
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocation of dopant dictates proton–coupled electron transfer mechanism in vanadium-substituted polyoxotungstates†
Zhou
Lu
 a,
Mamta
Dagar
a,
James R.
McKone
b and
Ellen M.
Matson
*a
a,
Mamta
Dagar
a,
James R.
McKone
b and
Ellen M.
Matson
*a
aDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA. E-mail: matson@chem.rochester.edu
bDepartment of Chemical and Petroleum Engineering and Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA
First published on 5th March 2025
Abstract
Heterometal doping in polyoxometalates (POMs) is a useful strategy to impart modular reactivity by leveraging control over the physicochemical properties of the resulting materials. The dopant can occupy different position(s) within the POM that may affect the mechanism and/or outcome of a desired reaction. In this work, we illustrate that substituting one tungsten atom with vanadium in [PVoutW11O40]4− (PVoutW11) modulates the basicity of a bridging μ2–O2− ligand, increasing the strength of the O–H bond formed upon addition of the first proton–electron pair to the cluster by >20 kcal mol−1 over that of its homometallic congener. The reaction of PVoutW11 with an H-atom donor of weaker bond dissociation free energy results in the successful isolation of singly reduced, singly protonated cluster 1e−/1H+-PVoutW11; kinetic analysis of the reaction of PVoutW11 with hydrazobenzene reveals that H-atom uptake proceeds via a concerted proton–electron transfer mechanism. By contrast, the centrally substituted [VinW12O40]3− (VinW12) decouples the proton from electron transfer, leading to differential reactivity of 5,10-hydrophenazine to give the products of electron transfer. These results highlight that the proton–coupled electron transfer reactivity of heterometal-substituted metal oxides critically depends on the physical accessibility of dopants to the hydrogen donor.
Introduction
The physicochemical properties of bulk transition metal oxides are of great interest, given the application of these materials in energy storage, optoelectronics, and electrocatalysis.1–3 One important feature of these redox-active transition metal oxides is their ability to facilitate the uptake and transfer of hydrogen atom (H-atom) equivalents (i.e., proton/electron pairs) via proton–coupled electron transfer (PCET).4–6 The underlying physics governing PCET processes at and in extended transition metal oxides remain the subject of debate, due to limited atomic insight into the binding motifs of protons.7–10 For example, it is possible that a small fraction of surface sites (e.g., defects) on surfaces of extended metal oxide materials are responsible for the majority of the observed activity of H-atom uptake and transfer. To address these fundamental questions, our research team is investigating molecular polyoxometalates (POMs) as models of transition metal oxide surfaces, with the goal of understanding the thermochemistry and dynamics of PCET.11–19One way to tune the thermochemistry of H-atom uptake/transfer at the surface of extended transition metal solids is to alter the physicochemical properties of the material.20 Doping is an attractive strategy to modify the electronic structure of extended transition solids. Similarly, heterometal doping of polyoxometalates (POMs) can influence their redox properties (Fig. 1). Several reports have illustrated the effect of the dopant identity21,22 and stoichiometry23 on the electrochemistry of POMs. However, the position of the dopant (central ion vs. addenda atoms) also becomes important when discussing the electronic properties of Keggin and Wells–Dawson anions.24 For instance, while the identity of the central heteroatom has a very limited effect on the energy gap between occupied and unoccupied molecular orbitals in fully oxidized Keggin-type POMs, addenda atom substitution contributes significantly to lower-lying unoccupied orbitals. These changes in both geometry and electronic structures further influence the reactivity of POM redox mediators.25–27
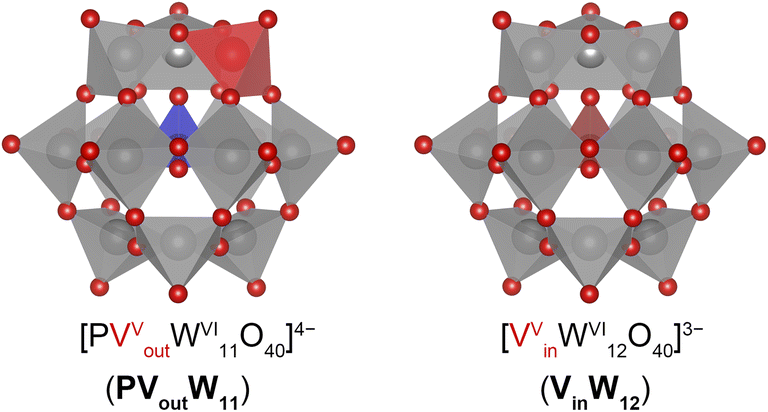 | ||
| Fig. 1 Molecular structure of vanadium-doped Keggin-type polyoxotungstates highlighting differences in accessibility of dopant ions – (left) surface V-substitution [PVoutW11O40]4−versus (right) internal V-substitution [VinW12O40]3− studied in this work. Key: O, red spheres; P, blue polyhedra; V, red polyhedra; W, grey polyhedra. | ||
Among heterometal-substituted POMs, vanadium doping in Keggin-type polyoxotungstates (POTs) has garnered significant attention in recent years. A series of reports from Ueda and coworkers describe the electrochemical properties of a series of vanadium-doped Keggin-type polyoxotungstates (POTs), [XVoutW11O40]4− (X = P, As, V).28–30 The V-doped POMs each features a VV/IV centred transition as the most positive reduction event. This reduction event was also found to shift positively upon the introduction of protons to the electrolyte, consistent with PCET reactivity at the surface of the cluster. However, explicit thermochemical studies of H-atom uptake and transfer at the surface of V-doped POT clusters have not been performed.
Given that the identity and position of a heteroatom can alter the structure, charge, electronic properties, and reactivity of POMs, we became interested in understanding the effects of confinement of aliovalent dopants on the thermochemical behaviour of Keggin POTs. Herein, we describe the PCET reactivity at a series of vanadium-substituted, Keggin-type POT clusters, namely [nBu4N]4[PVW11O40] (PVoutW11) and [nBu4N]3[VW12O40] (VinW12), in organic media (Fig. 1). Throughout this work, we refer to the V-doped POT clusters with external and internal V substitution as Vout and Vin, respectively. Our choice of these two clusters is rooted in the motivation to elucidate the impact of the differential location of the dopant on the resulting assemblies' ability to install H-equivalents on the surface. We demonstrate that the enhanced basicity of bridging O2− ligands associated with a surface V ion results in the formation of stronger O–H bonds. This high bond-dissociation free energy (BDFE(O–H)) value enables the chemical isolation of the singly reduced, singly protonated 1e−/1H+-PVoutW11. Our results further illustrate that electron transfer is decoupled from the transfer of the corresponding proton for the V-based redox event of centrally substituted VinW12; in this case, the cluster is only reduced and not protonated by molecular H-atom donors. The disparate reactivities of PVoutW11 and VinW12 highlight the dependency of H-atom uptake on the location (i.e., surface exposed or confined, respectively) of the dopant.
