
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNMRExtractor: leveraging large language models to construct an experimental NMR database from open-source scientific publications†
Qinggong Wang‡
ab,
Wei Zhang‡bc,
Mingan Chenbde,
Xutong Libc,
Zhaoping Xiongf,
Jiacheng Xiong*b,
Zunyun Fu*d and
Mingyue Zheng *abc
*abc
aNanjing University of Chinese Medicine, 138 Xianlin Road, Nanjing 210023, China. E-mail: myzheng@simm.ac.cn
bDrug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: fuzunyun@simm.ac.cn; s19-xiongjiacheng@simm.ac.cn
cUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
dShanghaiTech University, Shanghai 201210, China
eLingang Laboratory, Shanghai 200031, China
fProtonUnfold Technology Co., Ltd, Suzhou, China
First published on 28th May 2025
Abstract
Nuclear magnetic resonance (NMR) spectroscopy is crucial for elucidating molecular structures, but NMR data extraction remains largely manual and time-consuming. We developed NMRExtractor, a locally deployable tool using a fine-tuned large language model, to address this challenge. By processing 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 734869 open-source scientific publications, we created NMRBank, a dataset containing 225809 entries with compound IUPAC names, NMR conditions, 1H and 13C NMR chemical shifts, data confidence levels, and reference information. Our analysis reveals that NMRBank's chemical space significantly surpasses existing public NMR datasets. The extraction process is highly scalable, allowing automatic processing of new research papers and continuous updates to NMRBank. This approach not only expands the available open NMR data space but also provides a foundation for AI-based NMR predictions and related chemical research. By automating data extraction and creating a comprehensive, regularly updated NMR database, NMRExtractor and NMRBank address the scarcity of publicly available experimental NMR data, potentially accelerating progress in various fields of chemical research.
734869 open-source scientific publications, we created NMRBank, a dataset containing 225809 entries with compound IUPAC names, NMR conditions, 1H and 13C NMR chemical shifts, data confidence levels, and reference information. Our analysis reveals that NMRBank's chemical space significantly surpasses existing public NMR datasets. The extraction process is highly scalable, allowing automatic processing of new research papers and continuous updates to NMRBank. This approach not only expands the available open NMR data space but also provides a foundation for AI-based NMR predictions and related chemical research. By automating data extraction and creating a comprehensive, regularly updated NMR database, NMRExtractor and NMRBank address the scarcity of publicly available experimental NMR data, potentially accelerating progress in various fields of chemical research.
Introduction
In the era of artificial intelligence (AI)-driven scientific research, high-quality, large-scale datasets are crucial for the success of machine learning models.1,2 In chemistry, comprehensive databases like PubChem3 and ChEMBL4 provide extensive information on compound structures, properties, and biological activities. The USPTO dataset offers valuable data on chemical reactions. These resources have enabled researchers to apply machine learning techniques to discover new chemical insights, optimize reaction conditions, and accelerate drug discovery.5,6 However, despite the abundance of data on molecular structures and reactions, there remains a significant deficiency in publicly available spectral data,7 which is essential for understanding the properties of substances.8Nuclear magnetic resonance (NMR) spectroscopy is one of the most powerful and widely used techniques in chemical research for investigating molecular structures and dynamics.9 By measuring the magnetic properties of atomic nuclei, NMR provides detailed information about the molecular environment, which is sensitive to structure and atomic interactions.10 The most direct application of NMR spectral data is in the structural identification of unknown compounds.11 Recent advancements, such as the BART-based Conditional Molecular Generation Network (CMGNet) proposed by Yao et al., demonstrate the potential for automatic structural elucidation using 13C NMR data and prior knowledge.12 Beyond compound identification,12–15 NMR chemical shifts also reflect the electronic environment of atoms, revealing their electrophilic characteristics.16 This information has been applied to chemical reaction prediction studies, such as enhancing the performance of graph neural networks in predicting aldehyde oxidase (AOX) metabolic sites17 and predicting the functionalization likelihood of atoms.18
Over the past two decades, several databases have been developed to store 1H and 13C NMR spectra of molecules.19–28 Notable examples include the Human Metabolome Database (HMDB),19 NMRShiftDB2,21,22 and the Natural Products Magnetic Resonance Database (NP-MRD).24 However, the scale of these databases remains limited. NMRShiftDB2, the largest open NMR database, contains only 53954 experimentally measured spectra for about 44909 molecules. To address the issue of data scarcity, Jia et al. developed SRCV, a machine learning-based NMR spectrum recognition system.29 However, this system is limited by its reliance on standardized NMR images and inability to extract associated information such as compound structures and measurement conditions.
Recently, large language models (LLMs) like ChatGPT have demonstrated powerful text understanding and processing capabilities, making them promising tools for text mining in scientific literature.30–34 For instance, Zheng et al. used prompt engineering to guide ChatGPT in extracting information about metal–organic framework synthesis from scientific literature.30 W. Coley et al. fine-tuned LLMs to extract reaction information from organic synthesis texts into structured data conforming to the Open Reaction Database (ORD) model.33 Our previous work showed that fine-tuned large language models achieved excellent performance on five chemical text extraction tasks.34 Additionally, we found that fine-tuning open-source LLMs like Mistral-7b-instruct-v-0.2 provides a viable alternative for text mining, offering comparable performance with reduced computational costs and increased flexibility for private deployment.
The application of LLMs is actively being explored for data extraction in the field of materials science.35–46 Polak et al. developed ChatExtract by leveraging conversational LLMs and prompt engineering to extract structured material property data.38 MatSci-NLP demonstrated how instruction tuning enhances LLM performance,39 while LLaMat showed that continued pretraining on materials science literature, followed by instruction tuning, enables superior performance in specialized tasks compared to general-purpose LLMs.40 LLMs are also used for large-scale data extraction, Kim et al. developed L2M3, a system that uses a fine-tuned LLM to extract MOF data from over 40000 scientific articles and organize it into a structured format.41 Meanwhile, retrieval-augmented generation (RAG) and agent-based systems have gained attention, with HoneyComb integrating a high-quality materials science knowledge base (MatSciKB) and a specialized toolset (ToolHub) to significantly enhance LLM reasoning and computational capabilities.43 PaperQA, leveraging RAG techniques, has outperformed other LLMs and commercial products in answering scientific literature-related queries, even surpassing human experts in comparative evaluations.44 MaterialsBERT successfully identified and processed approximately 681000 polymer-related articles. The data of 24 properties of over 106000 unique polymers extracted has been made publicly available to the scientific community via the Polymer Scholar website.46
In this study, we present NMRExtractor, a high-precision and easily extensible NMR data extraction tool utilizing LLMs. As illustrated in Fig. 1, NMRExtractor extracts comprehensive information including the IUPAC name of compounds, NMR conditions, and 1H and 13C NMR chemical shifts. Using this tool, we have created NMRBank, a dataset containing 225809 experimental NMR data entries. This dataset encompasses a wide range of NMR chemical information and covers a chemical space significantly larger than existing open-source NMR libraries. Crucially, NMRExtractor's ability to automatically process new research papers ensures that NMRBank can be continuously updated and expanded, addressing the ongoing need for comprehensive and up-to-date NMR data in chemical research.
