
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIterative synthesis of stereodefined polyacetals and their domino-Coates–Claisen rearrangement†
Itai
Massad
* and
Ilan
Marek

Schulich Faculty of Chemistry and The Resnick Sustainability Center for Catalysis, Technion – Israel Institute of Technology, Haifa, 3200009, Israel. E-mail: itaimassad3@gmail.com
First published on 24th January 2025
Abstract
We report a fully stereocontrolled synthesis of previously unknown unsaturated polyacetals through an iteration of two steps: (1) Pd-catalyzed hydroalkoxylation of alkoxyallenes; and (2) base-mediated isomerization of propargyl ethers into alkoxyallenes. Site- and stereoselective alkene isomerization of the capping allyl group initiates a domino-Coates–Claisen rearrangement, which completely rearranges the iteratively assembled backbone with the stereochemistry of the newly formed stereocentres controlled by the configuration of the acetal centres in the starting materials. The resulting products feature multiple stereocentres in a 1,3-relationship, which map onto a broad range of bioactive natural products, including deoxypropionates.
Introduction
Iterative synthetic strategies can dramatically simplify the preparation of complex organic molecules, enabling studies into their properties and applications. A striking example is the development of polypeptide synthesis into a routine, automated process,1 facilitating de novo protein synthesis and tremendously advancing the field of chemical biology. In recent decades, a plethora of impressive iterative synthetic strategies have been developed, employing a diverse palette of chemical reactions to forge a vast scope of molecular architecture.2–4 Iterative syntheses of motifs featuring multiple neighbouring stereocentres are particularly rewarding, as they enable forays into unknown structure–property relationships. However, such approaches are also challenging, as pre-existing stereocenters in the substrate can complicate the selective formation of new ones.5 Aggarwal's pioneering synthesis of polymethylated alkanes exemplifies both the potential and challenges inherent to such strategies.6Herein, we present an efficient and completely stereocontrolled iterative synthesis of previously unknown acyclic polyacetals, formally derived from acrolein. Beyond their novel structural features, these polyacetals undergo a stereospecific domino-Coates–Claisen rearrangement,7–9 which entirely rearranges the iteratively assembled molecular backbone.
Building on our work on stereoselective transformations enabled by selective alkene isomerization,10–12 we explored the Coates–Claisen rearrangement of allylic vinyl acetals, generated in situ through the isomerization of readily available allyl acetals (Scheme 1a).13 We were soon intrigued by the prospect of engaging the enol ether moiety unique to the products of this rearrangement in a subsequent Coates–Claisen rearrangement, suggesting a domino process.14,15 This envisaged sequence would be initiated by the site-selective alkene isomerization of an allyl group (Scheme 1b), triggering a [3,3]-sigmatropic rearrangement that transposes an alkene for the next rearrangement, continuing the cascade. Ultimately, this domino process would convert a polyacetal backbone into an all-carbon skeleton, while transferring stereochemical information from the starting material to generate multiple aldehyde-bearing stereocentres in the product.
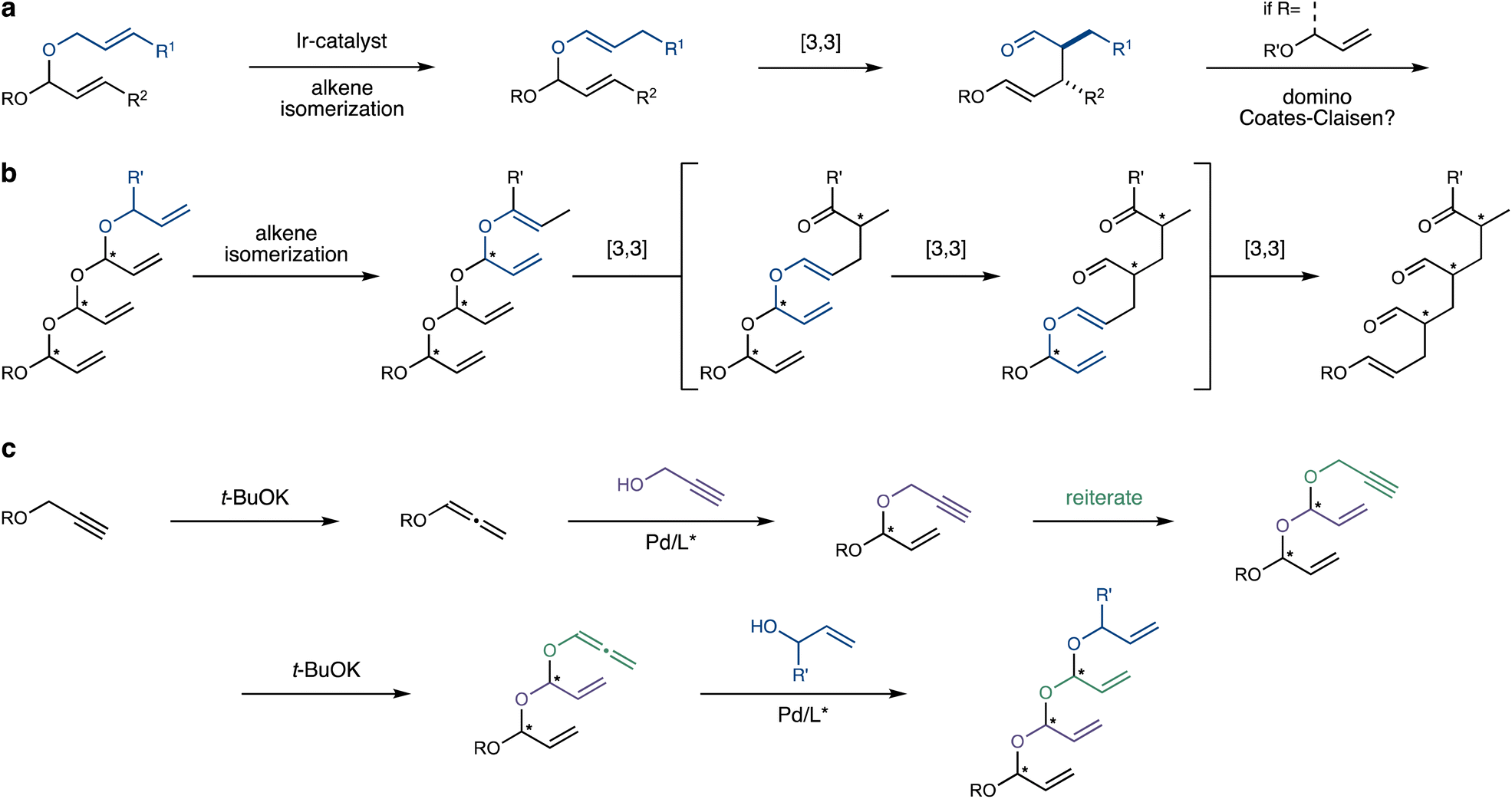 | ||
