
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances and developments in solar-driven photothermal catalytic CO2 reduction into multicarbon (C2+) products
Xiuting
Wu†
ab,
Senlin
Zhang†
a,
Shangbo
Ning
 *a,
Chuanyun
Yang
a,
Ling
Li
a,
Linjun
Tang
a,
Jing
Wang
*b,
Ruixiang
Liu
a,
Xingyu
Yin
a,
Ying
Zhu
a,
Shaohua
Chen
a and
Jinhua
Ye
*acd
*a,
Chuanyun
Yang
a,
Ling
Li
a,
Linjun
Tang
a,
Jing
Wang
*b,
Ruixiang
Liu
a,
Xingyu
Yin
a,
Ying
Zhu
a,
Shaohua
Chen
a and
Jinhua
Ye
*acd
aResearch Center for Solar Driven Carbon Neutrality, The College of Physics Science and Technology, Hebei University, Baoding 071002, China. E-mail: ningshangbo@hbu.edu.cn; jinhua.ye@hbu.edu.cn
bKey Laboratory of Analytical Science and Technology of Hebei Province, College of Chemistry and Materials Science, Hebei University, Baoding, 071002, China. E-mail: wangjing9804@163.com
cInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Tsukuba 305-0047, Japan. E-mail: jinhua.YE@nims.go.jp
dAdvanced Catalytic Materials Research Center, School of Material Science and Engineering, Tianjin University, Tianjin 300072, China
First published on 15th February 2025
Abstract
Solar-driven catalytic conversion of carbon dioxide (CO2) into value-added C2+ chemicals and fuels has attracted significant attention over the past decades, propelled by urgent environmental and energy demands. However, the catalytic reduction of CO2 continues to face significant challenges due to inherently slow reduction kinetics. This review traces the historical development and current state of photothermal CO2 reduction, detailing the mechanisms by which CO2 is transformed into C2+ products. A key focus is on catalyst design, emphasizing surface defect engineering, bifunctional active site and co-catalyst coupling to enhance the efficiency and selectivity of solar-driven C2+ synthesis. Key reaction pathways to both C1 and C2+ products are discussed, ranging from CO, CH4 and methanol (CH3OH) synthesis to the production of C2–4 products such as C2–4 hydrocarbons, ethanol, acetic acid, and various carbonates. Notably, the advanced synthesis of C5+ hydrocarbons exemplifies the remarkable potential of photothermal technologies to effectively upgrade CO2-derived products, thereby delivering sustainable liquid fuels. This review provides a comprehensive overview of fundamental mechanisms, recent breakthroughs, and pathway optimizations, culminating in valuable insights for future research and industrial-scale prospect of photothermal CO2 reduction.
 Shangbo Ning | Dr Shangbo Ning is associate professor of materials science and optical engineering in Hebei University, china and member of the Research Center for the Solar Driven Carbon Neutrality. He received his PhD degree from the Tianjin University under the supervision of professor Jinhua Ye and engaged in postdoctoral research at the China University of Mining and Technology from 2023 to 2025. His research interests focus on the research and development of novel photocatalytic materials and their applications in the fields of energy and environment photo-/photothermal-catalysis and solar to chemical energy conversion. |
 Jing Wang | Dr Jing Wang received her PhD from Huazhong University of Science and Technology (2009–2013) and was a visiting scholar at Georgia State University (2016). She is a professor and master's supervisor, recognized with the inaugural China Youth Science and Technology Innovation Award. She has published over 100 SCI papers, holds multiple patents, and has led national and provincial research projects. A recipient of several scientific awards, she was listed among the world's top 2% scientists in 2020. |
 Jinhua Ye | Professor Jinhua Ye received her PhD from the University of Tokyo in 1990, and joined National Research Institute for Metals (former organization of NIMS) in 1991. She is now the appointed director of Research Center for Solar Driven Carbon Neutrality, Hebei University, China. Her research interests focus on the research and development of photo functional materials and their applications in the fields of environment preservation and new energy production. She has been admitted as a Fellow of the Royal Society of Chemistry since 2016, and also selected as 2016, 2018–2024 Highly Cited Researcher (Clarivate Analytics), 2022 Pioneer in Energy Research (ACS Publications). She is currently serving as the Associate Editor of ACS Nano, and Science Advances (AAAS). |