 | ||
| Fig. 1 Schematic diagram of the NMR data extraction process and NMRBank construction using NMRExtractor. The process involves two main steps: (1) using regular expressions to identify NMR paragraphs, and (2) employing a fine-tuned large language model for batch extraction of NMR data. After eliminating entries with empty IUPAC names, we established the NMRBank dataset, comprising 225809 entries. Of these, 156621 entries successfully converted IUPAC names to SMILES notation. | ||
Methods
Workflow of NMR data extraction with NMRExtractor
The process of extracting NMR data using NMRExtractor and building NMRBank is shown in Fig. 2. In the first and second steps, we first access all open-access TXT documents in PubMed. To prevent potential parsing errors caused by encoding format mismatch, we first convert them to a unified UTF-8 encoding format and then use regular expressions to obtain all text paragraphs that mention NMR data. In the third step, after extracting NMR data from the NMR paragraphs using a large language model, we only retain data whose IUPAC names are not empty, and then further filter out data whose 1H and 13C NMR chemical shifts are not empty. In the fourth and fifth steps, we convert the IUPAC names of the compounds into SMILES and normalize the successfully converted SMILES. It is important to note that the term “IUPAC name” here is used broadly to include not only formal IUPAC names, but also commonly used names that can be recognized and interpreted by cheminformatics tools. A comprehensive example of NMR data extraction using NMRExtractor is provided in ESI and Fig. S1†, demonstrating the practical application of this streamlined process. | ||
| Fig. 2 The process of extracting NMR data from original research papers using NMRExtractor. Grey boxes represent starting and end points, purple parallelograms represent inputs and outputs, blue rectangles represent processing flows, and orange diamonds represent decision points in the pipeline. | ||
Extraction of NMR paragraphs
We downloaded open-access full-text articles in txt format from the PubMed database,47 using the latest version updated in June 2024, which contains 5734869 articles. All articles were first converted to UTF-8 encoding. In the literature containing NMR data, the IUPAC name is usually located in the first sentence of the paragraph or as an independent paragraph. 1H NMR and 13C NMR data are usually presented together, and 1H NMR data are usually located between the IUPAC name and 13C NMR data. Therefore, in order to obtain paragraphs containing NMR data and eliminate common writing differences such as 13C-NMR, 13CNMR, and 13C NMR, we use the regular expression 13C.{0,3}NMR for paragraph matching, means matching 13C followed by 0 to 3 arbitrary characters and then NMR. After the match is successful, we further splice the previous and next paragraphs to ensure that the obtained NMR paragraph contains the complete IUPAC name, 1H and 13C NMR data (Fig. S1†).
Fine-tuning and inference of LLMs
Based on our previous research,34 we found that fine-tuning the open-source Mistral-7b-instruct-v-0.2 model for NMR data extraction works almost as well as ChatGPT and can be deployed locally. We fine-tuned all parameters of Mistral-7b-instruct-v-0.2 and Llama3-8b-instruct on a 4 × 40GB A100, and Q-LoRA fine-tuned Llama2-13b-instruct on a 1 × 40GB A100.48 For inference phase, we used vLLM to boost speed,49 achieving an average inference speed of 2 records per second on a single 40GB A100. Examples of prompts used for fine-tuning and inference can be found in Fig. S2†. The hyperparameters used in this study are consistent with our previous work and provided in Table S1†. Details on hardware resources, memory cost, and runtime for both model fine-tuning and inference are provided in Table S2†.Evaluation of the performance of the LLMs
To enhance the performance of the model in extracting NMR data from millions of papers, we expanded our training set from 300 to 1000 samples. We added 700 NMR paragraphs from articles published before 2023 to the original Paragraph2NMR task dataset.34 For a more comprehensive evaluation, we created a new test set of 1022 unique texts, by randomly selecting 1–2 paragraphs from NMR articles published in 2023. All data are manually annotated by chemistry experts, the annotations include the compound IUPAC name, 1H/13C NMR measurement conditions and chemical shift. The 1H NMR chemical shift also retains information such as coupling constants, and N/A is used to fill in the non-existent content (Fig. S3†). We report results on two test sets: (1) test set 1, derived from our previous publication, serves as a benchmark for comparison with existing methods; (2) test set 2, newly constructed for this study, contains more diverse chemical structures and NMR data formats, including challenging cases not present in test set 1. The two test sets are completely non-overlapping, enabling a robust assessment across different data distributions and annotation styles. This dual evaluation provides both a direct comparison to prior work and an assessment of the model's generalization to real-world NMR data.We used exact match accuracy as our evaluation metric, and we also recognized that this is a particularly strict standard. Some failures under this metric were merely slight rephrasings by the model that would be considered correct by human evaluators (Fig. S4 and S5†).