| Scheme 1 An isomerization-domino-Coates–Claisen cascade and a projected synthesis of polyacetals: (a) alkene isomerization–Coates–Claisen rearrangement and the possibility of engaging the enol ether in the product in a subsequent rearrangement. (b) Site-selective alkene isomerization followed by a domino-Coates–Claisen rearrangement. E-Configured enol ethers are shown for clarity. (c) Proposed synthesis of polyacetals through an iteration of two steps: hydroalkoxylation of an alkoxyallene with propargyl alcohol and isomerization of a propargyl ether into an allenyl ether. | ||
The exciting prospect of this domino-Coates–Claisen rearrangement raised the challenge of preparing the prerequisite unsaturated polyacetals. Such polyacetals are completely unprecedented in the literature, with the closest analogues being the fused oligo-tetrahydrofurans reported by Werz.16 We thus recognized that their preparation might present a considerable challenge. In this vein, the impressive body of work by the Rhee group on the stereoselective synthesis of mixed acetals through Pd-catalyzed hydroalkoxylation of alkoxyallenes attracted our attention.17,18 We hypothesized that this reaction could serve as the foundation of an iterative process that assembles the polyacetal backbone (Scheme 1c). By adding propargyl alcohol to an alkoxyallene, the iteration could be completed through base-mediated isomerization19 of the propargyl group to the corresponding allene, primed for the next hydroalkoxylation step. Finally, the polyacetal chain would be capped by the addition of an allylic alcohol, providing the initiation site for the ensuing domino process.
Clearly, this iterative strategy entails significant challenges. First, the stability of each acetal intermediate is at risk, as acetals – and especially acyclic ones featuring unsaturation – are often susceptible to decomposition, even in the presence of weak acids like silica gel.20 In addition, intermediate allenyl acetals may be susceptible to premature Coates–Claisen rearrangement, promoted either thermally or by mild acids.13 If this daunting stability issue is successfully addressed, the next challenge lies in achieving precise stereocontrol, which requires a high degree of catalyst-control during the Pd-catalyzed hydroxylation steps. Finally, the fragile unsaturated polyacetal skeleton might engage in Pd-catalyzed Tsuji–Trost-type side-reactions, further complicating the process.21
Assuming the desired polyacetals can be reliably synthesized, we planned to employ an alkene isomerization catalyst to site- and stereoselectively isomerize the allylic system capping the polyacetal chain, thereby setting the stage for the first Coates–Claisen rearrangement in the cascade (Scheme 1b).11 Efficient, site- and stereoselective alkene isomerization of one out of several terminal alkenes thus represents another crucial part of the process.22,23 Following isomerization, the first [3,3]-sigmatropic rearrangement would shift the branched alkene into position for the next one, and so forth.
The final crucial aspect of this strategy is its overall stereospecificity. This hinges on the stereoselectivity of each [3,3]-sigmatropic rearrangement, which requires the stereochemical purity of the alkene and stereocentre in the starting material, and a strong preference for one of several possible chair- or boat-like transition states.24 Additionally, the resulting polyaldehyde products must maintain their configurational integrity, despite their potential lability.
Overall, this domino process would transform the iteratively assembled polyacetal backbone into an all-carbon skeleton featuring multiple 1,3-stereocentres bearing versatile aldehyde groups, with completely programable configurations.