Results and discussion
H-atom uptake at a surface-doped polyoxotungstate, PVoutW11
Initial studies focused on understanding the consequence of vanadium substitution at the surface of the Keggin-type POT. We opted to investigate a phosphate-centred, vanadium-substituted POT, PVoutW11, derivative, to minimize composition discrepancies between the systems studied here and the homometallic derivative [nBu4N]3[PW12O40] (PW12) reported previously by our group.17 The cyclic voltammogram (CV) of PVoutW11 recorded in acetonitrile (MeCN) exhibits three reversible redox processes, with ∼1.5 V separating the first (most positive) and second reduction events (red trace, Fig. 2). The CV of PVoutW11 is quite different from that of PW12 (black trace, Fig. 2), as the substitution of an addenda WVI atom with an aliovalent VV atom in PVoutW11 changes the charge of the polyoxoanion resulting in a negative shift in the redox potentials. As a result, only 3 redox events are observed for PVoutW11 in the employed potential window (−2.5 V to +0.5 V). The first reduction process, located at E1/2 = −0.30 V (vs. Fc+/0), is assigned to the vanadium dopant (e.g., VV/IV).31 This is consistent with the poor d-orbital overlap between W 5d and V 3d states. Hence, the redox chemistry of PVoutW11 is best described as consisting of sequential and largely independent, redox processes, wherein the W-oxide framework functions as a host for a single V-oxide center.21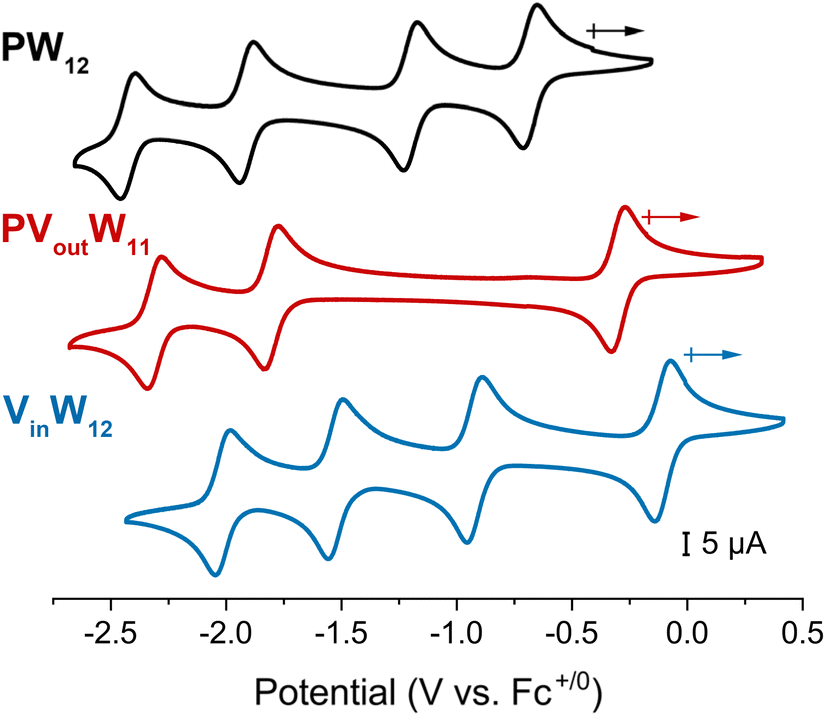 | ||
| Fig. 2 Cyclic voltammograms of 1 mM (top, black trace) PW12, (middle, red trace) PVoutW11, and (bottom, blue trace) VinW12 at a scan rate of 100 mV s−1 recorded in MeCN with 0.1 M [nBu4N]PF6. | ||
To further explore the PCET behaviour of PVoutW11, we analysed the electrochemical properties of the heterometallic assembly in the presence of organic acids (6 equiv.). Organic acids with pKa values ranging from 5 to 40 were selected (see ESI Fig. S7 and Table S1†). A potential-pKa diagram was constructed following the approach described by Dempsey and coworkers;32 a plot of the E1/2 values of each redox event versus the pKa of the added organic acid shows the proton-dependent redox properties of the vanadium-substituted POT (Fig. 3). In the presence of weak acids, the two tungsten-based 1e− reduction processes, originally beyond the electrochemical window of acetonitrile, collapse into a 2e−/2H+ process that shifts anodically (Fig. 3, black trace); a second 2e−/2H+ event is observed in the presence of acids with pKa values <25, indicating that association of protons with the surface of the cluster enables further reduction of the assembly (Fig. 3, grey trace). Fitting the acid dependent regions gives slopes of −62 ± 0.4 and −60 ± 1.7 mV pKa−1 units, consistent with values expected for equimolar nH+, ne− events (where n denotes the equivalents of electrons or protons involved in a given reaction). By locating the intersections of acid-dependent (diagonal lines, Fig. 3) and independent (horizontal lines, Fig. 3) regions, pKa values of the reduced and protonated species could be estimated to be 25.0 ± 0.80 and 33.4 ± 1.23, respectively.
 | ||
| Fig. 3 Potential-pKa diagram of PVoutW11. Each data point represents an individual CV collected using 1 mM PVoutW11 and 6 mM of the corresponding organic acid in MeCN. Reduction potentials are plotted against the pKa of the organic acid used in each experiment. The horizontal lines represent the acid-independent redox events: the red, grey, and black traces denote the redox event at E1/2 = −0.30 V, −1.80 V, and −2.31 V, respectively. The coloured diagonal lines represent the acid-dependent redox behaviour with the corresponding slopes; the red line is for the vanadium-based reduction process, while grey and black lines correspond to the tungsten-based reduction events. The potentials were calibrated using Fc+/0 as the internal standard. All relevant CVs are compiled in Fig. S7.† | ||
The average BDFE(O–H) values for the reduced, protonated forms of the cluster resulting from tungsten-based reduction processes are calculated using the Bordwell equation (eqn (1)).
| BDFE(E–H) = 1.37 pKa + 23.06E° + CG | (1) |
The one-electron reduction event assigned to the heterometal V dopant exhibits acid independent behaviour between pKa values of 16 and 40 (red trace, Fig. 3 and S7†). Upon addition of stronger organic acids, the redox event begins to shift in the positive direction, characteristic of PCET. The slope of the potential-pKa relationship is −59 mV pKa−1 units, following an n electron/n proton relationship (viz. one electron/one proton), and the pKa of the V-based protonation site is found to be 16.3 ± 1.3. From this information, we calculate a BDFE(O–H) value of 68.1 ± 1.8 kcal mol−1 for the reduced and protonated clusters using eqn (1).