Post-processing
Post-processing is essential for information extraction, particularly when dealing with inconsistent and diverse scientific data. In the research on standardized datasets in the field of materials science, after successfully extracting named entities related to materials science, Leigh Weston et al. used a normalization method combining rules and look-up tables to convert synonymous entities into a standardized name.50 Pranav Shetty et al. focused on the normalization of polymer named entities and trained a supervised clustering model with Word2Vec and fastText word vectors to classify named entities referring to the same polymer.51 Similarly, as an optimized multi-algorithm mapping method, ChemProps can unify polymer name expressions through API calls.52In our work, after extracting the NMR data using NMRExtractor, we standardized the output by: (1) to enhance the success rate of IUPAC-to-SMILES conversion, all non-standard characters in the IUPAC names were standardized. (2) Converting IUPAC names to SMILES using ChemDraw; (3) Using the Open Parser for Systematic IUPAC nomenclature (OPSIN) online service53 for IUPAC names ChemDraw couldn't convert; (4) standardizing SMILES with RDKit to ensure data consistency and usability (Fig. S6†).54
Results
Data preparation
We analyzed 5734869 open access articles from the PubMed database and identified 380220 paragraphs mentioning NMR passages from 58795 articles using rule-based method. The proportion of articles with NMR messages in the PubMed database is relatively small (Fig. 3a). Of these 380220 paragraphs, approximately 260000 paragraphs contain NMR data, originating from 35270 articles (Fig. 3b).
 | ||
| Fig. 3 Article counts and NMR data description formats. (a) Total open-access papers in PubMed (grey) versus papers mentioning NMR (blue) by year. (b) Papers mentioning NMR (blue) and those including NMR data (red) in PubMed. (c) Examples of standardized (test set 2_standard) and non-standardized NMR (test set 2_non-standard) data descriptions in papers. Note: while this data reflects PubMed's June 2024 update, papers from 2023 and 2024 are underrepresented due to incomplete updating. | ||
To enhance our evaluation, we expanded upon our previous work's 300 training and 300 test data items (test set 1) by creating a diverse test set of 1022 NMR data paragraphs (test set 2). This new set ensures comprehensive coverage across various journal types and allows for a thorough assessment of model performance. We identified a total of 10958 NMR paragraphs in NMR articles published in 2023.
From these, we randomly sampled 1022 paragraphs from 115 different journal types, which were then manually annotated by chemistry experts (Fig. S7†). We classified the 1022 NMR paragraphs into standardized and non-standardized descriptions based on the presentation format of the NMR data in the research paper (Fig. 3c). This classification resulted in 788 standardized NMR data descriptions (test set 2_standard) and 244 non-standardized descriptions (test set 2_non-standard). To further augment our dataset, we randomly selected and manually annotated 700 NMR paragraphs published before 2023, increasing our training set to 1000 samples.
Model performance
As shown in Fig. 4a, the performance of the fine-tuned Mistral-7b-instruct-v-0.2 model improves and gradually converges as the training set size increases. Optimal results across different test sets were achieved with a training set of 800 samples, outperforming both Llama3-8b-instruct and Llama2-13b-chat (Tables S3 and S4†). The model attained an accuracy exceeding 0.96 for various element extractions in test set 1 (300) and over 0.85 in test set 2 (1022). Further analysis of test set 2 (1022), categorized by text description methods, revealed an accuracy surpassing 0.9 for element extraction in test set 2_standard (778). However, test set 2_non-standard (244) posed challenges due to non-standardized NMR data descriptions. These natural language descriptions, characterized by diverse expressions and complex grammatical structures, often contained incomplete or missing information, hindering NMR data extraction and slightly reducing model performance. To further demonstrate the model's accuracy and data usability, we examined instances where both the IUPAC name and NMR chemical shift terms were correctly extracted. A prediction is deemed correct only when both components were accurate; otherwise, it was classified as incorrect. As the training set expanded to 800, the accuracy of correctly extracting both the IUPAC name and 1H NMR chemical shift could reach 0.78 in test set 2 (1022). And the accuracy of correctly extracting both the IUPAC name and 13C NMR chemical shift could reach 0.804 in test set 2 (1022) (Fig. 4b). | ||
| Fig. 4 (a) The performance of the fine-tuned Mistral-7b-instruct-v-0.2 on different test sets varies with the number of training sets changes. (b) The exact match accuracy of both the IUPAC name and 1H/13C NMR chemical shifts on different test sets with the number of training sets changes. Error bars represent the standard deviation across three runs with independently sampled training data. | ||
Data confidence and accuracy
To objectively evaluate NMRExtractor's performance, we assessed both the overall accuracy of extracted data and the model's confidence score for each data point. The confidence score ranges from 0 to 1. The confidence of the model predictions is computed based on the cumulative log probability of the predicted tokens. For detailed on how confidence levels are calculated, please refer to the ESI.† In test set 1 (300) and test set 2 (1022), when confidence exceeds 0.6, the accuracy of extracting all elements reaches over 86%. When confidence exceeds 0.8, the complete accuracy surpasses 97% (Fig. 5a and b). Results from three-fold cross-validation on the training set further confirm that model performance improves with increasing confidence (Table S5†). According to earlier studies,55 the error rate in manually curated bioactivity databases (such as ChEMBL and WOMBAT) is around 5%. This suggests that high-confidence predictions (confidence >0.8) of our model may achieve human-level accuracy, supporting its reliable large-scale deployment. | ||
| Fig. 5 NMRExtractor's performance across different test sets and confidence intervals. The number of data points in each confidence interval is shown below it. (a) The performance of test set 1 (300) with different confidence intervals. (b) The performance of test set 2 (1022) with different confidence intervals. (c) The performance of test set 2_standard (778) with different confidence intervals. (d) The performance of test set 2_non-standard (244) with different confidence intervals. Error bars represent the standard deviation across three runs with independently sampled training data. | ||
For test set 1 (300), 261 entities have a confidence score above 0.6, with 209 entities scoring above 0.8 (Fig. 5a). In the test set 2 (1022), 594 entities score above 0.6, and 386 entities above 0.8 (Fig. 5b). Test set 1 (300) and test set 2_standard (778) show a high number of high confidence values and good model performance (Fig. 5a and c). While test set 2_non-standard (244) shows poorer model performance overall, when the confidence score exceeds 0.8, the accuracy of element extraction still exceeds 88% (Fig. 5d). The consistently high accuracy of high-confidence data across different data description formats allow us to filter data based on model confidence after batch data extraction, thereby improving overall data quality.
