoms, while the conduction band is predominantly influenced by the ligands. As we examine Ag12-bpy, Ag12-bpa, and Ag12-azopy assemblies, the contribution from the ligands in the conduction band increases while the contribution from the Ag–S core decreases. Notably, in Ag12-pyz, the pyrazine ligand makes a substantial contribution to the conduction band, whereas in Ag12-py, both the pyridine ligand and the Ag–S core contribute to the conduction band.
Further, observing the theoretical band gap of the assemblies (Table S7†), Ag12-pyz manifests a shorter band gap (2.01 eV). This shortest band gap correlates with the favourable experimental conductivity of 4.1 × 10−6 S m−1 (Fig. S38†). Therefore, it indicates that pyrazine plays a substantial role in the stabilization of the cluster that appears from the lowest LUMOs of the Ag12-pyz CAM. These findings suggest that the electronic structure of the cluster can be effectively manipulated by changing the ligand structure.
From the experimental and theoretical analysis of the cluster structures, we made certain important observations. First, there is a systematic change in the bond distances of Ag–Ag and Ag–S upon variation of the ligand. The experimental crystal structure and theoretical bond length analyses show the Ag12-pyz cluster has a shorter average Ag⋯Ag interaction of 2.93 Å (2.89 Å theoretically), compared to the others (Table S5 and Fig. S35†). This might have resulted in a slightly longer Ag–S bond in Ag12-pyz (Table S5†). These structural differences corroborate the observed site preference. The apical S-sites in Ag12-pyz with a higher negative charge make it more prone to hydrogen adsorption compared to the others. It is also true that the basicity of the ligands in the series plays an important role in altering the active site based on the electron availability. The higher basicity of py, bpy, bpa, and azopy ligands increases the electron density on the Ag center, thereby favouring hydrogen adsorption at these sites. However, in the case of pyrazine, the reduced basicity alters the hydrogen adsorption site and maximizes HER activity through the S site. Second, the distribution of canonical orbitals changes, which varies with the ligand, affecting the structure–property relationship. Third, the inter-cluster interactions have a crucial effect on the conductivity of the materials. It has been observed that the material conductivity varies as a function of inter-cluster distance.53,54 The measured inter-cluster distances are as follows: Ag12-pyz at 7.40 Å, Ag12-bpa at 9.89 Å, Ag12-py at 10.1 Å, Ag12-bpy at 11.38 Å, and Ag12-azopy at 13.36 Å (considering the shortest distance, see Fig. S5, S8, S11, S14 and S16†). Ag12-pyz with the shortest Ag–Ag inter-cluster distance of 7.4 Å should possess the highest material conductivity, thereby reflecting faster charge transfer kinetics at the electrode–electrolyte interface. Moreover, the isotropic nature (equal along three crystallographic axes) of the Ag–Ag inter-cluster distance in Ag12-pyz allows the equivalent spatial distribution of the charge transfer process at the interface. In addition, the 3D structure of Ag12-pyz allows effective mass transfer at the interface. However, for the other four sets of cluster assemblies, the excessive increase in the Ag–Ag inter-cluster distance (by ∼3 Å) (Table S6†) does not significantly allow for altered electronic properties (such as conductivity) and therefore it can be considered that each Ag-cluster behaves independently. Moreover, the anisotropic behaviour of the Ag–Ag inter-cluster distance in different crystallographic directions might act as a barrier for improved electronic properties. Therefore, it can be said that for Ag12-pyz, the inter-cluster distance and dimensionality play a crucial role in the HER kinetics, while for the other clusters, the HER kinetics is mainly governed by the H-adsorption energetics. Thus, tuning the ligand can significantly impact the cluster arrangements and the catalytic properties. Moreover, we have compared our cluster assemblies with other silver-based NC catalysts reported in the literature, in terms of the overpotential required for reaching a current density of 10 mA cm−2 (Table S8†). As can be seen, Ag12-pyz displays the best performance among the series.