1. Introduction
The escalating impact of climate change and environmental degradation, primarily attributed to the widespread use of fossil fuels, highlights the necessity for effective approaches to curb.1,2 As the principal greenhouse gas, CO2 not only poses severe environmental threats but also presents a potential carbon feedstock for synthesizing high-value chemicals and fuels.3–7 Compared with C1 products such as CH4 and CO, multicarbon (C2+) hydrocarbon products not only have higher energy density, but also are convenient for transportation and storage, which is of great significance in the field of CO2 reduction.8 However, the thermodynamic and kinetic stability of CO2 renders its catalytic transformation into C2+ products particularly challenging, thereby requiring novel approaches capable of breaking the linear trade-off between high selectivity and high conversion.Photothermal catalysis has emerged as a promising avenue for transforming CO2 into valuable C2+ products by harnessing the dual benefits of solar irradiation and thermal energy.8–10 In contrast to purely thermal routes, photothermal catalysis leverages both the photonic energy (often manifested through photoexcited charge carriers in semiconductor or plasmonic materials) and the thermal energy generated from converting unabsorbed light (especially in the infrared region) into heat.11 This synergy offers the potential to accelerate reaction kinetics at relatively lower overall temperatures and to enhance reaction selectivity by modulating critical steps such as CO2 adsorption, intermediate formation (e.g. *CO, formate), and C–C coupling. 12–14 Directing the reaction pathway toward C2+ products calls for rational control of active sites, defect engineering, and local reaction environments, which can be made possible through an integrated photothermal approach.
Despite these advantages, achieving efficient and scalable photothermal CO2 reduction to C2+ products remains challenging. Catalysts must be meticulously engineered at multiple levels, encompassing nanoscale morphology, surface functionalization, and the precise tuning of electronic structures to activate and stabilize multi-carbon intermediates. Equally critical is the design of reactors to ensure optimal photon capture, uniform heat distribution, and accurate control of the reaction atmosphere. Addressing these challenges will require synergistic advancements in catalyst development, reactor engineering, and process optimization, paving the way for the practical realization of photothermal CO2 conversion technologies with high efficiency and scalability.
In this review, we focus on the rapid developments and emerging prospects for photothermal CO2 reduction, emphasizing the formation of C2+ products as a viable strategy for enhancing the economic and practical value of CO2 utilization. After outlining the historical context and current imperatives of photothermal CO2 reduction, we delve into the mechanistic insights that underpin the photothermal conversion of CO2 to C2+ products. We then discuss critical aspects of catalyst design, including surface defect engineering, bifunctional active site construction and co-catalyst coupling, which are instrumental in realizing efficient C2+ production. Finally, we provide an overview of key solar-driven CO2 reduction pathways, categorized according to product type, ranging from C1 to C2+ products. Furthermore, this review aims to highlight the pivotal role of C5+ products in shaping the future of photothermal CO2 upgrading, as these high-value hydrocarbons hold immense potential for sustainable fuel and chemical production. By addressing the challenges and identifying future directions, this review provides both theoretical and practical guidance for researchers and engineers striving to enhance the efficiency, selectivity, and scalability of C2+ products synthesis, ultimately paving the way for large-scale, sustainable implementations in industrial applications.