These data illustrate that substitution of one surface W atom in PW12 with a solvent-exposed vanadium centre increases the BDFE(O–H) of the most positive PCET event by ∼20 kcal mol−1 (from ∼48 to ∼68 kcal mol−1).17 The generation of a significantly stronger surface O–H bond can be rationalized on the basis of increased basicity and more modest reduction potential of the heterometallic dopant. Generally, tungsten is more electronegative than vanadium, resulting in an electron-deficient bridging oxide ligand.33,34 To support this argument, conceptual density functional theory (CDFT) calculations were conducted to differentiate the electronegativities of V and W atoms in PVoutW11. Results from these experiments reveal the higher electrophilicity index of V atoms than all other eleven W atoms (cf. 0.143 for V, 0.01 to 0.03 for W on varied positions; Table S2†).35 It is important to note that an intuitive relationship is expected between electronegativity and the atomic radius; however, the influence from discrepancies in the ionic radius is unclear in the calculations of the vanadium-substituted POT. To ignore the size effect on the reactivity, local hyper-softness derived from the CDFT results is adopted (Table S2†).36 The vanadium center and the bridging oxygen site that is directly coordinated to the V atom have the highest local hyper-softness index of 2.67 and 1.43, respectively. This finding indicates that this site is more basic than the bridging and terminal oxide moieties bound to tungsten atoms in the assembly.37 These results are consistent with those obtained by Johnson and coworkers, illustrating that V-doping results in more electron density on the adjacent O2− ligand to the heteroatom.34
Given that the BDFE(O–H) value of the 1e−/1H+ reduced PVoutW11 cluster is significantly higher than the BDFE(H–H) of H2,9,38 we postulated that it would be possible to isolate the reduced and protonated assembly, [PVoutW11O39(OH)]4- (1e−/1H+-PVoutW11). Indeed, addition of half an equivalent of dihydrophenazine (H2Phen, BDFE(N–H)avg = 58.7 kcal mol−1 in acetonitrile) to a solution of PVoutW11 in acetonitrile results in an immediate colour change from yellow to purple. Characterization of the products via1H NMR spectroscopy reveals quantitative formation of phenazine (Fig. S8†), consistent with successful delivery of a single H-atom equivalent to the surface of the V-doped POT.
The fully oxidized PVoutW11 cluster features an intense absorption at 363 nm (εmax ≈ 3150 M−1 cm−1) with a shoulder band at 424 nm (εmax ≈ 1850 M−1 cm−1), assigned to the ligand–to–metal charge transfer (LMCT, O → VV, Fig. 4b). Upon chemical reduction to 1e−-PVoutW11, these LMCT bands disappear, and a new feature, assigned to a VIV d–d transition, is observed at 500 nm (εmax ≈ 720 M−1 cm−1). An intervalence charge transfer (IVCT, VIV → WVI) band at 403 nm (εmax ≈ 570 M−1 cm−1, Fig. S9†) is also observed;39 the relatively low absorption coefficient is attributed to the poor orbital mixing between vanadium and tungsten atoms.40 Since the CV and bulk electrolysis (Fig. S10†) results indicate that the VV/IV redox couple occupies the most positive event, the lowest unoccupied molecular orbital (LUMO) of PVoutW11 or the highest singly occupied molecular orbital (HSOMO) of 1e−-PVoutW11 is dominated by the V 3d orbital, which in turn results in a lower energy of the VIV d–d transition than the VIV → WVI IVCT band. The electronic absorption spectrum of 1e−/1H+-PVoutW11 is similar to that of 1e−-PVoutW11 with absorbance features at 504 nm (εmax ≈ 648 M−1 cm−1) and 392 nm (εmax ≈ 442 M−1 cm−1, Fig. 4b; see the Experimental section for the preparation method). Infrared characterization of the reduced clusters and electron paramagnetic resonance (EPR) experiments provides further support for reduction and protonation of the assembly upon formation of 1e−/1H+-PVoutW11 (Fig. S12–S14†). The EPR results collected for 1e−/1H+-PVoutW11 reveal the partial electron delocalization over the VIV–Ob bond after the protonation to give the O–H bond, compared to localized electrons on the VIV centre in 1e−-PVoutW11. Attempts to obtain single crystals of the 1e−/1H+-PVoutW11 cluster were unsuccessful as the sample crystallized in high symmetry space groups, rendering identification of the protonated site challenging. However, elemental analysis supports the presumed composition of 1e−/1H+-PVoutW11.
