
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular charge transfer assisted multi-resonance thermally activated delayed fluorescence emitters for high-performance solution-processed narrowband OLEDs†
Zhi
Yang
a,
Shengyu
Li
a,
Lei
Hua
b,
Shian
Ying
 a,
Yuchao
Liu
*a,
Zhongjie
Ren
*c and
Shouke
Yan
*ac
a,
Yuchao
Liu
*a,
Zhongjie
Ren
*c and
Shouke
Yan
*ac
aDepartment Key Laboratory of Rubber-Plastics, Ministry of Education/Shandong Provincial Key Laboratory of Rubber-Plastics, School of Polymer Science and Engineering, Qingdao University of Science & Technology, Qingdao 266042, P. R. China. E-mail: liuyc@qust.edu.cn
bSchool of Materials Science & Engineering, Changzhou University, Changzhou 213164, P. R. China
cState Key Laboratory of Chemical Resource Engineering, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 29th January 2025
Abstract
Multi-resonance thermally activated delayed fluorescence (MR-TADF) emitters have been actively employed in high-resolution solution-processed organic light emitting diodes (OLEDs) due to their excellent color purity. Nonetheless, they are always confronted with intrinsic slow spin flip of triplet excitons, impeding the electroluminescence properties, especially in non-sensitized OLEDs. Herein, we constructed intramolecular charge transfer (ICT) assisted MR-TADF emitters by grafting donor–acceptor-type moieties with a meta- or para-substitution as a pendant on an organoboron multi-resonance core. The newly designed MR-TADF emitters not only maintain short range charge transfer characteristics in emissive states without sacrificing color purity but the accelerated spin flips facilitated by the ICT process at a high-lying state are also confirmed by ultrafast spectroscopy and theoretical calculation, achieving over a 10-fold increase in the reverse intersystem crossing rate compared with unsubstituted counterpart emitters. In sensitizer-free solution-processed OLEDs, a cutting-edge external quantum efficiency of 27.8% can be achieved together with reduced efficiency roll-offs and an attractive full width at half maximum of 29 nm, representing a breakthrough in efficiency for solution-processed MR-TADF based narrowband OLEDs.
Introduction
Thermally activated delayed fluorescence (TADF) molecules have attracted increasing attention for potentially fabricating high-quality organic light emitting diode (OLED) displays.1–5 The competitive advantage of TADF-based OLEDs is that theoretically 100% internal quantum efficiency can be achieved via a reverse intersystem crossing process (RISC) from triplet to singlet excited states for triplet excitons.6–8 In terms of molecular architecture, a twisted donor–acceptor (D–A) configuration should be thoughtfully constructed to induce the spatial separation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), thus attaining enough small energy splitting between triplet and singlet excited states (ΔEST) for fast spin flip of triplet excitons. Nevertheless, D–A-type TADF molecules always suffer from broad emission spectra with the full width at half maximum (FWHM) exceeding 60 nm due to the strong intramolecular charge transfer (ICT) effect, together with structural relaxation of the excited states.9–12 Consequently, the inferior color purity of conventional TADF materials will greatly limit their suitability for high-resolution OLED displays.13To mitigate this limitation, Hatakeyama and colleagues initially proposed an alternative approach to constructing TADF materials based on the multi-resonance (MR) effect.14 In MR-TADF emitters, the complementary resonance effects of boron and nitrogen/oxygen atoms enable alternate localization of the HOMO and LUMO at different positions on the fused aromatic skeleton. This kind of frontier molecular orbital (FMO) distribution can therefore minimize vibronic coupling and structural relaxation of the excited states, eventually resulting in high photoluminescence quantum yield (PLQY) and practicable color purity.15–25 Nonetheless, MR emitters are always confronted with intrinsic slow RISC rate constants (kRISC), which can be ascribed to the inherently tiny spin–orbit coupling (SOC) matrix element between singlet and triplet states and subsequently feeble spin flip of triplet excitons according to El-Sayed's rule.26,27 Thus the kRISC values of the majority of MR-TADF emitters, especially for 2,5,15,18-tetrakis(1,1-dimethylethyl)-indolo[3,2,1-de]indolo[3′,2′,1′:8,1][1,4]benzazaborino[2,3,4-kl] phenazaborine (BCzBN) based compounds, range from 103 to 104 s−1, leading to significant triplet accumulation and efficiency roll-off, especially at high brightness and impeding their extensive commercialization.
From the perspective of the chemical structure, the incorporation of boron with a vacant p-orbital endows the MR skeleton with electron deficient characteristics. In this case, the MR moiety can serve as an electron acceptor, and some D–A-type MR-TADF emitters can thus be constructed by appending an electron donor onto the boron-containing MR skeleton.28–30 After integrating D–A-type charge transfer tentatively defined as the long-range charge transfer singlet state (1LR-CT) with the intrinsic short-range charge transfer singlet state (1SR-CT) into the MR-TADF emitters, the kRISC value can be greatly boosted through an effective conversion channel of spin flip for triplet excitons.31 Consequently, extensive research has been conducted to construct MR-TADF emitters with hybridized 1LR-CT and 1SR-CT characteristics.32–36 For instance, Wang et al. have formed a novel molecule with twisted D–A and MR structural characteristics by attaching an extra t-butyl carbazole as an auxiliary donor to a MR skeleton (Scheme 1). Correspondingly, the target emitter (p/m-Cz-BCzBN) can achieve an extremely high kRISC of 1.0 × 106 s−1, leading to attractive maximum external quantum efficiency (EQEmax) but inferior efficiency roll-off in vacuum-deposited OLEDs without any sensitizer.34 Because of the extended HOMO distribution and unmanageable electron push–pull effect, 1LR-CT, however, might be highly degenerate with 1SR-CT, or even lie at a lower-energy level than 1SR-CT, particularly in high polarity matrices. Thus, the deteriorative color purities induced by dual emission behavior or a broadened emission bandwidth (FWHM > 40 nm) will emerge in electroluminescence (EL) spectra, substantially impeding their functionalization and further EL performance improvement.37 The emitters that simultaneously feature the MR effect and 1LR-CT character exhibit an enhanced RISC process and suppressed exciton quenching via a higher-order spin-vibronic mechanism; however, there are still two issues that need to be carefully considered: (i) the energy level of 1LR-CT must be moderately higher than that of 1SR-CT to guarantee narrowband emission; (ii) the higher-order long-range triplet excited state (3LR) is supposed to possess a different excited state nature compared to 1LR, ensuring efficient SOC between 1LR and 3LR according to Fermi's golden rule.38–40 To intentionally satisfy the above ideal situations, molecular design strategies to delicately manipulate the spin flip of triplet excitons at high-lying states are always in demand for high-efficiency narrowband MR-TADF emitters.