1.1 History and reality of photothermal CO2 reduction
Global population growth and rising living standards have led to increased energy consumption, with 85% of energy coming from fossil fuels, potentially causing an energy crisis and severe environmental challenges.15–19 A prominent issue is global warming, caused by the emission of greenhouse gases like CO2, methane (CH4).16,20–24Against the backdrop of escalating energy demands and environmental concerns, technologies for converting CO2 have garnered significant attention. CO2, as an abundant carbon source, holds promise not only for mitigating greenhouse gas emissions but also for serving as a resource in the synthesis of valuable chemicals and fuels. In the early 20th century, as catalysis emerged as a scientific discipline, researchers discovered the effectiveness of metallic and metalloid catalysts in reducing CO2.25 As shown in Fig. 1, in 1902, Paul Sabatier published seminal work, “New Synthesis of Methane”, documenting the catalytic conversion of H2 and CO2 into CH4 and H2O using a Ni catalyst at elevated temperatures. This process became known as the Sabatier reaction.25
 | ||
| Fig. 1 The history and recent advances of catalytic conversion of CO2 into C2+ products. | ||
The timeline of development continued until the late 1970s, highlighted by Fujishima and Honda's groundbreaking work in 1972, which laid the foundation for photocatalysis.26 Since then, interest in heterogeneous photocatalysts for converting solar energy into chemical energy has surged.27–33 Despite the development of various semiconductor photocatalysts, practical solar energy conversion efficiency has been unsatisfactory. This low efficiency is due to inadequate absorption of the solar spectrum, especially in the visible light and near-infrared (NIR) regions,34 and the rapid recombination of photoexcited charge carriers.35,36
Entering the 1970s and 1980s, as photocatalytic technology was studied more deeply, researchers sought more efficient photocatalysts. TiO2 became a popular research focus due to its stability and cost-effectiveness.37 Juraj Kizlink et al. reported using organotin(II) compounds to synthesize dimethyl carbonate from CO2 and CH3OH.38 Meanwhile, thermal catalysis also saw advancements with the development of various metal and metal oxide catalysts for CO2 reduction.39 By the 1990s, research on photocatalysis and thermal catalysis mechanisms deepened, focusing on catalyst active sites and reaction pathways, providing a theoretical basis for catalyst design and optimization.40,41
Since the early 21st century, the research in photocatalysis and thermal catalysis has been optimized. In photocatalysis, researchers have refined reaction parameters like temperature, light intensity, and developed more efficient reactors.42 In thermal catalysis, strategies like alloying, doping, and support modification have enhanced catalyst activity, selectivity, and stability.43 In 2014, Ye's group firstly demonstrated the effective use of sunlight and superior photothermal properties of VIII metal-based nanocatalysts, highlighting their unique activation capability for CO2 hydrogenation.44 Photothermal catalysis, a field that has gained considerable attention in recent years, is regarded as a promising avenue for maximizing the efficiency of solar energy utilization. This approach merges the benefits of both thermal and photocatalytic processes, offering excellent catalytic performance even under mild conditions.45 Rather than a mere combination, photothermal catalysis represents a synergistic interaction between photocatalysis and thermal catalysis.46–48 The light can convert to heat, which initiates CO2 reduction reaction, while the photo-excited charge carriers can also regulate reactivity and product selectivity, with reaction efficiency being further enhanced by the heat produced during thermochemical reaction. It optimizes the use of solar energy for the efficient transformation of CO2. The photothermal catalysis can reduce the activation energy required for CO2 conversion and fine-tune the reaction pathways by modulating the electronic structure of catalytic materials, thereby enhancing the selectivity of desired products.49 In 2017, Zeng's team reported the Au&Pt@ZIF catalyst, which combines photothermal effects with insulating properties, reducing the temperature needed for CO2 hydrogenation.50 In particular, the development of efficient new catalysts has become a research hotspot for converting photothermally-driven CO2 into C2+ products.51 For instance, in 2018, Zhang's group developed novel CoFe-based catalysts by reducing CoFeAl layered double hydroxide (LDH) nanosheets with hydrogen at 300–700 °C. These CoFe-x catalysts showed a shift in selectivity from CO to CH4 and then to C2+ under UV-visible light, with selectivity varying based on the reduction temperature of the LDH nanosheets.52
Our research group, dedicated to advancing photothermal CO2 conversion into C2+ products, has achieved significant progress. By altering the electronic structure, photoelectrons can enhance selectivity and prolong catalyst longevity.53 Photothermal catalysis, which combines photocatalysis and thermal catalysis, is a rapidly advancing technology. It utilizes light to generate heat and excite electron–hole pairs, providing an innovative method for CO2 reduction.54 We developed a photo-thermal synergistic catalytic system by integrating Co and Cu nanoparticles onto SrTiO3, demonstrating enhanced performance in Fischer–Tropsch synthesis.55 Meanwhile, we introduced a bifunctional catalyst with Co0–Coδ+ active centers on MgAl2O4 for photothermo-driven CO2 conversion into C2–4 olefins. These studies underscore the importance of carefully engineered metal/oxide interfaces for achieving selective CO2 hydrogenation into higher-value products, advancing both sustainable chemical processes and renewable energy utilization.9 In addition, we synthesized a Cu-modified CoFe alloy catalyst via an enhanced sol–gel method for solar-driven CO2 hydrogenation, which effectively promotes CO2 adsorption and hydrogenation, leading to high selectivity toward C2+ hydrocarbons and C2–4 olefins.56 Currently, photothermal-catalytic CO2 conversion predominantly yields C2–4 products, while C5+ products are still generated in relatively low quantities. There is a pressing need to develop more efficient catalysts and processes that can boost both the yield and selectivity of C5+ products.