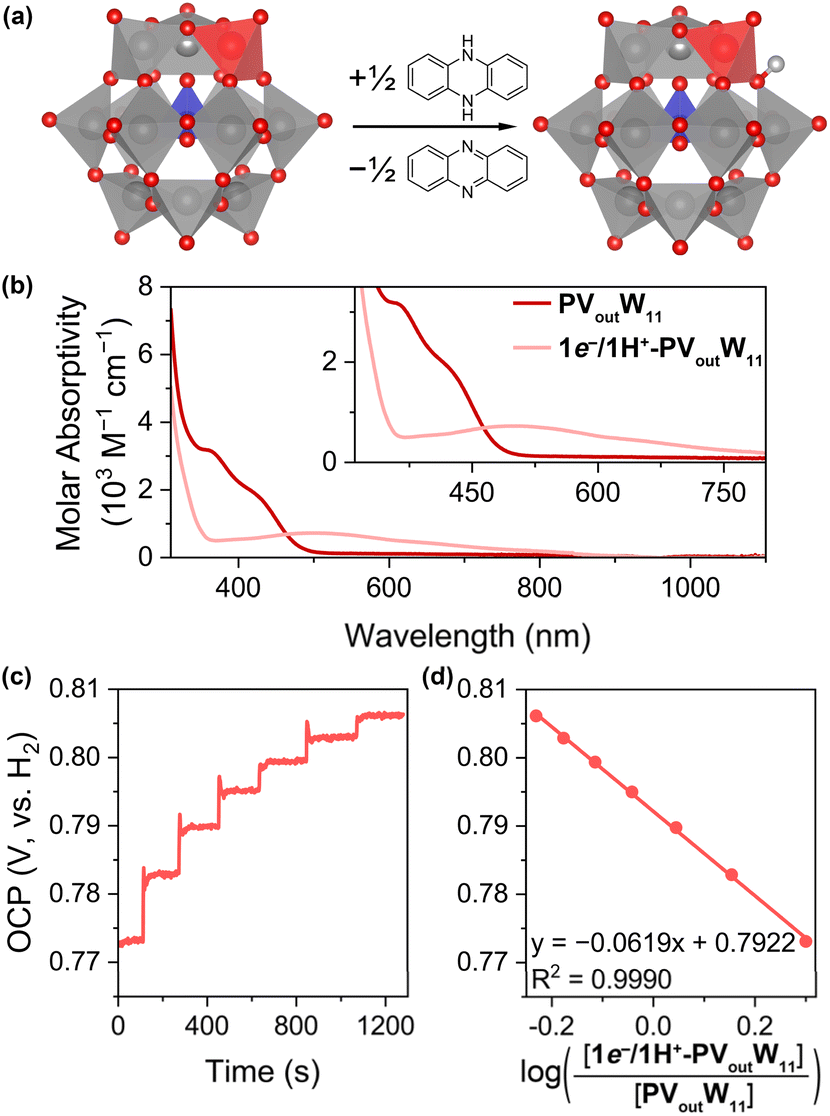 | ||
| Fig. 4 (a) Scheme showing formation of 1e−/1H+-PVoutW11 from PVoutW11. (b) Electronic absorption spectra of PVoutW11 and 1e−/1H+-PVoutW11 in MeCN. (c) Open circuit potential (OCP) measurement result for the cluster pair of 1e−/1H+-PVoutW11 with the injection of PVoutW11 stock solution in 100 μL aliquots. (d) Plots of the OCP values measured at various concentration ratios of 1e−/1H+-PVoutW11 and PVoutW11versus the log of the ratio of respective concentrations. | ||
With 1e−/1H+-PVoutW11 in hand, we performed additional experiments to verify the BDFE(O–H) value obtained from the construction of a potential-pKa diagram. Our group has established the use of a redox titration technique to obtain the same thermochemical parameter based on the measurement of open-circuit potential (OCP) across variable concentrations of the reduced/protonated and oxidized clusters.15 The expected relationship between the OCP and cluster concentrations is mentioned in eqn (2).
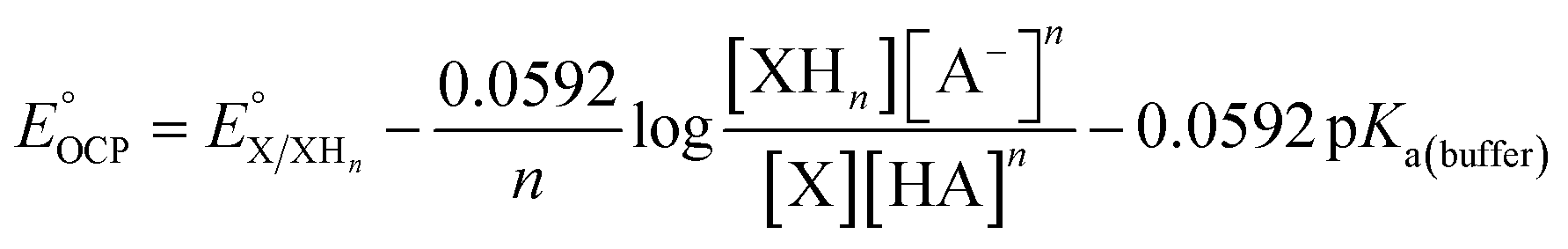 | (2) |
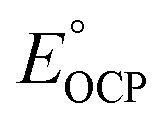 is the experimental OCP value,
is the experimental OCP value,  is the standard reduction potential of the cluster, n represents the number of proton–electron pairs transferred, [XHn] and [X] are the concentrations of two respective clusters, and [HA] and [A−] represent the concentrations of buffers in the forms of acid and conjugate base, respectively.41 The slope obtained from the linear correlation of OCP vs. the logarithm of the cluster concentration ratios yields the number of H-atom equivalents transferred between clusters. The
is the standard reduction potential of the cluster, n represents the number of proton–electron pairs transferred, [XHn] and [X] are the concentrations of two respective clusters, and [HA] and [A−] represent the concentrations of buffers in the forms of acid and conjugate base, respectively.41 The slope obtained from the linear correlation of OCP vs. the logarithm of the cluster concentration ratios yields the number of H-atom equivalents transferred between clusters. The 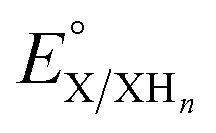 is used in eqn (3) to determine the BDFE(E–H) value of the reduced/protonated species, using the known value of ΔG°
is used in eqn (3) to determine the BDFE(E–H) value of the reduced/protonated species, using the known value of ΔG° as the constant in a given solvent.
as the constant in a given solvent.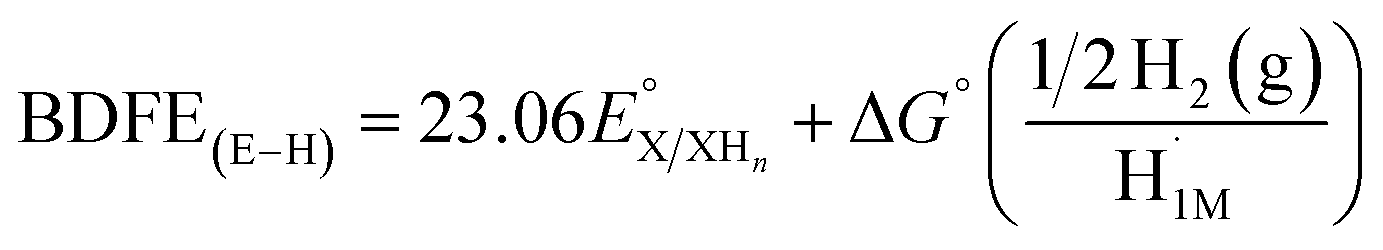 | (3) |
The OCP measurements for mixtures of PVoutW11 and 1e−/1H+-PVoutW11 are detailed in Fig. 4c, d, S15, and S16.† The resultant plot of  versus the natural log of the concentration ratio of [1e−/1H+-PVoutW11]/[PVoutW11] reveals an average slope of −64.3 ± 3.9 mV dec−1, suggesting a one-electron, one proton transfer process, consistent with the stoichiometric reactions performed between the oxidized cluster and H2Phen (vide supra). The average
versus the natural log of the concentration ratio of [1e−/1H+-PVoutW11]/[PVoutW11] reveals an average slope of −64.3 ± 3.9 mV dec−1, suggesting a one-electron, one proton transfer process, consistent with the stoichiometric reactions performed between the oxidized cluster and H2Phen (vide supra). The average 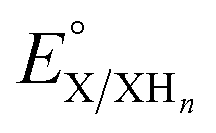 is found to be 0.790 ± 0.012 V (vs. SHE), yielding a BDFE(O–H) value of 70.2 ± 0.31 kcal mol−1, in agreement with the BDFE(O–H) value of 68.1 ± 1.78 kcal mol−1 derived from the potential-pKa diagram.
is found to be 0.790 ± 0.012 V (vs. SHE), yielding a BDFE(O–H) value of 70.2 ± 0.31 kcal mol−1, in agreement with the BDFE(O–H) value of 68.1 ± 1.78 kcal mol−1 derived from the potential-pKa diagram.
With the understanding of the stoichiometry and thermochemistry of H-atom uptake at PVoutW11, we shifted our focus to elucidating the mechanism of PCET to the surface of the vanadium-doped POT cluster (Fig. 5). The dynamics of the reaction between PVoutW11 and H2Phen was initially investigated; however, even at −30 °C, the rate of the reaction was too fast to be monitored (Fig. S17 and S18†). As such, another commonly used H-atom donor, hydrazobenzene (H2Azo, BDFE(N–H)avg = 60.5 kcal mol−1), was selected to determine the rate expression under pseudo-first-order conditions. Following addition of H2Azo, the LMCT band at 370 and 424 nm of PVoutW11 disappears with the emergence of a new transition centred at ∼460 nm, corresponding to the formation of both 1e−/1H+-PVoutW11 and azobenzene (Fig. 5a, 1H NMR spectroscopy confirms formation of azobenzene, Fig. S19†). To disentangle contributions to the absorption feature resulting from the reduced cluster and azobenzene at 460 nm, we monitored the decreasing absorption at 370 nm (Fig. S20–S26, see the ESI† for details). Exponential fitting of the resulting kinetic traces results in the pseudo-first-order decay rate constant, kobs, which is linearly correlated with the reductant concentration (Fig. 5b). This observation indicates the H-atom transfer is first order in the reductant (H2Azo).
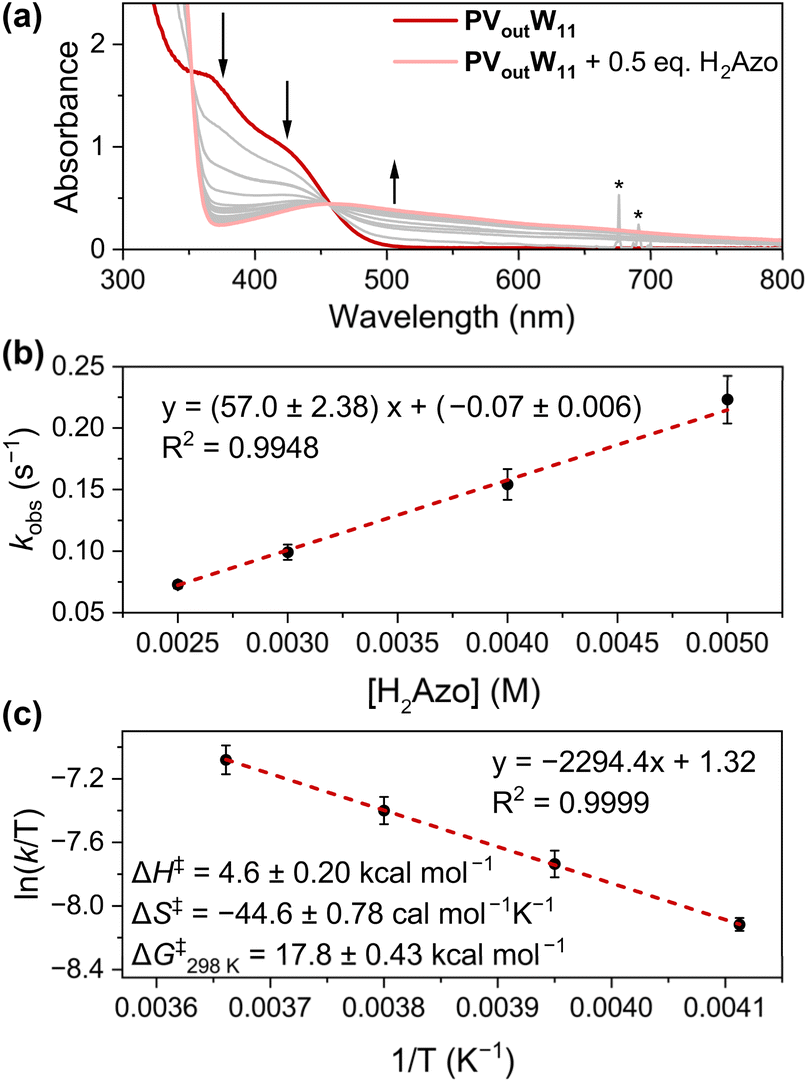 | ||
| Fig. 5 (a) Electronic absorption spectra of 0.5 mM PVoutW11 after addition of half an equivalent of H2Azo over a 5-min period at room temperature. The sharp absorption, denoted with asterisks at around 700 nm, indicates the injection of the H2Azo stock solution. (b) Plot of kobs as a function of [H2Azo] with the concentration of PVoutW11 held constant at 0.25 mM at −30 °C. The concentration of H2Azo is varied between 2.5 and 5 mM. (c) Eyring plot of the reaction of PVoutW11 (0.25 mM) and H2Azo (2.5 mM) in MeCN between −30 and 0 °C. The Y-axis adapts kobs values. | ||
As illustrated in Fig. 5b, the concentration of H2Azo was kept at least ten-fold excess to that of the cluster. As such, it is reasonable to approximate that the effective concentration of the reductant is held constant throughout the measurement. Thus, the observed linear trend between kobs (unit in s−1) and [H2Azo] (unit in M) reveals an overall second-order reaction (unit M−1 s−1).16 We therefore conclude that the H-atom transfer reaction from H2Azo to PVoutW11 is first order in both the reductant (H2Azo) and H-atom acceptor (PVoutW11) with the relevant rate expression depicted in eqn (4).
