
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and dynamics of photoswitchable flavoprotein charge-transfer complexes†
Bo
Zhuang
 *a,
Guangliu
Ran
b,
Wenkai
Zhang
*b and
Feng
Gai
*a
*a,
Guangliu
Ran
b,
Wenkai
Zhang
*b and
Feng
Gai
*a
aBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: zhuangbo@pku.edu.cn; fgai@pku.edu.cn
bSchool of Physics and Astronomy, Applied Optics Beijing Area Major Laboratory, Center for Advanced Quantum Studies, Beijing Normal University, Beijing, 100875, China. E-mail: wkzhang@bnu.edu.cn
First published on 7th March 2025
Abstract
Because of their diverse uses in biological science and engineering, continued effort has been made to expand the pool of photoswitchable protein systems. A recent study demonstrated that in monomeric sarcosine oxidase (MSOX), photoexcitation of a charge-transfer (CT) complex formed by a flavin cofactor and a nonreactive ligand (e.g., methylthioacetate) induces the ligand to reversibly change conformation, with implications for the development of flavin-dependent fast photochromic proteins. However, the factors that control the underlying switching mechanism and dynamics remain largely unexplored. Herein, combining extensive protein mutagenesis, ultrafast laser spectroscopic measurements and classical and quantum computational approaches, we assess those factors in a range of protein variants, including those of MSOX and another flavoenzyme, N-methyltryptophan oxidase (MTOX), where we find that a similar photoswitching cycle can occur. We demonstrate that (1) the kinetic behaviors of the photoswitching cycle are protein- and ligand-dependent; (2) the photoswitching and backward thermal recovery rates can be tuned by mutation of a specific active-site residue (Met245 and Thr239 in MSOX and MTOX, respectively), with recovery rates spanning over an order of magnitude, and (3) modifications of the protein environment alter the conformational energy landscape of the ligand–flavin complex, consequently regulating the photocycle. Taken together, these findings highlight the versatility of such photoswitchable systems, providing a molecular basis for fine-tuning their photophysical properties.
Introduction
The reception and transmission of light signals by organisms involve photoswitchable proteins or protein complexes, which, through reversible photoinduced transitions between different conformational, or light absorbing and emitting states,1 can regulate a wide variety of physiological light responses such as visual perception,2 phototropism and phototaxis,3,4 as well as responses to light–dark cycles such as the circadian rhythm and other photoperiodism.5,6 Typically, the actions of such photoswitchable proteins are derived from photoisomerization or other types of photochemical reactions of a protein-bound chromophore. These include retinal in rhodopsin and microbial rhodopsins,7–9 tetrapyrrole derivatives in phytochrome10 and cyanobacteriochromes,11 chromophores composed of post-translationally modified amino acids in the family of the green fluorescent proteins,12,13 coenzyme B12 in B12-dependent photo-responsive bacterial transcriptional repressors,14,15 as well as flavin cofactors in several flavin-dependent photoreceptors,16,17 namely![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) lue-
lue-![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ight-
ight-![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) sing-
sing-![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) lavin (BLUF) domain,18–20 ight-
lavin (BLUF) domain,18–20 ight-![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif) xygen-
xygen-![[v with combining low line]](https://www.rsc.org/images/entities/char_0076_0332.gif) oltage (LOV) domain,21–23 and cryptochromes (CRYs).24–26
oltage (LOV) domain,21–23 and cryptochromes (CRYs).24–26
Furthermore, photoswitchable proteins have also led to the design and development of various bioinspired photoswitchable complexes, with applications ranging from high-resolution microscopy12,27 to optogenetics.28–30 The biotechnological applicability of a photoswitchable protein is mainly determined by its photophysical properties, such as absorption, photoswitching quantum yield (QY), and thermal recovery rate. For example, fast photochromic proteins with photoproducts persisting on millisecond timescales can be used for biological imaging, while actuators require long-lived bistable photoproducts. In practice, it is crucial to suppress competitive decay pathways of the excited state(s) to improve the overall QY of the photoswitching reaction and to increase the lifetime of the desired photoproduct(s). Moreover, for optogenetic applications, it is preferable to have photoswitching reactions that can be initiated by radiation of long wavelengths, e.g., in the near-infrared (NIR) spectral region, so that better tissue penetration and lower phototoxicity can be achieved.31 However, despite the fast progress in this field, the currently available photoswitchable protein systems are still limited, due to the limited number of natural-occurring photoswitchable protein-bound chromophores, as well as the difficulty of fine-tuning their photophysical properties. Therefore, further expanding the number and variety of photoswitchable protein systems is highly desirable.
The vast majority of currently known proteins do not perform light-dependent physiological functions.32,33 For instance, photoreceptors CRYs, BLUF and LOV domains, as well as two photoenzymes DNA photolyase34–36 and fatty acid photodecarboxylase,37,38 are the only known photo-responsive flavoproteins thus far. However, many cofactor-containing proteins, including flavoproteins, while they do not directly participate in functional photochemical reactions, have diverse photophysical properties. Therefore, studying the photoinduced processes in proteins that are not utilized by nature can, on the one hand, deepen our understanding of the photophysics of complex biomolecules, and on the other hand, pave the way for harvesting such photochemical processes for new applications, as is the case with the use of a nonphotocatalytic flavoenzyme 'ene'-reductase to catalyze unnatural organic synthesis reactions.39,40 In this context, a recent study has demonstrated a novel type of reversibly photoswitchable protein system, based on photoexcitation of a noncovalently bound complex formed by a flavin adenine dinucleotide (FAD) cofactor and a small-molecule ligand in monomeric sarcosine oxidase (MSOX).41
MSOX is a nonphotocatalytic flavoprotein, which catalyzes the oxidative demethylation of sarcosine.44 Methoxyacetate (MOA), methylthioacetate (MSA), methylselenoacetate (MSeA) and methyltelluroacetate (MTeA), with the chemical structures of CH3XCH2CO2− (X = O, S, Se, and Te), are substrate analog inhibitors of MSOX that can bind to its active site (Fig. 1A). Among them, complexes formed by MSA, MSeA and MTeA with FAD in wild-type MSOX (MSOXWT) give rise to intense absorption bands in the visible-NIR spectra region (Fig. 1B), supposedly due to MXA–flavin (X = S, Se, and Te) charge-transfer (CT) interactions.42 Previously, it has been shown that the MSA:FAD and MSeA:FAD complexes in MSOX can undergo an unexpected photoswitching reaction that occurs on the sub-picosecond timescale (ca. 300 fs) with a high QY (ca. 0.8 for the MSA:FAD complex), which has been proposed to correspond a photoinduced conformational change of the ligands in the protein active site (Fig. 1C).41 While the photophysical properties of these systems are potentially useful for photocatalytic and photoswitching applications, the underlying mechanisms and the key determinants that regulate the photoswitching dynamics remain largely unexplored.
 | ||
| Fig. 1 (A) Chemical structures of the flavin ring and the MXA ligands, as indicated. (B) Steady-state absorption spectra of ligand-free MSOXWT and of ligand-bound complexes of MSOXWT and wild-type MTOX (MTOXWT). The spectra of the complexes (normalized to correspond to 100% complex formation) are reproduced from published data.42,43 (C) Active sites of MSOXWT from Bacillus sp. in complex with MSeA (PDB entry: 1EL8), as well as that of MTOXWT from E. coli without the ligand (PDB entry: 2UZZ), with both structures aligned on the flavin rings. | ||
In this work, we systematically investigated the photocycle kinetics of such CT complexes in several flavoprotein systems, including the structural homologue protein of MSOX, N-methyltryptophan oxidase (MTOX), which can also bind MSeA to form a CT complex (Fig. 1B)43 and of which the photochemistry has not been investigated before, as well as several MSOX and MTOX mutants. Specifically, we applied femtosecond transient absorption (TA) spectroscopy to examine the photoinduced dynamics in these systems, and employed molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) calculations to investigate the ligand–flavin interactions and evaluate the conformational distribution and energy landscape of the ligand in the protein active site. We demonstrated that the photocycle properties of MXA:FAD complexes in MSOX and MTOX can be regulated by the protein environment, in particular by a residue located at position 245 of MSOX and position 239 of MTOX in the protein active sites (Fig. 1C), providing detailed insight into the photoswitching mechanism and dynamics of such flavoprotein CT complexes.
Results and discussion
Characterization of MXA–FAD interactions
As evident from the steady-state absorption spectra of the MXA:FAD complexes (Fig. 1B), the electronic properties of the FAD cofactor are substantially perturbed due to strong noncovalent interactions between the flavin (isoalloxazine) ring of FAD and the X atom of the MXA ligand when X = S, Se, and Te. As the X atom gets heavier, the new absorption band arising from ligand binding shifts to a longer wavelength and becomes broader (Table S1†). To characterize the nature of MXA–FAD interactions from a theoretical standpoint, we employed a QM/MM approach to optimize the geometries of the MXA:FAD complexes in the active site of MSOXWT by taking into account the effect of the protein environment, based on the reported high-resolution MXA-bound structures of MSOXWT.42 Specifically, the QM part, which was described at a density functional theory (DFT) level, included the flavin ring of FAD, the MXA ligand, as well as the side chain of a close-lying methionine residue, Met245 (Fig. 1C), while the remaining part of the protein and water molecules were described by classical force fields.Geometry optimizations were based on the experimental crystal structures of MSOXWT complexed with MSeA (PDB entry: 1EL8), where MSeA binds to MSOXWT in two discrete conformations (Conf 1 and Conf 2).42 As shown in Fig. 1C, in Conf 1 the Se atom is located close to the flavin ring (3.3 Å to the flavin C4 atom), whereas in Conf 2 the Se atom rotates away from the flavin (4.8 Å to the flavin C4 atom), allowing it to interact with Met245 in the protein active site, with the positions of remaining ligand atoms essentially unchanged. We first optimized the geometries of the MXA:FAD (X = O, S, Se, and Te) complexes in Conf 1. As shown in Fig. 2, the obtained atomic charges (AC) of the interacting atoms of the flavin ring and ligands demonstrate that as the X atom becomes heavier, the negative charge on the X atom decreases, whereas those on the N5 atom of the flavin ring increase. For example, the O atom in MOA is the most negatively charged compared to the X atoms in the other ligands, and the corresponding flavin N5 atom is the least negatively charged, consistent with the fact that MOA binding only slightly perturbs the absorption spectrum of FAD, instead of forming a distinct CT band.42 When X = Se, there is barely any negative charge located on the X atom (ACSe3 = −0.048), and it becomes essentially positively charged when X = Te (ACTe3 = 0.042). This clearly reveals the CT nature of the MXA–FAD interactions as previously suggested,42,45 and confirms that a heavier X atom would lead to a stronger CT interaction. By contrast, as indicated in Fig. S1a,† when MSA is in Conf 2, the amount of negative charge localized on the N5 atom of the flavin is decreased compared to that in Conf 1 (i.e., ACN5 = −0.214 and −0.139 for Conf 1 and Conf 2, respectively). The intermolecular interactions between MSA (MSeA), FAD and Met245 visualized by the independent gradient model based on Hirshfeld partition (IGMH)46 are shown in Fig. S2,† which confirms a lack of MXA–FAD interaction involving the X atom and flavin ring in Conf 2.
 | ||
| Fig. 2 Atomic charges of the MOA:FAD (A), MSA:FAD (B), MSeA:FAD (C), and MTeA:FAD (D) complexes in Conf 1 in MSOXWT, computed at their QM/MM-optimized ground-state electronic structures. For clarity, the corresponding hydrogen atoms are not shown. | ||
Next, we computed the absorption spectra of the CT complexes in MSOXWT based on a QM/MM approach coupled with time-dependent DFT (TDDFT) calculations. As shown in Fig. 3A, despite a systematic offset originating from the limitations of TDDFT,47 the calculated spectra of these complexes reasonably match the general features of the experimental spectra, including the characteristic flavin absorption bands in the near-ultraviolet/visible range and those additional bands at longer wavelengths upon complex formation. In addition, the relative positions of the new bands obtained with different ligands not only agree with the experimental observations (Fig. 1B), i.e., with a heavier X atom the absorption maximum shifts to a longer wavelength, but also follow the trend of the degree of CT interaction of the complex (i.e., MTeA > MSeA > MSA; Fig. 2). Furthermore, the CT nature of the long-wavelength absorption band of MXA:FAD complexes in MSOX is further confirmed by a hole–electron analysis48 of the corresponding electronic transition. Specifically, as illustrated in Fig. 3B and S3,† in the excited electronic state of a MXA:FAD complex, the electron and hole are located separately on the flavin ring and the X atom of the ligand, respectively, indicating a well-defined CT from MXA to FAD upon photoexcitation.45 The calculated spectrum also demonstrates that when MSA or MSeA is bound in Conf 2, the CT excitation exhibits a much smaller oscillator strength (Fig. 3A), consistent with a diminished CT interaction due to the increased distance between the S or Se atom and the flavin ring in Conf 2 (Fig. 1C, S2b and d†). It should be noted that the relative amplitudes of the CT bands involving MSA, MSeA and MTeA in the experimentally measured (Fig. 1B) and calculated (Fig. 3A) spectra are reversed (Table S1†). This may be due to an overestimation of the calculated oscillator strengths, arising from the limitations of TDDFT calculations and, in particular, in the treatment of a heavy atom like Se and Te. More importantly, as mentioned above, the crystallography study suggested that parts of MSeA and MTeA populations were present in Conf 2 in the protein; there were also indications that MTeA was not stable and might gradually degrade in the protein active site.42 These fractions of protein-bound ligand molecules do not effectively contribute to the CT band (Fig. 3A). As estimating the experimental extinction coefficients requires extrapolating the ligand binding curves to obtain the absorption spectra corresponding to 100% complex formation,42 with partial ineffective ligand populations the obtained extinction coefficients would be smaller than the theoretically calculated values specific to Conf 1.
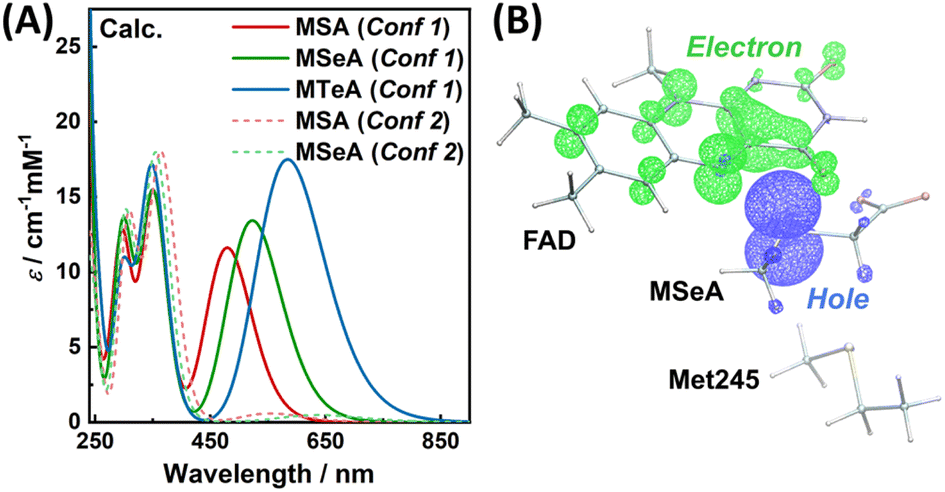 | ||
| Fig. 3 (A) TDDFT-calculated absorption spectra of the CT complexes in the active site of MSOXWT based on the QM/MM method described in the text. (B) Hole–electron distributions of the lowest excited electronic state of the MSeA:FAD complex in MSOXWT (isovalue: 0.004). | ||
As shown in Fig. 1B, the CT absorption band of the MSeA:FAD complex in MTOXWT is less intense and is situated at a shorter wavelength compared to that of the MSeA:FAD complex in MSOXWT. This suggests that the MSeA–FAD interaction in MTOXWT is weaker than that in MSOXWT. This also aligns with the fact that the binding of MSA to MTOXWT does not lead to a distinct CT band.43 As there is no experimental crystal structure of MTOXWT complexed with MXA available, the geometry of the MXA:FAD complex in the active site of MTOXWT is subjected to further investigation by MD simulations (vide infra).
Photocycle kinetics of protein-bound MXA:FAD CT complexes
We first performed TA measurements on the MSeA:FAD complexes in MSOXWT and MTOXWT, respectively, and results obtained for MSOXWT are similar to those reported in the previous study.41 As shown in Fig. S4a,† in the TA spectra, the ground-state bleaching (GSB) signal of the complex appears in the 430–700 nm range, as expected, whereas the initial excited-state absorption (ESA) spectrum is composed of an intense absorption band at ca. 400 nm and a broad band extending to the NIR region. As it is known that the spectral feature of FAD˙− in MSOXWT is characterized by a sharp absorption peak at ca. 400 nm,42,49 this TA result therefore confirms the CT nature of the underlying electronic transition as suggested by the excited-state hole–electron analysis (Fig. 3B). As time evolves, while the GSB signals persist, the ESA spectrum gradually adopts a shape that resembles that of uncomplexed FAD, and the entire TA spectrum is almost identical to the difference between the absorption spectra of uncomplexed FAD and the MSeA:FAD complex (Fig. 4A; the MSeA ligand does not absorb in the visible region). Furthermore, the room-temperature kinetic traces at ca. 590 nm in the region where the ESA and GSB signals are overlapped can be reasonably well fit with a biexponential function, with time constants of ca. 600 fs and 5.5 ns, respectively (Fig. 4C and Table S2†). Given the more pronounced coherent artifacts on the ultrafast timescale (i.e., <400 fs) in the wavelength range of <500 nm, we chose to analyze the decay kinetics using TA signals at a single wavelength measured in the range of >500 nm (Table S2†), which were relatively less affected by those artifacts. The thus obtained time constant of the ultrafast component (600 fs) is somewhat larger than the previously reported value (ca. 300 fs). The discrepancy may be attributed to differences in the pump–probe setups, the sources of the protein sample, and the data analysis methods used, where the accurate determination of ultrafast lifetimes was complicated by the artifacts presented in the TA kinetics in both studies.41 | ||
| Fig. 4 (A and B) TA spectra of the MSeA:FAD complexes in MSOXWT (A) and MTOXWT (B) recorded at selected time delays, as indicated, with 550 nm excitation. In each case, the steady-state difference spectrum that corresponds to the dissociation of the FAD:MSeA complex is shown as a dashed line for comparison. (C) Corresponding TA kinetics at representative probing wavelengths, as indicated. (D) Proposed photocycle upon photoexcitation of the protein-bound MXA:FAD complexes. | ||
Taken together, the TA results of MSeA:FAD in MSOXWT support a reaction scheme shown in Fig. 4D, which consists of an internal conversion process (k1), a photoswitching process (k2), and a back recovery process (k3), with (k1 + k2)−1 = 600 fs and (k3)−1 = 5.5 ns. On the picosecond–nanosecond timescales, the TA spectra are dominated by contribution from the photoproduct state (i.e., state B; eqn (S9)†). Therefore, the absolute QY, which equals to k2/(k1 + k2), was estimated to be 0.6 by using [Ru(bpy)3]2+ as a reference (see Section 4 of ESI† for more details), which is lower than that of the MSA:FAD complex in the same protein (ca. 0.8; Table S2†).41 As (k1 + k2)−1 = 600 fs, the values of k1 and k2 can be further estimated to be ca. (1.5 ps)−1 and (1.0 ps)−1, respectively.
As shown in Fig. S4b† and 4B, the TA results obtained with the MSeA:FAD complex in MTOXWT are similar to those in MSOXWT, suggesting that photoexcitation induces a similar photoswitching reaction in this system. Specifically, as observed in MSOXWT, the early-time ESA signal is characterized by an intense absorption band at ca. 400 nm (Fig. S4b†), which decays in ca. 800 fs to yield a spectrum resembling that of the FAD minus MSeA:FAD difference spectrum (Fig. 4B). The timescale of the fast decay component is comparable to that in MSOXWT (ca. 600 fs), while the slow component is significantly faster than that in MSOXWT. Specifically, at least three decay components, with time constants of 0.8, 37, and 690 ps, respectively, are required to satisfactorily fit the TA kinetics of MSeA:FAD in MTOXWT at 570 nm (Fig. 4C), with the 0.8 ps component being dominant (ca. 77%, Table S2†). This indicates that, in comparison to MSOXWT, MTOXWT provides a less favorable environment for the MSeA:FAD CT complex to dissociate upon photoexcitation. In addition, the more complex decay kinetics of the TA signals likely manifest conformational heterogeneity of MSeA in the active site of MTOXWT, which will be further discussed below.
As shown in Fig. 5A and B, as previously observed for the MSA:FAD complex in MSOXWT,41 for the MSeA:FAD complex in MSOXWT, although the rate (i.e., k1 + k2) of the fast decay component does not show appreciable temperature dependence (Fig. S5†), the decay rate (i.e., k3) of the photoproduct state markedly decreases with increasing temperature. The latter indicates that recovery of the ground-state CT complex from the dissociated state (i.e., state B in Fig. 4D) encounters an energetic barrier. This observation, in conjunction with structural studies of various MXA:MSOXWT complexes,42,44 suggests that the photoswitching process of the MXA:FAD complex involves a conformational change of the MXA ligand in the protein active site, most likely from Conf 1 to Conf 2, as this conformational transition would lead the X atom to rotate away from the flavin ring (Fig. 1C), consequently breaking up the CT interaction.41 This assessment is supported by the aforementioned spectral calculation (Fig. 3A), which demonstrates that switching of the ligand from Conf 1 to Conf 2 results in the diminishment of the CT band.
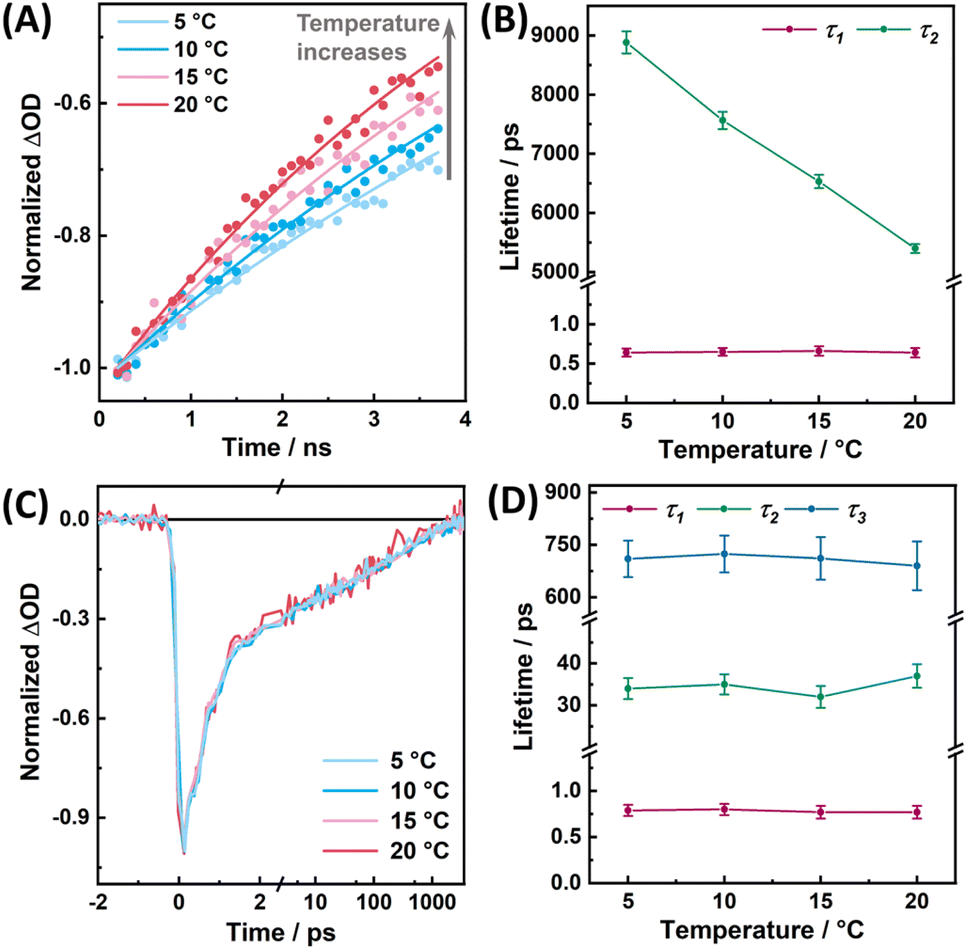 | ||
| Fig. 5 Temperature dependence of the TA results of the MSeA:FAD complex in MSOXWT (A and B) and MTOXWT (C and D), probed at 590 and 570 nm, respectively, with 550 nm excitation. (A and C) TA kinetics measured at different temperatures, as indicated. For MSOXWT, only those in the ps–ns timescales are shown for clarity, and the smooth lines represent the best fits of the data. (B and D) Temperature dependence of the time constants obtained from multiexponential fits of the TA kinetic traces. Errors are ±2σ estimated from multiple measurements. | ||
Moreover, it is noticeable that in both cases the GSB signal peaks at a wavelength that is blue-shifted from the respective CT band observed in the steady-state absorption spectrum of the complex (Fig. 4A and B), suggesting that a positive ESA band is present in this spectral region. This provides further evidence supporting the notion that the photoproduct (state B) corresponds to Conf 2 (Fig. 2A), as the theoretically calculated spectrum of this configuration (Fig. 3A) indicates that it gives rise to a small but noticeable (residual) CT absorption band.
Finally, as shown in Fig. 5C and D, all the time constants obtained from the TA kinetics of the MSeA:FAD in MTOXWT do not exhibit any measurable temperature dependence, suggesting that in this case the back recovery reaction is virtually a barrierless process. For comparison, we further investigated the excited-state dynamics of the CT complex formed by FAD and a rigid aromatic ligand, pyrrole-2-carboxylate (PCA, Fig. S6a†). As illustrated in Fig. S6b,† in addition to the GSB of the CT band, photoexcitation of this complex at 640 nm leads to the formation of an ESA band at <520 nm that appears to well correspond to a positive contribution of FAD˙− absorption,42,49 indicating the CT nature of the excited state, similar to what has been suggested for a CT complex formed by o-aminobenzoate and FAD in D-amino acid oxidase.50 However, the TA signals of this PCA:FAD complex disappear within ca. 1 ps, of which the kinetics can be fit with a monoexponential function (Fig. S6b†), indicating that the CT excited state directly decays back to the ground state by charge recombination on an ultrafast timescale, without the involvement of a photoswitched state, as observed for the MXA:FAD complexes in MSOXWT and MTOXWT. This finding indicates that for a rigid aromatic ligand like PCA, photoexcitation can only result in a redistribution of electron density and that a certain degree of conformational flexibility is required for the photoswitching process to occur.
Regulating the photocycle dynamics via protein engineering
As a close homologue of MSOX, the active-site structure of MTOXWT highly resembles that of MSOXWT.51 Therefore, it is intriguing that the photocycle dynamics of the MSeA:FAD complex in MTOXWT significantly differs from that in MSOXWT. However, a close inspection of their active-site environment indicates that a methionine residue, Met245, in MSOXWT, which can interact with the MSeA ligand in Conf 2 (Fig. S2d†), is replaced by a threonine (Thr239) in MTOXWT, which is located farther away from the ligand (Fig. 1C). Based on this observation, we hypothesized that the residue at position 245 of MSOX or at position 239 of MTOX plays a key role in determining the kinetic properties of these photoswitching systems, particularly the back recovery rate. To verify this hypothesis, we further studied the photoinduced conformational dynamics of the MSeA:FAD complex in the M245T and M245K variants of MSOX and in the T239M and T239K variants of MTOX (hereafter referred to as MSOXM245T, MSOXM245K MTOXT239M, and MTOXT239K, respectively). In MSOXM245T and MTOXT239M, we exchanged the two residues in question in the wild-type proteins with each other. In MSOXM245K and MTOXT239K, we introduced the lysine mutations for the following reasons: (1) methionine and lysine share a certain degree of structural similarity, and can behave similarly in molecular recognition52 and (2) unlike methionine that is neutral, lysine is positively charged, which may allow a stronger interaction with the carboxyl group of the ligand, hence affecting the binding modes.We found that all these mutant proteins can still bind MSeA to form CT complexes (Fig. S8†), although MTOXT239M and MSOXM245T exhibit a decrease in binding affinity (with Kd values estimated to be larger than 120 mM, compared to the 32.3 and 2.1 mM in MTOXWT and MSOXWT, respectively42,43). As indicated in Fig. S9,† TA measurements further confirm that the aforementioned photoswitching reaction can occur in these protein variants and as shown in Fig. 6, their respective TA kinetics in the range of 560–570 nm can be fit by either a biexponential or a triexponential function. For all the protein variants, the timescales of the initial excited-state decay component fall in the range of 0.6 to 0.9 ps, whereas the kinetics of the slow decay components differ significantly. Specifically, we found that the TA kinetics of MSOXM245T and MTOXT239M can be reasonably described by a biexponential function (Table S2†), with the slow kinetic component being temperature dependent (Fig. S10 and S11†). In comparison, in MTOXT239M, the rate of the back recovery reaction (ca. (2.8 ns)−1) is considerably slower than that in MTOXWT. This indicates that replacing Thr239 with methionine can indeed render MTOX ‘MSOX-like’ in terms of the overall photocycle kinetics. For the MSOXM245T variant, although the photoproduct has a shorter lifetime (τ2 = 4.0 ns) compared to that in MSOXWT (τ2 = 5.5 ns), it is still much more long-lived than that in MTOXWT. This indicates that the corresponding methionine-to-threonine mutation does not significantly alter the lifetime of the photoswitched state. By contrast, the MSeA:FAD complex in MSOXM245K, as well as in MTOXT239K, displays kinetic behaviors that resemble those of MTOXWT. Here, three decay components are required to obtain a satisfactory fit of the TA kinetics (Fig. 6A and Table S2†). Similar to MTOXWT, these kinetics were also found to be virtually temperature independent (Fig. S12†), although the slower decay components have a longer time constant in comparison (Table S2†).
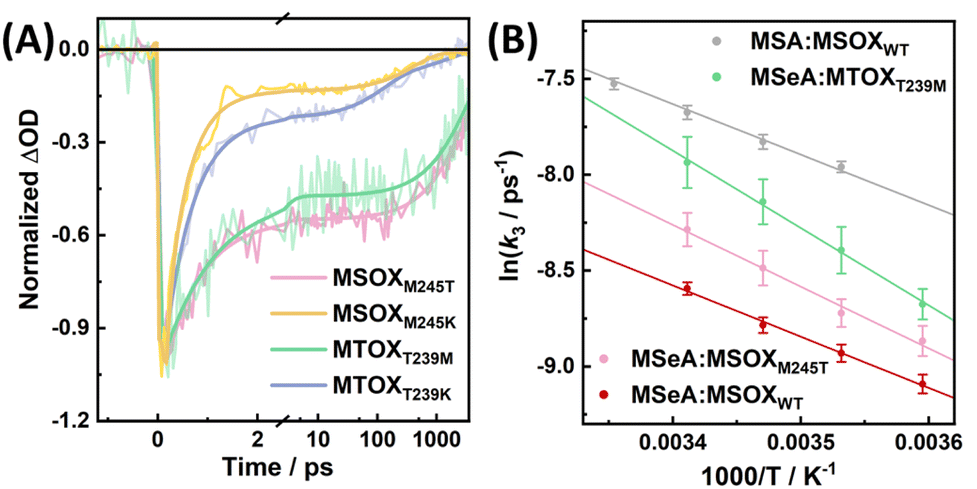 | ||
| Fig. 6 (A) TA kinetics of the MSeA:FAD complex in the MSOX and MTOX variants, as indicated, in the GSB region. (B) Arrhenius plots of the recovery rate constants (k3) obtained for different ligands and proteins, as indicated. Error bars are estimated from multiple measurements. | ||
As shown in Fig. 6B, for the MSeA:FAD complexes in MSOXWT, MSOXM245T, and MTOXT239M, the logarithm of k3 (i.e., (τ2)−1) exhibits a linear dependence on the reciprocal of the absolute temperature, allowing us to determine the activation energy (Ea) of the back recovery reaction in each case. As shown in Table S2,† the Ea values thus obtained indicate that the energetic barrier for this reaction is dependent on the protein, but not on the ligand, whereas the intrinsic electronic properties of the MXA:FAD complexes appear to mainly affect the pre-exponential factor of the reaction. This highlights the importance of the protein environment in controlling the underlying back-recovery dynamics.
Ligand conformational distribution
To gain further microscopic insight into the effect of protein active sites on ligand conformations, we performed all-atom MD simulations on various protein systems complexed with an MSA ligand (Fig. S13†). MSA was used as a representative ligand instead of MSeA, as classical additive force fields are usually unable to model molecules that contain heavy atoms such as Se (and Te) accurately.53 Specifically, we examined the distribution of the minimal distance between the S3 atom of MSA and any non-hydrogen atom of the flavin ring (dS3–flavin). As shown in Fig. 7A, in MTOXWT and MSOXM245K, the ligand was found to sample a broad conformational distribution, with two dominant conformations of comparable populations (dS3–flavin of ca. 3.6 and 4.8 Å in MTOXWT, and of ca. 3.6 and 5.0 Å in MSOXM245K, respectively), which qualitatively correspond to Conf 1 and Conf 2. In MSOXWT, MSOXM245T and MTOXT239M, however, the ligand predominantly samples a narrow conformational distribution with dS3–flavin ≈ 3.6 Å, indicating that the active sites of these proteins enable a tighter packing (i.e., a stronger interaction) between the ligand and FAD. Although the ligand in MTOXT239K also appears to sample only one conformational ensemble, the conformational distribution is nevertheless broader than those in MSOXWT, MSOXM245T and MTOXT239M. Taken together, these findings indicate a higher degree of active-site heterogeneity in MTOXWT, MSOXM245K and MTOXT239K.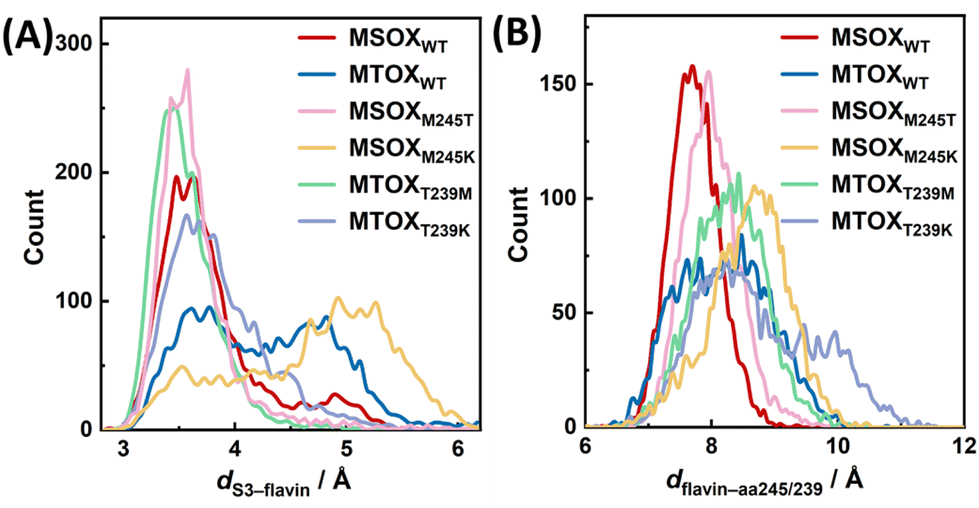 | ||
| Fig. 7 Distribution plots of the distance between the MSA ligand and the flavin ring (A) and that between the residue at position 245 (239) of the MSOX (MTOX) variants and the flavin ring (B), obtained from a 300 ns MD production run of different protein systems, as indicated. | ||
As shown in Fig. 7B, the distribution plots of the distance between the flavin ring and the amino acid residue at position 245 of MSOX or position 239 of MTOX (dflavin–aa245/239) obtained from these systems also support this notion. These results demonstrate that, although methionine, threonine and lysine differ in bulkiness, their effects on ligand conformations cannot simply be explained by steric hindrance; their interactions with other surrounding residues, as well as the overall scaffold of the protein, also need to be taken into consideration. The differences in ligand conformational distributions also correspond well with the steady-state absorption spectra of the protein-bound MSeA:FAD complexes, where in MSOXWT, MSOXM245T, and MTOXT239M the complexes exhibit a more distinct, red-shifted CT band compared to those in MTOXWT, MSOXM245K and MTOXT239K (Fig. 1B and S8b†). Moreover, these MD simulation results indicate that a more heterogeneous active-site configuration is correlated with a faster and near-barrierless back recovery process following the photoswitching reaction, as observed for MTOXWT, MSOXM245K and MTOXT239K.
As classical MD simulations lack the ability to accurately describe noncovalent interactions of non-electrostatic origin, to further evaluate the ground-state and excited-state potential energy surfaces (PESs), we performed QM/MM potential energy scans for the MSeA:FAD complex in the active sites of MSOXWT and MTOXWT, with the flavin ring of FAD, the MSeA ligand, as well as the side chain of Met245 (MSOXWT) or Thr239 (MTOXWT) included in the QM treatment. For these relaxed scans, the reaction coordinate was set to be the distance between the Se3 atom of the ligand and the C4 atom of the flavin ring (dSe3–C4). As shown in Fig. 8 and S14,† in both MSOXWT and MTOXWT, two energy minima that are separated by an energy barrier can be identified when MSeA:FAD is in its ground electronic state (S0). More specifically, in MSOXWT, the minimum corresponding to Conf 1 (I, dSe3–C4 = 3.5 Å) is considerably more stable than the other minimum where the S atom is located farther away from the flavin (III, dSe3–C4 = 4.1 Å). This suggests that at room temperature the MSeA ligand in MSOXWT is likely to assume the Conf 1 configuration (Fig. 8A). As the energy required for the conformer III to reach the transition state II is relatively small, it can be assumed that the conversion from III to I can easily occur. On the other hand, in MTOXWT, the two minima, VII and IX, with dSe3–C4 = 3.3 and 4.9 Å, respectively, are distinctly separated from each other by a sizable energy barrier (Fig. 8B). These findings are in general agreement with the MD simulation results (Fig. 7A).
 | ||
| Fig. 8 QM/MM potential energy scans of the MSeA:FAD complex in MSOXWT (A) and MTOXWT (B) in the electronic ground state (S0) and the lowest excited state (S1), with the distances between the C4 atom of FAD and the Se3 atom of MSeA (indicated in Fig. 2) as the reaction coordinate. Energy values are given relative to that of the global minimum of S0. Molecular geometries of the indicated minima and transition states are given in Fig. S14.† | ||
As indicated in Fig. 8, the excited-state (S1) PES of the MSeA:FAD complex in both proteins is different from the respective ground-state PES. For MSOXWT, two distinctly separated minima, IV and VI were identified along the scanned S1 PES, with dSe3–C4 = 3.4 and 4.5 Å, respectively (Fig. 8A and S14†), whereas for MTOXWT, essentially only one minimum (X), was identified, with dSe3–C4 = 3.6 Å (Fig. 8B). Although we did not directly identify the geometry points where the S0 and S1 PESs intersect in both cases, possibly due to the involvement of other reaction coordinates, as well as the presence of non-negligible effects from other surrounding residues that were not included in the QM region, the profiles of these energy surfaces can nevertheless provide a qualitative explanation for the differences between the decay pathways of the excited state of MSeA:FAD in MSOXWT and MTOXWT (Fig. 4C). Specifically, the result demonstrates that the S1 and S0 minima (X and VII, respectively) have similar molecular geometries (Fig. S14†), and the S1 PES of MSeA:FAD in MTOXWT is likely to intersect with the S0 PES at a geometry point located before the transition state (VIII) of the S0 PES (Fig. 8B). Therefore, excited-state decays of MSeA:FAD in MTOXWT are likely to be dominated by an ultrafast internal conversion process (X → VII). Moreover, due to the relatively shallow PESs, MSeA:FAD may adopt multiple conformations that are not well separated by energy barriers (cf.Fig. 7A, where the ligand assumes a broad conformational distribution in MTOXWT). As a result, a part of the excited-state populations can undergo additional deactivation processes involving changes in nuclear configurations, leading to relatively longer-lived photoswitched states, which may exhibit different back recovery dynamics depending on initial configurations. Taken together, for MSeA:FAD in MTOXWT, upon photoexcitation, full relaxation to the S0 minimum (i.e., reformation of the ground-state CT complex) does not require overcoming any energy barrier. These results support the ultrafast, multiphasic, and essentially temperature-independent TA kinetics as experimentally observed (Fig. 4C and Table S2†).
In MSOXWT, on the other hand, it is more reasonable to assume that upon photoexcitation, the system can first relax toward the S1 minimum (VI) that has a geometry resembling that of Conf 2 (Fig. S14†), then decays to the S0 state via a conical intersection, and eventually fully relaxes to the S0 minimum (I) by overcoming an energy barrier. Therefore, the full photochemical pathways of MSeA:FAD in MSOXWT are likely to comprise the following processes: (1) internal conversion (IV → I), (2) isomerization from Conf 1 to Conf 2 on the S1 PES (IV → VI), (3) excited-state deactivation via conical intersection (VI → III), and (4) back isomerization from Conf 2 to Conf 1 on the S0 PES (III → I). Taken together, this mechanism indicates that the photoswitching yield is governed by processes (1) and (2). Additionally, the present result also reveals a barrier separating the two energy minima on the S1 PES of MSeA:FAD in MSOXWT, seemingly at odds with the barrierless decay of the initial excited state observed in the TA measurements. However, it is possible that photoexcitation is able to directly promote the system to a point close to the transition state (V) on the S1 PES, allowing the geometry to relax to the more stable S1 minimum (VI) in a barrierless fashion.
Taken together, these results confirm that the residue at position 245 of MSOX or at position 239 of MTOX plays a key role in positioning an MXA ligand in the protein active site, as well as in regulating its conformational energy landscape. As the molecular structures of the MXA ligands resemble that of sarcosine, a substrate of MSOX and MTOX, it is reasonable to infer that the residue at position 245 of MSOX or at position 239 of MTOX can also affect the catalytic properties of these enzymes. This is indeed the case for MTOXT239M, which exhibits a 20-fold increase in the catalytic activity toward sarcosine with respect to MTOXWT, but is still seven times lower than that of MSOXWT.51 Interestingly, this appears to corroborate our findings that MTOXT239M exhibits an elongated lifetime of the photoswitched state compared to MTOXWT, which, however, is still shorter than that of MSOXWT (vide supra).
Conclusions
In this work, we combined comprehensive experimental and computational studies to assess the dynamics and mechanism of a recently demonstrated photoswitching reaction that involves a protein-bound complex formed by a FAD cofactor and an MXA ligand (where MXA represents CH3XCH2CO2− with X = S, Se, or Te). In particular, we investigated how the protein environment affects the interactions and photophysics of this type of complex and found that:(1) MXA and FAD form a CT complex in the protein binding site, giving rise to an intense CT absorption band in the red-to-NIR spectral region, whose maximum wavelength depends on the identity of the X atom.
(2) Photoexcitation of an MXA:FAD complex through its CT band initiates a photocycle that is composed of a photoswitching process, leading to the formation of a quasi-dissociated intermediate state (or photoswitched state) wherein the ligand most likely adopts a different conformation, and a back recovery process, reforming the initial complexed state.
(3) For the same ligand, the kinetics of the photocycle are protein-dependent. For instance, in MSOXWT, the back recovery process is thermally activated and occurs in a few nanoseconds near room temperature, whereas in MTOXWT, a structural homologue of MSOX, the kinetics of the back recovery is considerably faster (in tens to hundreds of picoseconds), and is virtually temperature-independent.
(4) For the same protein, the switching kinetics can be regulated through site-specific mutations. For example, in MTOX a T239M mutation elongates the lifetime of the photoswitched state, from hundreds of picoseconds in MTOXWT to a few nanoseconds in MTOXT239M. On the other hand, the reverse regulation, i.e., accelerating the back recovery process, can be achieved in MSOX through a M245K mutation.
(5) Modifications of the active-site environment allow the proteins to stabilize different conformers of the ligand by altering the conformational energy landscape of the MXA:FAD complex in the electronic ground and excited states, which consequently affects the energetics and dynamics of the switching processes.
In summary, our study demonstrated that the MXA:FAD complexes in MSOX and MTOX possess photophysical properties that are desirable for reversible molecular photoswitches, as their absorption spectra and photoswitching kinetics can be tuned by using different ligands or by modifying the ligand binding site via protein engineering. Moreover, the CT complexes are intrinsically nonfluorescent, but photoexcitation of these complexes generates uncomplexed FAD that can act as a fluorescent photoproduct, provided that there are no other quenchers, e.g., tyrosine or tryptophan residues, positioned close to the flavin ring in the protein.33,54 Therefore, one potential application of this type of photoswitching reaction is to engineer red-absorbing flavin-dependent fluorescence photoswitches, wherein the fluorescence can be selectively turned on by excitation of the corresponding CT complexes.
Data availability
The data supporting this study are available within the article and ESI.† The raw data are available from the corresponding authors upon reasonable request.Author contributions
BZ: conceptualization, methodology, investigation, data analysis, visualization, writing – original draft preparation, writing – reviewing and editing; GR: methodology, investigation, writing – reviewing and editing; WZ: conceptualization, investigation, writing – reviewing and editing; GF: conceptualization, investigation, data analysis, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Beijing Natural Science Foundation (IS23031 to FG and 2244070 to BZ), the National Natural Science Foundation of China (U2230203 to WZ and 22403003 to BZ), the China Postdoctoral Science Foundation (2024M750080 to BZ), and the Postdoctoral Fellowship Program of the China Postdoctoral Science Foundation (GZB20240067 to GR). We are grateful to Dr Marten Vos of École Polytechnique for stimulating discussion, and to Dr Ursula Liebl of École Polytechnique and Danqi Li of Peking University for their advice on protein production.Notes and references
- M. A. Van Der Horst and K. J. Hellingwerf, Acc. Chem. Res., 2004, 37, 13–20 CrossRef.
- R. C. Hardie, Trends Neurosci., 1986, 9, 419–423 CrossRef.
- M. Iino, Plant, Cell Environ., 1990, 13, 633–650 CrossRef.
- G. Jékely, Philos. Trans. R. Soc., B, 2009, 364, 2795–2808 CrossRef PubMed.
- F. Valverde, A. Mouradov, W. Soppe, D. Ravenscroft, A. Samach and G. Coupland, Science, 2004, 303, 1003–1006 CrossRef.
- K. N. Paul, T. B. Saafir and G. Tosini, Rev. Endocr. Metab. Disord., 2009, 10, 271–278 CrossRef PubMed.
- F. Gai, K. C. Hasson, J. C. McDonald and P. A. Anfinrud, Science, 1998, 279, 1886–1891 CrossRef CAS.
- S. O. Smith, Annu. Rev. Biophys., 2010, 39, 309–328 CrossRef CAS PubMed.
- K. Deisseroth and P. Hegemann, Science, 2017, 357, eaan5544 CrossRef.
- N. C. Rockwell, Y. S. Su and J. C. Lagarias, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS PubMed.
- M. Ikeuchi and T. Ishizuka, Photochem. Photobiol. Sci., 2008, 7, 1159–1167 CrossRef CAS.
- T. Grotjohann, I. Testa, M. Leutenegger, H. Bock, N. T. Urban, F. Lavoie-Cardinal, K. I. Willig, C. Eggeling, S. Jakobs and S. W. Hell, Nature, 2011, 478, 204–208 CrossRef CAS.
- J. Woodhouse, G. Nass Kovacs, N. Coquelle, L. M. Uriarte, V. Adam, T. R. M. Barends, M. Byrdin, E. de la Mora, R. Bruce Doak, M. Feliks, M. Field, F. Fieschi, V. Guillon, S. Jakobs, Y. Joti, P. Macheboeuf, K. Motomura, K. Nass, S. Owada, C. M. Roome, C. Ruckebusch, G. Schirò, R. L. Shoeman, M. Thepaut, T. Togashi, K. Tono, M. Yabashi, M. Cammarata, L. Foucar, D. Bourgeois, M. Sliwa, J. P. Colletier, I. Schlichting and M. Weik, Nat. Commun., 2020, 11, 1–11 CrossRef.
- M. Jost, J. Fernández-Zapata, M. C. Polanco, J. M. Ortiz-Guerrero, P. Y. T. Chen, G. Kang, S. Padmanabhan, M. Elías-Arnanz and C. L. Drennan, Nature, 2015, 526, 536–541 CrossRef CAS.
- A. R. Jones, Photochem. Photobiol. Sci., 2017, 16, 820–834 CrossRef CAS.
- A. Losi and W. Gärtner, Annu. Rev. Plant Biol., 2012, 63, 49–72 CrossRef CAS PubMed.
- K. S. Conrad, C. C. Manahan and B. R. Crane, Nat. Chem. Biol., 2014, 10, 801–809 CrossRef CAS.
- S. Y. Park and J. R. H. Tame, Biophys. Rev., 2017, 9, 169–176 CrossRef CAS PubMed.
- A. Lukacs, P. J. Tonge and S. R. Meech, Acc. Chem. Res., 2022, 55, 402–414 CrossRef CAS PubMed.
- Y. Zhou, S. Tang, Z. Chen, Z. Zhou, J. Huang, X. W. Kang, S. Zou, B. Wang, T. Zhang, B. Ding and D. Zhong, Nat. Commun., 2024, 15, 1–11 CAS.
- T. E. Swartz, S. B. Corchnoy, J. M. Christie, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, J. Biol. Chem., 2001, 276, 36493–36500 CrossRef CAS PubMed.
- A. Losi and W. Gärtner, Photochem. Photobiol., 2017, 93, 141–158 CrossRef CAS PubMed.
- J. N. Iuliano, J. T. Collado, A. A. Gil, P. T. Ravindran, A. Lukacs, S. Shin, H. A. Woroniecka, K. Adamczyk, J. M. Aramini, U. R. Edupuganti, C. R. Hall, G. M. Greetham, I. V. Sazanovich, I. P. Clark, T. Daryaee, J. E. Toettcher, J. B. French, K. H. Gardner, C. L. Simmerling, S. R. Meech and P. J. Tonge, ACS Chem. Biol., 2020, 15, 2752–2765 CrossRef CAS PubMed.
- B. Liu, H. Liu, D. Zhong and C. Lin, Curr. Opin. Plant Biol., 2010, 13, 578–586 CrossRef CAS PubMed.
- A. Czarna, A. Berndt, H. R. Singh, A. Grudziecki, A. G. Ladurner, G. Timinszky, A. Kramer and E. Wolf, Cell, 2013, 153, 1394–1405 CrossRef PubMed.
- Q. Wang and C. Lin, Annu. Rev. Plant Biol., 2020, 71, 103–129 CrossRef PubMed.
- T. J. Chozinski, L. A. Gagnon, J. C. Vaughan, E. M. Puchner, B. Huang, H. E. Gaub and W. Just, FEBS Lett., 2014, 588, 3603–3612 CrossRef PubMed.
- E. S. Boyden, F. Zhang, E. Bamberg, G. Nagel and K. Deisseroth, Nat. Neurosci., 2005, 8, 1263–1268 CrossRef PubMed.
- A. A. M. Fischer, M. M. Kramer, G. Radziwill and W. Weber, Curr. Opin. Chem. Biol., 2022, 70, 102196 CrossRef PubMed.
- L. Piccinini, S. Iacopino, S. Cazzaniga, M. Ballottari, B. Giuntoli and F. Licausi, Plant Physiol., 2022, 189, 1153–1168 CrossRef PubMed.
- K. Lehtinen, M. S. Nokia and H. Takala, Front. Cell. Neurosci., 2022, 15, 778900 CrossRef PubMed.
- A. Taylor, D. J. Heyes and N. S. Scrutton, Curr. Opin. Struct. Biol., 2022, 77, 102491 CrossRef CAS.
- B. Zhuang, U. Liebl and M. H. Vos, J. Phys. Chem. B, 2022, 126, 3199–3207 CrossRef CAS.
- K. Brettel and M. Byrdin, Curr. Opin. Struct. Biol., 2010, 20, 693–701 CrossRef CAS PubMed.
- D. Zhong, Annu. Rev. Phys. Chem., 2015, 66, 691–715 CrossRef CAS PubMed.
- J. Yamamoto, P. Plaza and K. Brettel, Photochem. Photobiol., 2017, 93, 51–66 CrossRef CAS PubMed.
- D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté, D. Nurizzo, P. Müller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903–907 CrossRef PubMed.
- D. Sorigué, K. Hadjidemetriou, S. Blangy, G. Gotthard, A. Bonvalet, N. Coquelle, P. Samire, A. Aleksandrov, L. Antonucci, A. Benachir, S. Boutet, M. Byrdin, M. Cammarata, S. Carbajo, S. Cuiné, R. B. Doak, L. Foucar, A. Gorel, M. Grünbein, E. Hartmann, R. Hienerwadel, M. Hilpert, M. Kloos, T. J. Lane, B. Légeret, P. Legrand, Y. Li-Beisson, S. L. Y. Moulin, D. Nurizzo, G. Peltier, G. Schirò, R. L. Shoeman, M. Sliwa, X. Solinas, B. Zhuang, T. R. M. Barends, J.-P. Colletier, M. Joffre, A. Royant, C. Berthomieu, M. Weik, T. Domratcheva, K. Brettel, M. H. Vos, I. Schlichting, P. Arnoux, P. Müller and F. Beisson, Science, 2021, 372, eabd5687 CrossRef PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 CrossRef CAS PubMed.
- H. Fu, J. Cao, T. Qiao, Y. Qi, S. J. Charnock, S. Garfinkle and T. K. Hyster, Nature, 2022, 610, 302–307 CrossRef CAS PubMed.
- B. Zhuang and M. H. Vos, J. Am. Chem. Soc., 2022, 144, 11569–11573 CrossRef CAS PubMed.
- M. A. Wagner, P. Trickey, Z. W. Che, F. S. Mathews and M. S. Jorns, Biochemistry, 2000, 39, 8813–8824 CrossRef CAS PubMed.
- P. Khanna and M. S. Jorns, Biochemistry, 2001, 40, 1441–1450 CrossRef CAS PubMed.
- P. Trickey, M. A. Wagner, M. S. Jorns and F. S. Mathews, Structure, 1999, 7, 331–345 CrossRef CAS.
- Y. Zheng, M. A. Wagner, M. S. Jorns and P. R. Carey, J. Raman Spectrosc., 2001, 32, 79–92 CrossRef CAS.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- Y. Shao, Y. Mei, D. Sundholm and V. R. I. Kaila, J. Chem. Theory Comput., 2020, 16, 587–600 CrossRef PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- B. Zhuang, R. Ramodiharilafy, U. Liebl, A. Aleksandrov and M. H. Vos, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2118924119 CrossRef CAS PubMed.
- S. Taniguchi, H. Chosrowjan, H. Tamaoki, Y. Nishina, A. Nueangaudom and F. Tanaka, J. Photochem. Photobiol., A, 2021, 420, 113448 CrossRef CAS.
- A. Ilari, A. Bonamore, S. Franceschini, A. Fiorillo, A. Boffi and G. Colotti, Proteins:Struct., Funct., Bioinf., 2008, 71, 2065–2075 CrossRef CAS PubMed.
- F. Forneris, C. Binda, A. Adamo, E. Battaglioli and A. Mattevi, J. Biol. Chem., 2007, 282, 20070–20074 CrossRef CAS PubMed.
- J. A. Lemkul, J. Huang, B. Roux and A. D. Mackerell, Chem. Rev., 2016, 116, 4983–5013 CrossRef CAS PubMed.
- A. Mukherjee and C. M. Schroeder, Curr. Opin. Biotechnol., 2015, 31, 16–23 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methods; electronic properties of MXA:FAD in MSOXWT; TA data of CT complexes in MSOXWT and MTOXWT; determination of the rate constants for the MSeA:FAD complex in MSOXWT; steady- and excited-state properties of MXA:FAD in other protein variants; additional results of MD simulations and QM/MM potential energy scans. See DOI: https://doi.org/10.1039/d4sc08614g |
| This journal is © The Royal Society of Chemistry 2025 |