
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlue light-induced diazo cross-coupling: synthesis of allyldiazo compounds through reshuffling of functionalities†
Jiabao
Tian
,
Jiahao
Ling
,
Yanan
Wang
and
Lei
Zhou
 *
*
Institute of Green Chemistry and Molecular Engineering, School of Chemistry, Sun Yat-sen University, Guangzhou, 510275, China. E-mail: zhoul39@mail.sysu.edu.cn
First published on 1st March 2025
Abstract
In this paper, we describe a new type of cross-coupling between simple diazo and vinyldiazo compounds that gives access to unusual allyldiazo products. Blue light discriminates two diazo compounds towards free carbene formation, triggering sequential cyclopropenation, (3+2) cycloaddition and ring opening rearrangement processes. This strategy involves an overall reshuffle of diazo functionality and olefinic carbons of vinyldiazo compounds with an extrusion of nitrogen. Mechanistic studies including a 15N-labelling experiment demonstrate that the diazo functionality of allyldiazo products derives from simple diazo compounds, while vinyldiazo reagents are selectively decomposed via energy transfer with thioxanthone photocatalyst. The obtained allyldiazo compounds can be efficiently converted into synthetically useful structures such as 1,3-dienes, gem-difluoro-1,4-diene, hydrazine, dihydropyrazole, pyridazine, and bicyclobutane.
Introduction
The chemistry of diazo compounds has been intensively studied since the first synthesis of ethyl diazoacetate by Curtius in 1883.1,2 The coupling of diazo compounds by complete decomposition of two diazo moieties is an alternative method for the synthesis of unsymmetrically substituted alkenes.3,4 However, this transformation has not found widespread applications due to the inevitable competition between homo- and cross-coupling processes.5 The challenge for the selective cross-coupling lies in the discrimination of diazo compounds toward carbenoids because most of them are readily decomposed by transition metals or under thermal and photochemical conditions. Diazo compounds are often coloured molecules, and their absorption is significantly affected by the substituents of the diazo carbon.6 In 2018, Jurberg and Davies pioneered the photolysis of aryldiazoacetates under blue light irradiation.7 Soon after this work, we reported a blue light-promoted selective diazo cross-coupling for the synthesis of trisubstituted alkenes by distinguishing aryldiazoacetates from other diazo compounds.8 Since then, various classical transition metal-catalyzed transformations of diazo compounds have been re-examined using visible light as the sole energy source, unlocking novel reaction pathways via free carbene intermediates.9As a result of its high reactivity, the diazo group is usually introduced onto a targeted product at the end of synthesis.2e Additional functionalization of an existing diazo compound without affecting the diazo functionality is also a promising method to prepare complex or even unconventional diazo compounds.10 Such manipulation usually employs relatively stable diazo compounds and relies on the transformations of molecular skeleton remote to the diazo functionality. Regarding the reactions of two diazo compounds, it is difficult to decompose one diazo compound while conserving the diazo functionality of another. In this context, our group developed a photoredox-enabled self-(3+2) cyclization of two vinyldiazo reagents, which provides cyclopentene derivatives with retention of one diazo functional group in the products.11
At this point, we became intrigued in the photochemical reaction of vinyldiazo with simple diazo compounds. Previous works by Davies and Sun disclosed the selective cross-coupling of vinyldiazoacetates and aryldiazoacetates for the generation of conjugated 1,3-dienes catalyzed by Rh(II) and Au(I) respectively (Scheme 1a-1).4 Switching the ligands, the Au(I)-catalyzed cross-coupling of these two diazo compounds can produce N-substituted pyrazoles.12 The selective diazo cross-coupling towards pyrazoles was also reported by Melen using a borane catalyst (Scheme 1a-2).13 Barluenga demonstrated the discrimination of simple and vinyldiazo systems towards carbenoids by a Cu(I) catalyst, allowing the formation of cyclobutenes via a sequential cyclopropanation/ring expansion process (Scheme 1a-3).14 Nevertheless, these transformations always result in the complete decomposition or functional loss of diazo groups. Herein, we report the use of blue light to discriminate vinyldiazo and simple diazo compounds, leading to the formation of allyldiazo compounds via a distinct diazo cross-coupling reaction. Both vinyldiazo and simple diazo compounds undergo structural reorganization in which the diazo functionality and olefinic carbons have been reshuffled with an extrusion of nitrogen. Due to the low energy of blue light, such allyldiazo products can be isolated and were used for further derivatization on the basis of the rich chemistry of the diazo functional group.