The experimentally obtained HER activity trend was further rationalized by the current density value at −0.2 V vs. RHE against the calculated free energy of H adsorption (Fig. 5). It is well known that for the HER, any electrocatalyst with a highly negative ΔGH value would bind the active H* very strongly and weakly with a positive ΔGH value. Very strong and weak H* adsorption would slow down the desorption and adsorption process, thereby reducing the catalytic activity. Hence, H* adsorption that is neither too strong nor too weak leads to the best catalytic activity according to the ‘Sabatier’ principle. Here, various nitrogen linkers with their characteristics of basicity and electronic structure play a significant role in H adsorption over the S site and the Ag12 cluster with a pyrazine stabilizer possesses optimum adsorption energy, facilitating a facile adsorption and desorption process.
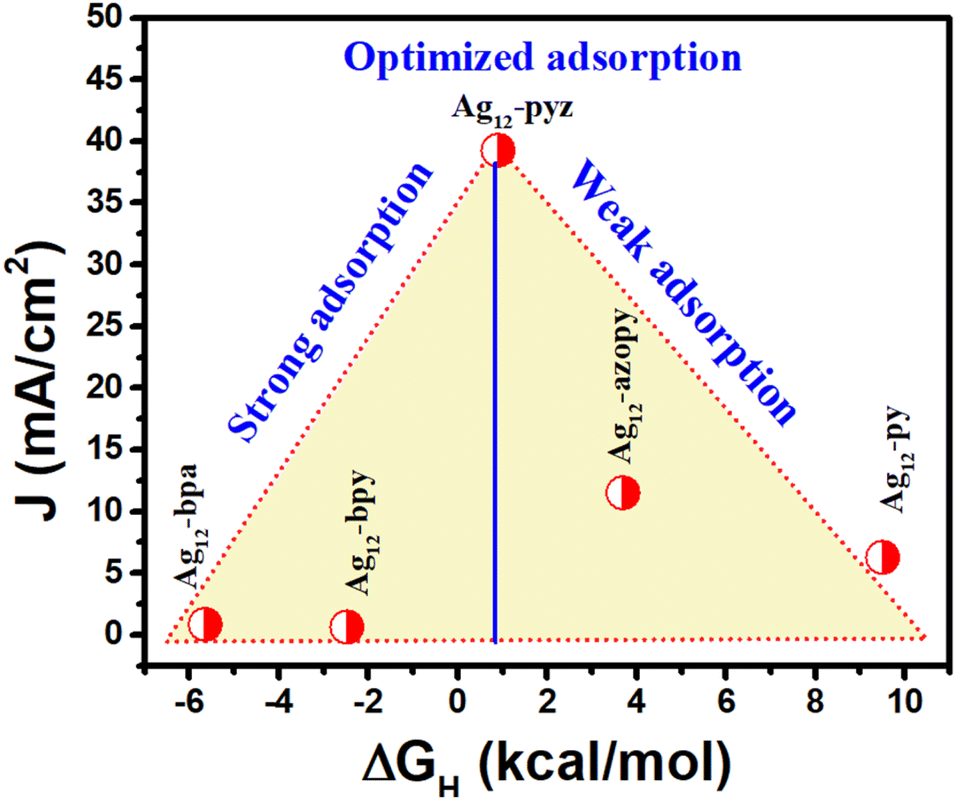 | ||
| Fig. 5 Volcanic HER activity trend for the five Ag12 cluster assemblies. | ||
Conclusions
In this study, five assemblies were studied: Ag12-py, Ag12-pyz, Ag12-bpy, Ag12-bpa, and Ag12-azopy. Ag12-pyz showed superior catalytic activity for the HER compared to the others in the series. Theoretical analysis on hydrogen adsorption free energy showed more favourable hydrogen adsorption for Ag12-pyz, enhancing its HER performance compared to the others. Further, the active center for catalytic hydrogen adsorption in Ag12-pyz was observed to be at the apical S site, while it is at the Ag–Ag site for the other 4 assemblies. This alteration of the active site is attributed to intercluster assembly and the basicity of the linker. The isotropic nature of the Ag–Ag inter-cluster distance in Ag12-pyz allows the equivalent spatial distribution of the charge transfer process. The relatively less basic pyrazine linker renders the silver more electropositive, a conclusion also supported by structural analysis, which showed the shortest Ag–Ag interactions in Ag12-pyz, thus favoring its positioning at the apical S site. Variations in catalytic activity and active site preference among these assemblies were achieved by systematically altering the ligands from pyridine to azopyridine. This work, offering a molecular-level correlation between linker-induced assembly, activity and active site switching in cluster assemblies, highlights a significant toolkit for selectively designing cluster assemblies as catalysts that hold broad implications for tailored catalytic activity in various applications.Data availability
The data will be available from the authors on request for academic purposes.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
