
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal synthesis of single-atom electrocatalysts via in situ fluoride ion etching for hydrogen evolution†
Peng
Liu
a,
Jiahui
Ye
a,
Kuan
Deng
a,
Xuesong
Liu
a,
Haohui
Dong
a,
He
Zhang
a,
Wen
Tian
a and
Junyi
Ji
 *ab
*ab
aSchool of Chemical Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: junyiji@scu.edu.cn
bState Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China
First published on 23rd January 2025
Abstract
Single-atom catalysts (SACs) have attracted considerable interest in the field of electrocatalysis due to their high efficiency of metal utilization and catalytic activity. However, traditional methods of SACs fabrication are often complex and time-consuming. Herein, F–Ru@TiOxNy was synthesized using a straightforward and universal approach via in situ surface etching and heteroatoms immobilization on a vacancies-rich hierarchical TiOxNy nanorods array. The fluorine ion-etched TiOxNy nanorods could produce abundant oxygen vacancies and F–Ti/F–C bonds, which could capture and stabilize Ru heteroatoms by strong host–guest electronic interactions. Due to the synergistic effect of oxygen vacancies anchoring and F–C bonds-assisted stabilization of single atoms, F–Ru@TiOxNy revealed excellent electrocatalytic hydrogen evolution performance, a low overpotential of 20.8 mV at 10 mA cm−2, a Tafel slope of 59.9 mV dec−1 and robust stability at 100 mA cm−2 over 48 h. Furthermore, this universal strategy could be applicable to various heterometals (Pd, Ir, Pt), which also exhibited high heteroatoms dispersity and high electrocatalytic HER activity/stability. This fabrication method is simple, easy-scalable and versatile, showcasing significant potential for electrocatalysts design and promising application prospects in electrocatalytic energy conversion.
1. Introduction
Hydrogen has been regarded as an alternative “clean” energy source due to its high energy density and non-pollution combustion product, which is a suitable substitute for fossil fuel-based energy sources.1,2 The coupling utilization of renewable energy to facilitate the water electrolysis for hydrogen production presents advantages in terms of environmental sustainability and process flexibility, which is a strategic focus for the future advancement of the hydrogen energy sector.3 Though precious metals-based electrocatalysts exhibit superior performance in the hydrogen evolution reaction (HER), their widespread applications are hindered by factors such as high cost, limited availability, and instability in alkaline environments.4 Consequently, it is imperative to develop highly efficient electrocatalysts with an ultralow amount of precious metals loading and high structural/electrocatalytic stability for alkaline hydrogen production to accelerate progress in the hydrogen energy industry.Highly dispersed precious metal catalysts demonstrate exceptional electrocatalytic activity and stability, which significantly diminishes the required quantity of precious metals. Especially, single atom catalysts (SACs)5 are extensively employed in electrocatalytic hydrogen evolution due to their high activity, favourable selectivity, and ultralow amount of precious metals consumed. However, traditional fabrication methods for SACs are usually complex and time-consuming, with weak interfacial force between heteroatoms and substrate, leading to an aggregation trend of precious metals and limitation of large-scale fabrication.6–8 Among various strategies for SACs fabrication, the defect engineering strategy is widely applied.9 It involves constructing defect sites on the surface of a support (e.g., carbon defects,10,11 oxygen defects,12 sulfur defects,13,14 and metal defects),15 then utilizing the support defects as “traps” to capture and separate the individual heteroatoms. Subsequently, the well dispersed single atoms can be stabilized via electronic modulation between the heterometal atoms and defect sites due to the different electron energy levels. This approach achieves uniform dispersion and local stabilization of heteroatoms to prevent aggregation.16 Therefore, it is crucial to effectively create and utilize defects on the substrate to capture and stabilize the heteroatoms, and further regulate their coordination environment to improve the electrocatalytic activity, selectivity, and stability.17–20 Thus, various approaches have been developed to optimize the electronic structure of Ru, such as introducing a cation/anion,21 designing a specific structure,22 and regulating the support structure,23 to achieve excellent electrocatalytic performances. However, little attention has been paid to the halogen effects on the ligands engineering. Fluoride ions (F−) possess the highest electronegativity and a highly malleable electronic structure, which can optimize the adsorption energy of critical key intermediates through enhanced electronic interactions. Meanwhile, the strong electronegativity of fluorine atoms can enhance the stability of anchored heteroatoms by effectively adsorbing positive-charged metal ions, thus further strengthening heteroatoms–support interactions.24–26 Moreover, the surface wettability of the electrode can also be modulated by adjusting the surface energy, thus facilitating the infiltration of the electrolyte and removal of the produced H2 gas.4,19,22 Therefore, it is meaningful and feasible to attempt oxygen vacancy anchoring and F− doping strategies to prepare highly stable SACs for electrocatalytic hydrogen evolution.
Herein, we presented a straightforward and universally applicable method for the preparation of heterometal single-atom catalysts on a free-standing hierarchical TiN/TiO2 heterostructure nanorods array (F–M@TiOxNy, M = Ru, Pd, Ir, Pt). The F–M@TiOxNy catalysts were synthesized using a simple and easy-scalable in situ etching and immobilization technique. Fluorine ion-etched TiOxNy nanorods with abundant oxygen vacancies and F–C/F–Ti bonds were utilized to capture and stabilize the heteroatoms. Detailed characterization indicated heteroatoms to be anchored within the oxygen vacancies in TiOxNy and F–C bonds could stabilize the heteroatoms, while the surface immobilization on hierarchical TiOxNy nanorods could effectively expose active sites and reduce the precious metal dosage. Therefore, due to the synergistic effect of oxygen vacancy anchoring and F–C assisted stabilization, F–Ru@TiOxNy revealed high electrocatalytic performance for hydrogen evolution with a low overpotential of 20.8 mV at 10 mA cm−2, a Tafel slope of 59.9 mV dec−1 and a robust stability at 100 mA cm−2 over 48 h. Furthermore, this universal method is applicable to various heterometals (Pd, Ir, Pt), which also exhibit good heteroatoms dispersity and high electrocatalytic HER activity/stability. Therefore, this simple and easy-scalable fabrication method showcases significant potential and promising application prospects due to its mild synthetic conditions and versatility.