Data extraction methods comparison
Before using a fine-tuned large language model for NMR data extraction, we explored a variety of methods to extract NMR data from text. Initially, we attempted to extract NMR data from research papers using a traditional rule-based approach. Based on the standardized description of NMR data in research papers, we developed regular expressions to locate NMR paragraphs, isolated the sections containing NMR data, and then applied additional rules to obtain the required NMR data (Fig. S8†). For IUPAC names, we compiled a list of 618 common compound group words (Fig. S9†) and scanned the text for these words to extract relevant information. However, the rule-based extraction of NMR data from research papers presented challenges. To accommodate the diversity of text, the rules required constant modification and refinement. We discovered that even with continuous addition of new rules, it was impossible to cover all scenarios. Moreover, the presence of reactants and solvents in the text interfered with the extraction of IUPAC names, resulting in both missed NMR data and errors in the extracted information. In comparison to NMRExtractor, which is based on large language models, the traditional rule-based method performed poorly on both test set 1 (300) and test set 2 (1022) (Fig. 6a and b). Furthermore, in batch data extraction, the large language model-based method can filter data according to the confidence level of each entry, whereas the rule-based method lacks the ability to assess the accuracy of the extracted NMR data. | ||
| Fig. 6 Performance comparison of rule-based method (blue) and NMRExtractor (red) on (a) test set 1 (300 samples) and (b) test set 2 (1022 samples). | ||
NMR data extraction and analysis
We employed NMRExtractor to process 380220 NMR paragraphs in batch. After removing entries with empty 13C NMR chemical shifts, we obtained approximately 260000 entries. Further filtering out entries with both empty IUPAC names and NMR chemical shifts yielded 225809 entries. To standardize these data, we converted IUPAC names to SMILES using ChemDraw and OPSIN, successfully converting 156621 entries. We then standardized the SMILES using RDKit (Fig. S6†), ultimately constructing the NMRBank dataset. During standardization, stereoisomer details are preserved in the SMILES string. Of these entries, the total number of unique SMILES strings is 149135, representing approximately 66% of our total records. This normalization process ensures that structural duplicates are properly accounted for, providing a more accurate comparison with existing databases.
Our detailed analysis of the 156621 records containing SMILES revealed 2906 compounds with multiple entries. The frequency distribution follows a power-law pattern, which is characteristic of chemical databases and reflects the prevalence of commonly studied compounds in the literature. We have visualized this distribution in Fig. S10† and provided a comprehensive list of the most frequently occurring compounds in Table S6†. For applications requiring the highest data quality, we identified 91707 unique SMILES records within our highest confidence interval (0.8–1.0). This subset represents our most reliable data points and is particularly valuable for experimental validation and machine learning applications.
We also analyzed the data distribution of the public NMRshiftDB2 dataset to compare it with NMRBank. As shown in Fig. 7, the distribution of physicochemical properties in NMRBank, including molecular weight, logP, and TPSA, significantly differs from that of NMRShiftDB2. Moreover, the property ranges observed in NMRShiftDB2 are fully contained within those of NMRBank, while NMRBank demonstrates a broader spread across all examined properties. These results indicate that NMRBank covers a more diverse and expansive chemical space. Based on the confidence level, we further assessed the accuracy of the data in NMRBank data. A rule-based approach was used to check whether the chemical shift values appeared sequentially in the paragraphs from which they were extracted and whether any modifications had occurred. Within the high-confidence interval, the 1H and 13C NMR chemical shift values and their order in NMRBank exhibited a high degree of consistency with the paragraphs in the original paper (Fig. S11†).
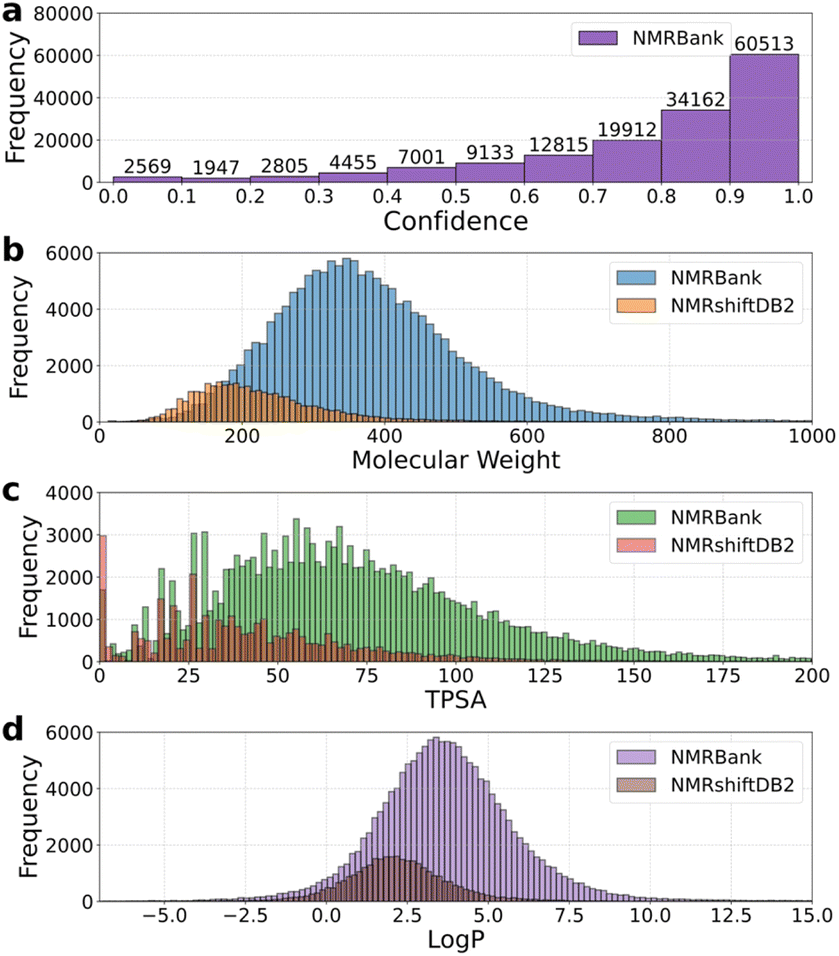 | ||
| Fig. 7 (a) Distribution of confidence values for data in the NMRBank. (b) Molecular weight distribution of molecules from NMRBank and NMRShiftDB2. (c) TPSA distribution of molecules from NMRBank and NMRShiftDB2. (d) Distribution of calculated logP of molecules from NMRBank and NMRShiftDB2. | ||
To illustrate typical extraction errors, we conducted case studies on missed NMR paragraphs caused by regex limitations. We employed the regular expression “13C.{0,3}NMR” as our primary method to identify NMR-relevant paragraphs. While this approach proved highly effective for standard NMR notation formats, we acknowledge its inherent limitations. Through comprehensive analysis, we identified several variant notations such as “C13 NMR”, “13 C-NMR”, or “C13-NMR” that could potentially be missed (Fig. S12†). Our thorough investigation of 5.7 million articles revealed that these variant notations appeared 1962 times, representing approximately 0.5% of the total 380220 identified NMR paragraphs. This small percentage demonstrates the robustness of our chosen regular expression pattern while acknowledging room for future optimization.