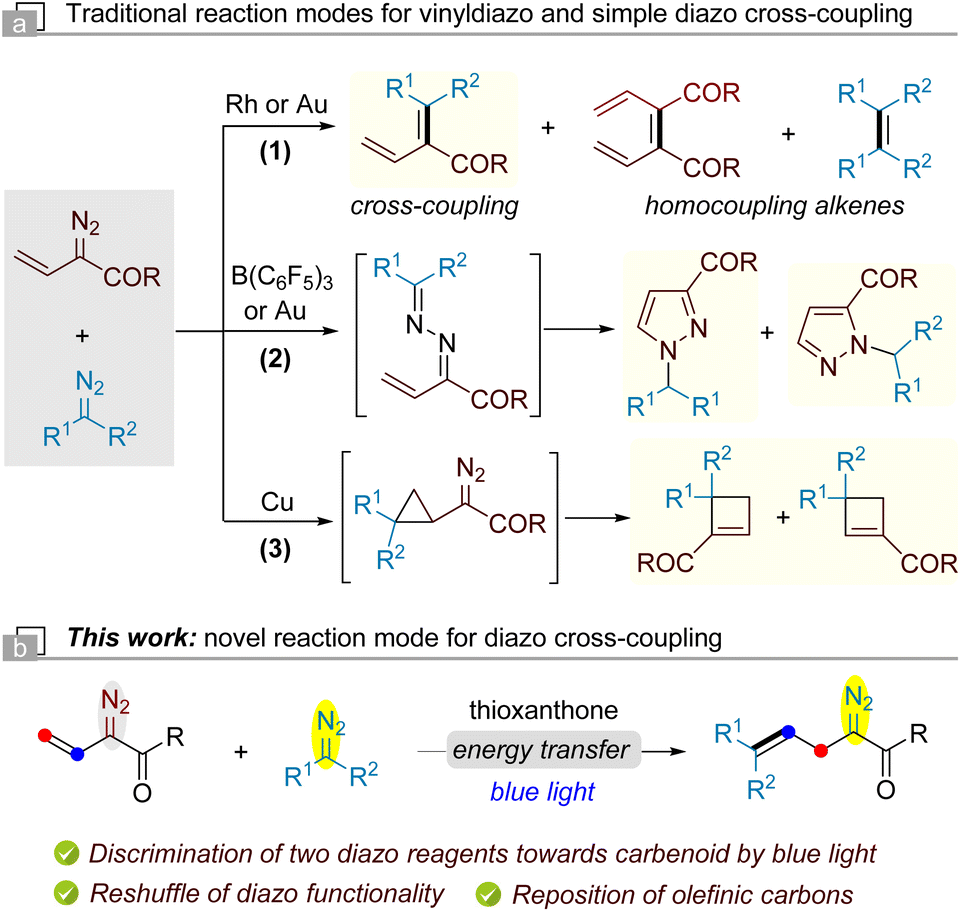 | ||
| Scheme 1 The reactions of simple diazo and vinyldiazo compounds. | ||
 | ||
| Scheme 2 Selective diazo cross-coupling toward allyldiazo compounds. | ||
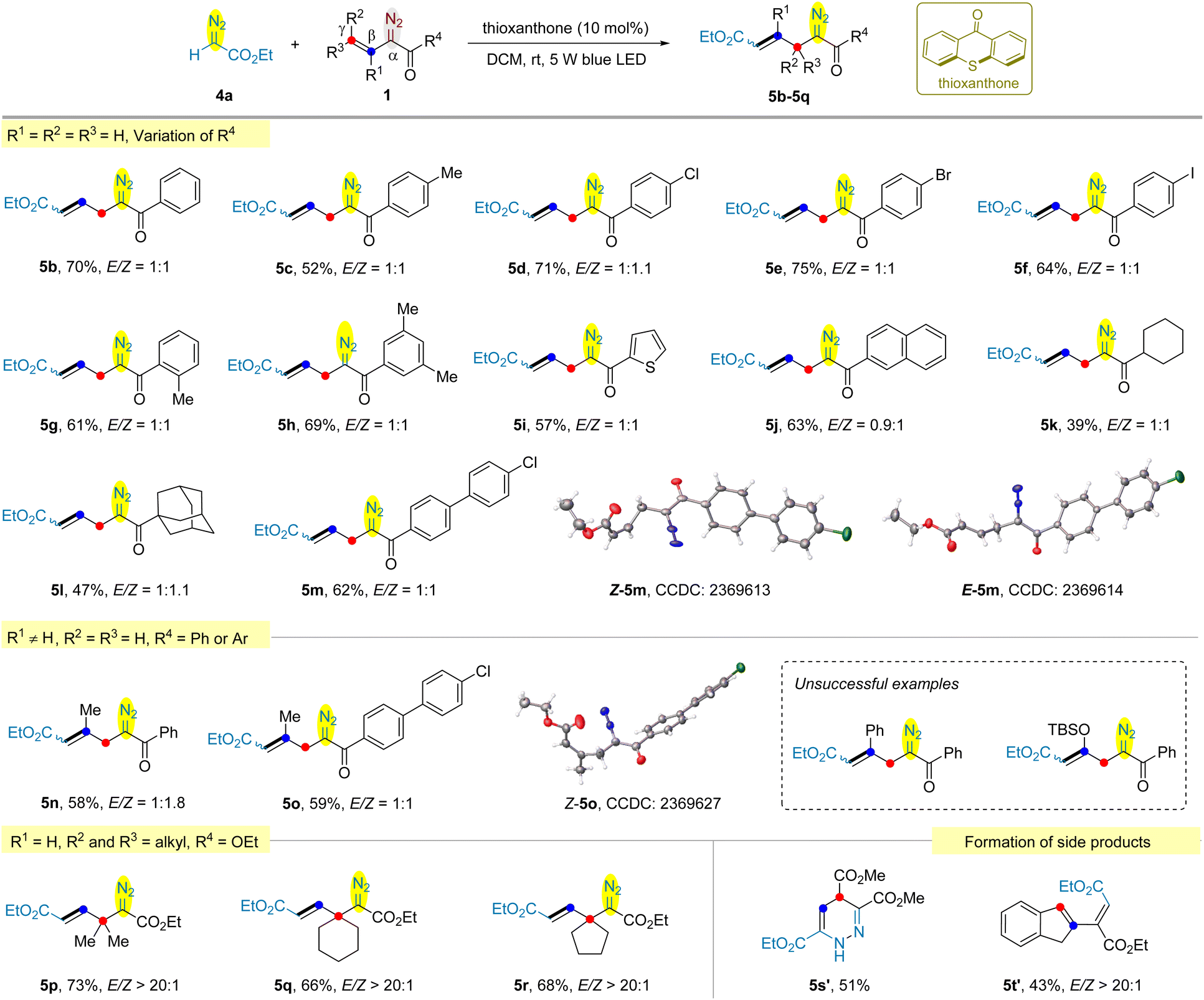 | ||
| Scheme 3 Visible light-promoted cross-coupling of ethyl diazoacetate 4a with various vinyldiazo compounds. Reaction conditions: vinyldiazo compound 1 (0.4 mmol, 2 equiv.), ethyl diazoacetate 4a (0.2 mmol, 1 equiv.), thioxanthone (10 mol%), DCM (2 mL), rt, 5 W blue LED, 16 h. Isolated yields of the products. | ||
Results and discussion
During our studies of photochemical reactions of vinyldiazo compounds,15 we need to examine the stability of typical substrates under light irradiation conditions in the presence or absence of photocatalysts. Irradiating the DCM solution of tert-butyl vinyldiazoacete 1a for 2 h without photocatalyst provided minor amount of pyrazole via 1,5-electrocyclization.16 After prolonging the irradiation time to 12 h, despite the yield of pyrazole 2 increased to 40%, an unexpected homo-coupling diazo product 3 was isolated in 8% yield (Scheme 2a). However, efforts to further improve the yield of 3 failed. We envisioned that the vinyl group of another molecule of 1a might only act as a substituent. To validate the hypothesis, the reaction of ethyl diazoacetate 4a with 1a was examined. Except for the homo-coupling product 3 (5% yield), the cross-coupling product 5a was obtained in 16% yield (Scheme 2b). We were delight to find that the irradiation of vinyldiazoketone 1b and 4a afforded allyldiazo 5b in 56% yield and the homo-coupling of 1b was not observed (Scheme 2c). This interesting result encouraged us to optimize the reaction conditions using 1b and 4a. It was found that the yields of 5b were not significantly affected by the solvents (see Table S1 in the ESI†). The addition of thioxanthone as the photocatalyst can increase the yield to 70%, while other photocatalysts such as benzophenone, riboflavin, Ru(bpy)3Cl2, fac-Ir(ppy)3 disfavoured the reactions. The best reaction time is 16 h because further prolonging the irradiation time led to the gradual decomposition of allyldiazo product.Under the above optimized reaction conditions, we explored the scope of the