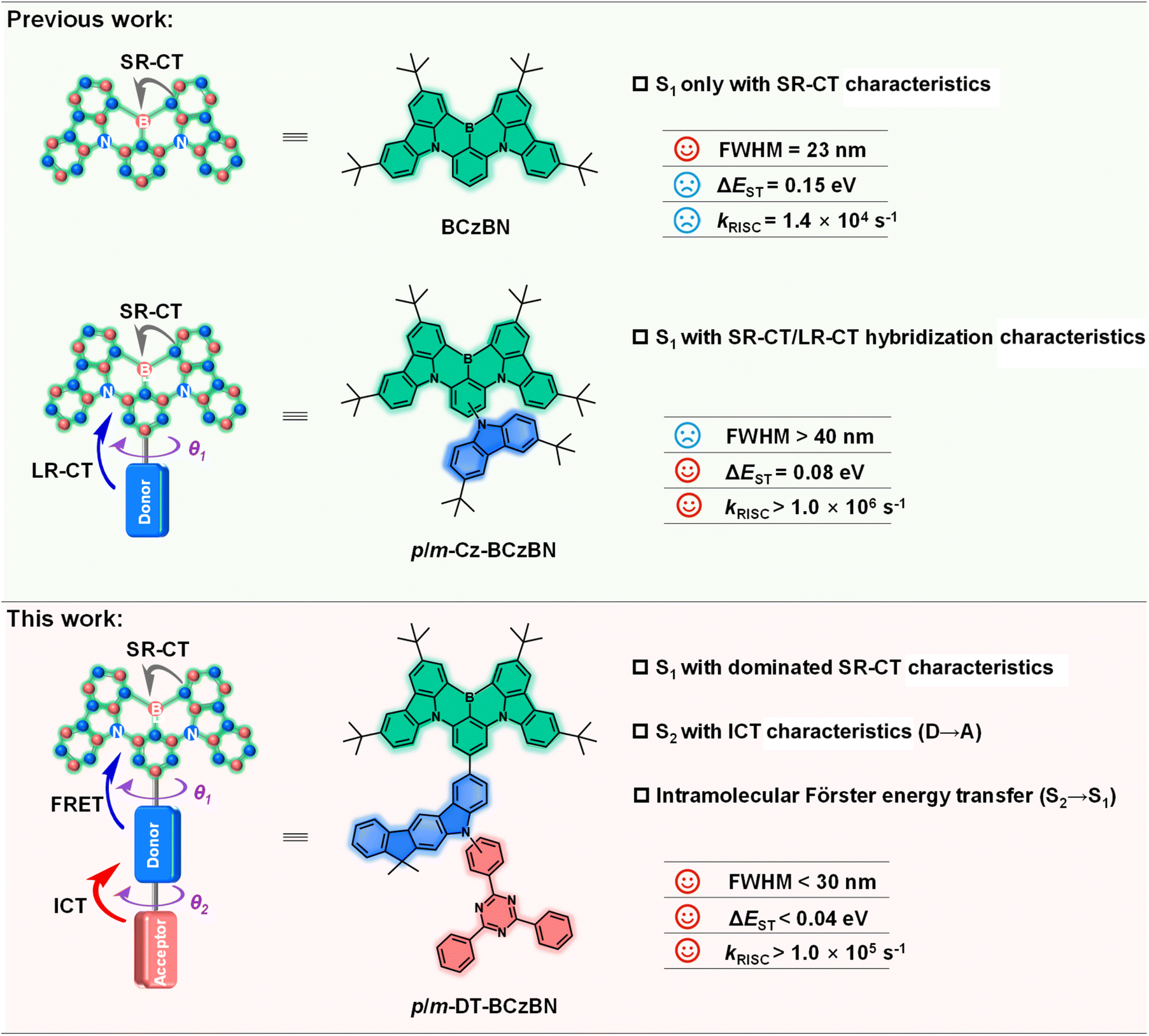 | ||
| Scheme 1 Schematic illustration of a molecular design strategy. Previous work: MR emitters with the structure of the isolated MR skeleton (BCzBN) and MR skeleton-donor moiety (p/m-Cz-BCzBN). This work: the proposed molecular design concept of MR skeleton-donor–acceptor moieties for high-efficiency narrowband MR-TADF emitters (p/m-DT-BCzBN). θ1: the dihedral angle between the MR skeleton and electron-donating units for the LR-CT process; θ2: the dihedral angle between electron-accepting and electron-donating units for the ICT process. | ||
As a proof-of-concept demonstration, we constructed ICT assisted MR-TADF emitters (denoted as m-DT-BCzBN and p-DT-BCzBN) by grafting a D–A-type substituent on the B/N MR skeleton, in which the electron-donating moiety of 5,7-dihydro-7,7-dimethylindeno[2,1-b]carbazole (DMIC) is covalently linked to the electron-withdrawing moiety of 2,4,6-triphenyl-1,3,5-triazine (TRZ) through meta- or para-substitution as a pendant (Scheme 1). Compared with unsubstituted counterpart emitters of BCzBN, two newly designed emitters not only manifest SR-CT dominated characteristics contributed by the MR effect, but also accelerate the exciton conversion and decay processes. As expected, two emitters exhibit a near-unity PLQY, together with dramatically optimized kRISC values of 1.4 × 105 and 1.0 × 105 s−1 for m-DT-BCzBN and p-DT-BCzBN, respectively, which are significantly higher than that of BCzBN (1.4 × 104 s−1). Theoretical calculations and transient absorption spectroscopy (TAS) studies unambiguously validated that the ICT process at high-lying states contributed by the DMIC-TRZ substituent can promote the spin flip of triplet excitons, so as to reduce the exciton aggregation quenching in the emission layer and overcome the efficiency roll-off drawback. Additionally, the merits of ICT assisted MR-TADF emitters are confirmed by relatively excellent performance in sensitizer-free solution-processed OLEDs, providing cutting-edge EQE values of 27.8% and 25.4% for m-DT-BCzBN and p-DT-BCzBN, and EL emission peaks of 492 nm and 494 nm with the same FWHM of 29 nm. More importantly, the efficiency roll-off in solution processed OLEDs is also dramatically suppressed, especially for m-DT-BCzBN. At a high luminance of 500 cd m−2, the EQEs can maintain a relatively high value of 21.5% for m-DT-BCzBN based OLEDs, representing a breakthrough in efficiency for solution-processed MR-TADF based narrowband OLEDs.
Results and discussion
The detailed synthetic routes are depicted in Schemes S1–S8.† The chemical structures of the resultant emitters were characterized by 1H NMR, 13C NMR, and mass spectrometry (Fig. S12–S32†). We conducted thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements (Fig. S1†), which show decomposition temperatures (Td) of 482 and 502 °C with favorable glass transition temperatures (Tg) of 117 and 148 °C for m-DT-BCzBN and p-DT-BCzBN, respectively, verifying good thermal stability that benefits the fabrication and operation of OLED devices. Density functional theory (DFT) calculations on m-DT-BCzBN and p-DT-BCzBN were carried out to investigate the FMO distribution and ground state configuration. For both emitters, the HOMO is principally localized on the nitrogen atoms and the carbon atoms at its ortho/para positions, while LUMO+2 is predominantly localized on the boron atom and the carbon atoms at its ortho/para positions (Fig. S2†). Moreover, the HOMO to LUMO+2 transition makes relatively dominating contributions of 97.4% and 88.3% to S1 → S0 excitations for m-DT-BCzBN and p-DT-BCzBN, respectively, signifying that the SR-CT characteristics can be effectively inherited into these ICT assisted emitters.41 As depicted in Scheme 1 and S3,† the dihedral angles of θ1 for the ICT process of the S2 state are found to be 44.5° and 39.2° for m-DT-BCzBN and p-DT-BCzBN, suggesting slightly distorted arrangements between the flat MR fragment and DMIC unit. The twist angles of θ2 for the ICT process are 58.0° and 51.4° for m-DT-BCzBN and p-DT-BCzBN, maintaining quasi-perpendicular conformations between DMIC and TRZ units, which are favorable for accelerating the spin flip of triplet excitons within high-lying excited states.42,43 In addition, it can be detected that p-DT-BCzBN might suffer from a more severe aggregation induced quenching effect due to a flatter optimized conformation.First, we determined the basic photophysical properties of the two D–A type sensitizing units. As shown in Fig. S4a and S4b,† both D–A type sensitizing molecules exhibit canonical absorption spectra with absorption bands at around 350–400 nm, indicating the ICT process as TADF emitters. The maximum emission peaks are at 446 and 462 nm for DMIC-m-TRZ and DMIC-p-TRZ, implying much higher singlet excited states than that of the BCzBN core. Importantly, DMIC-m-TRZ possesses a relatively smaller ΔEST of only 0.09 eV than DMIC-p-TRZ (0.22 eV), potentially enabling faster spin flip of triplet excitons from T1 to S1 for DMIC-m-TRZ. From Fig. S4c,† it can be detected that the overlaps between the absorption spectrum of BCzBN and the PL spectra of DMIC-m/p-TRZ emitters are favorable for an efficient FRET process from D–A units to the MR emission core. According to the transient PL decay curves of 2 wt% DMIC-m/p-TRZ doped in PhCzBCz (Fig. S4d†), it can be convinced that both D–A type sensitizing molecules can harness triplet excitons, and moreover, DMIC-m-TRZ (delayed lifetimes, τd = 9.9 μs/proportions of delayed component, φDF = 5%) features more efficient upconversion avenues of triplet excitons than DMIC-p-TRZ (τd = 13.4 μs/φDF = 1%), which might be accessible for superior exciton utilization and attractive photophysical properties. Then the photophysical properties of m/p-DT-BCzBN were evaluated in dilute toluene (1 × 10−5 M) using ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra at room temperature (Table 1). As shown in Fig. 1a and b, both emitters exhibit similar absorption profiles. The signals below 420 nm can be assigned to the n–π* and π–π* transitions of the molecular backbone, while the conspicuous signals peaking around 470 nm feature SR-CT transitions within the MR segment. According to the PL spectra of m-DT-BCzBN and p-DT-BCzBN in toluene, both emitters display greenish-blue emission peaking at 495 and 497 nm, respectively. Notably, the FWHM values of m-DT-BCzBN (21 nm) and p-DT-BCzBN (25 nm) are greatly consistent with the unsubstituted counterpart emitter of BCzBN (23 nm) without any spectral shoulder, manifesting that the dominant emissive states of the two emitters are still characterized by SR-CT contributed by the MR effect, and the introduction of the D–A-type substituent cannot produce detrimental vibrational modes. From the onset of low-temperature fluorescence and phosphorescence spectra, the lowest singlet excited state (S1) and triplet excited state (T1) energy levels can be determined to be 2.67/2.64 and 2.60/2.58 eV, providing the ΔEST values of 0.03 and 0.02 eV for m-DT-BCzBN and p-DT-BCzBN, respectively, which are even smaller than that of BCzBN (0.14 eV). Such small ΔEST values are supposed to be favorable for exciton upconversion from T1 to S1, thus showcasing efficient TADF characteristics for two emitters.