 | (4) |
Further kinetic experiments were conducted at varied temperatures to execute an Eyring analysis (Fig. 5c). An activation enthalpy (ΔH‡) of 4.6 ± 0.20 kcal mol−1 and entropy (ΔS‡) of −44.6 ± 0.78 cal mol−1 K−1 are measured. Overall, the activation Gibbs energy at 298 K (ΔG‡298K) is calculated to be 17.8 ± 0.43 kcal mol−1 (Fig. 5c). The large negative entropic contribution suggests that H-atom uptake proceeds through a highly ordered transition state, consistent with bimolecular PCET between H2azo and PVoutW11. Together with the small activation enthalpy ΔH‡, a transition state involving a H-bonded adduct between the V–O bond of the cluster and the N–H motif of the reductant is proposed.11,13,16,42–45 This supports the assignment of the mechanism of reduction of the assembly as concerted proton–electron transfer (CPET).
Finally, the reactivity of 1e−/1H+-PVoutW11 with a suitable H-atom acceptor, the 2,4,6-tBu3PhO˙ radical, was performed to model the ability of the reduced and protonated assembly to perform H-atom transfer reactions.8 As shown in Fig. S27,† the 2,4,6-tBu3PhO˙ radical exhibits characteristic absorption at ∼380, 400, and 626 nm in acetonitrile.46 After adding one equivalent of 1e−/1H+-PVoutW11, the characteristic absorption of the 2,4,6-tBu3PhO˙ radical disappeared along with the emergence of the absorption of fully oxidized PVoutW11 (Fig. S27†). The 1H NMR spectrum of the stoichiometric mixture of the 2,4,6-tBu3PhO˙ radical and 1e−/1H+-PVoutW11 was also collected, showing the existence of the –OH signal of 2,4,6-tBu3PhOH at 5.25 ppm in CD3CN (Fig. S28†). Taken together, it can be concluded that 1e−/1H+-PVoutW11 is able to undergo proton–coupled electron transfer to the 2,4,6-tBu3PhO˙ radical to give 2,4,6-tBu3PhOH in a thermodynamically favored pathway.
H-atom uptake at an internally doped polyoxotungstate, VinW12
The results described above demonstrate that incorporation of a vanadium dopant at the surface of the POT increases its affinity for H-atoms (i.e., BDFE(O–H) values increased by ∼20 kcal mol−1 in comparison to PW12). We next turned our attention to understanding the impact of an internal (i.e., lattice confined) vanadium dopant on PCET reactivity and mechanism.Upon substitution of the central, redox-innocent phosphorus atom of PW12 with a redox-active vanadium ion, the CV of VinW12 exhibits four 1e− redox events. As with PVoutW11, the first reduction event (−0.10 V vs. Fc+/0) is localized on the vanadium centre (VV + 1 e− → VIV). The remaining three events are WVI/V redox couples (−0.91, −1.51, and −2.01 V vs. Fc+/0, black trace, Fig. 2). We note that the potential difference between the first and second reductions (∼0.8 V, black trace, Fig. 2) is markedly smaller than that observed for PVoutW11 (∼1.5 V, red trace, Fig. 2). This behaviour can be ascribed to the different coordination geometries of heterometal atoms and corresponding molecular orbitals. When a redox-active tetrahedral [VO43−] occupies a central position in VinW12, contrary to the redox-innocent [PO43−] as in PW12, the first reducing equivalent in the doubly degenerate (eg) unoccupied orbitals undergoes a Jahn–Teller distortion, leading to increased mixing with W 5d orbitals and partial electron delocalization. Moreover, the addenda atoms in both PW12 and VinW12 are solely composed of tungstate ions, with the same overall charge on the cluster (i.e., −3), with slight changes in the HOMO–LUMO energy gap on internal vanadium substitution. As a result, the redox properties of VinW12 are more similar to those of PW12 than to those of PVoutW11, with a slight positive shift in the E1/2 values and retainment of the number of redox events in the studied voltage window. Notably, this increased mixing raises the question of whether electron transfer to the central V atom can be coupled with proton transfer to surface-exposed W–O electronic states.
Fig. 6 compiles the CV data collected to map the relationship between the E1/2 values of the reduction events of VinW12 upon addition of organic acids (see Fig. S29† for CV data from independent experiments). Using acids with pKa values <28, a 2e−/2H+ event is observed from the consolidation of two tungsten-based 1e− events (black trace, Fig. 6). A second 2e−/2H+ event is revealed by using acids with pKa values <12 (grey trace, Fig. 6). These multi-electron, multi-proton redox events are similar to those observed in PVoutW11 or PW12. Slopes derived from fitting the acid dependent regions are −59 ± 4.1 mV pKa−1, suggesting equimolar nH+, ne− events. The pKas of these tungsten-based events are found to be 12.6 ± 0.84, 22.3 ± 1.93, and 29.1 ± 2.63, respectively. BDFEs(O–H) of reduced, protonated clusters are further calculated to be ∼48, ∼48, and ∼46 kcal mol−1, resembling values reported for PW12.17
 | ||
| Fig. 6 Potential-pKa diagram of VinW12. Each data point represents an individual CV collected using 1 mM VinW12 and 4 mM of the corresponding organic acid in MeCN. Reduction potentials are plotted against the pKa of the organic acid used in each experiment. The horizontal lines represent the acid-independent redox events: the red, grey, and black traces denote the redox event at E1/2 = −0.30 V, −1.80 V and −2.31 V respectively. The coloured diagonal lines represent the acid-dependent redox behaviour with the corresponding slopes; the blue line is for the vanadium-based reduction process, while grey and black lines correspond to the tungsten based reduction events. The potentials were calibrated using Fc+/0 as the internal standard. All relevant CVs are compiled in Fig. S29.† | ||
In sharp contrast to the surface vanadium-substituted PVoutW11, the redox reactivity for the central vanadium site in VinW12 shows no evidence of PCET (blue trace, Fig. 6); the redox event remains at a constant potential across all organic acids studied. This result indicates that the tungsten cage shields the redox-active vanadium centre, inhibiting concerted proton binding upon reduction of the dopant. This can be further interpreted as inadequate electronic coupling between V- and W-centred redox chemistry, such that electron transfer to the central V(V) has very little impact on the basicity of peripheral oxygen sites.