Recent advances in this field underscore a sustained emphasis on solar-driven catalytic processes for CO2 conversion, which continues to be a focal area of research. Notably, the push toward producing higher-value C2+ hydrocarbons from CO2 has gained significant traction, highlighting the substantial promise and expanding prospects of CO2 high-added value conversion for sustainable energy applications. This progress demonstrates the potential of light-driven catalysis as a sustainable solution for both climate change mitigation and the production of high-value chemicals from CO2. The growing number of publications and citations reflects the increasing recognition of this technology's significance and its promise for both scientific and practical applications (Fig. 2). A key challenge remains optimizing selectivity and efficiency in C2+ hydrocarbon production, which is essential for scaling these systems. Ultimately, this research not only enhances our understanding of solar-driven catalysis but also contributes to the development of carbon-neutral technologies, with substantial implications for sustainable chemical manufacturing and global sustainability goals.
 | ||
| Fig. 2 The number of publications and citations from Web of Science on photo-driven catalytic CO2 reduction, encompassing all product types, has surged in recent years; the illustration focuses specifically on CO2 conversion into C2+ products. | ||
1.2 Mechanism of photothermal conversion of CO2 into C2+ products
Photo-driven direct conversion of CO2 to C2+ as major products, either saturated or unsaturated hydrocarbons or oxygenated products, such as light olefins (C2–4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), paraffins (C2–40), C2+ alcohol, carboxylic acid, and dimethyl carbonate, would be much more attractive, considering the higher market price per ton of these C2+ compounds, compared to the C1 products.57
), paraffins (C2–40), C2+ alcohol, carboxylic acid, and dimethyl carbonate, would be much more attractive, considering the higher market price per ton of these C2+ compounds, compared to the C1 products.57
Hydrogenation of CO2 to form various products is an exothermic process, where the exact is dependent on the product. Eqn (1) and (2) summarize the redox reactions of commonly reported C1 products of the photocatalytic CO2 reduction.
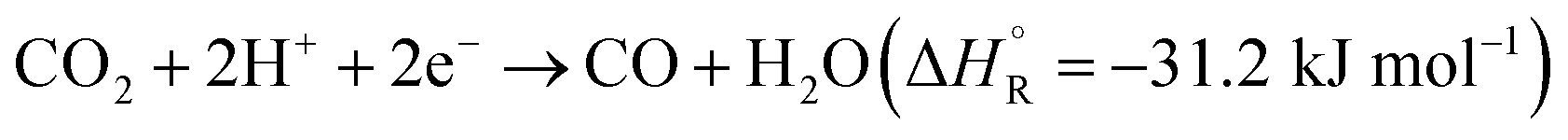 | (1) |
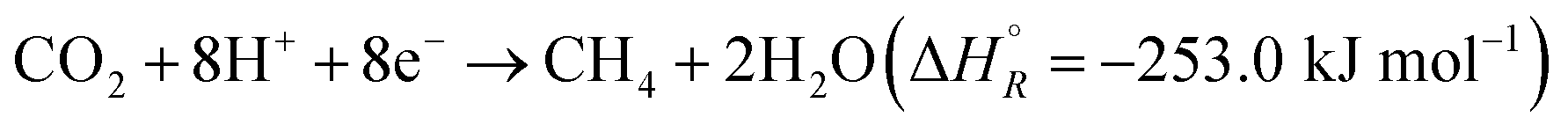 | (2) |
The high added C2+ chemicals with multiple carbon atoms are more attractive than C1 products because they have a higher energy density. Hydrogenation of CO2 to C2+ products can be achieved through two different paths.58 The first is the direct CO2 Fischer–Tropsch synthesis pathway; the second is the indirect methanol-mediated pathway, where CO2 is first converted to CH3OH, which is subsequently transformed into C2+ products through reactions such as dehydration, oligomerization, and hydrogenation. The direct pathway is the CO2-Fischer–Tropsch (CO2-FT) path: CO2 and H2 undergo reverse water gas conversion (RWGS) reaction to form the intermediate CO, followed by Fischer–Tropsch synthesis reaction (FTS). In the following CO2-FT reaction, C1 intermediates undergo C–C coupling and hydrogenation to form C2+ products. C–C coupling intermediates play a crucial role in the synthesis of C2+ hydrocarbons. Due to the complexity of intermediates and diverse reaction pathways, uncovering the reaction mechanisms of C2+ hydrocarbons remains a significant challenge.59–61 However, the chain growth pathway in FTS remains controversial. Two proposed mechanisms are CHx insertion (carbide type) 62 and “CO” insertion.63 The CHx insertion assumes CO activation either by direct reaction with chemisorbed hydrogen or via an H-assisted pathway, possibly through formyl (HCO*), formaldehyde (H2CO*), or hydroxymethylene (HCOH*) species, to form surface 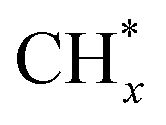 .