| Emitter | λ abs [nm] | λ PL [nm] | ΔESTc [eV] | Φ PL [%] | τ p [ns] | τ d [μs] | ϕ DF [%] | k Sr [107 s−1] | k RISC [105 s−1] |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption peaks of emitters diluted in toluene. b Maximum emission peaks of emitters diluted in toluene. c Energy splitting between S1 and T1 determined from fitting the line of temperature, kRISC. d Photoluminescence quantum yields of the doped films. e The lifetimes of prompt fluorescence components. f The lifetimes of delayed fluorescence components. g The proportions of delayed fluorescence components. h The rate constants of nonradiative decay for singlet excitons determined using the equation kSr = (1 − φDF)/τp. i The rate constants of the reverse intersystem crossing process calculated using the equation kRISC = ∅PL/[τd(1 − φDF)]. | |||||||||
| m-DT-BCzBN | 270, 468 | 495 | 0.03 | 99 | 9.0 | 18.2 | 59 | 4.5 | 1.4 |
| p-DT-BCzBN | 271, 469 | 497 | 0.02 | 98 | 9.8 | 20.5 | 52 | 4.9 | 1.0 |
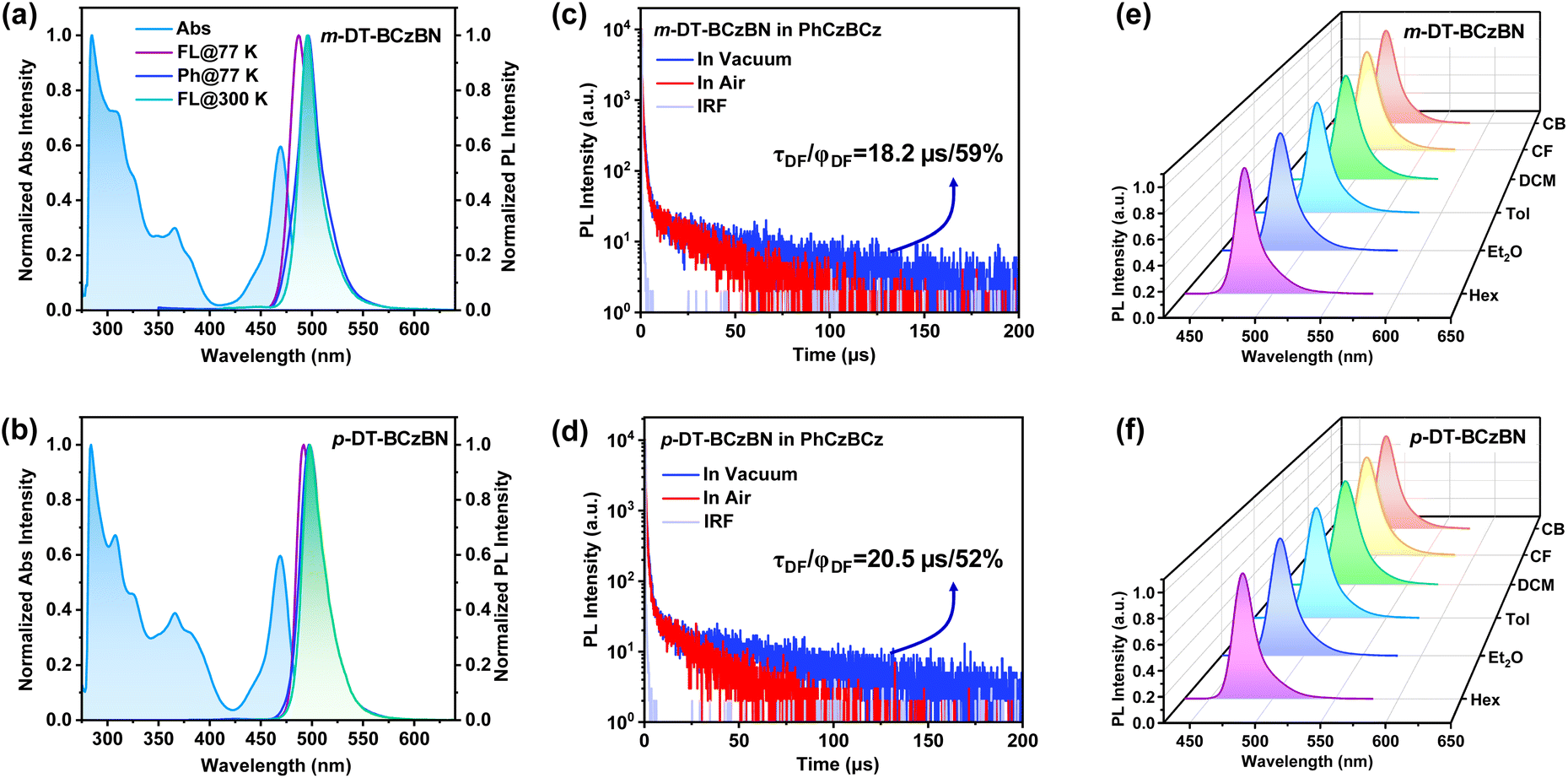 | ||
| Fig. 1 Normalized UV-vis absorption and PL spectra of (a) m-DT-BCzBN and (b) p-DT-BCzBN with an excitation wavelength of 375 nm in 1 × 10−5 M toluene solution. Transient PL decay curves of the emitters diluted in the PhCzBCz matrix for (c) m-DT-BCzBN and (d) p-DT-BCzBN under vacuum/air. Solvatochromism effect on the PL spectra of (e) m-DT-BCzBN and (f) p-DT-BCzBN in 1 × 10−5 M solution. Hex: n-hexane, Et2O: diethyl ether, Tol: toluene, DCM: dichloromethane, CF: chloroform, and CB: chlorobenzene. | ||
To evaluate the delayed fluorescence properties of the two emitters, we recorded transient PL decay spectra of m/p-DT-BCzBN blended in the 9-(2-(9-phenyl-9H-carbazole-3-yl)phenyl)-9H-3,9′-bicarbazole (PhCzBCz) matrix under vacuum and air. As shown in Fig. 1c and d, the PL decay curves of the doped films feature clear biexponential decay, consisting of prompt fluorescence (PF) regimes and subsequently delayed fluorescence (DF) components. The corresponding prompt (τPF) and delayed (τDF) lifetimes are fitted to be 9.0 ns/18.2 μs for m-DT-BCzBN and 9.8 ns/20.5 μs for p-DT-BCzBN under vacuum, respectively. It is noteworthy that both emitters exhibit significantly shortened τDF values compared to that of BCzBN (102 μs), credibly verifying that there exists a concealed pathway to accelerate the triplet exciton decay at higher-lying states for m/p-DT-BCzBN. Comparatively, the doped films in air exhibit much shorter τDF values of 13.9 μs for m-DT-BCzBN and 16.0 μs for p-DT-BCzBN, as shown in Fig. 1c and d, and furthermore, the ϕDF values of 45% and 44% for m-DT-BCzBN and p-DT-BCzBN are slightly lower than those under vacuum (59% and 52% for m-DT-BCzBN and p-DT-BCzBN). Thus, it can be preliminarily concluded that the insusceptible RISC process at higher energy levels is the dominant function in the upconversion of triplet excitons. According to the temperature-dependent transient decay curves (Fig. S5a and S5b†), the delayed components are gradually intensified as the temperatures increase, affording compelling evidence of typical TADF characteristics for two MR-type emitters. We further studied the temperature dependence of kRISC for the doped films by using the Arrhenius equation, as shown in Fig. S5c.† By fitting Arrhenius plots, the experimental activation energy (ERISCa) values of MR-TADF emitters can be evaluated to be 0.30–0.35 eV. The ERISCa values are much higher than the corresponding adiabatic ΔEST, which apparently indicates multiple upconversion channels for triplet excitons from triplet to singlet excited states.40,44
Moreover, two MR-TADF emitters exhibit not only significantly shortened exciton lifetimes, but also satisfactory absolute photoluminescence quantum yields (ΦPLs) of 99% and 98% for m-DT-BCzBN and p-DT-BCzBN, respectively. Combined with the exciton lifetimes and the ΦPL values, the exciton decay kinetic constants can be extrapolated. First, the kRISC values of m-DT-BCzBN and p-DT-BCzBN are determined to be 1.4 × 105 and 1.0 × 105 s−1, which are significantly higher than that of BCzBN (1.4 × 104 s−1), further validating that exciton conversion and decay are relatively accelerated in m/p-DT-BCzBN decorated with a D–A-type substituent. Then, the rate constant of singlet exciton radiative decay (kSr) can also be calculated to be 4.5 × 107 and 4.9 × 107 s−1 for m-DT-BCzBN and p-DT-BCzBN, respectively. Since the kSr values are significantly faster than the rate constants of singlet exciton nonradiative decay (kSnr) of 4.5 × 105 s−1 and 10.0 × 105 s−1 for m-DT-BCzBN and p-DT-BCzBN, nonradiative loss and exciton concentration quenching effects can be suppressed here, which is of paramount importance to achieve efficient triplet exciton dynamics in these two MR-TADF emitters. Besides simplifying device fabrication, the ICT sensitizing molecular design strategy can also enhance the energy transfer and exciton upconversion from D–A units to the emitting moiety. To verify the validity of the ICT sensitizing molecular design strategy, we also performed transient PL spectroscopy using DMIC-m/p-TRZ, BCzBN, and PhCzBCz based ternary blended films under vacuum (Fig. S6†). Compared with the ICT sensitizing system, these blended films exhibit relatively poor decay efficiency of triplet excitons (τd > 70 μs), implying unsatisfactory sensitization processes in ternary blended films.