We conducted a systematic analysis of the conversion gap between IUPAC names and SMILES strings. We implemented a multi-tool approach, first using ChemDraw and then OPSIN for failed cases, to maximize conversion success. However, some structures remained challenging for both tools. Among the approximately 226000 records, around 70000 (∼31%) could not be successfully converted to SMILES strings. This gap can be attributed to several factors: (1) complex molecular structures: some compounds, particularly natural products and complex synthetic molecules, contain challenging structural features that exceed the current capabilities of conversion tools. As shown in Fig. S13,† even accurately extracted IUPAC names like 2-[2-(2,6-dichlorophenyl)amino]benzyl-3-(2-hydroxyphenylacrylamido)-6,8-dibromoquinazolin-4(3H)ones could not be converted due to their structural complexity; (2) common names: some compounds are referred to by common or trade names rather than systematic IUPAC names, such as Telisatin A. These names often lack the structural detail required by conversion tools like ChemDraw and OPSIN (Fig. S13†); we are actively working on improving our conversion pipeline and exploring additional chemical structure parsing tools to reduce this gap in future updates.
Existing NMR database
In addition to analyzing statistical properties, we compared NMRBank to the most common NMR databases (Table 1). Our findings revealed that NMRBank contains significantly more entries than any existing open-source NMR database. Moreover, our NMR extraction process offers excellent scalability. By utilizing our NMRExtractor, we can quickly and automatically process new literature, greatly facilitating the continuous updating of the NMRBank database.| Dataset name | Number of NMR spectra | Number of compounds | Compound types | NMR technique | URL | Available |
|---|---|---|---|---|---|---|
| HMDB5.0 (ref. 19) | 4149 | — | Metabolites | 1H/13C | https://hmdb.ca/ | Open-source |
| BMRB20 | 1200+ | — | Small molecule metabolites | 1H/13C | https://bmrb.io/ | Open-source |
| NMRShiftDB2 (ref. 21 and 22) | 53954 |
— | Not specified | 1H/13C | https://nmrshiftdb.nmr.uni-koeln.de/ | Open-source |
| SDBS23 | 15218 (1H) 13457 (13C) |
900+ | Natural products | 1H/13C | https://sdbs.db.aist.go.jp/Disclaimer.aspx | Open-source |
| NP-MRD24 | 1290 | — | Natural products | 1H/13C | https://np-mrd.org/ | Open-source |
| NAPROC-13 (ref. 25) | 6000+ | — | Natural products | 13C | https://c13.materia-medica.net/ | Open-source |
| CH-NMR-NP26 | 30500 |
926 | Natural products | 1H/13C | https://ch-nmr-np.jeol.co.jp/en/nmrdb/ | Open-source |
| Spektraris-NMR27 | 466 | 250 | Taxanes | 1H/13C | http://langelabtools.wsu.edu/nmr/ | Open-source |
| C6H6 (ref. 28) | — | 506 | Not specified | 1H/13C | https://www.c6h6.org/ | Open-source |
| Ilm-NMR-P31 (ref. 56) | 14250 |
13730 |
Not specified | 31P | https://github.com/clacor/Ilm-NMR-P31 | Open-source |
| KnowItAll NMR57 | 1280000+ |
— | Not specified | 1H/13C | https://sciencesolutions.wiley.com/solutions/technique/nmr/knowitall-nmr-collection/ | Commercial |
| Micronmr58 | 1000000+ |
— | Not specified | 13C | https://www.nmrdata.com | Commercial |
| NMRBank | 225809 |
149135 |
Not specified | 1H/13C | https://github.com/eat-sugar/NMRExtractor | Open-source |
Data records
The NMRBank dataset constructed in this work is available at https://github.com/eat-sugar/NMRExtractor, a public data repository providing open code and data for researchers, research projects/teams, journals, institutions, universities, etc. Each data entry in the NMRBank dataset includes: article PMID, NMR paragraph, compound IUPAC name, SMILES, 1H NMR conditions, 1H NMR chemical shifts, 13C NMR conditions, 13C NMR chemical shift, confidence, and other metadata such as article information. Table 2 provides an overview of the information items in the NMRBank dataset, while Table 3 presents examples from the NMRBank dataset.| Data description | Data value |
|---|---|
| Dataset name | NMRBank |
| Data introduction | Contains NMR data from 5.7 million PubMed articles |
| Types of NMR technique covered | 1H/13C NMR |
| Total number of NMR data | 225809 |
| The number of NMR data with SMILES | 156621 |
| The number of NMR data with SMILES and confidence greater than 0.8 | 94675 |
| The number of NMR data with SMILES and confidence greater than 0.6 | 127402 |
| The number of NMR data with unique SMILES | 149135 |
| The number of NMR data with unique SMILES and confidence greater than 0.8 | 91707 |
| The number of NMR data with unique SMILES and confidence greater than 0.6 | 123174 |
| URL | https://github.com/eat-sugar/NMRExtractor |
| Available | Open-source |
| Data description | Data value |
|---|---|
| PubMed ID (PMID) | 35601446 |
| PubChem CID | 35960 |
| NMR paragraph | 4.2.1.4. 2,6-Dimethoxy-4-vinylphenol 2d yellow oil, yield 94%. IR (KBr plate): νmax 3144, 2938, 2844, 1605, 1462, 1213, 1115, 837. 1H NMR (600 MHz, CDCl3) δ 6.65 (s, 2H), 6.61 (dd, J = 17.5, 10.9 Hz, 1H), 5.60 (d, J = 17.5 Hz, 1H), 5.56 (s, 1H), 5.15 (d, J = 10.8 Hz, 1H), 3.90 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 147.06, 136.83, 134.76, 129.18, 111.87, 102.9, 56.26 |
| IUPAC name of compound | 2,6-Dimethoxy-4-vinylphenol |
| SMILES | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cc1cc(OC)c(O)c(OC)c1 Cc1cc(OC)c(O)c(OC)c1 |
| 1H NMR conditions | 600 MHz, CDCl3 |
| 1H NMR chemical shift | 6.65 (s, 2H), 6.61 (dd, J = 17.5, 10.9 Hz, 1H), 5.60 (d, J = 17.5 Hz, 1H), 5.56 (s, 1H), 5.15 (d, J = 10.8 Hz, 1H), 3.90 (s, 6H) |
| 13C NMR conditions | 151 MHz, CDCl3 |
| 13C NMR chemical shift | 147.06, 136.83, 134.76, 129.18, 111.87, 102.9, 56.26 |
| Confidence in data given by large language models | 0.927 |
| Article citation | R. Soc. Open Sci.; 9(4): 220014 |
Conclusions
NMR data contains crucial chemical properties, yet it is scattered across complex scientific literature in various forms. Extracting experimental NMR data from these documents is a challenging but vital task. In this study, we leveraged large language model technology to mine and construct an NMR database. We developed NMRExtractor and used it to batch-extract NMR data from over 5.7 million public documents in the PubMed database. This effort resulted in the NMRBank dataset, containing 225809 NMR entries, with 156621 entries including SMILES descriptors. Each entry comprises the compound's IUPAC name, SMILES descriptor, 1H NMR, 13C NMR, model-assigned confidence score, and source metadata such as article number and journal name.