reaction. As shown in Scheme 3, the cross-coupling of ethyl diazoacete 4a with vinyldiazo bearing distinct aryl ketones gave the desired allyldiazoketones 5c–5h in 52–75% yields. Many substituents, including methyl and halogen (Cl, Br, and I) at the ortho- (5g), meta- (5h) and para- (5c–5f) are well tolerated. The reaction was also compatible with 2-thienyl ketone (5i) and 2-naphthyl ketone (5j). We prepared vinyldiazo alkyl ketones (R4 = cyclohexyl and adamantyl), and their reactions with 4a afforded allyldiazo compounds 5k and 5l in moderate yields. Allyldiazoketone 5m is a solid and both structures of its Z- and E-isomers were confirmed by X-ray diffraction. Subsequently, the cross-coupling of β-methyl substituted vinyldiazoketone 1n with ethyl diazoacetate 4a was examined. Surprisingly, the β-methyl group of 1n relocated at γ-position in the product 5n, indicating a rearrangement occurred in this reaction. The X-ray diffraction of Z-isomer of allyldiazoketone 5o confirmed our proposed structure. Switching the methyl on the β-position to phenyl or OTBS led to complex mixtures. Although we detected the formation of allyldiazoketones by GC-MS, the attempt to obtain pure products failed. To further verify the reposition, γ,γ-dimethyl substituted vinyldiazoacetate 1p was employed to react with 4a, providing 5p in 73% yield with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z selectivity. It was found that both methyl groups were presented on the β-position of product 5p. Using exocyclic vinyldiazoacetates 1q and 1r as the substrates, no ring expansion was observed and the corresponding allyldiazoacetates 5q and 5r were obtained in 66% and 68%, respectively.17 However, 1,4-dihydropyridazine 5s′ was isolated as the major product in the reaction of 4a with vinyldiazoacete 1s bearing an ester motif at the γ-position. We speculate that the additional electron-withdrawing ester group further destabilized the cyclopropane of 2,3-diazabicyclo[3.1.0]hex-2-ene intermediate, thus facilitating the direct cleavage of weakened fused [c] bond to form 1,4-dihydropyridazine.18 The cross-coupling of 2-indenyldiazoacetate 1t with 4a provided carbene dimerization product 5t′ presumably due to the difficulty in generating indene-fused cyclopropene.