Furthermore, a solvatochromic test was also performed to analyze the S1 state properties of m-DT-BCzBN and p-DT-BCzBN. As depicted in Fig. 1e and f, the two emitters exhibit insignificant bathochromic shifts (<15 nm) when solvent polarities increase. In particular, the emission maxima of m-DT-BCzBN and p-DT-BCzBN are at 478/476 nm with FWHMs of 19/20 nm in non-polar n-hexane (Hex) and then shift to 490/489 nm with FWHMs of 26/26 nm in relatively higher-polarity chloroform (CF). It should be notable that both MR-TADF emitters display only one emission band even in high-polarity solvents, well consistent with the unsubstituted MR core of BCzBN, while other previously reported MR-TADF emitters decorated with donor moieties always exhibit distinct dual emission in polar solvent due to overly close energy levels or even excited state degeneracy within singlet excited states.31 By fitting the experimental data using the Lippert–Mataga equation (Fig. S7†), the excited-state dipole moments (μe) are estimated to be 5.5 D and 5.2 D for m-DT-BCzBN and p-DT-BCzBN, respectively, resembling that of BCzBN (6.1 D) as well. It can be inferred that the energy level of the ICT state of the D–A-type substituent is much higher than that of the 1SR-CT state, and thus the excitons formed at a high-lying ICT state, presumably S2, are thoroughly transferred to 1SR-CT, which possesses relatively weak CT characters through efficient intramolecular Förster energy transfer (FRET).41,43,45
To elucidate the excited-state nature and upconversion pathway of excitons, excited-state energy level and natural transition orbital (NTO) analyses were theoretically performed through time-dependent DFT (TD-DFT) calculations by adopting the PBE1PBE functional with the 6-311G** basis set. The SOC matrix elements (SOCMEs) of 〈Sn|ĤSO|Tn〉 between the Tn and Sn states were calculated using the ORCA program.46–48 As evidenced by the NTO analysis (Fig. 2), the S1 states of both emitters maintain pronounced SR-CT character, in which the highest occupied natural transition orbital (HONTO) and lowest unoccupied natural transition orbital (LUNTO) are localized on separate atoms induced by nitrogen and boron atoms with extremely limited involvement of the donor units. Thus, the suppressed vibronic coupling and vibrational relaxation jointly achieve exceptionally narrowed emission. Due to the significant electron–hole overlaps within the MR skeleton, both emitters demonstrate favorable f values of 0.510–0.534, enabling high kr values that are well consistent with photophysical properties. Notably, the estimated ΔEST values between S1 and T1 are 0.39 eV for m-DT-BCzBN and 0.40 eV for p-DT-BCzBN, both of which are usually regarded as too large to implement favorable spin flip of triplet excitons. Moreover, the S1 and T1 states of the two emitters are all accompanied by SR-CT features with a similar overlap integral of the HONTO and LUNTO (OIH/L), and the corresponding SOCME values of 〈S1|ĤSO|T1〉 are as low as 0.011–0.032 cm−1.
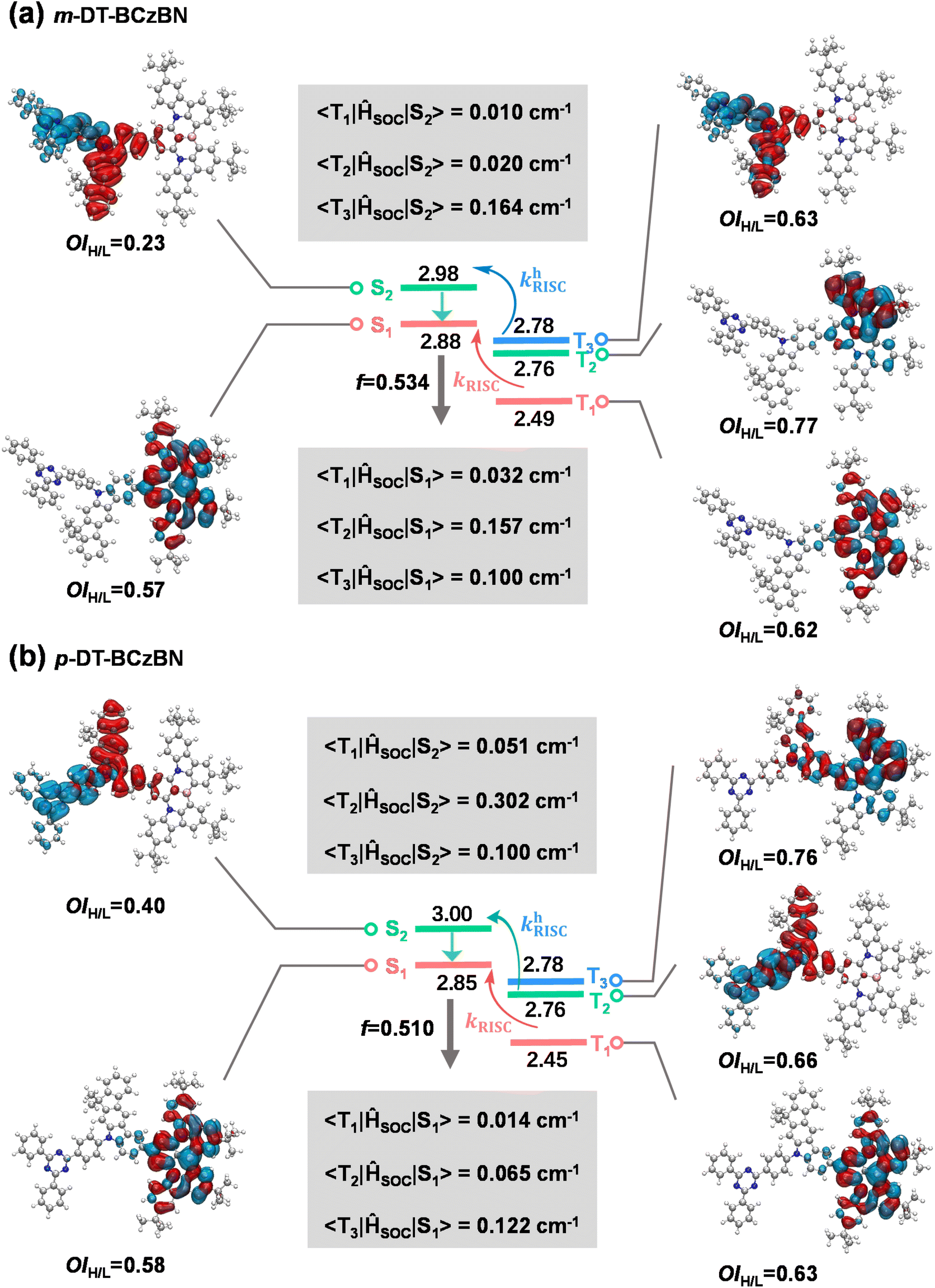 | ||
| Fig. 2 Simulated energy-level diagrams, NTO distributions, overlap of the HONTO and LUNTO, and associated SOC matrix elements for (a) m-DT-BCzBN and (b) p-DT-BCzBN. | ||
The closely high-lying S2 states with moderate energy differences between S1 and S2 should also be noted. The energy gaps are only 0.10 and 0.15 eV for m-DT-BCzBN and p-DT-BCzBN, respectively, small enough for provoking consecutive and fast internal conversion and FRET processes. For the S2 states of both emitters, the HONTOs are mainly distributed on DMIC moieties, while the LUNTOs are concentrated on TRZ units, accompanied by small OIH/L values of 0.23 and 0.40 for m-DT-BCzBN and p-DT-BCzBN, respectively. As a result, the S2 states exhibit CT features resembling those of D–A type TADF compounds due to the approximately orthogonal dihedral angles between DMIC and TRZ moieties and the inherent ICT effect. The calculated T2 and T3 energy levels of m-DT-BCzBN are 2.76 and 2.78 eV, respectively, and thus the theoretical ΔEST values of S2–T2 (0.22 eV) and S2–T3 (0.20 eV) become dramatically smaller than that of S1–T1. The NTO distributions of the T2 state are mainly located on the MR skeleton, proving a pronounced SR-CT nature with an OIH/L value of 0.77. Compared with 〈S1|ĤSO|T1〉, the SOCME value of 〈S1|ĤSO|T2〉 increases to 0.157 cm−1, caused by the synergistic effect of small energy splitting and more different transition characteristics between S1 and T2 states. Likewise, the T3 state is accompanied by the localized excited (LE) feature (OIH/L: 0.63) because of the delocalized HONTO and LUNTO on the m-DMIC-TRZ fragment. In line with the large difference between the excited natures of the S2 and T3 states on the m-DMIC-TRZ moiety, the corresponding SOCME 〈S2|ĤSO|T3〉 of 0.164 cm cm−1 is more than five times larger than that of 〈S1|ĤSO|T1〉. Considering the energetic proximity of S2 and T3, it can be anticipated that the T3 states serve as intermediaries in the SOC interaction between singlet and triplet excited states, and thus the higher-lying conversion channels (T3 → S2) may synergistically accelerate the spin flip of triplet excitons, thereby contributing to boosted RISC dynamics.