Results and discussion
We began our experimentation by testing the Pd-catalyzed addition of propargyl alcohol to benzyloxyallene 1 (prepared in one step from commercial benzyl propargyl ether19), using a modification of the conditions reported by Rhee.17 Pleasingly, the reaction afforded propargyl acetal 2 with excellent yield and enantioselectivity (Scheme 2a). Allyl alcohol and (R)-phenyl vinyl carbinol25 also performed well under similar conditions, leading to acetals 3 and 4, respectively.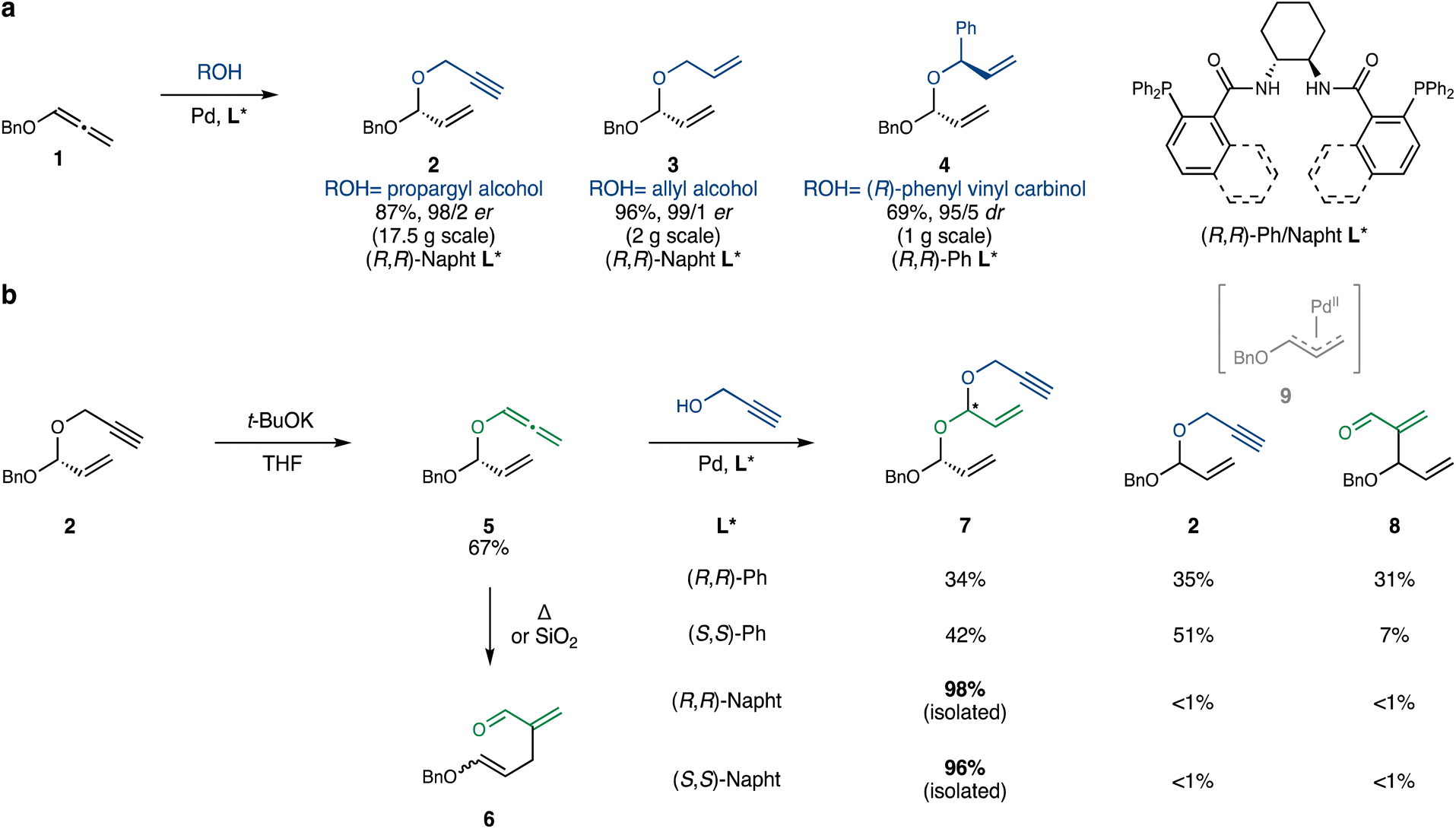 | ||
| Scheme 2 Synthesis of monoacetals and establishment of the second iteration. (a) Pd-catalyzed hydroalkoxylation of benzyloxyallene forming monoacetals. Conditions: 2/3: alcohol (1.5 equiv.), Pd2dba3 (2 mol%), (R,R)-naphthyl Trost ligand (4 mol%), DCM (0.5 M), 0 °C, 5 hours. 4: alcohol (1.1 equiv.), Pd2dba3 (5 mol%), (R,R)-phenyl Trost ligand (10 mol%), PhMe (0.5 M), 40 °C, 2 hours. (b) Isomerization of propargyl acetal 2 and hydroalkoxylation of allenyl acetal 5 en route to double acetals. The yields of products 2, 7 and 8 were determined by 1H-NMR, unless otherwise noted. Conditions: Pd2dba3 (2 mol%), Trost ligand (4 mol%), PhMe (0.5 M), 40 °C, 2 hours. | ||
Next, 2 was exposed to t-BuOK in THF to induce propargyl-to-allenyl isomerization (Scheme 2b). Although the reaction proceeded relatively cleanly, it plateaued after several hours, even with the addition of extra t-BuOK. This seemingly straightforward isomerization is complicated by the inherent propensity of allenyl acetal 5 to undergo a thermal Coates–Claisen rearrangement, requiring careful control of reaction time and temperature. Furthermore, isolation of 5via column chromatography proved challenging, as silica gel also promotes the Coates–Claisen rearrangement of this highly reactive acetal. We ultimately resorted to purification on silica gel neutralized with triethylamine and packed in a column equipped with a cooling jacket circulating ice-water.20 This technique minimized the formation of premature Claisen product 6 and afforded pure 5 in 67% yield on a 3 g scale. On a 20 g scale, 5 could also be obtained in 80% yield, contaminated with ∼10% of remaining starting material 2, which can be removed following the subsequent hydroalkoxylation step.