2. Experimental section
The comprehensive preparation process for a hierarchically aligned TiOxNy nanorods array has been reported in our previous work.27 The synthesis of F–Ru@TiOxNy was achieved via a mild fluoride ion etching technique. The precursor solution comprised potassium fluoride (KF, 10 mg) and ruthenium chloride trihydrate (RuCl3·3H2O, 1.5 mM), along with ethanol and water at a volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (3 mL in total). Initially, the as-prepared TiOxNy was immersed in the precursor solution for 10 min. Then, the mixture was agitated on a shaker for another 50 min to ensure a complete surface reaction. After washing and drying, the F–Ru@TiOxNy composite was obtained. Furthermore, the same method was also used to immobilize Ru ions onto the TiO2 surface for comparison (F–Ru@TiO2). The universality of this in situ etching strategy was investigated by fabricating various F–M@TiOxNy electrocatalysts by replacing the corresponding precious metal precursors, where M represents palladium (Pd), iridium (Ir), or platinum (Pt), respectively.
:1 (3 mL in total). Initially, the as-prepared TiOxNy was immersed in the precursor solution for 10 min. Then, the mixture was agitated on a shaker for another 50 min to ensure a complete surface reaction. After washing and drying, the F–Ru@TiOxNy composite was obtained. Furthermore, the same method was also used to immobilize Ru ions onto the TiO2 surface for comparison (F–Ru@TiO2). The universality of this in situ etching strategy was investigated by fabricating various F–M@TiOxNy electrocatalysts by replacing the corresponding precious metal precursors, where M represents palladium (Pd), iridium (Ir), or platinum (Pt), respectively.
The experimental details pertaining to the characterization of materials and the electrochemical testing of these composites are provided in the ESI experimental section in the ESI.†
3. Results and discussion
3.1 Characterization of F–Ru@TiOxNy
The preparation process of SACs was illustrated in Fig. 1. Initially, TiOxNy hollow nanorods, characterized by a relatively high density of oxygen vacancies, were synthesized using a seed-assisted hydrothermal method in conjunction with a dicyandiamide (DCDA) nitriding approach, as described in our previous work.27,28 This synthetic procedure provides a hierarchical and interconnected framework with large accessible surface area for the anchoring of heteroatoms. Subsequently, the F–M@TiOxNy composite was formed by immersing the TiOxNy nanorods into a precious metal precursor solution, which consisted of an ethanol–water mixture along with KF and MClx·yH2O salts. Meanwhile, the continuous agitation provided by a shaker facilitated the uniform dispersion of metal single atoms and prevented surface agglomeration. During the reaction, ethanol served as a reducing agent to inhibit the hydrolysis or oxidation of metal salts, while KF functioned as a surface etchant to the TiOxNy nanorods. Upon immersion of the TiOxNy composite into the reaction solution, many bubbles could be formed rapidly on the composite surface to form extra surface vacancies (Fig. S1†). This phenomenon led to the effective immobilization of metal atoms at the as-formed oxygen vacancies to serve as active sites for electrocatalytic reactions.29 Furthermore, it is noteworthy that fluoride ions could react with the amorphous carbon layer presented on the nanorods surface to form specific F–C bonds, which is beneficial for regulating the electronic structure of Ru heteroatoms and, thus, enhancing the structural/catalytical stability.24 Consequently, single-atom catalysts supported by TiOxNy nanorods were prepared through a straightforward and mild soaking method based on oxygen vacancies defect anchoring and a fluorocarbon bonding stabilization strategy. Furthermore, the optimized operation parameters were also studied. The influence of the type and amount of the etching salts, the composition of the nanorods support, and the concentration of precious metal salts on the morphology and electrocatalytic activity were investigated (Fig. S1–S13†), which are described in detail in the ESI† (except for a special description, the 10 mg KF, TiOxNy composite, and 1.5 mM RuCl3·3H2O were chosen for the optimized parameters in subsequent experiments, respectively). | ||
| Fig. 1 An in situ fluoride ion etching and heteroatoms immobilization strategy for the synthesis of F–M@TiOxNy (M = Ru, Pd, Ir, Pt) (schematic). | ||
Scanning electron microscopy (SEM) was employed to examine the morphology evolution of the composites. Initially, TiO2 nanorod arrays with a smooth surface anchored onto the carbon fiber surface were synthesized using a seed-assisted hydrothermal method (Fig. 2a). To induce oxygen vacancy defects and a carbon coating layer onto the nanorods surface, TiOxNy was produced by nitridation of TiO2 through pyrolysis of DCDA under a high temperature, which resulted in the rough surface and hollow structure of the nanorods (Fig. 2b). During solution immersion, the morphology of the nanorod arrays was well-preserved (Fig. 2c). The rough surface with a carbon coating layer enhanced the number of immobilization points for heteroatoms and increased the number of accessible active sites for the HER. Importantly, significant heterometal agglomeration was not observed on the TiOxNy nanorods surface, demonstrating the uniform distribution of the heteroatoms.
 | ||
| Fig. 2 SEM images of (a) TiO2, (b) TiOxNy and (c) F–Ru@TiOxNy composites. TEM images of (d) TiOxNy (inset is a TEM image and lattice fringe of TiOxNy) and (e and f) F–Ru@TiOxNy. (g) HRTEM image and related lattice pseudo-colour surface intensity plot and X–Y intensity profiles of F–Ru@TiOxNy. (h) STEM-EDS line scan and mapping images of F–Ru@TiOxNy. | ||
Transmission electron microscopy (TEM) was employed to evaluate the microstructure of the composites. As illustrated in Fig. 2d, TiOxNy nanorods exhibited a porous and hollow structure, while the rough surface was consistent with the SEM image. The lattice spacing of 0.211 nm corresponded to the (200) crystal plane of TiN (Fig. 2d), demonstrating the successful nitridation of the nanorods. The in situ phase transformation could lead to the formation of abundant crystalline dislocations and defects to facilitate subsequent heteroatoms immobilization. After surface etching and heteroatoms immobilization, the microstructure of the TiOxNy nanorods remained almost unchanged (Fig. 2e and f), indicating the mild surface etching process and robust structure stability of the nanorods. Furthermore, the immobilization and uniform dispersion of Ru single atoms was corroborated by the red spots observed in the high-resolution image. The high-resolution TEM (HRTEM) image of F–Ru@TiOxNy nanoparticles revealed a bright and ordered arrangement of Ti atoms along the (111) crystal direction (Fig. 2g). Both the three-dimensional pseudo-colour surface intensity plot and X–Y intensity profiles of the F–Ru@TiOxNy lattice exhibited the atomically dispersed feature of Ru element. Furthermore, the additional bright atomic columns on the TiOxNy surface could be identified as Ru atoms due to the lower atomic number of Ti compared with that of Ru.30 The introduction of Ru heteroatoms could introduce new active sites and consequently enhance the surface activity of the electrocatalyst for the HER.31 Additionally, the energy-dispersive X-ray (EDX) elemental line scan and mapping images indicated the homogeneous distribution of Ti, O, N, C, F, and Ru elements without obvious discernible aggregation of Ru species (Fig. 2h and S14†), indicating the successful introduction and stable immobilization of Ru and F species. Therefore, these results demonstrated the synthesis of the TiOxNy composite with Ru single-atoms loading via immersion in a precursor solution.