:1 E/Z selectivity. It was found that both methyl groups were presented on the β-position of product 5p. Using exocyclic vinyldiazoacetates 1q and 1r as the substrates, no ring expansion was observed and the corresponding allyldiazoacetates 5q and 5r were obtained in 66% and 68%, respectively.17 However, 1,4-dihydropyridazine 5s′ was isolated as the major product in the reaction of 4a with vinyldiazoacete 1s bearing an ester motif at the γ-position. We speculate that the additional electron-withdrawing ester group further destabilized the cyclopropane of 2,3-diazabicyclo[3.1.0]hex-2-ene intermediate, thus facilitating the direct cleavage of weakened fused [c] bond to form 1,4-dihydropyridazine.18 The cross-coupling of 2-indenyldiazoacetate 1t with 4a provided carbene dimerization product 5t′ presumably due to the difficulty in generating indene-fused cyclopropene.
Next, we used vinyldiazoketone 1b to examine its cross-coupling with a series of acceptor-type diazo compounds (Scheme 4). Alkyl diazoacetates, such as n-pentyl (5u), cyclohexyl (5v) and cyclobutyl (5w) were found to react with 1b smoothly to give the anticipated coupling products 5u–5w in 47–69% yields. The C–C double of allyl diazoacetate remained intact, providing 5x in 63% yield. Benyzl (5y) and its analogues such as thiophenylmethyl (5z) and benzodioxolylmethyl (5aa) in the ester motif of diazoacetates were compatible. Encouraged by these results, diazoacetates deriving from naturally occurring alcohols, including geraniol (5ab), L-menthol (5ac), borneol (5ad), diacetone-D-glucose (5ae), and pregnenolone (5af) were employed for the cross-coupling with vinyldiazoketone 1b; and they were all converted into the corresponding allyldiazoketones in satisfactory yields. The electron-withdrawing group of acceptor-type diazo compounds is not limited to ester, as exemplified by the use of trimethylsilyl (5ag), trifluoromethyl (5ah), cyano (5ai), and tosyl (5aj) substituteddiazomethanes as the substrates. The reaction of donor–acceptor aryldiazoacetate with 1b afforded the product 5ak in 61% yield. However, acceptor–acceptor diethyl diazomalonate was not suitable substrate for this reaction. Replacing the terminal hydrogen of ethyl diazoacetate 4a by deuterium led to allyldiazoketone 5al bearing a vinyl C–D bond in identical yield as compared to 5b. Finally, we were delighted to find that the cross-coupling of 1b with unstable diazomethane proceeded smoothly, providing allyldiazoketone 5am in 74% yield. In most of cases, the E- and Z-isomers were separable, except for 5ah, 5ai and 5ak.
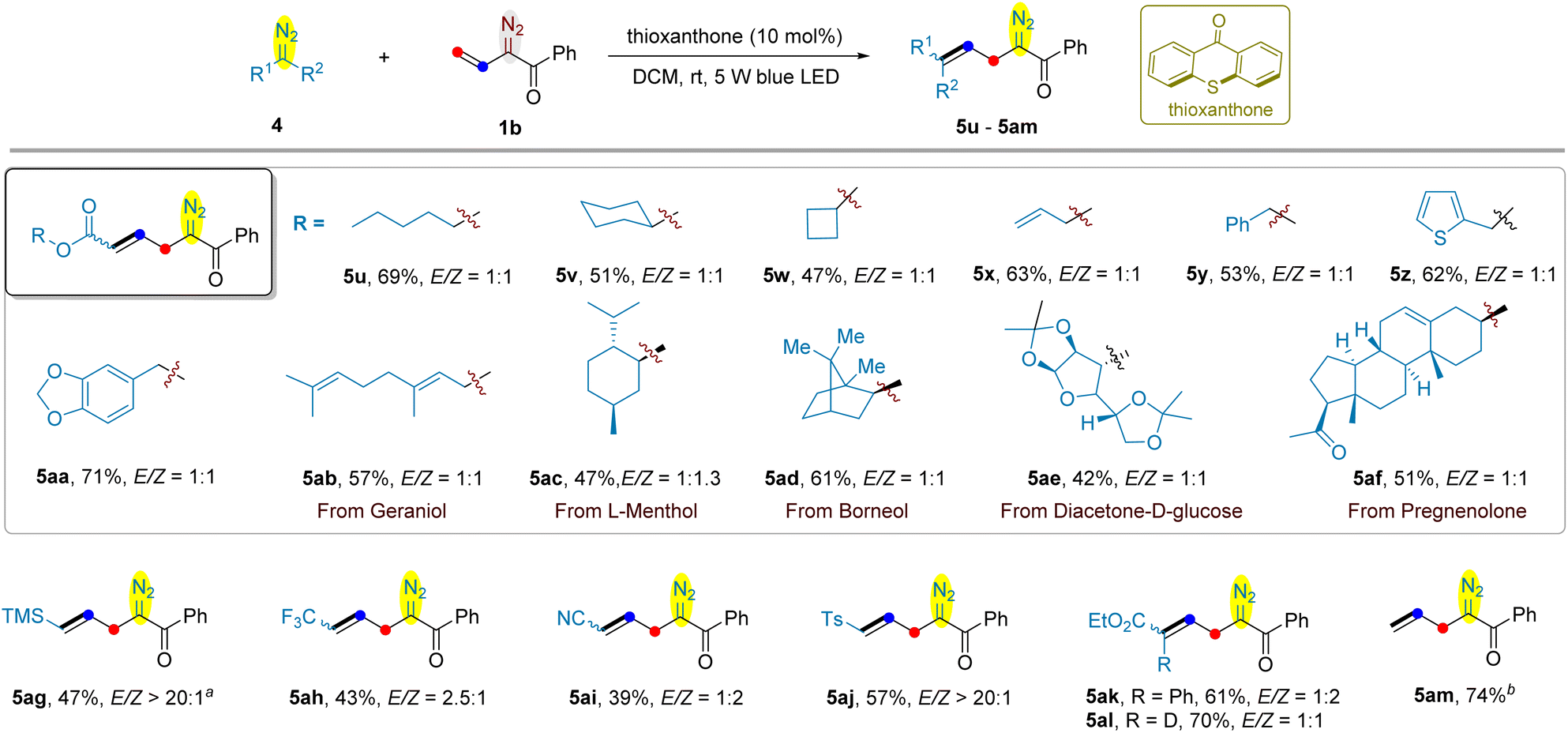 | ||
| Scheme 4 Visible light-promoted cross-coupling of vinyldiazoketone 1b and simple diazo compounds. Reaction conditions: vinyldiazo ketone 1b (0.4 mmol, 2 equiv.), simple diazo compound 4 (0.2 mmol, 1 equiv.), thioxanthone (10 mol%), DCM (2 mL), rt, 5 W blue LED, 16 h. Isolated yields of the products. a The ratio of TMSCHN2 and vinyldiazoketone 1b is 2:1. b The reaction was carried out using 20 equiv. of CH2N2 in 3 mL of DCM/Et2O (v:v = 1:2). | ||
To demonstrate the synthetic applications of allyldiazo compounds, the cross-coupling of vinyldiazoketone 1b and ethyl diazoacetate 4a was performed on 5 mmol scale (Scheme 5-1). Both E- and Z-isomers of 5b were isolated in 62% total yield in a ratio of 1:1. Typical transformations of allyldiazoketone 5b were shown in Scheme 5-2. Using Cu(MeCN)4PF6 as the catalyst, E-5b underwent selective 1,2-H migration to give conjugated 1,3-diene 6 in 87% yield. gem-Difluoroalkene 7 was obtained in 61% yield via gem-difluoroolefination of the diazo group of E-5b. Both the diazo group and C–C double were reduced by hydrogen gas (1 atm), affording hydrazine 8 in 74% yield. Cu-catalyzed N–H insertion of E-5b onto p-anisidine produced the corresponding product 9 in 53% yield. In the presence of PnBu3, the reduced 5b (1:1 E/Z mixture) further underwent intramolecular Michael addition to form dihydropyrazole 10 in 80% yield. Treatment of 5b (1:1 E/Z mixture) with DABCO led to pyridazine 11 in the air. Although intermolecular cyclopropanation of 5b with styrene was unsuccessful due to the competitive 1,2-H migration, we were delighted to find that Rh2(OAc)4-catalyzed intramolecular cyclopropanation of allyldiazoketone 5am produced bicyclo[1.1.0]butane 12 in 84% yield (Scheme 5-3).19
 | ||
| Scheme 5 Synthesis of 5b on 5 mmol scale and synthetic applications of the products. (a) E-5b (0.1 mmol), Cu(MeCN)4PF6 (10 mol%), DCM, rt; (b) E-5b (0.1 mmol), TMSCF3 (1.2 equiv.), NaI (2 equiv.), THF, 65 °C; (c) 5b (E/Z = 1:1, 0.1 mmol), PtO2 (10 mol%), H2 (1 atm), HOAc (1 equiv.), EtOAc, rt; (d) E-5b (0.1 mmol), Cu(MeCN)4PF6 (5 mol%), p-anisidine (1 equiv.), DCM, rt; (e) 5b (E/Z = 1:1, 0.1 mmol), PnBu3 (3 equiv.), iPr2O, rt; (f) 5b (E/Z = 1:1, 0.1 mmol), DABCO (1 equiv.), DCM, rt. | ||
In this cross-coupling reaction of vinyldiazo and simple diazo system, only one diazo group is decomposed while another is preserved in the products. To figure out the origin of diazo functionality of the products, we prepared 15N-labeling ethyl diazoacetate 4a and its cross-coupling with vinyldiazoketone 1b provided 15N-5b in 71% yield (Scheme 6a). This result indicated that the reaction was initiated by the decomposition of vinyldiazo compounds. Very recently, the conversion of vinyldiazo compounds to unstable cyclopropenes under blue light irradiation has been reported by several groups.20 To verify cyclopropene is involved as an intermeidate in this reaction, a stable cyclopropene 13 was prepared and its reactions with ethyl diazoacetate 4a provided allyldiazo compound 5p with or without the irradiation of blue light (Scheme 6b).21 Indeed, such a 1,3-dipolar addition of stable cyclopropenes with diazo compounds to give 2,3-diazabicyclo[3.1.0]hex-2-enes has been reported, which usually underwent rapid rearrangement to form 1,4-dihydropyridazines under the reaction conditions or on treatment with a trace of acid or base.22 The thermal or photolytic rearrangement of 2,3-diazabicyclo[3.1.0]hex-2-enes to form diazo compounds is also viable, but in most cases bicyclo[1.1.0]butanes or complex mixture were obtained due to the decomposition of diazo compound under reaction conditions.23 The luminescence quenching experiments demonstrated that vinyldiazoketone 1b displayed luminescence quenching of the excited state of thioxantone*, while a decrease was not observed by adding ethyl diazoacetate 4a (Fig. S1–S3 in the ESI†). Vinyldiazoketone 1b has a slightly higher oxidation potential (E1/2 = + 1.35 V vs. SCE) than thioxantone (EPC*/PC1/2− = + 1.18 V vs. SCE),24 suggesting the single electron transfer between 1b and the excited thioxantone is less likely (Fig. S4 in the ESI†). As shown in Scheme 2c, moderate yield of 5b was obtained without photocatalyst, while the use of thioxanthone of high triplet energy (ET = 63.4 kcal mol−1) increased the yield.24 Therefore, the current reaction process may go through an energy-transfer mechanism. The conjugated vinyldiazoketones are better quenching reagents than other diazo compounds. Using slightly excess amounts of vinyldiazo starting materials can suppress the further decomposition of allyldiazo products (for the reaction time profile, see Fig. S5 in the ESI†).
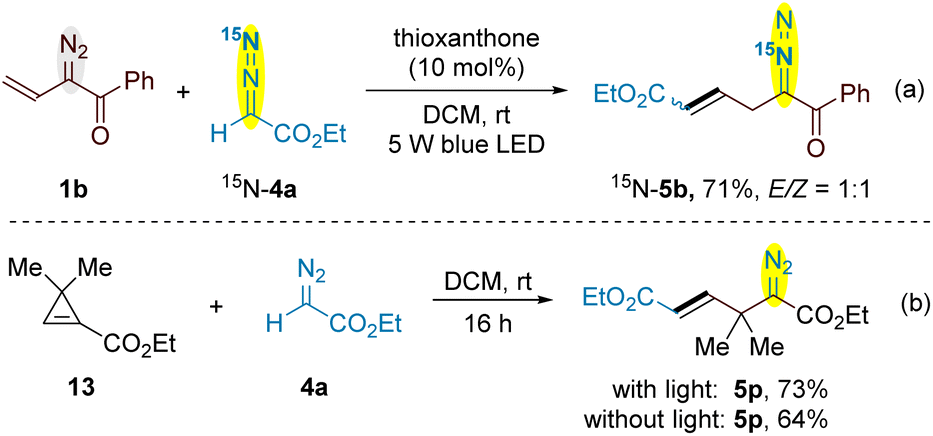 | ||
| Scheme 6 (a) The cross-coupling of 1b with 15N-4a; (b) the reaction of stable cyclopropene 13 with ethyl diazoacetate 4a. | ||
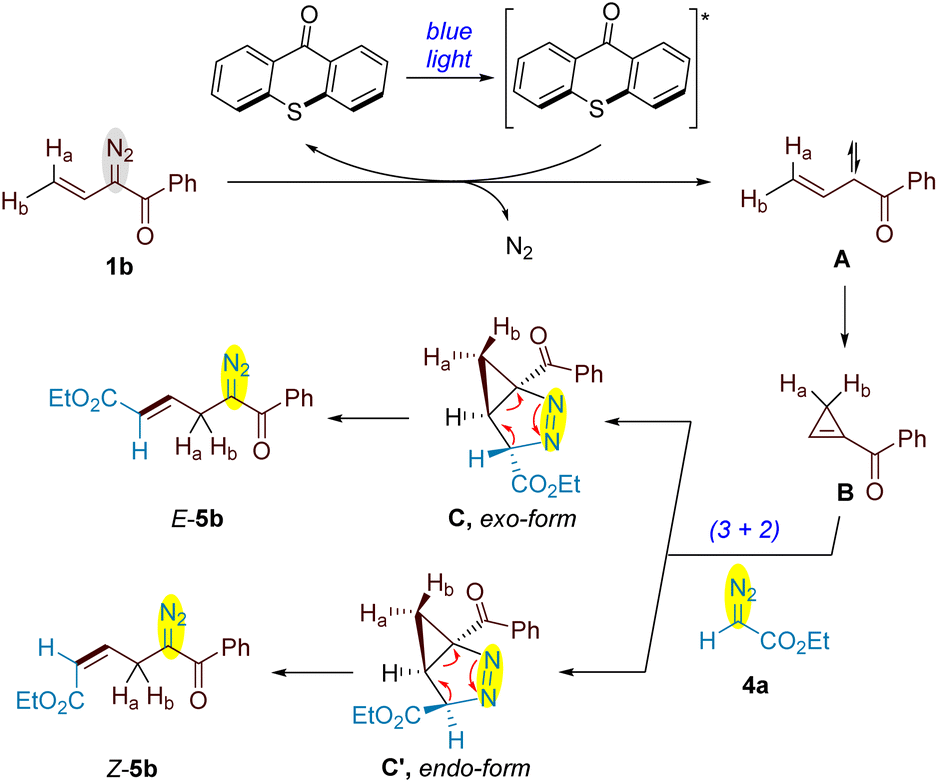
A plausible mechanism for this new type of diazo cross-coupling reaction has been depicted in Scheme 7. Under the irradiation of blue light, the energy transfer between photoexcited thioxanthone* and vinyldiazoketone 1b generates free vinyl carbene A with a nitrogen extrusion. Intramolecular cyclization of A produces unstable cyclopropene B, which undergoes (3+2) cycloaddition with ethyl diazo acetate 4b to give a mixture of exo- and endo- 2,3-diazabicyclo[3.1.0]hex-2- ene C and C′ respectively. The rearrangement of exo-C by opening both cyclopropane and pyrazoline rings forms the allyldiazo product E-5b, while Z-5b is generated via ring-opening of endo-form C′. In the cases of γ,γ-disubstituted vinyldiazoacetates 1p–1r, the replacement of Ha and Hb by alkyl groups significantly increases the steric hindrance of cyclopropane motif in 2,3-diazabicyclo[3.1.0]hex-2-enes, thus favours the formation of exo-intermediate C. The current results indicated that the bulky groups attached to carbonyl group of vinyldiazoketones or in the ester motif have little effect on the diastereoselectivity of (3+2) cycloaddition.
 | ||
| Scheme 7 A plausible reaction pathway. | ||
Conclusions
In summary, we have developed a new type of cross-coupling reaction between simple diazo and vinyldiazo systems, affording allyldiazo products through sequential cyclopropenation, (3+2) cycloaddition and ring opening rearrangement processes. Unlike classical transition metal-catalyzed diazo coupling reactions, which often lead to the complete decomposition of two diazo moieties, the present photochemical protocol discriminates two diazo compounds towards free carbene formation. The reaction reshuffles another diazo functionality into the products and repositions olefinic carbons as well as substituents on them. The method depicted herein provides a distinct route to access complex or even unconventional allyldiazo compounds, which themselves are ideal precursors for advanced carbene transfer reactions.Data availability
The data underlying this study are available within the article and in the ESI.†Author contributions
JT, JL and YW performed the experiments and analysed the data. LZ conceived and directed the project. LZ prepared the manuscript while JT prepared the ESI.† All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22271318).Notes and references
- T. Curtius, Ber. Dtsch. Chem. Ges., 1883, 16, 2230–2231 CrossRef.
- For selected reviews: (a) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS; (b) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 CrossRef CAS PubMed; (c) M. M. Déaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed; (d) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed; (e) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed; (f) J. Wang, Tetrahedron Lett., 2022, 108, 154135 CrossRef CAS.
- For examples of intramolecular diazo cross-couplings, see: (a) J. Font, F. Serratosa and J. Valls, J. Chem. Soc. D, 1970, 721–722 RSC; (b) S. Kulkowit and M. A. McKervey, J. Chem. Soc. Chem. Commun., 1978, 1069–1070 RSC; (c) M. P. Doyle, W. Hu, I. M. Phillips and A. G. H. Wee, Org. Lett., 2000, 2, 1777–1779 CrossRef CAS PubMed; (d) G.-Y. Li and C.-M. Che, Org. Lett., 2004, 6, 1621–1623 CrossRef CAS PubMed; (e) D. M. Hodgson and D. Angrisha, Chem. Commun., 2005, 4902–4904 RSC; (f) Y. Xia, Z. Liu, Q. Xiao, P. Qu, R. Ge, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 5714–5717 CrossRef CAS PubMed; (g) C. Zhu, G. Xu, K. Liu, L. Qiu, S. Peng and J. Sun, Chem. Commun., 2015, 51, 12768–12770 RSC.
- For examples of intermolecular diazo cross-coupling, see: (a) J. H. Hansen, B. T. Parr, P. Pelphrey, Q. Jin, J. Austbach and H. M. L. Davies, Angew. Chem., Int. Ed., 2011, 50, 2544–2548 CrossRef CAS PubMed; (b) Z. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070–11074 CrossRef PubMed.
- (a) D. M. Hodgson and D. Angrish, Chem. - Eur. J., 2007, 13, 3470–3479 CrossRef CAS PubMed; (b) I. Rivilla, W. M. C. Sameera, E. Alvarez, M. M. Díaz-Requejo, F. Maseras and P. J. Pérez, Dalton Trans., 2013, 42, 4132–4138 RSC; (c) Z. Zhu, X. Xu, D. Ding, L. Qiu and J. Sun, Org. Lett., 2015, 17, 4244–4247 CrossRef PubMed.
- S. Jana, C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 60, 13271–13279 CrossRef CAS PubMed.
- I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC.
- T. Xiao, M. Mei, Y. He and L. Zhou, Chem. Commun., 2018, 54, 8865–8868 RSC.
- (a) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC; (b) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS; (c) C. Empel, C. Pei and R. M. Koenigs, Chem. Commun., 2022, 58, 2788–2798 RSC; (d) R. D. C. Gallo, G. Cariello, T. A. C. Goulart and I. D. Jurberg, Chem. Commun., 2023, 59, 7346–7360 RSC; (e) Z.-Y. Xie and J. Xuan, Chem. Commun., 2024, 60, 2125–2136 RSC; (f) Z. Zhang and V. Gevorgyan, Chem. Rev., 2024, 124, 7214–7261 CrossRef CAS PubMed; (g) F. Li, C. Pei and R. M. Koenigs, Angew. Chem., Int. Ed., 2022, 61, e202111892 CrossRef CAS PubMed.
- G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186–8195 CrossRef CAS PubMed.
- W. Li, S. Li, C. Empel, R. M. Koenigs and L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202309947 CrossRef CAS PubMed.
- G. Xu, C. Zhu, W. Gu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 883–887 CrossRef CAS PubMed.
- A. Dasgupta, S. Pahar, R. Babaahmadi, L. Gierlichs, B. F. Yates, A. Ariafard and R. L. Melen, Adv. Synth. Catal., 2022, 364, 773–780 CrossRef CAS.
- J. Barluenga, L. Riesgo, L. A. López, E. Rubio and M. Tomás, Angew. Chem., Int. Ed., 2009, 48, 7569–7572 CrossRef CAS PubMed.
- (a) S. Li and L. Zhou, Org. Lett., 2024, 26, 3294–3298 CrossRef CAS PubMed; (b) S. Li and L. Zhou, Org. Lett., 2023, 25, 8700–8705 CrossRef CAS PubMed; (c) W. Li, X. Zhou, T. Xiao, Z. Ke and L. Zhou, CCS Chem., 2022, 4, 638–649 CrossRef CAS; (d) W. Li and L. Zhou, Green Chem., 2021, 23, 6652–6658 RSC; (e) W. Li and L. Zhou, Org. Lett., 2021, 23, 4279–4283 CrossRef CAS PubMed.
- (a) E. C. Taylor and I. J. Turchi, Chem. Rev., 1979, 79, 181–231 CrossRef CAS; (b) M. B. Supurgibekov, D. Cantillo, O. Kappe, G. K. S. Prakashd and V. A. Nikolaev, Org. Biomol. Chem., 2014, 12, 682–689 RSC.
- In the cases of, 5p–5r trace amount of 1,4-dihydropyridazines were observed as the side products. However, 1,4-dihydropyridazines were not detected in all the reactions of vinyldiazoketones and simple diazo compounds, even in the presence of weak bases such as K2CO3 and NaOAc.
- W. R. Dolbier and M. A. Merle, Chem. Rev., 2003, 103, 1071–1098 CrossRef CAS PubMed.
- (a) J. C. Sharland and H. M. L. Davies, Org. Lett., 2023, 25, 5214–5219 CrossRef CAS PubMed; (b) C. Qin and H. M. L. Davies, Org. Lett., 2013, 15, 310–331 CrossRef CAS PubMed.
- (a) A. Dasgupta, S. Bhattacharjee, Z. Tong, A. Guin, R. E. McNamee, K. E. Christensen, A. T. Biju and E. A. Anderson, J. Am. Chem. Soc., 2024, 146, 1196–1203 CrossRef CAS PubMed; (b) M. Bao, K. Łuczak, W. Chaładaj, M. Baird, D. Gryko and M. P. Doyle, Nat. Commun., 2024, 15, 4574 CrossRef CAS PubMed; (c) M. Bao, A. R. R. Bohórquez, H. Arman and M. P. Doyle, Chem. Sci., 2024, 15, 12042–12046 RSC; (d) Y. Hussain, P. R., C. Empel, D. Sharma, L. Kloene, W. F. Zhu, A. Kaiser, L. Weizel, E. Proschak, R. M. Koenigs and P. Chauhan, Angew. Chem., Int. Ed., 2024, 63, e202416956 Search PubMed.
- (a) M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed; (b) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- (a) G. Tran, D. Gomez Pardo, T. Tsuchiya, S. Hillebrand, J.-P. Vors and J. Cossy, Org. Lett., 2015, 17, 3414–3417 CrossRef CAS PubMed; (b) V. V. Razin, M. E. Yakovlev, K. V. Shataev and S. I. Selivanov, Russ. J. Org. Chem., 2004, 40, 1027–1032 CrossRef CAS; (c) A. R. Al Dulayymi and M. S. Baird, Tetrahedron, 1998, 54, 12897–12906 CrossRef; (d) M. S. Baird and H. H. Hussain, Tetrahedron, 1987, 43, 215–224 CrossRef CAS; (e) D. H. Aue, R. B. Lorens and G. S. Helwig, J. Org. Chem., 1979, 44, 1202–1207 CrossRef CAS.
- (a) A. Padwa, T. Kumagai and M. Tohidi, J. Org. Chem., 1983, 48, 1834–1840 CrossRef CAS; (b) M. Schneider and B. Csacsko, Angew. Chem., Int. Ed., 1977, 16, 867–869 CrossRef; (c) D. F. Aton, R. G. Bergman and G. S. Hammon, J. Am. Chem. Soc., 1972, 94, 1351–1353 CrossRef CAS.
- Y. Zheng, Q.-X. Dong, S.-Y. Wen, H. Ran and H.-M. Huang, J. Am. Chem. Soc., 2024, 146, 18210–18217 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2369613, 2369614 and 2369627. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00277j |
| This journal is © The Royal Society of Chemistry 2025 |