Similar to m-DT-BCzBN, the T2 and T3 energy levels of p-DT-BCzBN lie close to each other and are slightly lower than its S2 state, and the gaps of S2–T2 and S2–T3 are only 0.22 and 0.24 eV, small enough for competent upconversion of triplet excitons. For the T2 state, the HONTO and LUNTO are entirely located on the whole p-DMIC-TRZ moiety with a relatively large OIH/L value of 0.66, validating a relatively high LE component. Due to more different transition characteristics between S2 and T2 states, the SOCME 〈S2|ĤSO|T2〉 can reach 0.302 cm−1, suggesting that the higher-lying excited states can potentially contribute to fast RISC from triplet to singlet states. Additionally, the T2 state of m-DT-BCzBN exhibits a greatly enlarged SR-CT feature (OIH/L = 0.77), while the T3 state of p-DT-BCzBN displays an approximately mixed SR-CT (OIH/L = 0.76) and LR-CT nature as the NTO distributions extend to the DMIC moiety, and therefore, the SOCME values of 〈S1|ĤSO|T2〉 for m-DT-BCzBN (0.157 cm−1) and 〈S1|ĤSO|T3〉 for p-DT-BCzBN (0.122 cm−1) are also dramatically higher than the corresponding 〈S1|ĤSO|T1〉 values. Consequently, it can be concluded that these triplet states will also serve as an assistant channel for the RISC process. It also should be pointed out that the determined kRISC value of m-DT-BCzBN is higher than that of p-DT-BCzBN, although p-DT-BCzBN possesses cutting-edge SOCME, particularly 〈S2|ĤSO|Tn〉, indicative of favorable motivation for spin flip of triplet excitons. It is widely recognized that junction sites can induce extra flexibility of excited-state structures in isomerized components, resulting in pronounced structural deformation during electronic transitions.49 According to the description of Fermi's golden rule, kRISC depends on not only ΔEST and SOC but also the reorganization energy (λ).37,50,51 Thus, the unexpectedly higher kRISC value of m-DT-BCzBN might be induced by limited structural changes introduced by meta-substituted conformation. To investigate the influence of junction sites on structural changes for both ICT assisted MR-TADF emitters, Huang–Rhys factors of the vibrational modes were first calculated to elucidate the spectral progression between S0 and S1 states. As depicted in Fig. 3a, the primary vibrational modes of m-DT-BCzBN arising from the twisting vibrations of the TRZ units are at a frequency of 10.22 cm−1, and the high-frequency vibrational modes are quite limited. In contrast, p-DT-BCzBN reveals a significantly higher frequency of 268.84 cm−1 for the twisting vibrations of the TRZ units (Fig. 3b), and additional vibrations introduced by para-substituted conformation are also predominantly located in the high-frequency region with wavenumbers over 500 cm−1. The root mean squared displacement (RMSD) between S0 and S1 states is only 0.2661 Å for m-DT-BCzBN, which is much smaller than that of p-DT-BCzBN (1.0286 Å), as shown in Fig. 3c and d. The restrained high-frequency twisting vibrations and limited structural changes, coupled with the decreased RMSD between S0 and S1, contribute to the smaller λ value of m-DT-BCzBN (0.3546 eV) than that of p-DT-BCzBN (0.4833 eV), responsible for the boosted kRISC values of m-DT-BCzBN.52 Furthermore, the aggregation structures of both MR-TADF emitters were further simulated by molecular dynamics (MD), as shown in Fig. 3e and f. The details of MD simulation are described in the Computational methods of the ESI.† According to the bimolecular packing configuration extracted from the resultant box, both MR-TADF molecules exhibit misaligned parallel arrangements, especially for m-DT-BCzBN with a centroid distance of 6.2 Å, indicative of significantly suppressed intermolecular aggregation quenching. In comparison, the centroid distance of the p-DT-BCzBN based dimer decreases to 4.5 Å, displaying an apparent face-to-face packing mode in the fused MR skeleton. Therefore, the nonradiative loss and exciton concentration quenching effect can be restricted in m-DT-BCzBN, well consistent with the relatively lower kSnr value determined from photophysical analysis. Concerning the smaller λ resulting from the favorable meta-substituted conformation and weakened intermolecular interaction, m-DT-BCzBN is anticipated to achieve much higher exciton utilization and luminous efficiency.
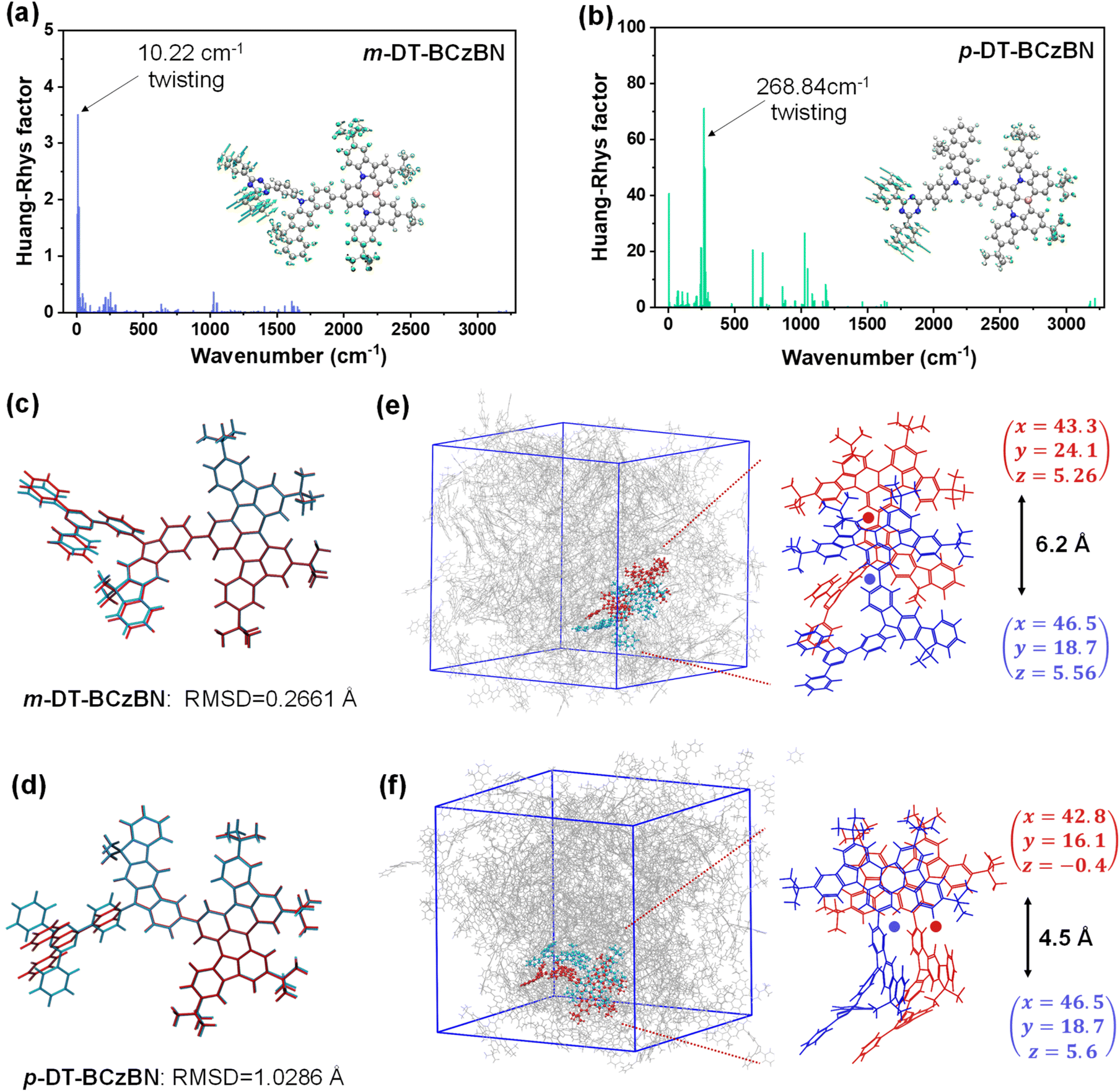 | ||
| Fig. 3 Calculated Huang–Rhys factors from the S1 to S0 transition for (a) m-DT-BCzBN and (b) p-DT-BCzBN (inset: twisting vibration modes with the most significant contribution to the Huang–Rhys factor). The RMSD between optimized S0 (red) and S1 (blue) geometries for (c) m-DT-BCzBN and (d) p-DT-BCzBN. MD simulation results of (e) m-DT-BCzBN and (f) p-DT-BCzBN. Centroid coordinates and the corresponding centroid distances are also presented. | ||
To gain further insight into the triplet exciton dynamics of these two MR-TADF emitters and especially confirm the experimental presence of upconversion channels of triplet excitons at higher energy levels, we performed pump-probe transient absorption (TAS) spectroscopy for the MR-TADF emitters diluted in an insulating PMMA matrix. Measurements were conducted with a pump wavelength of 340 nm in the detected range of 490–910 nm at room temperature. From the TAS contour maps as shown in Fig. 4a and b, both emitters exhibit broad photo-induced absorption (PIA) signals at 600–900 nm. Coupled with the abovementioned conclusion that a concealed pathway at higher energy levels indeed exists in these MR-TADF emitters, we can preliminarily infer that this PIA signal belongs to the absorption band of the higher-lying S2 (ICT) state.40,53,54 The kinetic decay processes of excitons for both emitters detected at 750–780 nm are also studied, as shown in Fig. 4c and d. Intriguingly, the exciton dynamics feature double exponential decay, potentially implying that the presence of S1 (1SR-CT) → S2 and subsequent S2 → Tn during excited-state absorption. Although the spectral evolution of m-DT-BCzBN resembles that of p-DT-BCzBN, m-DT-BCzBN exhibits different exciton kinetics (τ1 = 10 ps and τ2 = 302 ps) with p-DT-BCzBN (τ1 = 17 ps and τ2 = 367 ps), disclosing that m-DT-BCzBN exhibits a relatively quicker internal conversion between S1 and S2via the FRET process and a lower intersystem crossing (ISC) rate between S2 and Tn than that of p-DT-BCzBN. The accelerated internal conversion process is well consistent with the relatively smaller calculated energy gap of m-DT-BCzBN (0.10 eV) than that of p-DT-BCzBN (0.15 eV) for S1 → S2, and the more limited excited-state structural relation of m-DT-BCzBN. The enhanced SOCME values of 〈S2|ĤSO|Tn〉 for p-DT-BCzBN than that of m-DT-BCzBN can induce more competent ISC and RISC processes. In consequence, these experimental results demonstrate that the higher-lying excited states are fundamentally involved in facilitating the spin flip process of triplet excitons, thus enhancing the dynamics of RISC.