Comparative analysis reveals that NMRBank is currently the largest open-source experimental NMR dataset available, which covers a wide range of chemical space. This comprehensive resource is poised to significantly advance NMR data-driven deep learning and its applications in chemistry. NMRExtractor's ability to automatically process new research papers for NMR data extraction ensures efficient updates to the NMRBank dataset. Moving forward, we plan to continually refine NMRExtractor and expand the NMRBank dataset. This research not only broadens the scope and accessibility of experimental NMR data but also introduces a versatile data extraction method applicable to chemical research and drug design. Ultimately, these advancements will accelerate the discovery and development of new drugs.
Data availability
All data and code of this work are available at GitHub: https://github.com/eat-sugar/NMRExtractor. The model weights of NMRExtractor can be downloaded from https://huggingface.co/sweetssweets/NMRExtractor. We also provide an online demo of NMRExtractor on https://huggingface.co/spaces/sweetssweets/NMRExtractor.Author contributions
M. Y. Z., Z. Y. F., and J. C. X. conceived the idea. Q. G. W., W. Z. and M. Y. Z. designed the research. Q. G. W., W. Z. implemented the codes. Q. G. W., W. Z., M. A. C. collected, annotated, and processed training data. Q. G. W., W. Z., Z. Y. F. checked the data. Q. G. W., W. Z., Z. P. X. benchmarked the models. Q. G. W. wrote the initial draft. M. Y. Z., Z. Y. F., J. C. X., and X. T. L. reviewed and refined the article. All authors contributed to the analysis of the results. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We extend our gratitude to PubMed for offering a wealth of open literature, as well as to the open-source communities and tools like LLM and OPSIN for their invaluable contributions. This work was supported by the National Natural Science Foundation of China (T2225002 and 82273855 to M. Y. Z. and 82204278 to X. T. L.), the National Key Research and Development Program of China (2022YFC3400504 to M. Y. Z.), the SIMM-SHUTCM Traditional Chinese Medicine Innovation Joint Research Program (E2G805H to M. Y. Z.), the Shanghai Post-doctoral Excellence Program (2023693 to Z. Y. F. and 2024707 to J. C. X.) and the Shanghai Municipal Science and Technology Major Project.References
- D. Zha, Z. P. Bhat, K.-H. Lai, F. Yang, Z. Jiang, S. Zhong and X. Hu, Data-centric Artificial Intelligence: A Survey, ACM Comput. Surv., 2025, 57, 1–42 CrossRef.
- L. Budach, M. Feuerpfeil, N. Ihde, A. Nathansen, N. Noack, H. Patzlaff, F. Naumann and H. Harmouch, arXiv, 2022, preprint, arXiv:2207.14529, DOI:10.48550/arXiv.2207.14529.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, PubChem Substance and Compound databases, Nucleic Acids Res., 2015, 44, D1202–D1213 CrossRef PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, ChEMBL: a large-scale bioactivity database for drug discovery, Nucleic Acids Res., 2011, 40, D1100–D1107 CrossRef PubMed.
- M. Staszak, K. Staszak, K. Wieszczycka, A. Bajek, K. Roszkowski and B. Tylkowski, Machine learning in drug design: Use of artificial intelligence to explore the chemical structure–biological activity relationship, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1568 Search PubMed.
- A. C. Mater and M. L. Coote, Deep Learning in Chemistry, J. Chem. Inf. Model., 2019, 59, 2545–2559 CrossRef CAS PubMed.
- N. Norori, Q. Hu, F. M. Aellen, F. D. Faraci and A. Tzovara, Addressing bias in big data and AI for health care: A call for open science, Patterns, 2021, 2, 100347 CrossRef PubMed.
- I. V. Tetko, O. Engkvist, U. Koch, J.-L. Reymond and H. Chen, BIGCHEM: Challenges and Opportunities for Big Data Analysis in Chemistry, Mol. Inform., 2016, 35, 615–621 CrossRef CAS PubMed.
- P. R. L. Markwick, T. Malliavin and M. Nilges, Structural Biology by NMR: Structure, Dynamics, and Interactions, PLoS Comput. Biol., 2008, 4, e1000168 CrossRef PubMed.
- H. Günther, NMR spectroscopy: basic principles, concepts and applications in chemistry, John Wiley & Sons, 2013 Search PubMed.
- X. Xue, H. Sun, M. Yang, X. Liu, H.-Y. Hu, Y. Deng and X. Wang, Advances in the Application of Artificial Intelligence-Based Spectral Data Interpretation: A Perspective, Anal. Chem., 2023, 95, 13733–13745 CrossRef CAS PubMed.
- L. Yao, M. Yang, J. Song, Z. Yang, H. Sun, H. Shi, X. Liu, X. Ji, Y. Deng and X. Wang, Conditional Molecular Generation Net Enables Automated Structure Elucidation Based on 13C NMR Spectra and Prior Knowledge, Anal. Chem., 2023, 95, 5393–5401 CrossRef CAS PubMed.