To validate the independent electron transfer to the central V(V) site from the potential-pKa diagram, the reactivity of VinW12 with organic HAT reagents was explored. Addition of H2Phen to VinW12 results in a kinetic trace that is strikingly different from that observed in the case of PVoutW11 (Fig. 7a). Upon addition of the substrate, an immediate colour change to green is observed (Fig. 7b). The absorption spectrum possesses a series of intense transitions between 400–500 and 550–750 nm, which correspond to the oxidized H2Phen cation radical (H2Phen˙+).47 This observation can be rationalized by estimating a ΔGET of −5.3 kcal mol−1 between H2Phen and VinW12. The optical data further indicates that the cation radical remains stable in solution over days,48 indicating that the 1e− reduced VinW12 is insufficiently basic to accept a proton from H2Phen˙+.
 | ||
| Fig. 7 Electronic absorption spectra of 0.5 mM (a) PVoutW11 and (b) VinW12 after adding half an equivalent of H2Phen at room temperature. (c) Illustration of different reactivities of H2Phen towards V-doped POTs to generate phenazine or H2Phen˙+, respectively. | ||
Executing the same reaction between H2Phen and PVoutW11 yields phenazine and 1e−/1H+-PVoutW11 (Fig. 7c). Notably, transitions corresponding to the formation of the oxidized H2Phen˙+ are absent from the kinetic trace, despite a negative ΔGET of −0.7 kcal mol−1. This observation is surprising, as the ΔGET is negative to afford ET-only products. Collectively, these data indicate that the presence of the surface V dopant in PVoutW11 plays a key role in H-equivalent installation; the availability of a basic, proton-accessible site at the cluster surface promotes hydrogen bonding interactions between the H-atom donor and the POM, facilitating a concerted proton–electron transfer pathway.
As a final confirmation of the interplay between reduction potential and availability of basic surface sites, we sought to identify an H-atom donor with a reduction potential that is sufficiently positive to inhibit electron transfer to either cluster. Hence, we investigated the reactivity of PVoutW11 and VinW12 with 1,4-dihydroxynaphthalene (H2NQ, E1/2 = 0.00 V vs. Fc+/0, Fig. S30†), as ΔGET for both V-doped POTs is positive (ΔGET of +2.3 kcal mol−1, VinW12; ΔGET of +6.9 kcal mol−1, PVoutW11). As expected, the addition of H2NQ to VinW12 results in no reaction; neither electron transfer nor PCET is observed by NMR and electronic absorption spectroscopies (Fig. S31 and S32†). In contrast, the addition of H2NQ to PVoutW11 results in a gradual colour change from yellow to purple, consistent with cluster reduction (Fig. S33†). The BDFE(O–H)avg value of H2NQ is 63.3 kcal mol−1,49 somewhat weaker than that measured for 1e−/1H+-PVoutW11. This results in a favourable driving force for H-atom transfer to the surface of the cluster from the substrate, enabled only by a CPET-type mechanism. Hence, H2NQ constitutes a reagent whose thermochemistry enables complete differentiation between the two clusters: full PCET for PVoutW11 and no reaction with VinW12.
Conclusions
In sum, this work discusses the PCET chemistry of two vanadium-doped Keggin-type polyoxotungstates and thermodynamics of H-atom uptake. The heterometal dopant location plays an essential role in controlling the basicity of surface oxide ligands. The central V(V) site of VinW12 has limited influence on the peripheral oxygen sites, leading to comparable thermodynamics of surface O–H bonds with PW12. By contrast, electron transfer to a solvent-exposed VV/IV redox centre in PVoutW11 is proton–coupled over a wide range of acid pKa values. With the basicity enhanced by the V dopant, the O–H bond at the surface of PVoutW11 is strengthened by >20 kcal mol−1 relative to that of the all-tungsten surface in VinW12 or PW12.The reactivities between V-doped clusters and H-atom transfer reagents are explored; the singly reduced, singly protonated 1e−/1H+-PVoutW11 can be isolated due to its comparatively high BDFE(O–H), which prevents bimolecular H–H bond formation. Kinetic experiments and Eyring analyses nonetheless suggest bimolecular reactivity between H-atom donors and PVoutW11, revealing a concerted proton–electron transfer mechanism whose transition state energy is dominated by entropic contributions. By contrast, VinW12 readily undergoes V-centred reduction, but lacks a suitably basic surface site for deprotonation of H2Phen˙+. Collectively, these results strongly imply that catalytic H-transfer reactions with heterobimetallic POMs, and perhaps with analogous doped WOx solids, fundamentally depend on the ability to install H-equivalents onto sites that are directly exposed to the external medium.
Data availability
All experimental data are provided in the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Z. L. – experiment design, investigation, writing – original draft; M. D. – writing; J. R. M. – project administration, funding acquisition, writing; E. M. M. . – conceptualization, experiment design, project administration, funding acquisition, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work was provided by the Department of Energy under Award No. DE-SC0023465.Notes and references
- S. Yuan, X. Duan, J. Liu, Y. Ye, F. Lv, T. Liu, Q. Wang and X. Zhang, Energy Storage Mater., 2021, 42, 317–369 CrossRef.
- Z. Zhang, J. Liu, J. Gu, L. Su and L. Cheng, Energy Environ. Sci., 2014, 7, 2535–2558 RSC.
- J. Desilvestro and O. Haas, J. Electrochem. Soc., 1990, 137, 5C–22C CrossRef CAS.
- Y. Qiu, D. Ray, L. Yan, X. Li, M. Song, M. H. Engelhard, J. Sun, M.-S. Lee, X. Zhang, M.-T. Nguyen, V.-A. Glezakou, Y. Wang, R. Rousseau and Y. Shao, J. Am. Chem. Soc., 2023, 145, 26016–26027 CrossRef CAS PubMed.
- J. Castillo-Lora, M. F. Delley, S. M. Laga and J. M. Mayer, J. Phys. Chem. Lett., 2020, 11, 7687–7691 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- K. R. Proe, E. Schreiber and E. M. Matson, Acc. Chem. Res., 2023, 56, 1602–1612 CrossRef CAS PubMed.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- A. A. Fertig, S. E. Cooney, R. L. Meyer, W. W. Brennessel and E. M. Matson, Chem. Commun., 2022, 58, 6004–6007 RSC.
- S. E. Cooney, E. Schreiber, B. M. Ferrigno and E. M. Matson, Chem. Commun., 2024, 60, 5610–5613 RSC.
- S. E. Cooney, A. A. Fertig, M. R. Buisch, W. W. Brennessel and E. M. Matson, Chem. Sci., 2022, 13, 12726–12737 RSC.
- A. A. Fertig and E. M. Matson, Inorg. Chem., 2023, 62, 1958–1967 CrossRef CAS PubMed.
- K. R. Proe, A. Towarnicky, A. Fertig, Z. Lu, G. Mpourmpakis and E. M. Matson, Inorg. Chem., 2024, 63, 7206–7217 CrossRef CAS PubMed.
- S. E. Cooney, E. Schreiber, W. W. Brennessel and E. M. Matson, Inorg. Chem. Front., 2023, 10, 2754–2765 RSC.
- Z. Lu, S. E. Cooney, J. R. McKone and E. M. Matson, JACS Au, 2024, 4, 1310–1314 CrossRef CAS PubMed.
- A. A. Fertig, W. W. Brennessel, J. R. McKone and E. M. Matson, J. Am. Chem. Soc., 2021, 143, 15756–15768 CrossRef CAS PubMed.
- S. E. Cooney, M. R. A. Walls, E. Schreiber, W. W. Brennessel and E. M. Matson, J. Am. Chem. Soc., 2024, 146, 2364–2369 CrossRef CAS PubMed.
- P. Mondal, I. Ishigami, E. F. Gérard, C. Lim, S.-R. Yeh, S. P. de Visser and G. B. Wijeratne, Chem. Sci., 2021, 12, 8872–8883 RSC.
- E. N. Glass, J. Fielden, Z. Huang, X. Xiang, D. G. Musaev, T. Lian and C. L. Hill, Inorg. Chem., 2016, 55, 4308–4319 CrossRef CAS PubMed.
- K. Uehara, T. Miyachi, T. Nakajima and N. Mizuno, Inorg. Chem., 2014, 53, 3907–3918 CrossRef CAS PubMed.
- W. Sun, D. Yao, Y. Tai, L. Zhou, W. Tian, M. Yang and C. Li, J. Colloid Interface Sci., 2023, 650, 121–131 CrossRef CAS PubMed.
- X. López, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2002, 124, 12574–12582 CrossRef PubMed.
- M. Wang, J. Pang, J. Wang and J. Niu, Coord. Chem. Rev., 2024, 508, 215730 CrossRef CAS.
- M. J. da Silva, P. H. de A. da Silva, S. O. Ferreira, R. C. da Silva and C. G. O. Brusiquezi, ChemistrySelect, 2022, 7, e202104174 CrossRef CAS.
- T. Ma, R. Yan, X. Wu, M. Wang, B. Yin, S. Li, C. Cheng and A. Thomas, Adv. Mater., 2024, 36, e2310283 CrossRef PubMed.
- J. Nambu, T. Ueda, S.-X. Guo, J. F. Boas and A. M. Bond, Dalton Trans., 2010, 39, 7364–7373 RSC.
- S. Yokoyama, S. Azuma, Y. Eguchi, K. Kodani, T. Hasegawa, S. Ogo, H. Ota, S.-X. Guo, J. F. Boas, J. Zhang, A. M. Bond and T. Ueda, Inorg. Chem., 2024, 63, 117–128 CrossRef CAS PubMed.
- T. Ueda, ChemElectroChem, 2018, 5, 823–838 CrossRef CAS.
- J. M. Maestre, X. Lopez, C. Bo, J.-M. Poblet and N. Casañ-Pastor, J. Am. Chem. Soc., 2001, 123, 3749–3758 CrossRef CAS PubMed.
- B. D. McCarthy and J. L. Dempsey, Inorg. Chem., 2017, 56, 1225–1231 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Angew Chem. Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- O. M. Primera-Pedrozo, S. Tan, D. Zhang, B. T. O'Callahan, W. Cao, E. T. Baxter, X.-B. Wang, P. Z. El-Khoury, V. Prabhakaran, V.-A. Glezakou and G. E. Johnson, Nanoscale, 2023, 15, 5786–5797 RSC.
- T. Lu and Q. Chen, in Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory, WILEY-VCH GmbH, Weinheim, 2022, vol. 2, pp. 631–647 Search PubMed.
- C. Cárdenas, N. Rabi, P. W. Ayers, C. Morell, P. Jaramillo and P. Fuentealba, J. Phys. Chem. A, 2009, 113, 8660–8667 CrossRef PubMed.
- M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354–7367 RSC.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef CAS PubMed.
- T. Yamase, Chem. Rev., 1998, 98, 307–326 CrossRef CAS PubMed.
- D. M. D'Alessandro and F. R. Keene, Chem. Rev., 2006, 106, 2270–2298 CrossRef PubMed.
- C. F. Wise, R. G. Agarwal and J. M. Mayer, J. Am. Chem. Soc., 2020, 142, 10681–10691 CrossRef CAS PubMed.
- E. Schreiber, W. W. Brennessel and E. M. Matson, Chem. Sci., 2022, 14, 1386–1396 RSC.
- J. Amtawong, B. B. Skjelstad, D. Balcells and T. D. Tilley, Inorg. Chem., 2020, 59, 15553–15560 CrossRef CAS PubMed.
- T. G. Carrell, P. F. Smith, J. Dennes and G. C. Dismukes, Phys. Chem. Chem. Phys., 2014, 16, 11843–11847 RSC.
- N. Kindermann, C.-J. Günes, S. Dechert and F. Meyer, J. Am. Chem. Soc., 2017, 139, 9831–9834 CrossRef CAS PubMed.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC.
- T. Kwon, J. Y. Koo and H. C. Choi, Cryst. Growth Des., 2019, 19, 551–555 CrossRef CAS.
- J. E. Coffield, G. Mamantov, S. P. Zingg and G. P. Smith, J. Electrochem. Soc., 1991, 138, 2543–2549 CrossRef CAS.
- D. Shiels, Z. Lu, M. Pascual-Borràs, N. Cajiao, T. V. Marinho, W. W. Brennessel, M. L. Neidig, R. J. Errington and E. M. Matson, Inorg. Chem., 2024, 63, 23304–23316 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterization data of reported materials, details about electrochemical studies, kinetics data, and results of reactivity investigations. See DOI: https://doi.org/10.1039/d4sc08452g |
| This journal is © The Royal Society of Chemistry 2025 |