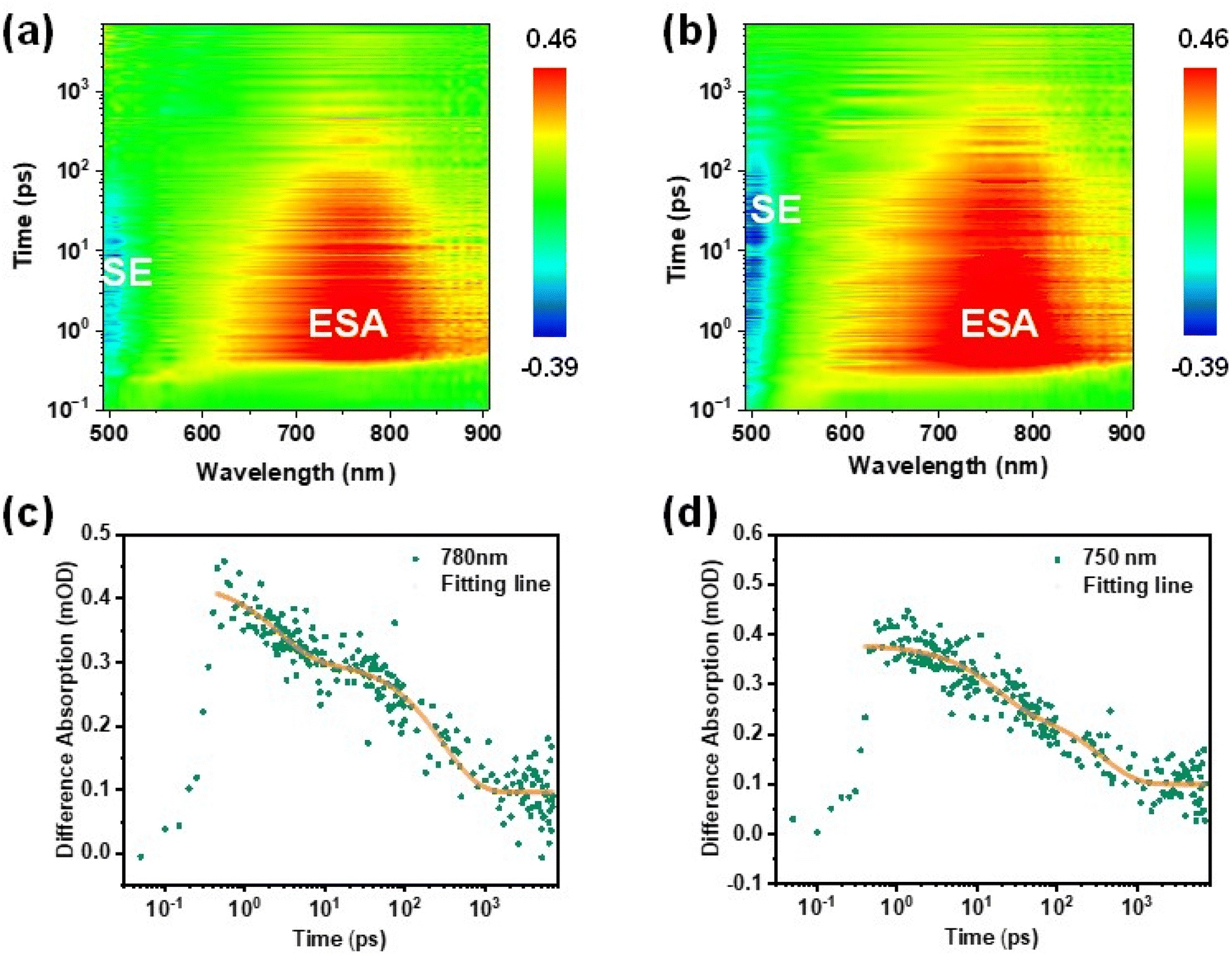 | ||
| Fig. 4 The TAS spectra of emitters diluted in the PMMA matrix for (a) m-DT-BCzBN and (b) p-DT-BCzBN. The kinetic decay processes of excitons for (c) m-DT-BCzBN detected at 780 nm and (d) p-DT-BCzBN detected at 750 nm. SE: stimulated emission and ESA: excited state absorption. | ||
To evaluate the potential of both emitters in electroluminescence (EL) devices, we fabricated solution-processed OLEDs with the architecture of ITO/PSS:PEDOT 4083 (30 nm)/PVK (10 nm)/emitting layer (40 nm)/TmPyPB (45 nm)/LiF (0.9 nm)/Al (110 nm), where PSS:PEDOT, PVK and TmPyPB are poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate), poly(N-vinyl carbazole), and 1,3,5-tri(m-pyridin-3-ylphenyl)benzene, respectively. Herein, ITO and Al were the anode and cathode, respectively. PSS:PEDOT 4083 was adopted as the hole injecting and transferring layer, while LiF served as the electron injecting layer. PVK functioned as the electron blocking layer, and TmPyPB was applied as the electron transferring layer. According to the cyclic voltammetry results, the HOMO values of both emitters can be determined to be around −5.6 eV, and then by combining with absorption spectra (the optical gaps are 2.5 eV for both emitters), the LUMO values can be calculated to be around −3.1 eV for both emitters (Fig. S8†). The molecular structures of the functional materials are given in Scheme S9.† For the emitting layer, the MR-TADF emitters were embedded into the host of PhCzBCz with different doping concentrations (Fig. S9 and S10†). The most optimized doping concentration is 2 wt%, which could not only restrain the π–π aggregation caused exciton quenching, but also improve the film-forming ability and morphology of the emitting layer. The device configuration and corresponding energy–level diagram of the solution-processed OLEDs are shown in Fig. 5a, and the detailed device parameters are summarized in Table 2.
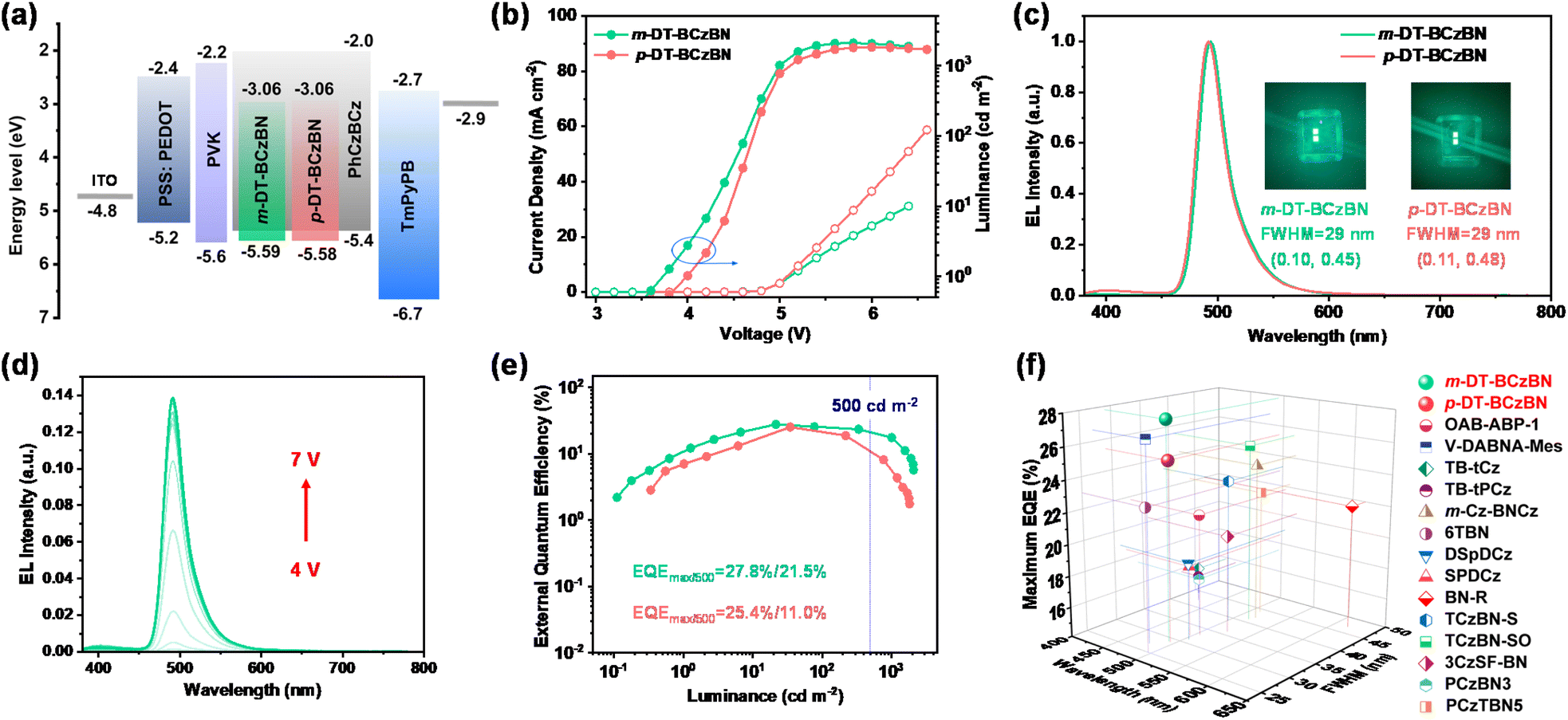 | ||
| Fig. 5 (a) Device architectures and energy diagrams of functional layers for the solution-processed OLEDs. (b) Luminance–voltage–current density curves. (c) The EL spectra of both emitters and corresponding photographs of OLEDs together with FWHMs and CIE coordinates. (d) The EL spectra at different driving voltages from 4 to 7 V. (e) EQE versus luminance curves. The blue dashed lines indicate the EQE values at 500 cd m−2. (f) The EQEmax and FWHM of all reported MR-TADF based solution-processed nonsensitized OLEDs, as a function of wavelength from 400 to 650 nm.42,56–65 | ||
| Devicea | V on [V] | ELc [nm] | FWHMd [nm] | L max [cd m−2] | CEmaxf [cd A−1] | PE max [lm W−1] | EQEmax/500h [%] | CIEi |
|---|---|---|---|---|---|---|---|---|
| a Device 1: 1 wt% m-DT-BCzBN in PhCzBCz; Device 2: 2 wt% m-DT-BCzBN in PhCzBCz; Device 3: 3 wt% m-DT-BCzBN in PhCzBCz; Device 4: 5 wt% m-DT-BCzBN in PhCzBCz; Device 5: 1 wt% p-DT-BCzBN in PhCzBCz; Device 6: 2 wt% p-DT-BCzBN in PhCzBCz; Device 7: 3 wt% p-DT-BCzBN in PhCzBCz; Device 8: 5 wt% p-DT-BCzBN in PhCzBCz b The electroluminescence spectral peaks. c The turn-on voltages at 1 cd m−2. d The maximum luminances. e The maximum current efficiencies. f The maximum power efficiencies. g The maximum external quantum efficiencies (EQEs). h The EQE values at 1000 cd m−2. i The coordinates of the Commission Internationale de L'Eclairage. | ||||||||
| 1 | 4.0 | 492 | 29 | 2863 | 58.6 | 36.8 | 27.2/21.7 | (0.10, 0.44) |
| 2 | 4.0 | 492 | 29 | 2079 | 62.7 | 44.7 | 27.8/21.5 | (0.10, 0.45) |
| 3 | 4.0 | 492 | 29 | 2321 | 50.8 | 31.9 | 23.6/15.5 | (0.10, 0.45) |
| 4 | 4.0 | 492 | 29 | 2051 | 41.0 | 26.4 | 19.6/14.9 | (0.12, 0.46) |
| 5 | 4.0 | 494 | 29 | 1949 | 53.8 | 36.8 | 24.5/10.4 | (0.10, 0.48) |
| 6 | 4.0 | 494 | 29 | 1820 | 55.0 | 37.6 | 25.4/11.0 | (0.11, 0.48) |
| 7 | 3.9 | 494 | 29 | 1760 | 45.6 | 31.2 | 20.8/11.7 | (0.11, 0.48) |
| 8 | 4.0 | 495 | 29 | 1773 | 38.2 | 26.1 | 19.2/9.8 | (0.12, 0.49) |
From the current density–voltage–luminance curves shown in Fig. 5b, the turn-on voltages (Von) of devices are comparable to each other at 4.0 V. The maximum luminance (Lmax) can exceed 2000 cd m−2 at a driving voltage of 6 V for the m-DT-BCzBN based OLED. As shown in Fig. 5c, the EL spectra of these devices are in agreement with the PL profiles despite slight blueshift due to restricted molecular structural relaxation in the PhCzBCz matrix. Both emitters display greenish-blue emission bands peaking at 492–494 nm with small FWHMs of 29 nm, which are in nearly full accord with those of BCzBN based OLEDs,55 further confirming that the formation of higher-order ICT states can enhance exciton utilization without disturbing EL spectra and color purity. At different driving voltages, the EL spectra remain stable (Fig. 5d). The maximum current efficiency (CEmax) and power efficiency (PEmax) are 62.7 cd A−1/44.7 lm W−1 for m-DT-BCzBN, which are relatively higher than those of 55.0 cd A−1/37.6 lm W−1 for p-DT-BCzBN (Fig. S11†). Correspondingly, the maximum external quantum efficiency (EQEmax) value of m-DT-BCzBN (27.8%) also surpasses that of p-DT-BCzBN (25.4%) without any additional sensitizer (Fig. 5e), in line with the faster kRISC values of the m-DT-BCzBN based doping films. Notably, the EQEmax value of 27.8% represents the champion EQE value of MR-TADF based nonsensitized solution-processed OLED devices (Fig. 5f). More importantly, the efficiency roll-off in solution-processed OLEDs is also dramatically suppressed. At a high luminance of 500 cd m−2, the EQEs can maintain a relatively high value of 21.5%, representing a breakthrough in efficiency for solution-processed MR-TADF based narrowband OLEDs. Thus, the reserved narrowband emission and relatively enhanced device performance of m/p-DT-BCzBN based solution-processable OLEDs compared with those of BCzBN based device (EQEmax = 16.3% with TADF molecules as the host) clearly demonstrate the effectiveness of our molecular design tactic to construct upconversion channels of triplet excitons at higher energy levels.55
Conclusions
In this work, we proposed an ICT assisted MR-TADF approach by grafting a D–A-type substituent on the B/N MR skeleton. The newly designed emitters not only manifest SR-CT dominated characteristics contributed by the MR effect, but also accelerate the exciton conversion and decay processes, exhibiting a near-unity PLQY, together with a dramatically optimized kRISC value of exceeding 1.0 × 105. Theoretical calculations and TAS studies unambiguously validated that the ICT process at high-lying states contributed by the D–A-type substituent can facilitate the spin flip of triplet excitons, so as to reduce the exciton aggregation quenching in the emission layer and overcome the efficiency roll-off drawback. Thanks to the ICT assisted MR-TADF strategy, relatively excellent EL performance in sensitizer-free solution-processed OLEDs is achieved, providing a record-high EQE value of 27.8% together with EL emission peaks at 490 nm and a small FWHM of 29 nm. More importantly, the efficiency roll-off in solution-processed OLEDs is also dramatically suppressed. At a high luminance of 500 cd m−2, the EQEs can maintain a relatively high value of 21.5%, representing a breakthrough in efficiency for solution-processed MR-TADF based narrowband OLEDs.Data availability
The data (experimental instrumentation, synthetic procedures, structural characterization data including NMR and MS spectra, theoretical calculations, thermodynamics data, and device performance data) that support this article are available in the article itself and its ESI.†Author contributions
Z. Yang, S. Li, L. Hua, and Y. Liu – conceptualization, investigation, OLED devices, and writing – original draft; Z. Yang, and S. Li – theoretical calculation, and investigation; Z. Yang, and S. Li– formal analysis and OLED devices; Z. Yang – investigation (thermal properties); Z. Yang, S. Li, and L. Hua– investigation (transient PL properties); Y. Liu, Z. Ren, S. Ying, and S. Yan – project administration and supervision; Y. Liu, Z. Ren, and S. Yan – writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of the National Natural Science Foundation of China (52103220 and 52273164), the Shandong Provincial Natural Science Foundation (ZR2022ZD37 and ZR2023QE078), the Science and Technology Support Plan for Youth Innovation of Colleges and Universities in Shandong Province (2023KJ097), and the Natural Science Foundation of Qingdao (23-2-1-75-zyyd-jch) is gratefully acknowledged.Notes and references
- L. Hua, Y. Liu, B. Liu, Z. Zhao, L. Zhang, S. Yan and Z. Ren, Nat. Commun., 2022, 13, 7828 CrossRef CAS PubMed.
- Y. Chen, D. Zhang, Y. Zhang, X. Zeng, T. Huang, Z. Liu, G. Li and L. Duan, Adv. Mater., 2021, 33, 2103293 CrossRef CAS PubMed.
- Z. Li, D. Yang, C. Han, B. Zhao, H. Wang, Y. Man, P. Ma, P. Chang, D. Ma and H. Xu, Angew. Chem., Int. Ed., 2021, 60, 14846–14851 CrossRef CAS PubMed.