.
Direct CO activation removes O* from CO2, while H-assisted CO dissociation forms H2O exclusively. In the CO insertion mechanism, -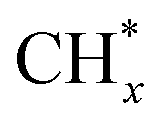 species are believed to couple with CO* to form –CH2CO*, facilitated by surface OH groups, followed by deoxyhydrogenation of the oxygen intermediate.63 The main reaction is as follows eqn (3)–(5). Considering the optimal catalyst strategy, the different electron distribution and valence state of two different metal active sites at the interface may cause the charge distribution of the C1 intermediate to be significantly different, thus reducing the C1 electrostatic repulsion, promoting the C–C coupling reaction. Ning et al. have successfully developed a Co0–Coδ+ double active centre catalyst supported on MgAl2O4 (Co–CoOx/MAO) for the efficient conversion of CO2 into C2–4 olefins by photothermocatalysis.9
species are believed to couple with CO* to form –CH2CO*, facilitated by surface OH groups, followed by deoxyhydrogenation of the oxygen intermediate.63 The main reaction is as follows eqn (3)–(5). Considering the optimal catalyst strategy, the different electron distribution and valence state of two different metal active sites at the interface may cause the charge distribution of the C1 intermediate to be significantly different, thus reducing the C1 electrostatic repulsion, promoting the C–C coupling reaction. Ning et al. have successfully developed a Co0–Coδ+ double active centre catalyst supported on MgAl2O4 (Co–CoOx/MAO) for the efficient conversion of CO2 into C2–4 olefins by photothermocatalysis.9
| CO2 + H2 ⇌ CO + H2O | (3) |
| nCO + 2nH2 ⇌ CnH2n + nH2O | (4) |
| nCO + (2n + 1)H2 ⇌ CnH2n+2 + nH2O | (5) |
The indirect pathway is methanol-mediated, where CO2 and hydrogen are first converted to CH3OH, followed by methanol-to-olefin reaction (MTO). The stoichiometric reaction of CO2 hydrogenation to CH3OH is shown in eqn (6) and (7). This reaction pathway has been recognized over bifunctional systems comprising a CH3OH synthesis catalyst and a methanol-to-hydrocarbon (MTH) catalyst. Li et al.64 proposed the use of a ZnZrO/SAPO tandem catalyst for the direct hydrogenation of CO2 to produce light olefins. In mechanistic studies, in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) was used to detect surface species formed on ZnZrO and the tandem catalyst. Quantitative analysis using chemical trapping-mass spectrometry revealed that the intensity of CH3O* on the tandem catalyst was lower than on ZnZrO, but chemical trapping produced strong mass signals for CD3CHO and CH3OCD3, indicating that CHO* and CH3O* species were more actively involved in the reaction (Fig. 3a and b). These results suggest that these derived CHxO species migrate into the zinc-modified SAPO-34 zeolite, where they are converted into lower olefins (Fig. 3c).
| CO2 + 3H2 ⇌ CH3OH + H2O ΔH25°C = −49.5 kJ mol−1 | (6) |
| 2CH3OH → CH3OCH3 + H2O → CnH2n + H2O | (7) |
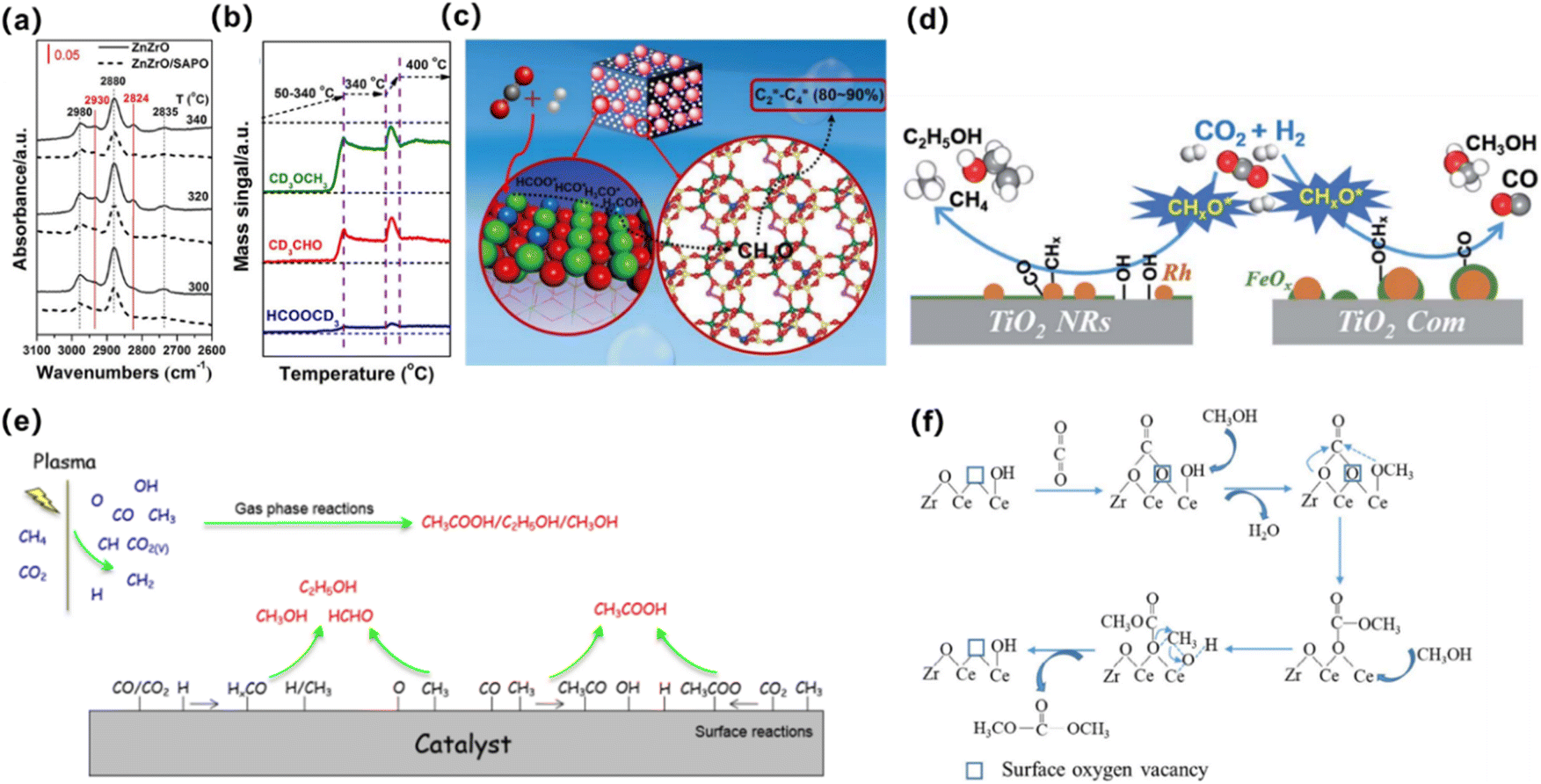 | ||
| Fig. 3 (a) In situ DRIFT spectrafrom CO2 hydrogenation over ZnZrO and ZnZrO/SAPO at different temperatures. (b) Chemical trapping-mass spectrometry results with a trapping reagent CD3OD during the CO2 hydrogenation over ZnZrO at reaction temperatures of 50–400 °C. (c) Schematic for the proposed reaction mechanism of CO2 hydrogenation on the tandem catalyst, ZnZrO/SAPO.64 Copyright 2017, American Chemical Society. (d) Schematic of CO2 hydrogenation over the Rh-based catalyst with or without hydroxyl groups on TiO2. The hydroxyls play an important role in accelerating the scission of CHxO* and promote the formation of ethanol.65 Copyright 2019, Royal Society of Chemistry. (e) Possible reaction mechanisms for the formation of CH3COOH, CH3OH, C2H5OH and HCHO using the plasma-catalysis approach.66 Copyright 2017, Royal Society of Chemistry. (f) Oxygen vacancy promoting dimethyl carbonate synthesis from CO2 and CH3OH over Zr-doped CeO2 nanorods.67 Copyright 2018, Royal Society of Chemistry. | ||
Ethanol is arguably available CO2 hydrogenation C2 product due to its safe transport and higher calorific value.68 The process of CO2 hydrogenation to ethanol includes CO2 to produce CO by reverse water-gas conversion reaction, partial CO dissociation to produce 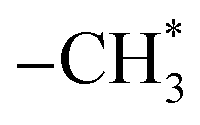 group, CO embedding into
group, CO embedding into 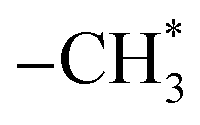 to complete carbon chain growth, and further hydrogenation to produce ethanol.69,70 Therefore, the ethanol synthesis catalyst requires the following functional sites, H2 dissociation, CO dissociation adsorption site, CO non-dissociation adsorption site, and C–C coupling site. The reaction formula of CO2 catalytic hydrogenation to ethanol is shown in eqn (8).