- Z. Huang, M. S. Chen, C. P. Woroch, T. E. Markland and M. W. Kanan, A framework for automated structure elucidation from routine NMR spectra, Chem. Sci., 2021, 12, 15329–15338 RSC.
- Z. Yang, J. Song, M. Yang, L. Yao, J. Zhang, H. Shi, X. Ji, Y. Deng and X. Wang, Cross-Modal Retrieval between 13C NMR Spectra and Structures for Compound Identification Using Deep Contrastive Learning, Anal. Chem., 2021, 93, 16947–16955 CrossRef CAS PubMed.
- M. Alberts, F. Zipoli and A. C. Vaucher, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-8wxcz.
- C. P. Gordon, C. Raynaud, R. A. Andersen, C. Copéret and O. Eisenstein, Carbon-13 NMR Chemical Shift: A Descriptor for Electronic Structure and Reactivity of Organometallic Compounds, Acc. Chem. Res., 2019, 52, 2278–2289 CrossRef CAS PubMed.
- J. Xiong, R. Cui, Z. Li, W. Zhang, R. Zhang, Z. Fu, X. Liu, Z. Li, K. Chen and M. Zheng, Transfer learning enhanced graph neural network for aldehyde oxidase metabolism prediction and its experimental application, Acta Pharm. Sin. B, 2024, 14, 623–634 CrossRef CAS PubMed.
- E. King-Smith, F. A. Faber, U. Reilly, A. V. Sinitskiy, Q. Yang, B. Liu, D. Hyek and A. A. Lee, Predictive Minisci late stage functionalization with transfer learning, Nat. Commun., 2024, 15, 426 CrossRef CAS PubMed.
- D. S. Wishart, A. Guo, E. Oler, F. Wang, A. Anjum, H. Peters, R. Dizon, Z. Sayeeda, S. Tian, B. L. Lee, M. Berjanskii, R. Mah, M. Yamamoto, J. Jovel, C. Torres-Calzada, M. Hiebert-Giesbrecht, V. W. Lui, D. Varshavi, D. Varshavi, D. Allen, D. Arndt, N. Khetarpal, A. Sivakumaran, K. Harford, S. Sanford, K. Yee, X. Cao, Z. Budinski, J. Liigand, L. Zhang, J. Zheng, R. Mandal, N. Karu, M. Dambrova, H. B. Schiöth, R. Greiner and V. Gautam, HMDB 5.0: the Human Metabolome Database for 2022, Nucleic Acids Res., 2021, 50, D622–D631 CrossRef PubMed.
- J. C. Hoch, K. Baskaran, H. Burr, J. Chin, H. R. Eghbalnia, T. Fujiwara, M. R. Gryk, T. Iwata, C. Kojima, G. Kurisu, D. Maziuk, Y. Miyanoiri, J. R. Wedell, C. Wilburn, H. Yao and M. Yokochi, Biological Magnetic Resonance Data Bank, Nucleic Acids Res., 2022, 51, D368–D376 CrossRef PubMed.
- S. Kuhn, H. Kolshorn, C. Steinbeck and N. Schlörer, Twenty years of nmrshiftdb2: A case study of an open database for analytical chemistry, Magn. Reson. Chem., 2024, 62, 74–83 CrossRef CAS PubMed.
- C. Steinbeck, S. Krause and S. Kuhn, NMRShiftDBConstructing a Free Chemical Information System with Open-Source Components, J. Chem. Inf. Comput. Sci., 2003, 43, 1733–1739 CrossRef CAS PubMed.
- T. Saito and S. Kinugasa, Development and release of a spectral database for organic compounds-key to the continual services and success of a large-scale database, Synthesiology, 2011, 4, 35–44 CrossRef.
- D. S. Wishart, Z. Sayeeda, Z. Budinski, A. Guo, B. L. Lee, M. Berjanskii, M. Rout, H. Peters, R. Dizon, R. Mah, C. Torres-Calzada, M. Hiebert-Giesbrecht, D. Varshavi, D. Varshavi, E. Oler, D. Allen, X. Cao, V. Gautam, A. Maras, E. F. Poynton, P. Tavangar, V. Yang, J. A. van Santen, R. Ghosh, S. Sarma, E. Knutson, V. Sullivan, A. M. Jystad, R. Renslow, L. W. Sumner, R. G. Linington and J. R. Cort, NP-MRD: the Natural Products Magnetic Resonance Database, Nucleic Acids Res., 2021, 50, D665–D677 CrossRef PubMed.
- J. L. López-Pérez, R. Therón, E. del Olmo and D. Díaz, NAPROC-13: a database for the dereplication of natural product mixtures in bioassay-guided protocols, Bioinformatics, 2007, 23, 3256–3257 CrossRef PubMed.
- K. Asakura, A NMR spectral database of natural products "CH-NMR-NP", J. Synth. Org. Chem., 2015, 73, 1247–1252 CrossRef CAS.
- J. T. Fischedick, S. R. Johnson, R. E. B. Ketchum, R. B. Croteau and B. M. Lange, NMR spectroscopic search module for Spektraris, an online resource for plant natural product identification – Taxane diterpenoids from Taxus×media cell suspension cultures as a case study, Phytochemistry, 2015, 113, 87–95 CrossRef CAS PubMed.
- L. Patiny, M. Zasso, D. Kostro, A. Bernal, A. M. Castillo, A. Bolaños, M. A. Asencio, N. Pellet, M. Todd, N. Schloerer, S. Kuhn, E. Holmes, S. Javor and J. Wist, The C6H6 NMR repository: An integral solution to control the flow of your data from the magnet to the public, Magn. Reson. Chem., 2018, 56, 520–528 CrossRef CAS PubMed.
- W. Jia, Z. Yang, M. Yang, L. Cheng, Z. Lei and X. Wang, Machine Learning Enhanced Spectrum Recognition Based on Computer Vision (SRCV) for Intelligent NMR Data Extraction, J. Chem. Inf. Model., 2021, 61, 21–25 CrossRef CAS PubMed.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT Chemistry Assistant for Text Mining and the Prediction of MOF Synthesis, J. Am. Chem. Soc., 2023, 145, 18048–18062 CrossRef CAS PubMed.