The crystallinity and phase structures of the as-prepared composites were analyzed by X-ray diffraction (XRD). As depicted in Fig. 3a, the nitridation process resulted in a weakening of the TiO2 peaks and the presence of new peaks associated with TiN, while the crystalline phases of TiO2 and TiN corresponded closely with rutile TiO2 (JCPDS 77-0440) and osbornite TiN (JCPDS 38-1420), respectively. In contrast, for F–Ru@TiOxNy, the peak intensity remained almost unchanged and no new peaks could be observed, suggesting weak surface damage during the etching process and ultralow amount of Ru loading. Furthermore, as shown in electron paramagnetic resonance (EPR) curves, the TiOxNy exhibited an obvious higher oxygen vacancies intensity compared with that of TiO2 (Fig. 3b), which was attributed to the nitridation process that induces many crystalline interfaces, dislocations and vacancies. It is worth noting that fluoride ion etching led to further enhancement of the oxygen vacancies strength in F–TiOxNy (Fig. S15a†), demonstrating the strong surface reaction/interaction during surface etching and the introduction of extra vacancies.32 Meanwhile, the oxygen vacancies intensity revealed a significant decrease with the presence of Ru ions (Fig. S15b†), which may have been due to the anchoring of Ru heteroatoms into oxygen vacancies. Moreover, as the concentration of Ru salt in the etching solution increased, the peak intensity of oxygen vacancies decreased accordingly (Fig. S15c†), demonstrating the accurate location and strong interaction of the Ru heteroatoms with surface vacancies.33
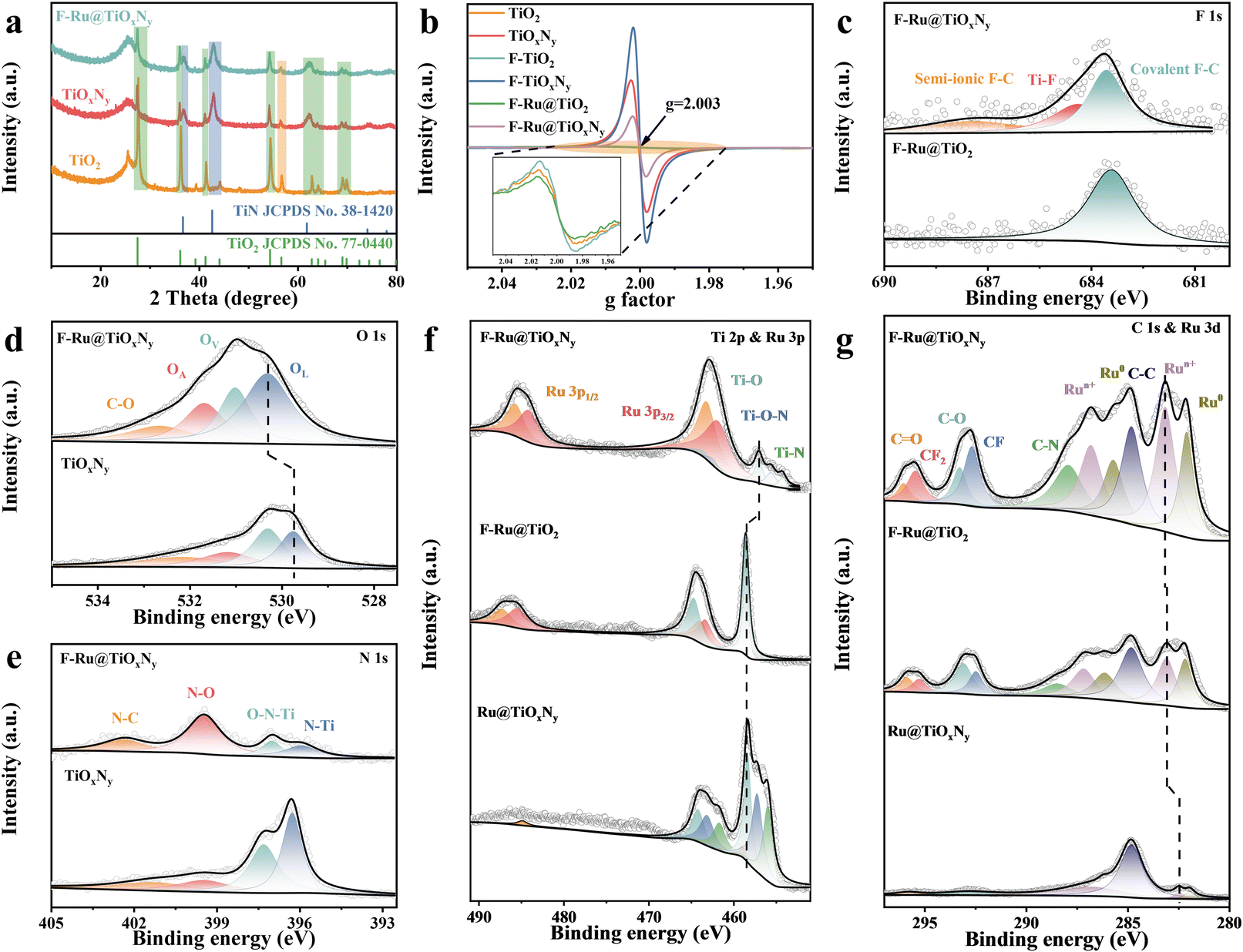 | ||
| Fig. 3 (a) XRD patterns of TiO2, TiOxNy, and F–Ru@TiOxNy. (b) EPR spectra of TiO2, TiOxNy, F–TiO2, F–TiOxNy, F–Ru@TiO2 and F–Ru@TiOxNy (inset is a partially enlarged image). XPS deconvoluted (c) F 1s, (d) O 1s, (e) N 1s, (f) Ti 2p and Ru 3p and (g) C 1s and Ru 3d spectra of TiOxNy, F–Ru@TiO2 and F–Ru@TiOxNy. | ||
Moreover, the oxidation valence states of the surface elements were investigated by X-ray photoelectron spectroscopy (XPS). The full XPS survey revealed that F–Ru@TiOxNy displayed two additional peaks compared with TiOxNy, indicating the presence of F and Ru elements (Fig. S16†). The F 1s spectra of F–Ru@TiOxNy could be deconvoluted into three peaks located at 687.78, 684.78 and 683.78 eV (Fig. 3c), which corresponded to semi-ionic F–C,25 F–Ti,34 and covalent F–C,35 respectively. In comparison, F–Ru@TiO2 revealed only covalent F–C, demonstrating the high surface reactivity and strong interaction of F ions with the defect-rich Ti sites and amorphous carbon. It is worth noting that the formation of semi-ionic C–F bonds tends to adsorb H2O molecules onto the hydrophilic end F–C through hydrogen bonding, so the introduction of F enhances the wettability of the electrolyte on the electrode surface.25 The presence of F–Ti bonds is beneficial for regulating the electronic structure and coordination environment of Ru heteroatoms,25 which provides an extra interaction force to guarantee stability during electrocatalytic hydrogen evolution. Furthermore, O 1s in F–Ru@TiOxNy could be deconvoluted into four peaks centred at binding energies of 533.03, 532.08, 531.38 and 530.73 eV (Fig. 3d), which were composed to C–O, adsorbed oxygen (OA), oxygen vacancy (Ov) and lattice oxygen (OL), respectively.36–39 The O 1s spectra shift towards higher binding energy after F− doping, so the electronic environment around O/N is highly weakened due to the high electronegativity of F atoms.40 Meanwhile, the strong positive peak shift in O 1s may also due to the significant electron transfer from Ti to Ru heteroatoms.41 Moreover, the proportion of the Ov peak area significantly decreased after immobilization of Ru, indicating that Ru atoms were anchored within the oxygen vacancies to avoid aggregation, which is in line with the EPR results. As illustrated in Fig. 3e, the N 1s spectra of TiOxNy and F–Ru@TiOxNy consisted of similar peaks located at 402.78, 399.88, 397.38 and 396.33 eV, which corresponded to N–C, N–O, O–N–Ti, and N–Ti, respectively. Obviously, the peak intensities of O–N–Ti and N–Ti in F–Ru@TiOxNy revealed a significantly decreased trend, possibly due to the formation of F–Ti bonds.