The next challenge was the hydroalkoxylation of the sensitive allenyl acetal 5. In addition to issues of stereoselectivity (catalyst versus substrate control), 5 might undergo oxidative addition in the presence of Pd0, in a classical Tsuji–Trost fashion.21 When 5 was reacted with propargyl alcohol under the conditions developed by Rhee, the desired double acetal 7 was obtained with high catalyst-controlled stereoselectivity. However, as anticipated, two additional byproducts, propargyl acetal 2 and aldehyde 8, were formed in equal amounts. These are presumably derived from the allylpalladium intermediate 9, which can react with the added propargyl alcohol or recombine with the excised allenyloxy fragment, respectively. Remarkably, substituting the phenyl DACH-Trost ligand with its naphthyl congener resulted in a dramatic shift in selectivity: the desired double acetal 7 was isolated almost quantitatively, with only trace amounts of 2 and 8 detectable by 1H-NMR. This unprecedented switch in site-selectivity – distinct from the more common subtle change in enantioselectivity26,27 – observed between the Ph- and naphthyl-DACH-Trost ligands will be investigated in detail in future studies.28
Under the optimized conditions, we successfully prepared multi-gram quantities of double acetals 10–15 (Scheme 3). A particularly attractive feature of this iterative hydroalkoxylation strategy is its high level of catalyst-controlled diastereoselectivity. For example, diastereomers 12 and 13 were cleanly obtained using ligands of opposite handedness, despite the presence of potentially interfering stereocentres near the reaction site in both the alcohol and allene partners. Notably, double acetals 10–15 are stable during chromatography on silica gel. Further chain extension of double acetals 14 and 15 with a third iteration proved successful. Isomerization using t-BuOK proved experimentally simpler with double acetals 14 and 15 than with monoacetal 2, as the resulting allenyl acetals 16 and 17 rearrange significantly slower than 5, allowing for easier handling and purification. This initially counterintuitive observation is attributed to the more electron-poor nature of the central oxygen in 16 and 17 (compared to the benzyloxy oxygen in 5), which attenuates its rate-accelerating effect on the Claisen rearrangement. Pleasingly, double allenyl acetals 16 and 17 smoothly underwent hydroalkoxylation with (R)-phenyl vinyl carbinol, efficiently and selectively affording an array of four triple acetals 18–21, featuring all possible diastereomeric relationships between the acetal centers. Our strategy towards these previously unknown polyacetals has thus proven highly successful: challenges of stability and selectivity were overcome, affording an efficient, flexible and selective iterative approach that paves the way for their application in synthesis.