- Q. Chen, H. Sun, H. Liu, Y. Jiang, T. Ran, X. Jin, X. Xiao, Z. Lin, H. Chen and Z. Niu, An extensive benchmark study on biomedical text generation and mining with ChatGPT, Bioinformatics, 2023, 39, btad557 CrossRef CAS PubMed.
- J. Dagdelen, A. Dunn, S. Lee, N. Walker, A. S. Rosen, G. Ceder, K. A. Persson and A. Jain, Structured information extraction from scientific text with large language models, Nat. Commun., 2024, 15, 1418 CrossRef CAS PubMed.
- Q. Ai, F. Meng, J. Shi, B. Pelkie and C. W. Coley, Extracting structured data from organic synthesis procedures using a fine-tuned large language model, Digit. Discov., 2024, 3, 1822–1831 RSC.
- W. Zhang, Q. Wang, X. Kong, J. Xiong, S. Ni, D. Cao, B. Niu, M. Chen, Y. Li, R. Zhang, Y. Wang, L. Zhang, X. Li, Z. Xiong, Q. Shi, Z. Huang, Z. Fu and M. Zheng, Fine-tuning large language models for chemical text mining, Chem. Sci., 2024, 15, 10600–10611 RSC.
- S. Miret and N. M. Krishnan, arXiv, 2024, preprint, arXiv:2402.05200, DOI:10.48550/arXiv.2402.05200.
- K. Hira, M. Zaki, D. Sheth, Mausam and N. M. A. Krishnan, Reconstructing the materials tetrahedron: challenges in materials information extraction, Digit. Discov., 2024, 3, 1021–1037 RSC.
- M. Schilling-Wilhelmi, M. Ríos-García, S. Shabih, M. V. Gil, S. Miret, C. T. Koch, J. A. Márquez and K. M. Jablonka, From text to insight: large language models for chemical data extraction, Chem. Soc. Rev., 2025, 54, 1125–1150 RSC.
- M. P. Polak and D. Morgan, Extracting accurate materials data from research papers with conversational language models and prompt engineering, Nat. Commun., 2024, 15, 1569 CrossRef CAS PubMed.
- Y. Song, S. Miret and B. Liu, arXiv, 2023, preprint, arXiv:2310.08511, DOI:10.48550/arXiv.2310.08511.
- V. Mishra, S. Singh, D. Ahlawat, M. Zaki, V. Bihani, H. S. Grover, B. Mishra, S. Miret and N. M. Krishnan, arXiv, 2024, preprint, arXiv:2412.09560, DOI:10.48550/arXiv.2412.09560.
- Y. Kang, W. Lee, T. Bae, S. Han, H. Jang and J. Kim, Harnessing Large Language Models to Collect and Analyze Metal–Organic Framework Property Data Set, J. Am. Chem. Soc., 2025, 147, 3943–3958 CrossRef CAS PubMed.
- Y. Song, S. Miret, H. Zhang and B. Liu, arXiv, 2023, preprint, arXiv:2310.08511, DOI:10.48550/arXiv.2310.08511.
- H. Zhang, Y. Song, Z. Hou, S. Miret and B. Liu, arXiv, 2024, preprint, arXiv:2409.00135, DOI:10.48550/arXiv.2409.00135.
- J. Lála, O. O'Donoghue, A. Shtedritski, S. Cox, S. G. Rodriques and A. D. White, arXiv, 2023, preprint, arXiv:2312.07559, DOI:10.48550/arXiv.2312.07559.
- J. Choi and B. Lee, Accelerating materials language processing with large language models, Comput. Mater., 2024, 5, 13 Search PubMed.
- S. Gupta, A. Mahmood, P. Shetty, A. Adeboye and R. Ramprasad, Data extraction from polymer literature using large language models, Comput. Mater., 2024, 5, 269 Search PubMed.
- J. White, PubMed 2.0, Med. Ref. Serv. Q., 2020, 39, 382–387 CrossRef PubMed.
- T. Dettmers, A. Pagnoni, A. Holtzman and L. Zettlemoyer, Adv. Neural Inf. Process. Syst., 2023, 36, 10088–10115 Search PubMed.
- W. Kwon, Z. Li, S. Zhuang, Y. Sheng, L. Zheng, C. H. Yu, J. Gonzalez, H. Zhang and I. Stoica, presented in part at the Proceedings of the 29th Symposium on Operating Systems Principles, Koblenz, Germany, 2023 Search PubMed.
- L. Weston, V. Tshitoyan, J. Dagdelen, O. Kononova, A. Trewartha, K. A. Persson, G. Ceder and A. Jain, Named Entity Recognition and Normalization Applied to Large-Scale Information Extraction from the Materials Science Literature, J. Chem. Inf. Model., 2019, 59, 3692–3702 CrossRef CAS PubMed.
- P. Shetty and R. Ramprasad, Machine-Guided Polymer Knowledge Extraction Using Natural Language Processing: The Example of Named Entity Normalization, J. Chem. Inf. Model., 2021, 61, 5377–5385 CrossRef CAS PubMed.
- B. Hu, A. Lin and L. C. Brinson, ChemProps: A RESTful API enabled database for composite polymer name standardization, J. Cheminf., 2021, 13, 22 CAS.
- D. M. Lowe, P. T. Corbett, P. Murray-Rust and R. C. Glen, Chemical Name to Structure: OPSIN, an Open Source Solution, J. Chem. Inf. Model., 2011, 51, 739–753 Search PubMed.
- RDKit: Open-source cheminformatics Software, https://www.rdkit.org Search PubMed.
- P. Tiikkainen, L. Bellis, Y. Light and L. Franke, Estimating Error Rates in Bioactivity Databases, J. Chem. Inf. Model., 2013, 53, 2499–2505 CrossRef CAS PubMed.
- J. Hack, M. Jordan, A. Schmitt, M. Raru, H. S. Zorn, A. Seyfarth, I. Eulenberger and R. Geitner, Ilm-NMR-P31: an open-access 31P nuclear magnetic resonance database and data-driven prediction of 31P NMR shifts, J. Cheminf., 2023, 15, 122 CAS.
- KnowItAll, https://www.knowitall.com, accessed Dec 8, 2024.
- Micronmr Database, https://www.nmrdata.com, accessed Dec 8, 2024.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08802f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |