
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A focus on phosphinophosphination of apolar bonds by a structurally constrained P–P bonded system
Tyler J.
Hannah
* and
Saurabh S.
Chitnis
 *
*
Department of Chemistry, Dalhousie University, 6243 Alumni Crescent, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: tyler.hannah@dal.ca; saurabh.chitnis@dal.ca
First published on 6th January 2025
Abstract
In this article, we highlight the recent report of Greb et al. on the use of a structurally constrained P–P bonded system for phosphinophosphination of alkenes, alkynes, and carbonyls with high regio- and stereoselectivity (https://doi.org/10.1039/D4SC06581F).
Functionalization of organic substrates by main-group systems is a rapidly advancing field with the potential to complement or exceed established transition metal chemistry for valuable transformations.1 An emergent strategy in this pursuit is the use of multi-dentate ligands with p-block elements. Detailed studies involving pnictogen complexes have shown that such ligands can distort molecules to expose vacant orbitals for coordination and template the ensuing activation steps by enforcing the proximity of reactive centres.2–7
Cation 1, reported by Greb et al.,8 is in some ways similar to known intramolecularly-stablized phospheniums, whose high electrophilicity and geometrically-constrained nature allow activation of various E–H (E = N, C, H, O) bonds via a stepwise mechanism.9–14 But due to their very polar P–X (X = O, N, C) bonds, these stabilized phospheniums are not well-suited to activate weakly-polar or non-polar π-systems. In contrast, 1 features a homoatomic P–P bond that readily adds to non-activated alkynes, alkenes, and carbonyls. This diphosphination reaction was shown to proceed through a concerted mechanism, which enabled high regio- and diastereoselectivity (Scheme 1). Furthermore, this system demonstrates the first diphosphination of an unactivated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Interestingly, 1 exhibits low reactivity towards very polar alkenes, setting the stage for selective transformations when multiple reactive groups are present. The products of phosphinophosphination, 2 and 3, are 7-membered phosphaheterocycles that are in most cases obtained with a high preference for one set of racemic diastereomers. These compounds are of interest for their photophysical properties, which were demonstrated by absorption in the visible region for a derivative of 2. Additionally, a derivative of 3 was able to coordinate to a rhodium complex, demonstrating an approach to access unusual chiral phosphine ligands.
C double bond. Interestingly, 1 exhibits low reactivity towards very polar alkenes, setting the stage for selective transformations when multiple reactive groups are present. The products of phosphinophosphination, 2 and 3, are 7-membered phosphaheterocycles that are in most cases obtained with a high preference for one set of racemic diastereomers. These compounds are of interest for their photophysical properties, which were demonstrated by absorption in the visible region for a derivative of 2. Additionally, a derivative of 3 was able to coordinate to a rhodium complex, demonstrating an approach to access unusual chiral phosphine ligands.