C. P. acknowledges the Council of Scientific & Industrial Research India for the fellowship. We acknowledge funding from a grant by the Italian Ministry of Foreign Affairs and International Cooperation and the Indian Department of Science & Technology. K. B. acknowledges the IITG start-up grant and PARAM KAMRUPA for computational facilities. The authors acknowledge Dr A. Mukherjee and S. K. Singh for their assistance during I–V measurements. The CSIR-CECRI MS reference number is CECRI/PESVC/Pubs/2024-045. The authors acknowledge the Science Engineering and Research Board through the grants CRG/2022/000984. The authors acknowledge V. Nandana, N. Jithya, M. Arunima, N. Avani, V. Dayona and M. Rahul for their help during synthesis.References
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- R. Jin and T. Higaki, Commun. Chem., 2021, 4, 6–9 CrossRef PubMed.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- O. J. H. Chai, Z. Liu, T. Chen and J. Xie, Nanoscale, 2019, 11, 20437–20448 RSC.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- H. Yan, H. Xiang, J. Liu, R. Cheng, Y. Ye, Y. Han and C. Yao, Small, 2022, 18, 1–21 Search PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- Z. Liu, H. Tan, B. Li, Z. Hu, D. Jiang, Q. Yao, L. Wang and J. Xie, Nat. Commun., 2023, 14, 3374 CrossRef CAS PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- J. Ding, H. Yang, S. Zhang, Q. Liu, H. Cao, J. Luo and X. Liu, Small, 2022, 18, 2204524 CrossRef CAS PubMed.
- D. K. Jangid, S. G. Dastider, R. Biswas, S. Khirid, S. Meena, P. Kumar, S. C. Sahoo, V. P. Verma, R. D. Makde, A. Kumar, R. Jangir, K. Mondal, K. K. Haldar and R. S. Dhayal, Inorg. Chem., 2022, 61, 13342–13354 CrossRef CAS PubMed.
- S. Dai, T. Kajiwara, M. Ikeda, I. Romero-Muñiz, G. Patriarche, A. E. Platero-Prats, A. Vimont, M. Daturi, A. Tissot, Q. Xu and C. Serre, Angew. Chem., Int. Ed., 2022, 61, e202211848 CrossRef CAS PubMed.
- H. Wang, X. Zhang, W. Zhang, M. Zhou and H.-L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202401443 CrossRef CAS PubMed.
- J. V. Rival, P. Mymoona, K. M. Lakshmi, Nonappa, T. Pradeep and E. S. Shibu, Small, 2021, 17, 1–33 Search PubMed.
- Z. Wang, X.-Y. Li, L.-W. Liu, S.-Q. Yu, Z.-Y. Feng, C.-H. Tung and D. Sun, Chem.–Eur. J., 2016, 22, 6830–6836 Search PubMed.
- S.-S. Zhang, X. Wang, H.-F. Su, L. Feng, Z. Wang, W.-Q. Ding, V. A. Blatov, M. Kurmoo, C.-H. Tung, D. Sun and L.-S. Zheng, Inorg. Chem., 2017, 56, 11891–11899 Search PubMed.
- Y. Jin, C. Zhang, X. Y. Dong, S. Q. Zang and T. C. W. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- A. Ebina, S. Hossain, H. Horihata, S. Ozaki, S. Kato, T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 1105 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- S. A. Claridge, A. W. Castleman, S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 Search PubMed.
- S. Bonacchi, S. Antonello, T. Dainese and F. Maran, Chem.–Eur. J., 2021, 27, 30–38 CrossRef CAS PubMed.
- P. Chandrashekar, G. Sardar, T. Sengupta, A. C. Reber, P. K. Mondal, D. Kabra, S. N. Khanna, P. Deria and S. Mandal, Angew. Chem., Int. Ed., 2023, 63, e202317345 CrossRef PubMed.
- R. W. Huang, Y. S. Wei, X. Y. Dong, X. H. Wu, C. X. Du, S. Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- S. Wang, A. Lu and C.-J. Zhong, Nano Convergence, 2021, 8, 4 CrossRef CAS PubMed.
- T. B. Ferriday, P. H. Middleton and M. L. Kolhe, Energies, 2021, 14, 8535 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- J. Cai, R. Javed, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2020, 8, 22467–22487 RSC.
- L. Chang, D. Cheng, L. Sementa and A. Fortunelli, Nanoscale, 2018, 10, 17730–17737 RSC.
- X. Yuan and M. Zhu, Inorg. Chem. Front., 2023, 10, 3995–4007 RSC.
- R. P. Brocha Silalahi, Y. Jo, J. Liao, T. Chiu, E. Park, W. Choi, H. Liang, S. Kahlal, J. Saillard, D. Lee and C. W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301272 CrossRef CAS PubMed.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- X. Wang, L. Zhao, X. Li, Y. Liu, Y. Wang, Q. Yao, J. Xie, Q. Xue, Z. Yan, X. Yuan and W. Xing, Nat. Commun., 2022, 13, 1596 Search PubMed.
- Y. Tang, F. Sun, X. Ma, L. Qin, G. Ma, Q. Tang and Z. Tang, Dalton Trans., 2022, 51, 7845–7850 RSC.
- G. Ma, Y. Tang, L. Chen, L. Qin, Q. Shen, L. Wang and Z. Tang, Eur. J. Inorg. Chem., 2022, 21, e202200176 CrossRef.
- W. Choi, G. Hu, K. Kwak, M. Kim, D. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- H. Shen, Q. Zhu, J. Xu, K. Ni, X. Wei, Y. Du, S. Gao, X. Kang and M. Zhu, Nanoscale, 2023, 15, 14941–14948 RSC.
- M. Ghosal Chowdhury, L. Sahoo, S. Maity, D. Bain, U. K. Gautam and A. Patra, ACS Appl. Nano Mater., 2022, 5, 7132–7141 CrossRef CAS.
- M. A. Abbood, R. H. Althomali, F. Al-dolaimy, R. M. Portilla, S. S. Abdullaev, M. Del Carmen Delgado Laime, Z. F. Hassan, A. H. R. Abbas and A. H. Alsaalamy, Ionics, 2024, 30, 433–444 Search PubMed.
- Z. Wang, R. K. Gupta, F. Alkan, B.-L. Han, L. Feng, X.-Q. Huang, Z.-Y. Gao, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2023, 145, 19523–19532 CrossRef CAS PubMed.
- Y. Wang, X.-K. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Häkkinen, B. K. Teo, Q.-M. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS PubMed.
- G. Hu, Q. Tang, D. Lee, Z. Wu and D. Jiang, Chem. Mater., 2017, 29, 4840–4847 CrossRef CAS.
- X. Cai, Y. Sun, J. Xu and Y. Zhu, Chem.–Eur. J., 2021, 27, 11539–11547 CrossRef CAS PubMed.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- M. H. Naveen, R. Khan and J. H. Bang, Chem. Mater., 2021, 33, 7595–7612 CrossRef CAS.
- O. López-Estrada, N. Mammen, L. Laverdure, M. M. Melander, H. Häkkinen and K. Honkala, ACS Catal., 2023, 13, 8997–9006 CrossRef.
- F. Sun, Q. Tang and D. Jiang, ACS Catal., 2022, 12, 8404–8433 CrossRef CAS.
- A. Karmakar, M. Durairaj, R. Madhu, A. De, H. N. Dhandapani, M. J. S. Spencer and S. Kundu, ACS Mater. Lett., 2024, 6, 3050–3062 Search PubMed.
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC.
- R. Tsunashima, I. Nakamura, R. Oue, S. Koga, H. Oki, S. Noro, T. Nakamura and T. Akutagawa, Dalton Trans., 2017, 46, 12619–12624 RSC.
- C. Bansal, S. G. Praveen, J. T. T. Kumaran and A. Chatterjee, Sci. Rep., 2015, 5, 7685 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis, crystallographic information, optical images, crystal structure details, PXRD, SEM, XPS, TGA, post-catalytic analysis, DFT results, I–V characteristics, comparison table, references and optimized coordinates. CCDC 2342609, 2379538, 2379540 and 2379541. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08408j |
| This journal is © The Royal Society of Chemistry 2025 |