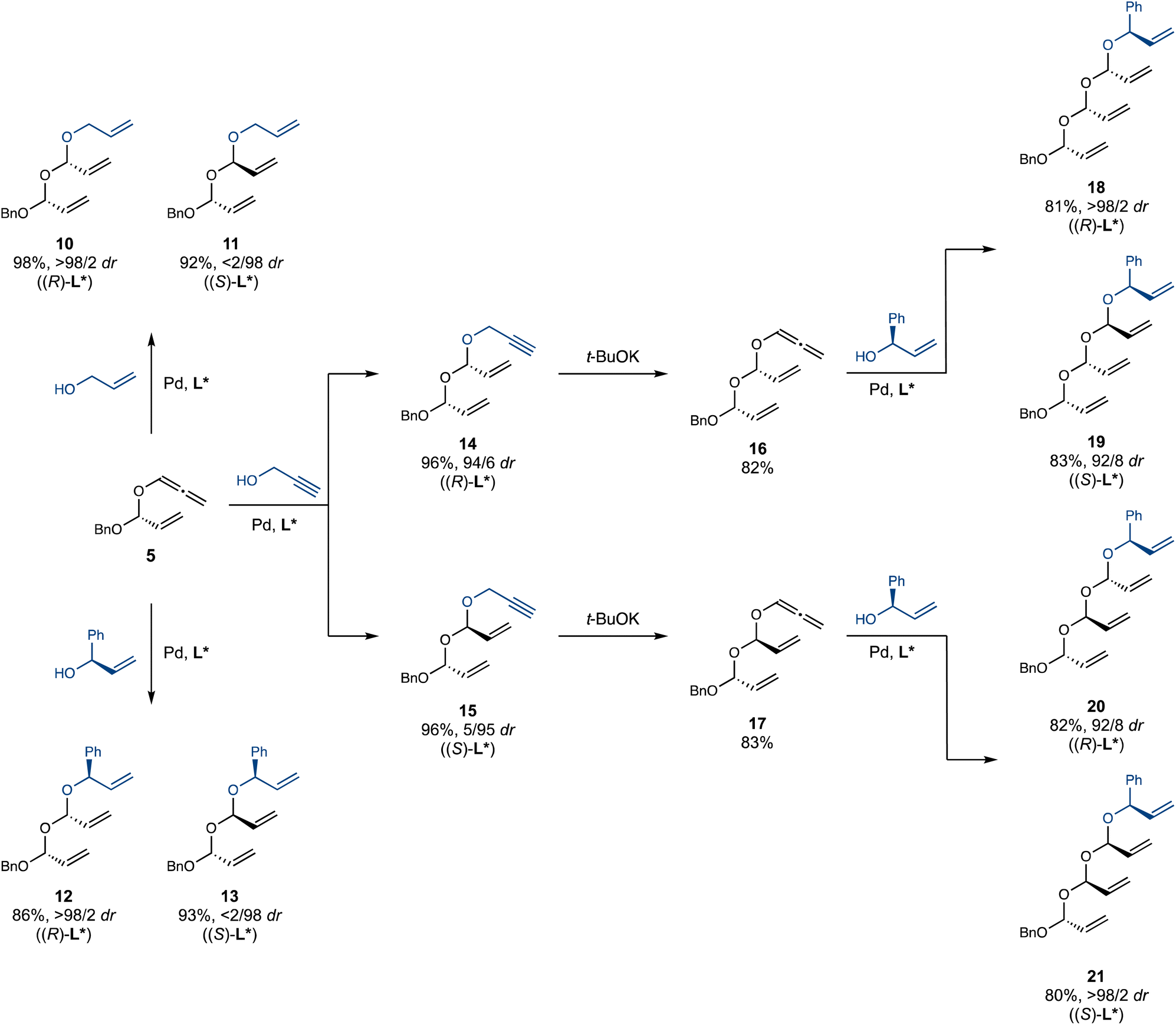 | ||
| Scheme 3 Synthesis of double- and triple acetals. Conditions: allyl/propargyl alcohol: alcohol (1.5 equiv.), Pd2dba3 (1 mol%), naphthyl Trost ligand (2 mol%), PhMe (0.5 M), 40 °C, 1 hour. (R)-Phenyl vinyl carbinol: alcohol (1.1 equiv.), Pd2dba3 (3 mol%), naphthyl Trost ligand (6 mol%), PhMe (0.5 M), 40 °C, 4–12 hours. | ||
With an assortment of stereodefined polyacetals in hand, we set out to explore their Coates–Claisen rearrangement. We first tested the isomerization and rearrangement of single acetal substrates 3 and 4 (Scheme 4). Pleasingly, allyl acetal 3 underwent Ir-catalyzed alkene isomerization smoothly and with complete site- and stereoselectivity. This high selectivity is a hallmark of the underlying 1,3-hydride shift mechanism, which preferentially targets unbranched allylic systems.29,30 Subsequent exposure of the isomerized acetal 22 to MgBr2 as a Lewis-acid, along with LiAlH4 to reduce the nascent aldehyde prior to opportune epimerization, yielded alcohol 23 with high Z-selectivity13 and enantiospecificity (Scheme 4a). The slight erosion in enantiopurity is attributed to one or more possible factors: epimerization of the aldehyde prior to reduction, the intervention of a boat-like transition state,31,32 or a dissociative mechanism.33–36
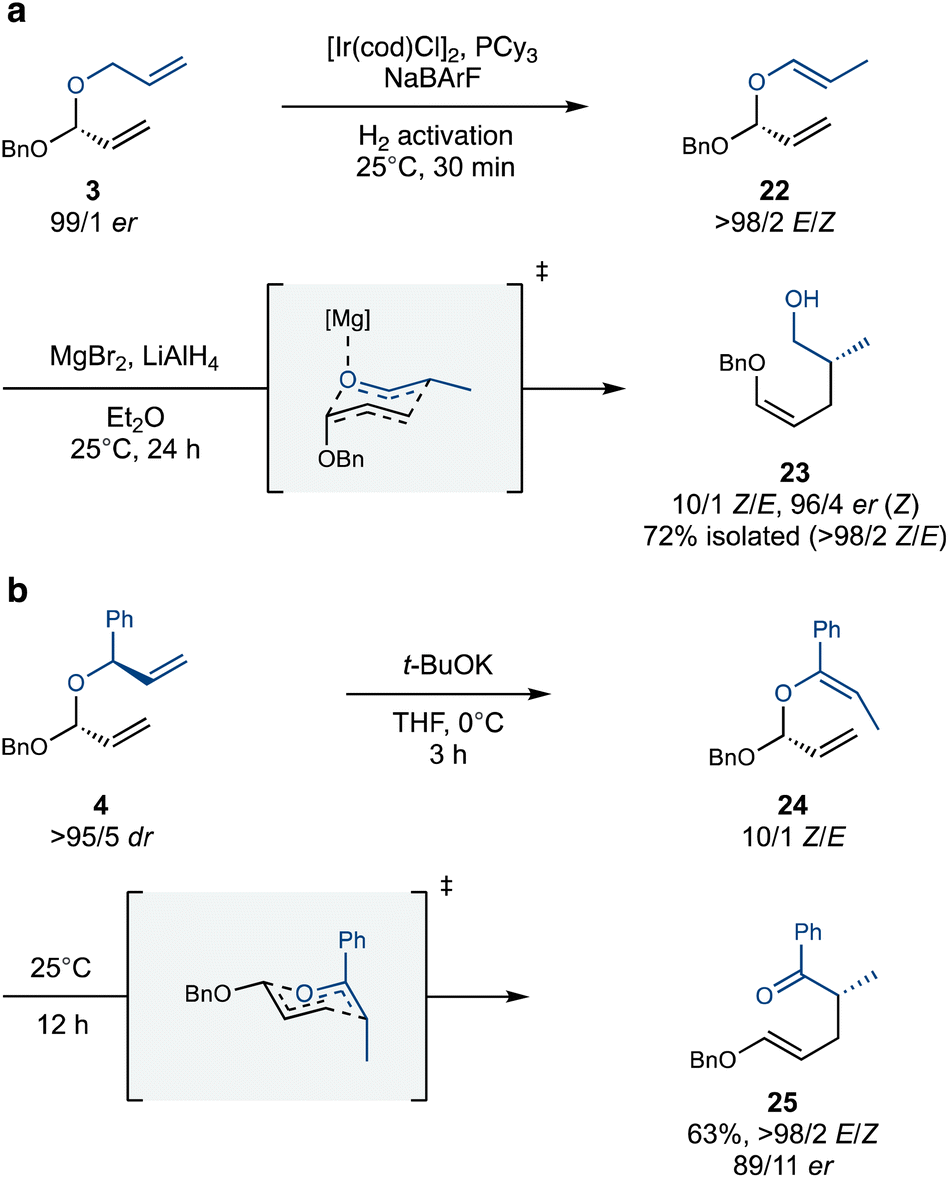 | ||
| Scheme 4 Isomerization of monoacetals with either a cationic iridium catalyst (a) or t-BuOK (b) followed by a Coates–Claisen rearrangement. | ||
The branched allyl group of acetal 4 also serves as an effective initiation site for the isomerization–Coates–Claisen rearrangement (Scheme 4b). Simply exposing 4 to t-BuOK in THF leads to isomerization with a Z/E ratio of 10/1, presumably through the intermediacy of a chelated allylpotassium intermediate.37 The resulting isomerized acetal 24 rearranges with striking facility, producing rearrangement product 25 with high enantiospecificity and complete E-selectivity. This outcome is especially advantageous for a domino Claisen process, where the global stereoselectivity depends on the E/Z selectivity of each step. The observed E-selectivity is likely due to the Ph group occupying the pseudoaxial position in the chair-like transition state, which disfavours the pseuodoaxial positioning of the OBn group. Furthermore, the aromatic ketone in the product enhances the synthetic versatility of this rearrangement, providing opportunities for selective derivatization and expanding its scope.