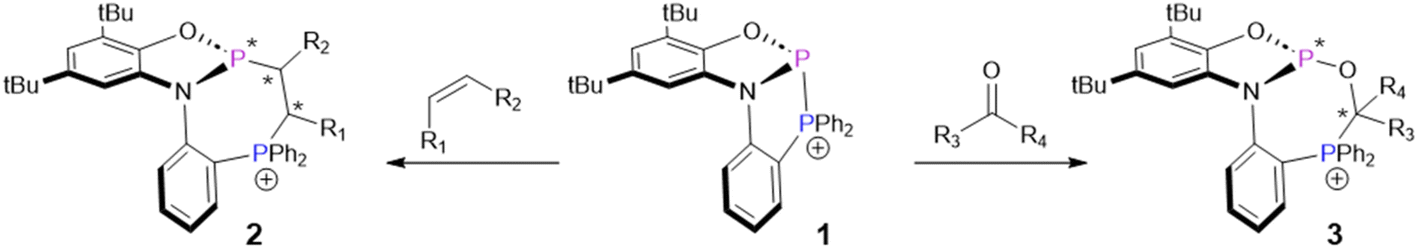 | ||
| Scheme 1 Diphosphination of alkenes and carbonyls by compound 1. Asterisks denote newly formed chiral centres. Counterions have been omitted for simplicity. | ||
The unprecedented reactivity and selectivity of 1 can be understood via a closer look at the P–P bond in this cation relative to other phosphino–phosphoniums. Scheme 2 shows the calculated partial charges on phosphorus in 1 and other P–P bonded systems that have been utilized for diphosphination. Compound A features a very polarized P–P bond despite being neutral. This is due to strong resonance stabilization of the phosphenium ion resulting from P–P heterolysis and the attachment to two electronegative nitrogen atoms at phosphorus. The very polar bond in this species is able to react with polar substrates including nitriles and alkenes containing one electron withdrawing group.15 Compound B, with a slightly less polar P–P bond, is able to react with activated terminal alkynes through a frustrated Lewis pair mechanism, but only under forcing conditions.16 The intramolecular phosphino–phosphonium C features a less polar P–P bond than B, but due to the strained three-membered ring, it can nevertheless diphosphinate some polar substrates such as nitriles, while no addition across less polar unsaturated groups was reported.17,18 Although phosphino–phosphoniums, as in B and C, have been shown to have similar barriers for both homolytic and heterolytic bond cleavage, their reactivity is dominated by polar, stepwise addition due to the considerable bond polarization.19,20
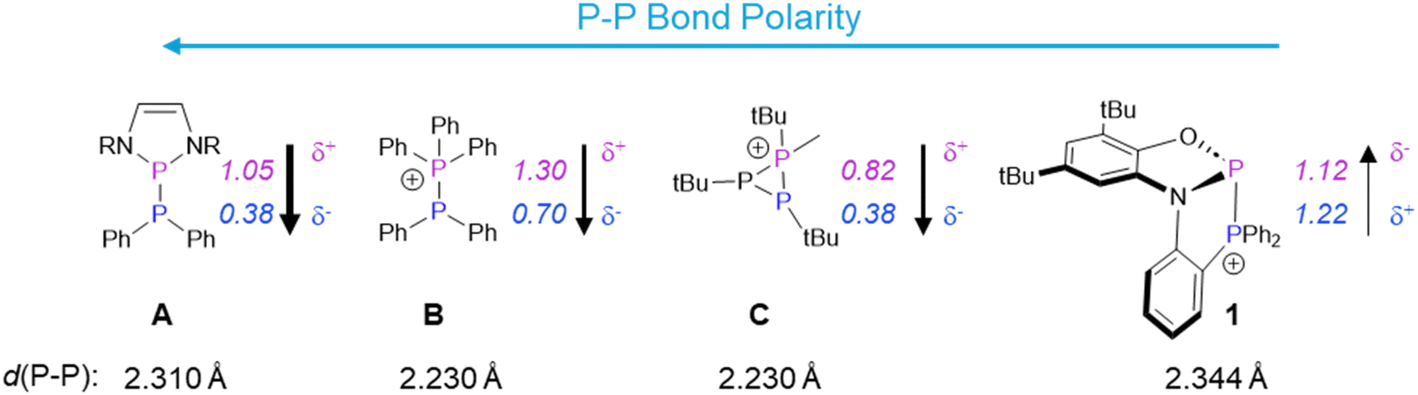 | ||
| Scheme 2 P–P bonded systems utilized for diphosphination and a comparison of their P–P bond polarities and lengths with those of 1.21,22 Calculated partial charges are denoted in italics. These values have been taken from the relevant literature.18,8,23 Counterions have been omitted for simplicity. For C, only the bond length involving the phosphino–phosphonium unit is given. | ||
Compared to A–C, compound 1 features a minimally polarized P–P bond: the high charge at its donor phosphorus is comparable to the values for B and C, and the high charge at its acceptor phosphorus arises from attachment to σ-withdrawing, π-donating groups (as in the case of A). The π-donor groups on the acceptor phosphorus also result in a very long P–P bond (even longer than that of A). This unique combination of a long and low-polarity P–P bond is well-suited for weak or non-polar π-bond activation via a concerted pathway that is not available to polar (A) or strongly-bonded (B, C) analogues. As a bonus, the lack of epimerizable intermediates in a concerted mechanism results in the stereoselectivity observed in transformations starting from 1. The thermodynamic driving force is provided by replacement of a weak P–P bond and a strong CC or CO bond with two strong P–C (or P–O) bonds.
The multidenticity of the ligand is also a key enabling feature of this system. Indeed, the P–P bond in 1 is very unusual, since phosphenium cations with strongly π-donating N-substituents do not generally coordinate to triarylphosphines. They are instead readily isolated as stable two-coordinate species.24,25 In 1, the pincer ligand reinforces the bonding interaction between the two phosphorus centres to generate a functional group that may not persist if the interaction were intermolecular. In this manner, the chemistry of 1 is also reminiscent of intramolecular frustrated Lewis pairs.26 Besides templating the P–P interaction, the ligand constraint is also expected to lower the energy of the disphenoidal transition state (TSconc) of the concerted addition pathway, as planarization of three out of four substituents is facilitated by their tethering (Fig. 1).27 Finally, the use of a weakly-coordinating [Al(OC(CF3)3)4] counterion ensures that poor donors such as non-polar π-systems can out-compete the anion for coordination to the cation, enabling the subsequent diphosphination.
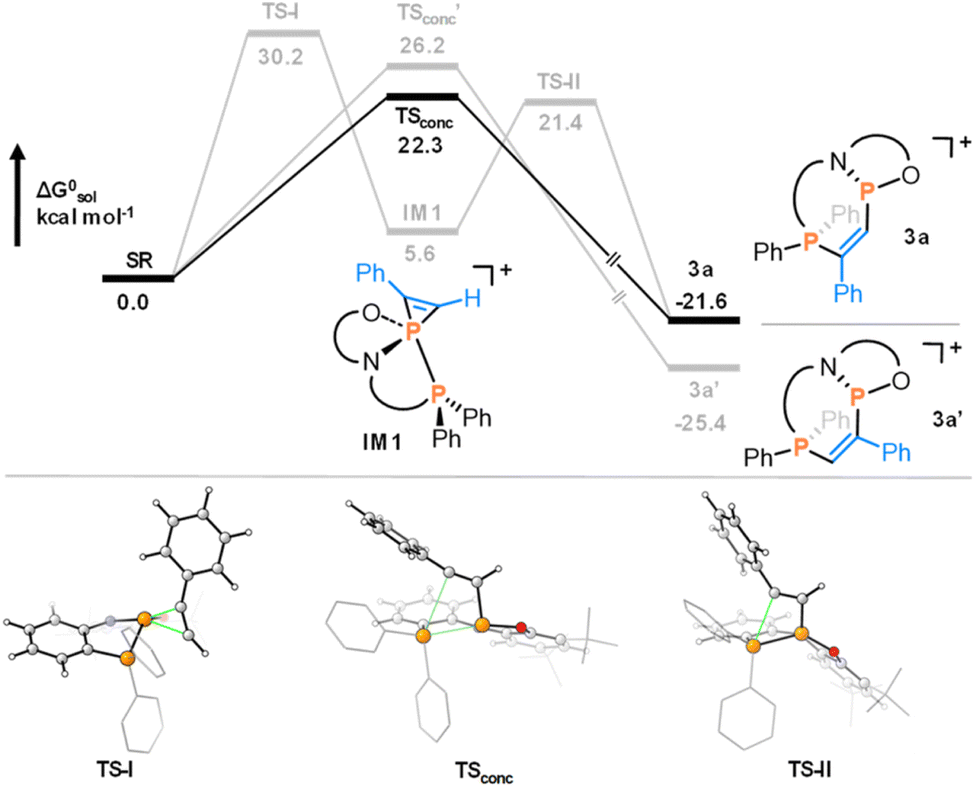 | ||
| Fig. 1 Calculated mechanism for the concerted reaction of phenylacetylene to 1. Reproduced from ref. 8. | ||
Combining unusual electronics, ligand constraints, and weakly-coordinating anions within a single molecular system is a powerful and creative strategy for challenging bond activation at main-group centres. A slew of recent contributions,11,14,28–30 including the subject of this Focus article, illustrate how bridging the established fields of pincer coordination chemistry and Lewis acid chemistry represents a rich vein for both fundamental and applied discoveries. Although the reactivity of 1 reported so far is stoichiometric, the fundamental principles it illustrated may lead to new catalytic strategies for transforming weakly-polar bonds.
Author contributions
TJH and SSC co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TJH acknowledges the Natural Science & Engineering Research Council of Canada (NSERC) and the Walter C. Sumner Foundation for graduate scholarships. SSC acknowledges NSERC, Dalhousie University, and the Alfred P. Sloan Foundation for funding in support of this work.References
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC.
- S. Kundu, Chem.–Asian J., 2020, 15, 3209–3224 CrossRef CAS PubMed.
- T. J. Hannah and S. S. Chitnis, Chem. Soc. Rev., 2024, 53, 764–792 RSC.
- A. Maiti, R. Yadav and L. Greb, in Advances in Inorganic Chemistry, ed. K. Meyer and R. van Eldik, Academic Press, 2023, vol. 82, pp. 261–299 Search PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- L. You, D. Roth and L. Greb, Chem. Sci., 2024 10.1039/D4SC06581F.
- K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786–3794 CrossRef CAS PubMed.
- D. Bawari, S. Volodarsky, Y. Ginzburg, K. Jaiswal, P. Joshi and R. Dobrovetsky, Chem. Commun., 2022, 58, 12176–12179 RSC.
- D. Bawari, D. Toami, K. Jaiswal and R. Dobrovetsky, Nat. Chem., 2024, 16, 1261–1266 CrossRef CAS PubMed.
- S. Volodarsky, D. Bawari and R. Dobrovetsky, Angew. Chem., Int. Ed., 2022, 61, e202208401 CrossRef CAS PubMed.
- S. Volodarsky and R. Dobrovetsky, Chem. Commun., 2018, 54, 6931–6934 RSC.
- D. Roth, A. T. Radosevich and L. Greb, J. Am. Chem. Soc., 2023, 145, 24184–24190 CrossRef CAS PubMed.
- S. Burck, D. Gudat and M. Nieger, Angew. Chem., Int. Ed., 2004, 43, 4801–4804 CrossRef CAS PubMed.
- H. Kim, Z.-w. Qu, S. Grimme, N. Al-Zuhaika and D. W. Stephan, Angew. Chem., Int. Ed., 2023, 62, e202312587 CrossRef CAS PubMed.
- S. S. Chitnis, H. A. Sparkes, V. T. Annibale, N. E. Pridmore, A. M. Oliver and I. Manners, Angew. Chem., Int. Ed., 2017, 56, 9536–9540 CrossRef CAS PubMed.
- S. S. Chitnis, R. A. Musgrave, H. A. Sparkes, N. E. Pridmore, V. T. Annibale and I. Manners, Inorg. Chem., 2017, 56, 4521–4537 CrossRef CAS PubMed.
- K. L. Bamford, S. S. Chitnis, R. L. Stoddard, J. S. McIndoe and N. Burford, Chem. Sci., 2016, 7, 2544–2552 RSC.
- S. S. Chitnis, J. M. Whalen and N. Burford, J. Am. Chem. Soc., 2014, 136, 12498–12506 CrossRef CAS PubMed.
- C. A. Dyker, N. Burford, G. Menard, M. D. Lumsden and A. Decken, Inorg. Chem., 2007, 46, 4277–4285 CrossRef CAS PubMed.
- D. Gudat, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2018, pp. 1–23, DOI:10.1002/9781119951438.eibc2642.
- K. G. Pearce, A. M. Borys, E. R. Clark and H. J. Shepherd, Inorg. Chem., 2018, 57, 11530–11536 CrossRef CAS PubMed.
- D. Gudat, in Phosphorus Heterocycles II, ed. R. K. Bansal, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 63–102, DOI:10.1007/7081_2009_5.
- A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367–382 CrossRef CAS.
- G. Kehr, S. Schwendemann and G. Erker, in Frustrated Lewis Pairs I: Uncovering and Understanding, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 45–83, DOI:10.1007/128_2012_373.
- T. J. Hannah, T. Z. Kirsch and S. S. Chitnis, Chem.–Eur. J., 2024, 30, e202402851 CrossRef CAS PubMed.
- K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786–3794 CrossRef CAS PubMed.
- W. Lv, Y. Dai, R. Guo, Y. Su, D. A. Ruiz, L. L. Liu, C.-H. Tung and L. Kong, Angew. Chem. Int. Ed., 2023, 62, e202308467 CrossRef CAS PubMed.
- S. G. Kim, D. Kim, J. Oh, Y. J. Son, S. Jeong, J. Kim and S. J. Hwang, J. Am. Chem. Soc., 2024, 146, 11440–11449 CAS.
| This journal is © The Royal Society of Chemistry 2025 |