As shown in the Ti 2p and Ru 3p spectra in Fig. 3f, the Ru 3p peak was obviously weak without surface F− etching, which demonstrated the necessity of the presence of F species and extra defects to anchor sufficient Ru atoms. In comparison, with the addition of F− etching, a small number of Ru atoms could be introduced into TiO2 lattices. After F− etching of TiOxNy, the loading amount of Ru heteroatoms was greatly increased, while an obvious shift in Ti 2p orbitals and a significant decrease in peak intensity could be observed, indicating the doping of fluoride ions. The Ti 2p and Ru 3p spectra in F–Ru@TiOxNy exhibited 10 prominent peaks located at 485.74/463.21, 484.19/461.91, 463.51/457.07, 462.55/455.64, 461.76/454.29 eV, which aligned well with Ru 3p1/2, Ru 3p3/2, Ti–O, Ti–N–O and Ti–N,42,43 respectively. Moreover, the C 1s and Ru 3d orbitals also revealed similar situations; no F–C bond could be observed in Ru@TiO2 without the addition of the KF etchant (Fig. 3g). With the presence of KF, F–C bonds could be clearly observed on F–Ru@TiO2, while the carbon species may have originated from carbon fiber. In comparison, the peak intensity of F–C bonds and Ru 3d peaks of F–Ru@TiOxNy were significantly enhanced, while the peaks around 295.46 and 292.68 eV demonstrated the formation of CF2 and CF, respectively.44 The formation of F–C bonds and many oxygen vacancies during KF etching could enhance the host–guest interaction force to immobilize Ru heteroatoms, thereby increasing the loading amount and stability of single atoms. The binding energies located at 296.04, 293.28, 287.91 and 284.80 eV were associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, C–N and C–C, respectively. Meanwhile, the peaks centred at 286.80/283.13 and 285.70/282.08 eV were attributed to Run+ and Ru0,23 respectively. Both the Run+ and Ru0 peaks shifted to lower binding energies, suggesting the enhanced electron-acquiring behaviour of Ru sites.25 Moreover, the incorporated fluorine could also enhance the surface hydrophilicity of the electrocatalyst, significantly improving the bubble diffusion kinetics at the catalyst surface and thus boosting the catalytic stability (Fig. S17†). Therefore, the formation mechanism of the SACs by etching TiOxNy hollow nanorods with fluoride ions could be attributed to the immobilization of Ru atoms within the oxygen vacancy defects, as well as the electronic structure regulation of Ru atoms by Ru–Ti interactions and F–C/F–Ti bonds.
O, C–O, C–N and C–C, respectively. Meanwhile, the peaks centred at 286.80/283.13 and 285.70/282.08 eV were attributed to Run+ and Ru0,23 respectively. Both the Run+ and Ru0 peaks shifted to lower binding energies, suggesting the enhanced electron-acquiring behaviour of Ru sites.25 Moreover, the incorporated fluorine could also enhance the surface hydrophilicity of the electrocatalyst, significantly improving the bubble diffusion kinetics at the catalyst surface and thus boosting the catalytic stability (Fig. S17†). Therefore, the formation mechanism of the SACs by etching TiOxNy hollow nanorods with fluoride ions could be attributed to the immobilization of Ru atoms within the oxygen vacancy defects, as well as the electronic structure regulation of Ru atoms by Ru–Ti interactions and F–C/F–Ti bonds.
3.2 Electrocatalytic hydrogen evolution performance
The HER of the as-synthesized free-standing composites was assessed using a standard three-electrode electrochemical system in 1 M KOH solution. As illustrated in Fig. 4a, the F–Ru@TiOxNy catalyst demonstrated superior HER activity in comparison with other composites. The overpotential of F–Ru@TiOxNy was measured to be 20.8 mV at a current density of 10 mA cm−2, which was significantly lower than that of F–Ru@TiO2 (51.4 mV), TiOxNy (360.9 mV), TiO2 (364.5 mV) and commercial Pt/C (36.3 mV). Cyclic voltammetry (CV) curves were obtained in the non-Faraday region at varying scan rates (Fig. S18a–e†), while the plot of double-layer capacitances (Cdl) was directly proportional to the electrochemically active surface area (ECSA). The calculated ECSA of F–Ru@TiOxNy (260.7 mF cm−2) was substantially greater than that of TiOxNy (98.4 mF cm−2), F–Ru@TiO2 (8.3 mF cm−2), TiO2 (3.3 mF cm−2) and Pt/C (21.9 mF cm−2) (Fig. S18f†), indicating that F–Ru@TiOxNy possessed higher exposure of catalytic active sites. Moreover, the Nyquist plots plotted from electrochemical impedance spectroscopy (EIS) revealed the lowest charge transfer resistance (Rct) for the F–Ru@TiOxNy composite (Fig. S19†), suggesting that the Ru nanoparticles supported on TiOxNy could effectively enhance proton and charge transfer at the electrode/electrolyte interface. Furthermore, Tafel slope curves were calculated to provide further insight into the HER reaction kinetics. As shown in Fig. 4b, F–Ru@TiOxNy exhibited the smallest Tafel slope (59.9 mV dec−1), indicating the fastest HER reaction kinetics, which was significantly lower than that of TiO2 (558.2 mV dec−1), TiOxNy (670.4 mV dec−1), F–Ru@TiO2 (85.7 mV dec−1) and commercial Pt/C (102.6 mV dec−1). To gain thorough insight into the high electrocatalytic activity of F–Ru@TiOxNy in alkaline media, the hydrogen binding energy (HBE)45 was measured by CV in the potential range of −1.0 to 0 V (Fig. 4c). The various response current densities of the composites demonstrated different accessible surface area and different active sites density due to N/F doping.46,47 The underpotentially deposited hydrogen (Hupd) desorption peak of F–Ru@TiOxNy moved to a more negative potential, verifying its favourable hydrogen desorption step due to the relatively weak chemisorption strength of H*. Therefore, it is expected that the strong interactions between the surface of catalysts and *OH species can facilitate water dissociation.48 | ||
| Fig. 4 (a) LSV polarization curves, (b) Tafel slope, and (c) CV curves at 20 mV s−1 from −1.0 to 0 V of TiO2, TiOxNy, F–Ru@TiO2, F–Ru@TiOxNy and Pt/C catalysts in 1 M KOH. Nyquist plots and related fitting results of (d) Pt/C, (e) F–Ru@TiO2 and (f) F–Ru@TiOxNy under different applied potentials. Bode phase plots of (g) Pt/C, (h) F–Ru@TiO2, and (i) F–Ru@TiOxNy under different applied potentials. (j) Equivalent circuit models of F–Ru@TiOxNy for the HER and correlation of the equivalent resistances (R1 and R2) and potentials for catalysts during the HER. (k) Durability test of F–Ru@TiOxNy, F–Ru@TiO2 and Pt/C at 10 mA cm−2 (inset is the durability test of F–Ru@TiOxNy at 100 mA cm−2 and LSV curves of F–Ru@TiOxNy before/after the 48 h test). | ||
Moreover, operando electrochemical impedance spectroscopy (EIS) at various voltages was conducted to elucidate the surface adsorption and desorption kinetics of reaction intermediates on Ru active sites. The fitting results of relevant EIS are shown in Fig. 4d–f and S19.† Generally, the equivalent impedance gradually decreases with an increased applied potential. The Nyquist plots of F–Ru@TiOxNy revealed the smallest semicircular trend than that of Pt/C and F–Ru@TiO2 under high applied potentials (Fig. 4d–f). This suggested that the F–Ru@TiOxNy surface was favorable for the adsorption of reactants and intermediates during the HER, which was attributed to the optimized low-valence state Ru sites with electronic structures matching the reaction intermediates.49 Compared with Pt/C, F–Ru@TiO2 also exhibited a smaller semicircle, indicating that the slight surface etching of TiO2 by fluoride ions could also load a small number of Ru sites and favor the surface adsorption/reaction. In addition, F–Ru@TiOxNy revealed the smallest surface transfer resistance (Rs) and charge transfer resistance (Rct) under a high potential, which is beneficial for surface adsorption and the electrocatalytic HER. As shown in the Bode plots in Fig. 4g–i, the response of phase transition peaks was derived from the interfacial charge transfer and inner-layer electron transport of the electrocatalyst, which is consistent with the Volmer step and Heyrovsky reaction, respectively.50 The first step of the HER is the Volmer reaction with H2O in an alkaline medium to generate H*. The second step is the rate-determining step (RDS), which depends on the activity of the catalyst. For Ru-based catalysts, the second step is the Heyrovsky reaction, where H* reacts with water to produce H2.51 Compared with Pt/C and F–Ru@TiO2, in situ EIS indicated a faster decrease trend of the phase angle for F–Ru@TiOxNy, indicating the faster Volmer step and Heyrovsky step. Therefore, the Ru single atom can accelerate the hydrolysis dissociation rate and rapidly generate H2 through the Heyrovsky step.52 Moreover, the rapid decrease of the phase angle peak of F–Ru@TiOxNy in the low/high-frequency region verified the facilitated charge transfer efficiency at the electrolyte/catalyst interface and the rapid transfer of electrons within the bulk body, which could effectively stimulate the HER reaction kinetics. Moreover, the equivalent circuit models of F–Ru@TiOxNy for the operando EIS measured in 1 M KOH are shown in Fig. 4j. Generally, the equivalent resistance of these electrocatalysts gradually decreased with an increment in applied potential, indicating the accelerated charge transfer efficiency with the increasing potential. Below −0.944 V, the equivalent impedance of all catalysts was relatively similar, but that of the F–Ru@TiOxNy decreased rapidly to the lowest values above −0.964 V, indicating the rapidly decreased charge/electron transfer resistance of F–Ru@TiOxNy. After achieving −1.104 V, the equivalent resistance of R2 for F–Ru@TiOxNy reflecting the HER reaction dropped to near zero, indicating the optimized surface adsorption and reaction kinetics towards hydrogen production.53 Furthermore, F–Ru@TiOxNy demonstrated high long-term stability due to the strong interface electronic interaction, and a negligible decrease in current density around 10 mA cm−2 at constant applied potential for over 24 h could be achieved (Fig. 4k), which outperformed F–Ru@TiO2 and Pt/C with a relatively high attenuation trend. Even under a high current density of ∼100 mA cm−2, F–Ru@TiOxNy revealed excellent stability over 48 h, while the LSV curves before/after 48 h exhibited almost complete overlap (Fig. 4k inset), further demonstrating the robust and stable electrocatalytic HER activity. Moreover, F–Ru@TiOxNy retained its nanorod array structure after stability testing, but the surface underwent reconstruction under long-term reductive potential, resulting in the formation of nanoneedles or nanosheets arrays (Fig. S20†).54 Furthermore, no significant change could be observed for crystalline phases after the i–t test (Fig. S21a†). However, the peak intensity of N 1s, F 1s and C 1s and Ru 3d decreased obviously, while the O 1s peak showed a significant shift and the Ti 2p peak tended towards a non-nitridation state (Fig. S21b–f†), demonstrating that the surface reconstruction structure may have originated from amorphous TiOx. Therefore, F–Ru@TiOxNy revealed high HER activity and Ru heteroatoms immobilization stability during the long-term test.
3.3 Versatility of the fabrication strategy
To further illustrate the versatile applicability of this synthetic strategy for the preparation of precious metal-based SACs, various precious metal-loaded composites were synthesized: palladium (Pd), iridium (Ir), and platinum (Pt). As depicted in Fig. S22,† the morphology of the TiOxNy nanorods remained unchanged after in situ surface etching and heteroatoms doping, which provided stable substrates with many accessible oxygen vacancies and F–C bonds for the deposition of heterometals. HRTEM images (Fig. 5a, c and e) revealed the presence of individual heterometal single atoms (Pd, Ir, and Pt, which are highlighted by red dashed circles), demonstrating the immobilization of the heteroatoms on the surface of TiOxNy nanorods. Moreover, both the three-dimensional pseudo-colour surface plot and X–Y intensity profiles for these F–M@TiOxNy composites exhibited single-atomic features with well-dispersed heteroatoms. The intensity of lattice fringes at various locations was also illustrated. The peaks exhibited higher intensity corresponding to the single atoms of the precious metals with a larger atomic radius. This observation indicated that the precious metals had been incorporated into the TiOxNy lattice in the form of single atoms. Additionally, EDX also demonstrated that the precious metals were uniformly distributed on the entire surface of TiOxNy nanorods (Fig. 5b, d, f and S23†). Therefore, the anchoring of precious metals within the TiOxNy lattices confirmed the robustness and versatility of this preparation method through an oxygen vacancy defects anchoring and F–C bonds stabilizing strategy. | ||
| Fig. 5 HRTEM images and related lattice pseudo-colour surface intensity plot and X–Y intensity profiles of (a) F–Pd@TiOxNy, (c) F–Ir@TiOxNy and (e) F–Pt@TiOxNy. STEM-EDS mapping images of (b) F–Pd@TiOxNy, (d) F–Ir@TiOxNy and (f) F–Pt@TiOxNy. | ||
To further illustrate the electrocatalytic performance of F–M@TiOxNy (M = Pd, Ir, and Pt) synthesized via this universal synthetic method, the electrocatalytic HER activity was evaluated in 1 M KOH solution. As depicted in Fig. 6a, all these F–M@TiOxNy composites demonstrated remarkable electrocatalytic activity for hydrogen production. Specifically, F–Pd@TiOxNy, F– Ir@TiOxNy, and F–Pt@TiOxNy exhibited low overpotential of 18.9, 13.2, and 22.3 mV at 10 mA cm−2, and 47.5, 53.9, and 59.5 mV at 100 mA cm−2, respectively. Moreover, the ECSA for F–Pd@TiOxNy, F–Ir@TiOxNy, and F–Pt@TiOxNy was calculated to be 248.5, 127.6, and 214.5 mF cm−2, respectively (Fig. 6b and S24a–c†), which was significantly higher than that of TiOxNy (98.4 mF cm−2). The underpotentially deposited hydrogen (Hupd) desorption peaks of F–M@TiOxNy (M = Pd, Ir, and Pt) moved to negative potential (Fig. S24d†), verifying a favourable hydrogen desorption step due to the relatively weak chemisorption strength of H*. Furthermore, the F–M@TiOxNy (M = Pd, Ir, Pt) composites exhibited relatively low charge transfer impedance (Fig. 6c), which was obviously lower than that of commercial Pt/C (Fig. 4d). The Tafel slopes for F–Pd@TiOxNy, F–Ir@TiOxNy, and F–Pt@TiOxNy were measured to be 41.5, 48.6, and 46.1 mV dec−1, respectively (Fig. 6d), together with the favourable hydrogen desorption kinetics (Fig. S24d†), demonstrating fast surface adsorption/desorption efficiency and reaction kinetics. As shown in Fig. S25,† the F–M@TiOxNy electrocatalysts revealed an obviously larger enclosed area for CV curves compared with the other contrast composites, demonstrating a distinct difference in accessible surface area. Meanwhile, the turnover frequency (TOF) of F–M@TiOxNy (M = Ru, Pd, Ir, Pt) was close to or better than that of commercial Pt/C (Fig. 6e). The potentials required for F–Ir@TiOxNy, F–Pd@TiOxNy, F–Pt@TiOxNy, F–Ru@TiOxNy and Pt/C to achieve a TOF of 1.5 s−1 were 86.98, 121.37, 209.48, 151.88 and 164.36 mV, respectively, which illustrated the high intrinsic electrocatalytic activity of F–M@TiOxNy. Furthermore, the long-term stability of F–M@TiOxNy composites was also evaluated at a constant applied potential of ∼100 mA cm−2, and relatively high long-term stability could be maintained over 24 h (Fig. 6f). The LSV curves before and after 24 h revealed almost complete overlap (Fig. S26†), indicating stable reaction kinetics and heteroatoms interface immobilization stability. Furthermore, compared with the recently reported high-end Ru-based HER electrocatalysts, the F–M@TiOxNy (Ru, Pd, Ir, and Pt) composites demonstrated significantly lower onset overpotentials and Tafel slopes, and excellent stability (Fig. 6g and Table S1†), demonstrating superior HER activity and stability, as well as fast reaction kinetics due to the high dispersity and stability of the heteroatoms.
 | ||
| Fig. 6 (a) LSV polarization curves, (b) calculated ECSA, (c) Nyquist plots, (d) Tafel slope, (e) calculated TOF curves and (f) long-term durability at 100 mA cm−2 of F–Pd@TiOxNy, F–Ir@TiOxNy and F–Pt@TiOxNy. (g) Comparison of the overpotentials at 10 mA cm−2 and Tafel values of F–M@TiOxNy (M = Ru, Pd, Ir, Pt) with state-of-the-art Ru-based electrocatalysts in an alkaline electrolyte. | ||
4. Conclusions
A universal strategy was designed to fabricate single-atom catalysts on TiOxNy nanorods surface via a straightforward in situ fluoride ion etching and heteroatoms doping process. The TiOxNy hollow nanorod arrays with many oxygen vacancy defects and high accessible surface area could effectively immobilize Ru atoms. This action, combined with the surface stabilizing effect of the as-formed F–C/F–Ti bonds, ensured the uniform dispersion and stable immobilization of the heteroatoms with strong electronic interactions. Therefore, F–Ru@TiOxNy required an overpotential of only 20.8 mV to achieve 10 mA cm−2 for the HER, as well as fast reaction kinetics and remarkable long-term stability. An operando EIS test demonstrated the optimized surface adsorption and reaction kinetics on the F-doped heteroatom catalyst. Moreover, this in situ heteroatoms immobilization method possessed significant universality. F–M@TiOxNy (M = Pd, Ir, Pt) electrocatalysts with high heteroatoms dispersity and good electrocatalytic HER performance could also be obtained. The advantages of this approach include broad applicability and exceptional simplicity, indicating substantial potential for the large-scale production of single-atom catalysts towards electrocatalytic energy conversion.Data availability
The data supporting the conclusions reached in this study have been included as a part of ESI.†Author contributions
Peng Liu: investigation, data curation, and writing (original draft). Jiahui Ye: data analysis. Kuan Deng: data analysis. Xuesong Liu: data analysis and visualization. Haohui Dong: visualization. He Zhang: data analysis. Wen Tian: methodology, funding acquisition, and writing (review and editing). Junyi Ji: conceptualization, methodology, writing (review and editing), funding acquisition, and supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors appreciate the financial support from National Natural Science Foundation of China (22278282), Sichuan Science and Technology Program (2023NSFSC1915, 2024NSFSC1137). The authors thank Yanping Huang and Yuanlong Wang (Center of Engineering Experimental Teaching) for SEM, TEM and EPR characterization, and Yingming Zhu (Institute of New Energy and Low Carbon Technology) for XRD characterization.References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- K. Deng, X. Liu, P. Liu, X. Lv, W. Tian and J. Ji, Angew. Chem., Int. Ed., 2024, e202416763 Search PubMed.
- A. Pareek, R. Dom, J. Gupta, J. Chandran, V. Adepu and P. H. Borse, Mater. Sci. Energy Technol., 2020, 3, 319–327 CAS.
- F. Guo, T. J. Macdonald, A. J. Sobrido, L. Liu, J. Feng and G. He, Adv. Sci., 2023, 10, 2301098 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef CAS.
- H. Zhang, K. Kawashima, M. Okumura and N. Toshima, J. Mater. Chem. A, 2014, 2, 13498–13508 RSC.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- S. Kaushik, D. Wu, Z. Zhang, X. Xiao, C. Zhen, W. Wang, N.-Y. Huang, M. Gu and Q. Xu, Adv. Mater., 2024, 36, 2401163 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- S. Wei, Y. Sun, Y.-Z. Qiu, A. Li, C.-Y. Chiang, H. Xiao, J. Qian and Y. Li, Nat. Commun., 2023, 14, 7549 CrossRef CAS PubMed.
- J. Liu, X. Qiu, S. Sun, B. Liu, Y. Tian, Y. Qin and X. Lin, Green Chem., 2024, 26, 8020–8029 RSC.
- X. Kang, J. Liu, Y. Xie, D. Wang, Q. Liu, P. Yu, C. Tian and H. Fu, Sci. China Mater., 2024, 67, 3579–3588 CrossRef CAS.
- F. Gong, Y. Liu, Y. Zhao, W. Liu, G. Zeng, G. Wang, Y. Zhang, L. Gong and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308091 CrossRef CAS PubMed.
- D. Guo, X.-X. Xue, M. Jiao, J. Liu, T. Wu, X. Ma, D. Lu, R. Zhang, S. Zhang, G. Shao and Z. Zhou, Chem. Sci., 2024, 15, 16281–16290 RSC.
- L. Wang, L. Zhang, W. Ma, H. Wan, X. Zhang, X. Zhang, S. Jiang, J. Y. Zheng and Z. Zhou, Adv. Funct. Mater., 2022, 32, 2203342 CrossRef CAS.
- Y. Zhao, H.-C. Chen, X. Ma, J. Li, Q. Yuan, P. Zhang, M. Wang, J. Li, M. Li, S. Wang, H. Guo, R. Hu, K.-H. Tu, W. Zhu, X. Li, X. Yang and Y. Pan, Adv. Mater., 2024, 36, 2308243 CrossRef CAS PubMed.
- W. Zhang, Y. Zhao, W. Huang, T. Huang and B. Wu, Coord. Chem. Rev., 2024, 515, 215952 CrossRef CAS.
- W. Song, C. Xiao, J. Ding, Z. Huang, X. Yang, T. Zhang, D. Mitlin and W. Hu, Adv. Mater., 2024, 36, 2301477 CrossRef CAS PubMed.
- C. Tang, Y. Jiao, B. Shi, J.-N. Liu, Z. Xie, X. Chen, Q. Zhang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed.
- Y. Song, Y. Zhang, W. Gao, C. Yu, J. Xing, K. Liu and D. Ma, Chem. Sci., 2024, 15, 9851–9857 RSC.
- Y. Zhao, X. Wang, G. Cheng and W. Luo, ACS Catal., 2020, 10, 11751–11757 CrossRef CAS.
- Y. Yang, Y. Yu, J. Li, Q. Chen, Y. Du, P. Rao, R. Li, C. Jia, Z. Kang, P. Deng, Y. Shen and X. Tian, Nano-Micro Lett., 2021, 13, 160 CrossRef CAS PubMed.
- W. Xu, H. Xie, F. Cao, S. Ran, Y. Duan, B. Li and L. Wang, J. Mater. Chem. A, 2022, 10, 23751–23759 RSC.
- S.-M. Wu, I. Hwang, B. Osuagwu, J. Will, Z. Wu, B. B. Sarma, F.-F. Pu, L.-Y. Wang, Z. Badura, G. Zoppellaro, E. Spiecker and P. Schmuki, ACS Catal., 2023, 13, 33–41 CrossRef CAS.
- W. Liu, Q. Chen, Y. Shang, F. Liu, R. He, J. Zhang, Q. Li, H. Chai, Y. Tan and S.-J. Bao, Adv. Funct. Mater., 2024, 34, 2410325 CrossRef CAS.