Double acetal 10 was also isomerized efficiently and selectively (Scheme 5a), a noteworthy result given the presence of three different terminal alkenes in the system, which could have introduced selectivity issues. Furthermore, cationic iridium complexes are often prone to chelation and deactivation by polyenes, potentially inhibiting reactivity altogether. Exposure of the crude isomerized acetal 26 to MgBr2 and LiAlH4 led to double rearrangement product 27, which was isolated in a decent 50% yield in an enantioenriched and diastereomerically pure form, with exclusive Z stereochemistry of the enol ether. An intriguing byproduct, 28, was also observed, likely formed by attack of the alkoxide from the first Claisen-reduction step onto the aldehyde generated in the second rearrangement, thereby preventing its reduction. The diastereomeric acetal 11 performed similarly well and led to the complementary diastereomer of the product, 29. This demonstrates that the ability to control the stereochemistry of the polyacetal substrates translates into access to all possible stereoisomers of the rearrangement products.
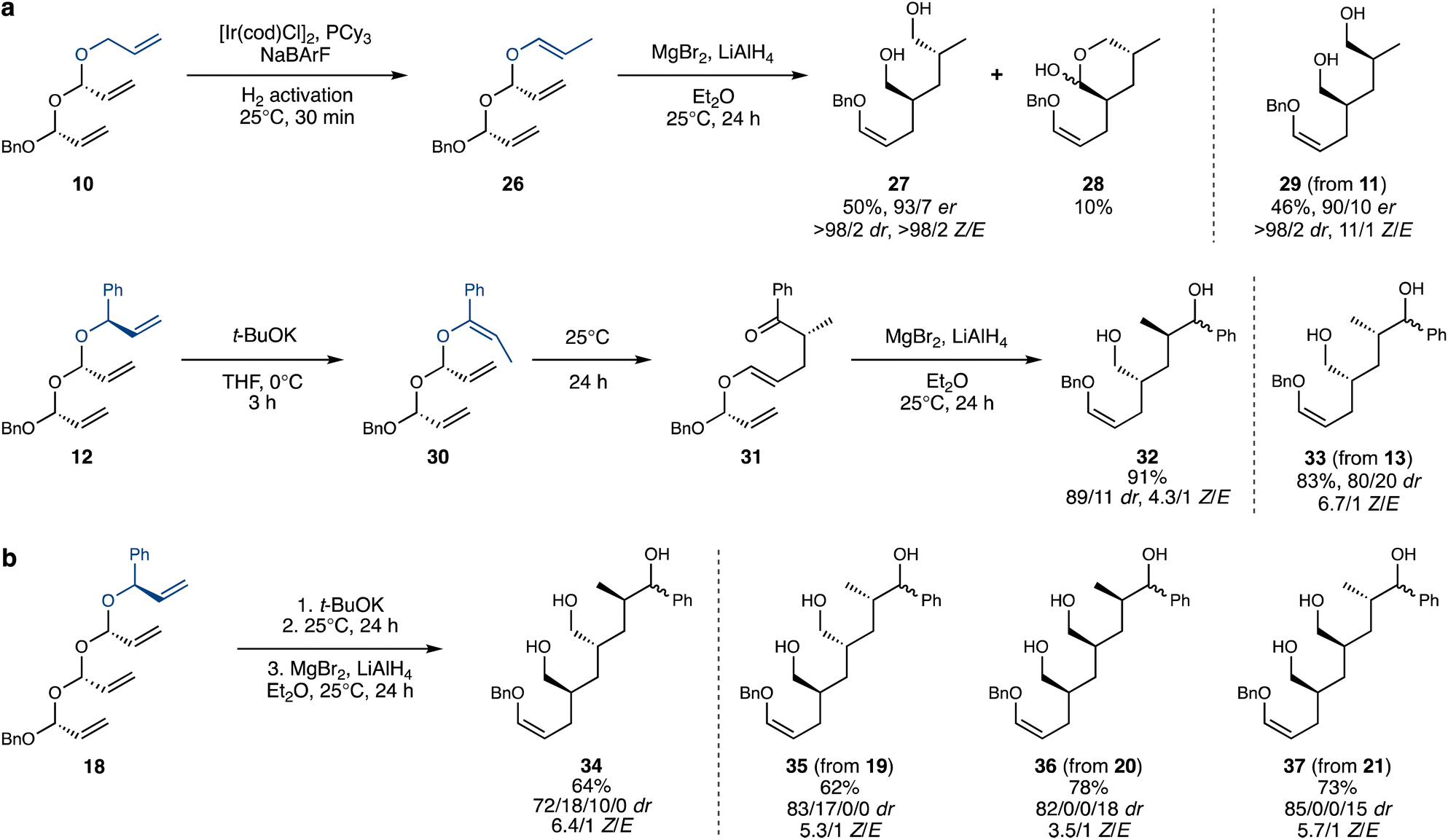 | ||
| Scheme 5 Isomerization–double (a) /triple (b) Coates–Claisen sequence. dr values for products 32–37 were assigned following acetylation and oxidation (see the ESI†). | ||
Branched double acetal 12 (and its diastereomer, 13) also successfully underwent isomerization with t-BuOK. Following an aqueous workup, 30 rearranged spontaneously at room temperature, leading to 31, again as a pure E enol ether. The crude 31 was then subjected to Coates–Claisen-reduction conditions, producing 32 in high yield. The stereochemical purity of 32 and 33 was confirmed by the acetylation of the primary alcohol38 and oxidation of the non-stereocontrolled secondary alcohol (see the ESI† for details).
Finally, we exposed triple acetals 18–21 to the isomerization–thermal rearrangement–Lewis-acid-mediated rearrangement sequence described above (Scheme 5b), aiming to achieve an isomerization–triple Coates–Claisen sequence. Pleasingly, triol products 34–37 were obtained in satisfactory yields and decent stereoselectivities, considering the complexity of the transformation. The attainment of triple Coates–Claisen products 34–37 completes the proof-of-concept for this project – encompassing the stereocontrolled iterative synthesis of polyacetals, their site-selective alkene isomerization and the stereospecific domino-Coates–Claisen rearrangement.
The carbonyl groups present in the rearrangement products (or the alcohols in the reduced products) represent versatile handles for further modification and functionalization. In this initial study, we chose the relatively conservative reduction of the alcohols into methyl groups, to demonstrate the synthetic relevance of the rearrangement products as precursors for deoxypropionate natural products.39 In particular, we targeted primary alcohol 41, a key intermediate in the total synthesis of several natural products including supellapyrone40 and tumescenamide C41 (Scheme 6).
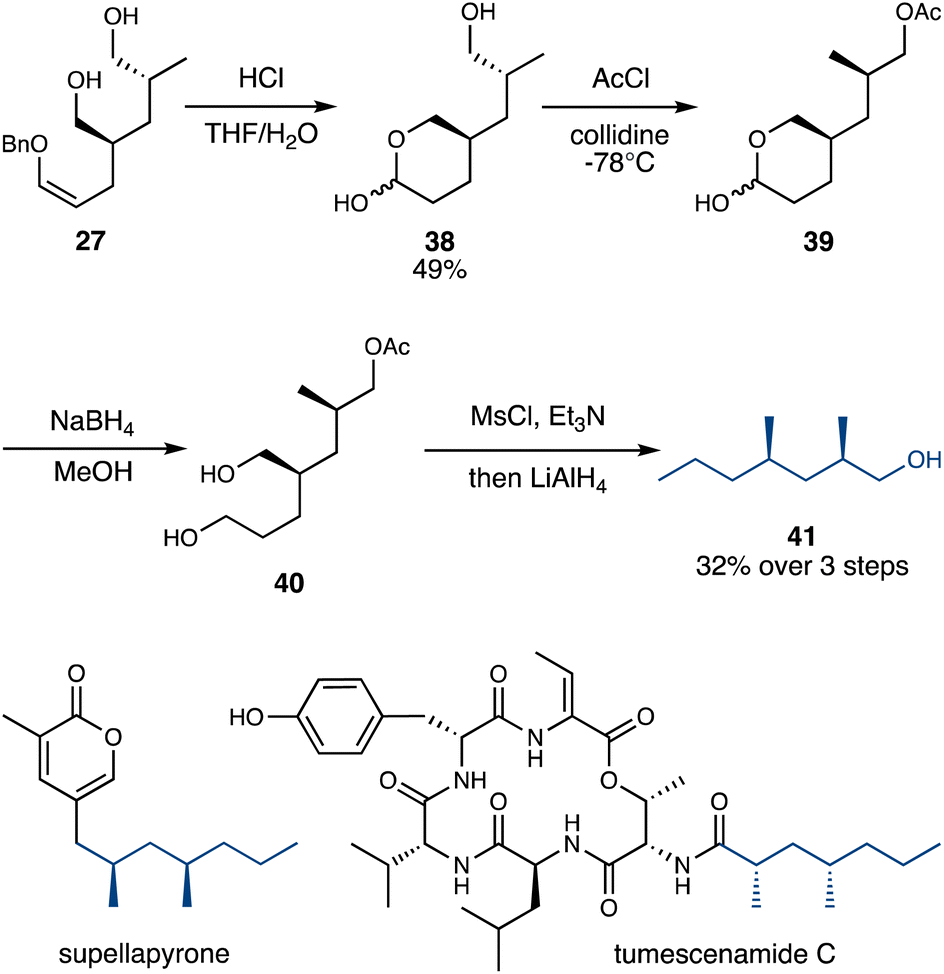 | ||
| Scheme 6 Application to the formal synthesis of deoxypropionates. | ||
The enol ether in double Claisen product 27 was hydrolyzed under acidic conditions, forming lactol 38. This hydrolysis thus selectively achieved the intramolecular protection of one primary alcohol in 27, allowing the remaining primary alcohol in 38 to be selectively acetylated.38 The resulting crude 39 was then treated with NaBH4 to reduce the lactol, after which 40 was doubly mesylated and then treated with LiAlH4, which reductively cleaved both alkyl mesylates and the acetate protecting group, furnishing alcohol 41 in 32% yield from 38 (Scheme 6).
Conclusions
In conclusion, we have developed an efficient and stereocontrolled approach for the synthesis of novel unsaturated polyacetals, featuring up to three consecutive allylic acetal linkages, through an iterative strategy. Beyond their intrinsic interest, these polyacetals were then utilized for their originally intended purpose: an isomerization-triggered domino-Coates–Claisen process. Remarkably, this transformation performed successfully, despite the multitude of potential pitfalls. The synthetic utility of the rearrangement products was preliminarily demonstrated through the synthesis of a deoxypropionate fragment.Overall, this study provides two significant contributions to the rapidly developing field of iterative synthesis:
(1) The efficient iterative assembly of a previously unexplored acyclic polyacetal backbone.
(2) The first example of the global rearrangement of an iteratively assembled skeleton, rather than a modification of functional groups at the periphery.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
I. Massad conceived the project, carried out the experiments and wrote the manuscript. I. Marek co-directed the project, obtained funding, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Notes and references
- R. B. Merrifield, Angew. Chem., Int. Ed., 1985, 24, 799–810 CrossRef.
- J. W. Lehmann, D. J. Blair and M. D. Burke, Nat. Rev. Chem, 2018, 2, 0115 CrossRef PubMed.
- K. Zheng, C. Xie and R. Hong, Front. Chem., 2015, 3, 32 Search PubMed.
- K. Yeung and V. K. Aggarwal, Homologation Reactions: Reagents, Applications, and Mechanisms, 2023, vol. 2, pp. 549–574 Search PubMed.
- I. P. Beletskaya, C. Najera and M. Yus, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS PubMed.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- R. M. Coates, S. K. Shah and R. W. Mason, J. Am. Chem. Soc., 1982, 104, 2198–2208 CrossRef CAS.
- R. M. Coates and S. J. Hobbs, J. Org. Chem., 1984, 49, 140–152 CrossRef CAS.
- R. M. Coates, B. D. Rogers, S. J. Hobbs, D. P. Curran and D. R. Peck, J. Am. Chem. Soc., 1987, 109, 1160–1170 CrossRef CAS.
- I. Massad, H. Sommer and I. Marek, Angew. Chem., Int. Ed., 2020, 59, 15549–15553 CrossRef CAS PubMed.
- R. Suresh, I. Massad and I. Marek, Chem. Sci., 2021, 12, 9328–9332 RSC.
- L. Segura, I. Massad, M. Ogasawara and I. Marek, Org. Lett., 2021, 23, 9194–9198 CrossRef CAS PubMed.
- I. Massad and I. Marek, Angew. Chem., Int. Ed., 2021, 60, 18509–18513 CrossRef CAS PubMed.
- A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 2556–2591 CrossRef CAS PubMed.
- D. Fiorito, S. Scaringi and C. Mazet, Chem. Soc. Rev., 2021, 50, 1391–1406 RSC.
- T. F. Schneider, J. Kaschel, B. Dittrich and D. B. Werz, Org. Lett., 2009, 11, 2317–2320 CrossRef CAS PubMed.
- W. Lim, J. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2014, 136, 13618–13621 CrossRef CAS PubMed.
- Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee and L. E. Overman, J. Am. Chem. Soc., 2017, 139, 7192–7195 CrossRef CAS PubMed.
- B. M. Trost, J. Xie and J. D. Sieber, J. Am. Chem. Soc., 2011, 133, 20611–20622 CrossRef CAS PubMed.
- D. Boese, M. Luebcke and J. Pietruszka, Synthesis, 2013, 729–733 CAS.
- N. Vicart, J. Goré and B. Cazes, Tetrahedron, 1998, 54, 11063–11078 CrossRef CAS.
- B. M. Trost and T. Zhang, Org. Lett., 2006, 8, 6007–6010 CrossRef CAS PubMed.
- I. Massad and I. Marek, ACS Catal., 2020, 10, 5793–5804 CrossRef CAS.
- A. M. Martín Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef PubMed.
- J. Stambasky, A. V. Malkov and P. Kocovsky, J. Org. Chem., 2008, 73, 9148–9150 CrossRef CAS PubMed.
- M. Kim and Y. H. Rhee, Org. Lett., 2021, 23, 3584–3587 CrossRef CAS PubMed.
- H. Luo, M. Zhang, Z.-Q. Xing and X.-C. Wang, J. Am. Chem. Soc., 2025, 147, 104–110 CrossRef PubMed.
- A recent example of a change in site-selectivity on the alcohol rather than allene partner: H. Guo, J. Kirchhoff, C. Strohmann, B. Grabe and C. C. Loh, Angew. Chem., Int. Ed., 2024, 63, e202400912 CrossRef CAS PubMed.
- D. Baudry, M. Ephritikhine and H. Felkin, J. Chem. Soc., Chem. Commun., 1978, 694–695 RSC.
- S. G. Nelson, C. J. Bungard and K. Wang, J. Am. Chem. Soc., 2003, 125, 13000–13001 CrossRef CAS PubMed.
- K. Maruoka and H. Yamamoto, Synlett, 1991, 793–794 CrossRef CAS.
- T. J. Fulton, A. Q. Cusumano, E. J. Alexy, Y. E. Du, H. Zhang, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2020, 142, 21938–21947 CrossRef CAS PubMed.
- N. F. O'Rourke and J. E. Wulff, Org. Biomol. Chem., 2014, 12, 1292–1308 RSC.
- L. Yao, K. Takeda, K. Ando and K. Ishihara, Chem. Sci., 2023, 14, 2441–2446 RSC.
- J. M. Burns, E. H. Krenske and R. P. McGeary, Synthesis, 2018, 50, 1750–1772 CrossRef CAS.
- B. Maryasin, D. Kaldre, R. Galaverna, I. Klose, S. Ruider, M. Drescher, H. Kählig, L. González, M. N. Eberlin and I. D. Jurberg, Chem. Sci., 2018, 9, 4124–4131 RSC.
- M. Murakami, H. Minamikawa and T. Mukaiyama, Chem. Lett., 1987, 16, 1051–1052 CrossRef.
- K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1993, 58, 3791–3793 CrossRef CAS.
- B. ter Horst, B. L. Feringa and A. J. Minnaard, Chem. Commun., 2010, 46, 2535–2547 RSC.
- X. Shi, W. S. Leal, Z. Liu, E. Schrader and J. Meinwald, Tetrahedron Lett., 1995, 36, 71–74 CrossRef CAS.
- N. Takahashi, K. Kaneko and H. Kakeya, J. Org. Chem., 2020, 85, 4530–4535 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08790a |
| This journal is © The Royal Society of Chemistry 2025 |