- W. Zeng, H. Lai, W. Lee, M. Jiao, Y. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. Wong, C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef.
- K. Shi, Y. Xie, L. Hua, S. Li, Z. Yang, Y. Yin, Z. Wang, S. Ying, Y. Liu, Z. Ren and S. Yan, ACS Mater. Lett., 2024, 6, 1491–1503 CrossRef CAS.
- J. Lee, N. Aizawa, M. Numata, C. Adachi and T. Yasuda, Adv. Mater., 2017, 29, 1604856 CrossRef PubMed.
- K. Kumar, React. Chem. Eng., 2024, 9, 496–527 RSC.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- J. Bian, S. Chen, L. Qiu, R. Tian, Y. Man, Y. Wang, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Adv. Mater., 2022, 34, 2110547 CrossRef CAS PubMed.
- R. Zhong, M. Wang, X. Wang, S. Wang, S. Shao and L. Wang, Chem. Sci., 2024, 15, 13290–13298 RSC.
- N. Kwon, H. Kwak, H. Kim, S. Park, J. Park, M. Kang, C. Koh, S. Park, M. Cho and D. Choi, Chem. Sci., 2024, 15, 12361–12368 RSC.
- J. Wang, D. Chen, J. Moreno-Naranjo, F. Zinna, L. Frédéric, D. Cordes, A. McKay, M. Fuchter, X. Zhang and E. Zysman-Colman, Chem. Sci., 2024, 15, 16917–16927 RSC.
- T. Huang, L. Yuan, X. Lu, Y. Qu, C. Qu, Y. Xu, Y.-X. Zheng and Y. Wang, Chem. Sci., 2024, 15, 15170–15177 RSC.
- L. Li, J. Li, L. Guo, Y. Xu, Y. Bi, Y. Pu, P. Zheng, X. –K. Chen, Y. Wang and C. Li, Chem. Sci., 2024, 15, 11435–11443 RSC.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912 CrossRef CAS PubMed.
- Y. Qu, D. Zhou, F. Kong, Q. Zheng, X. Tang, Y. Zhu, C. Huang, Z. Feng, J. Fan, C. Adachi, L. Liao and Z. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201886 CrossRef CAS PubMed.
- F. Liu, Z. Cheng, Y. Jiang, L. Gao, H. Liu, H. Liu, Z. Feng, P. Lu and W. Yang, Angew. Chem., Int. Ed., 2022, 61, e202116927 CrossRef CAS PubMed.
- X. Fan, K. Wang, Y. Shi, Y. Cheng, Y. Lee, J. Yu, X. Chen, C. Adachi and X. Zhang, Nat. Photonics, 2023, 17, 280–285 CrossRef CAS.
- J. Liu, Y. Zhu, T. Tsuboi, C. Deng, W. Lou, D. Wang, T. Liu and Q. Zhang, Nat. Commun., 2022, 13, 4876 CrossRef CAS PubMed.
- G. Meng, H. Dai, Q. Wang, J. Zhou, T. Fan, X. Zeng, X. Wang, Y. Zhang, D. Yang, D. Ma, D. Zhang and L. Duan, Nat. Commun., 2023, 14, 2394 CrossRef CAS PubMed.
- X. Luo, X. Xiao and Y. Zheng, Chem. Commun., 2024, 60, 1089–1099 RSC.
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC.
- L. Wan, Z. Cheng, F. Liu and P. Lu, Mater. Chem. Front., 2023, 7, 4420–4444 RSC.
- Z. Huang, H. Xie, J. Miao, Y. Wei, Y. Zou, T. Hua, X. Cao and C. Yang, J. Am. Chem. Soc., 2023, 145, 12550–12560 CrossRef CAS PubMed.
- J. Jin, D. Liu, W. Chen, C. Shi, G. Chen, X. Wang, L. Xing, W. Ying, S. Ji, Y. Huo and S. Su, Angew. Chem., Int. Ed., 2024, 63, e202401120 CrossRef CAS PubMed.
- Y. He, F. Xie, K. Zhang, D. Yang, Y. Shen, H. Li, D. Ma, Y. Li and J. Tang, Adv. Funct. Mater., 2023, 33, 2304006 CrossRef CAS.
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 17442–17446 CrossRef CAS PubMed.
- G. Chen, J. Wang, W. Chen, Y. Gong, N. Zhuang, H. Liang, L. Xing, Y. Liu, S. Ji, H. Zhang, Z. Zhao, Y. Huo and B. Tang, Adv. Funct. Mater., 2023, 33, 2211893 CrossRef CAS.
- Y. Zou, M. Yu, Y. Xu, Z. Xiao, X. Song, Y. Hu, Z. Xu, C. Zhong, J. He, X. Cao, K. Li, J. Miao and C. Yang, Chem, 2024, 10, 1485–1501 CAS.
- L. Xing, J. Wang, W. Chen, B. Liu, G. Chen, X. Wang, J. Tan, S. Chen, J. Chen, S. Ji, Z. Zhao, M. Tang and Y. Huo, Nat. Commun., 2024, 15, 6175 CrossRef CAS PubMed.
- M. Etherington, J. Gibson, H. Higginbotham, T. Penfold and A. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- P. Samanta, D. Kim, V. Coropceanu and J. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- Y. Liu, L. Hua, Z. Zhao, S. Ying, Z. Ren and S. Yan, Adv. Sci., 2021, 8, 2101326 CrossRef CAS PubMed.
- S. Li, Y. Xie, Y. Yin, J. Chen, Y. Cao, S. Ying, Y. Liu, Z. Ren and S. Yan, Macromolecules, 2024, 57, 5253–5261 CrossRef CAS.
- T. Wang, Y. Zou, Z. Huang, N. Li, J. Miao and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202211172 CrossRef CAS PubMed.
- Y. Liu, Y. Xie, L. Hua, X. Tong, S. Ying, Z. Ren and S. Yan, CCS Chem., 2023, 5, 1005–1017 CrossRef CAS.
- C. Li, A. Harrison, Y. Liu, Z. Zhao, C. Zeng, F. Dias, Z. Ren, S. Yan and M. Bryce, Angew. Chem., Int. Ed., 2022, 61, e202115140 CrossRef CAS PubMed.
- Y. Liu, X. Tong, X. Chen, Y. Wang, S. Ying, Z. Ren and S. Yan, ACS Appl. Mater. Interfaces, 2021, 13(7), 8997–9005 CrossRef CAS PubMed.
- R. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Pei, Q. Ou, Y. Mao, J. Yang, A. Lande, F. Plasser, W. Liang, Z. Shuai and Y. Shao, J. Phys. Chem. Lett., 2021, 12, 2712–2720 CrossRef CAS PubMed.
- X. Chen, S. Zhang, J. Fan and A. Ren, J. Phys. Chem. C, 2015, 119, 9728–9733 CrossRef CAS.
- H. Gao, S. Shen, Y. Qin, G. Liu, T. Gao, X. Dong, Z. Pang, X. Xie, P. Wang and Y. Wang, J. Phys. Chem. Lett., 2022, 13, 7561–7567 CrossRef CAS PubMed.
- G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- H. Noda, X. K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J. L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- Y. Liu, L. Hua, S. Yan and Z. J. Ren, Nano Energy, 2020, 73, 104800 CrossRef CAS.
- S. Xu, Q. Yang, Y. Zhang, H. Li, Q. Xue, G. Xie, M. Gu, J. Jin, L. Huang and R. Chen, Chin. Chem. Lett., 2021, 32, 1372–1376 CrossRef CAS.
- F. Xie, H. Li, K. Zhang, H. Wang, Y. Li and J. Tang, ACS Appl. Mater. Interfaces, 2023, 15, 39669–39676 CrossRef CAS PubMed.
- X. Cai, Y. Xu, Y. Pan, L. Li, Y. Pu, X. Zhuang, C. Li and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216473 CrossRef CAS PubMed.
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda and T. Hatakeyama, Adv. Mater., 2020, 32, 2004072 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 CrossRef CAS PubMed.
- H. J. Kim, H. Kang, J.-E. Jeong, S. H. Park, C. W. Koh, C. W. Kim, H. Y. Woo, M. J. Cho, S. Park and D. H. Choi, Adv. Funct. Mater., 2021, 31, 2102588 CrossRef CAS.
- R. Ma, Z. Ma, X. Wang, Z. Si, Q. Duan and S. Shao, Chem. Eng. J., 2022, 447, 137517 CrossRef CAS.
- B. Du, K. Zhang, P. Wang, X. Wang, S. Wang, S. Shao and L. Wang, J. Mater. Chem. C, 2023, 11, 9578–9585 RSC.
- F. Huang, Y. Cheng, H. Wu, X. Xiong, J. Yu, X. Fan, K. Wang and X. Zhang, Chem. Eng. J., 2023, 465, 142900 CrossRef CAS.
- X. Zhuang, B. Liang, C. Jiang, S. Wang, H. Bi and Y. Wang, Adv. Opt. Mater., 2024, 12, 2400490 CrossRef CAS.
- T. Wang, Z. Huang, H. Zhang, J. Miao and C. Yang, Adv. Funct. Mater., 2024, 240811 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08708a |
| This journal is © The Royal Society of Chemistry 2025 |