- X. Shao, A. Maibam, F. Cao, H. Jin, S. Huang, M. Liang, M. Gyu Kim, K. My Tran, A. R. Jadhav, H. Seung Jung, R. Babarao and H. Lee, Angew. Chem., Int. Ed., 2024, 63, e202406273 CrossRef CAS PubMed.
- P. Liu, K. Deng, Y. Huang, X. Liu, X. Lv, W. Tian and J. Ji, Chem. Eng. Sci., 2023, 281, 119148 CrossRef CAS.
- Y. Du, P. Liu, H. Zhang, L. Zou, K. Deng, X. Li, W. Tian and J. Ji, Adv. Funct. Mater., 2024, 34, 2309830 CrossRef CAS.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- H. Peng, T. Yang, H. Lin, Y. Xu, Z. Wang, Q. Zhang, S. Liu, H. Geng, L. Gu, C. Wang, X. Fan, W. Chen and X. Huang, Adv. Energy Mater., 2022, 12, 2201688 CrossRef CAS.
- R. Wang, Q. Chen, X. Liu, Y. Hu, L. Cao and B. Dong, Small, 2024, 20, 2311217 CrossRef CAS PubMed.
- Z. Zhang, S. Lu, G. Huang, W. Wang, D. He, Y. Liu, F. Gao, Y. Chen, H. Zhan, J. Mei, M. Terrones, Y. Wang and X. Chen, Carbon, 2024, 221, 118885 CrossRef CAS.
- X. Liu, Y. Zhou, J. Lin, X. Xiao, Z. Wang, L. Jia, M. Li, K. Yang, J. Fan, W. Yang and G. Li, Angew. Chem., Int. Ed., 2024, 63, e202406650 CrossRef CAS PubMed.
- D. Lv, D. Wang, N. Wang, H. Liu, S. Zhang, Y. Zhu, K. Song, J. Yang and Y. Qian, J. Energy Chem., 2022, 68, 104–112 CrossRef CAS.
- J. Cui, P. Yin, A. Xu, B. Jin, Z. Li and M. Shao, Nano Energy, 2022, 93, 106837 CrossRef CAS.
- Y. Yao, Y. Zhu, C. Pan, C. Wang, S. Hu, W. Xiao, X. Chi, Y. Fang, J. Yang, H. Deng, S. Xiao, J. Li, Z. Luo and Y. Guo, J. Am. Chem. Soc., 2021, 143, 8720–8730 Search PubMed.
- K. Deng, P. Liu, X. Liu, H. Li, W. Tian and J. Ji, Green Chem., 2023, 25, 9837–9846 Search PubMed.
- T. You, K. Deng, P. Liu, X. Lv, W. Tian, H. Li and J. Ji, Chem. Eng. J., 2023, 470, 144348 CrossRef CAS.
- X. Liu, K. Deng, P. Liu, X. Lv, W. Tian, K. Ma, H. Li and J. Ji, Appl. Catal., B, 2024, 343, 123470 CrossRef CAS.
- X. Xu, H. Ullah, M. Humayun, L. Li, X. Zhang, M. Bououdina, D. P. Debecker, K. Huo, D. Wang and C. Wang, Adv. Funct. Mater., 2023, 33, 2303986 CrossRef CAS.
- S. Zhang, X. Chang, L. Zhou, X. Liu and J. Zhang, ACS Sens., 2024, 9, 2101–2109 CrossRef CAS PubMed.
- Y. Yang, D. Wu, R. Li, P. Rao, J. Li, P. Deng, J. Luo, W. Huang, Q. Chen, Z. Kang, Y. Shen and X. Tian, Appl. Catal., B, 2022, 317, 121796 CrossRef CAS.
- J. Li, L. Zhang, X. An, K. Feng, X. Wang, J. He, Y. Huang, J. Liu, L. Zhang, B. Yan, C. Li and L. He, Angew. Chem., Int. Ed., 2024, 63, e202407025 CrossRef CAS PubMed.
- J. Liang, L. Liang, B. Zeng, B. Feng, L. Du, X. Qiu, Y. Wang, H. Song, S. Liao, M. Shao and Z. Cui, Angew. Chem., Int. Ed., 2024, 63, e202412825 CrossRef CAS PubMed.
- Q. Wang, C.-Q. Xu, W. Liu, S.-F. Hung, H. Bin Yang, J. Gao, W. Cai, H. M. Chen, J. Li and B. Liu, Nat. Commun., 2020, 11, 4246 Search PubMed.
- C. Yang, X. Wang, W. Dong, I. W. Chen, Z. Wang, J. Xu, T. Lin, H. Gu and F. Huang, Sci. China Mater., 2020, 63, 1227–1234 Search PubMed.
- Z. Liu, H. Li, X. Liu, J. Guo, S. Zhang and Y. Lu, J. Energy Storage, 2023, 67, 107527 CrossRef.
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 Search PubMed.
- P. Yan, T. Yang, M. Lin, Y. Guo, Z. Qi, Q. Luo and X.-Y. Yu, Adv. Funct. Mater., 2023, 33, 2301343 Search PubMed.
- W. Gou, Z. Xia, X. Tan, Q. Xue, F. Ye, S. Dai, M. Zhang, R. Si, Y. Zou, Y. Ma, J. C. Ho and Y. Qu, Nano Energy, 2022, 104, 107960 Search PubMed.
- Z. Li, B. Li, M. Yu, C. Yu and P. Shen, Int. J. Hydrogen Energy, 2022, 47, 26956–26977 Search PubMed.
- D. Li, M. Chen, D. Liu, C. Shen, H. Sun, Y. Zhang, T. He, Q. Lu, B. Li, T. Zhou, B. Wang, Y. Wu, G. Na, Y. Chen, J. Zhao, Y. Zhang, J. Zhang, F. Liu, H. Cui and Q. Liu, Adv. Energy Mater., 2024, 2404714 CrossRef.
- H. C. Fu, X. H. Chen, B. Yang, Y. H. Luo, T. Li, X. H. Wang, Q. Zhang, X. L. Li, N. B. Li and H. Q. Luo, Appl. Catal., B, 2023, 332, 122739 Search PubMed.
- W. Yuan, B. Chen, Z.-K. Han, R. You, Y. Jiang, R. Qi, G. Li, H. Wu, M. V. Ganduglia-Pirovano and Y. Wang, Nat. Commun., 2024, 15, 1616 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08603a |
| This journal is © The Royal Society of Chemistry 2025 |