to complete carbon chain growth, and further hydrogenation to produce ethanol.69,70 Therefore, the ethanol synthesis catalyst requires the following functional sites, H2 dissociation, CO dissociation adsorption site, CO non-dissociation adsorption site, and C–C coupling site. The reaction formula of CO2 catalytic hydrogenation to ethanol is shown in eqn (8).
| 2CO2 + 6H2 → C2H5OH + 3H2O | (8) |
In a continuous reactor, Yang et al. explored the key role of hydroxyl in Rh-based catalysts supported on TiO2 nanorods (Fig. 3b). The results show that the ethanol selectivity of RhFeLi/TiO2 nanorods is 32% in the catalytic CO2 hydrogenation reaction. The promotion effect can be attributed to the highly dispersed Rh and high density *CHx on the TiO2 nanorods, and then CO (generated by RWGS reaction) is inserted into *CHx to form *CH3CO, and *CH3CO is hydrogenated to produce ethanol.65
Acetic acid is an important commodity chemical, mainly used as a raw material for the production of vinyl acetate monomer, ethyl acetate, propyl acetate, n-butyl acetate, isobutyl acetate and acetic anhydride, or as a solvent in the synthesis of terephthalic acid.71
It is generally believed that the catalytic mechanism of direct synthesis of acetic acid by coupling CH4 and CO2 consists of 5 steps (eqn (9)–(13)): (1) CH4 dissociates on the catalyst surface to form methylated species (M–CH3) and a Brønsted proton; (2) CO2 is adsorbed on the surface of the catalyst to produce surface adsorbed species CO2 (on some catalysts, the gas phase CO2 can be directly inserted into the M–CH3 bond, without this step); (3) 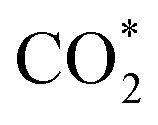 is inserted into M–CH3 bond (C–C coupling) to generate surface acetate species; (4) Brønsted protons on the surface acetate species extraction carrier form acetic acid adsorption species; (5) acetic acid adsorbed species desorption to produce acetic acid.72–76
is inserted into M–CH3 bond (C–C coupling) to generate surface acetate species; (4) Brønsted protons on the surface acetate species extraction carrier form acetic acid adsorption species; (5) acetic acid adsorbed species desorption to produce acetic acid.72–76
 | (9) |
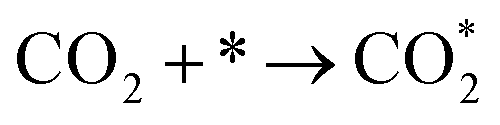 | (10) |
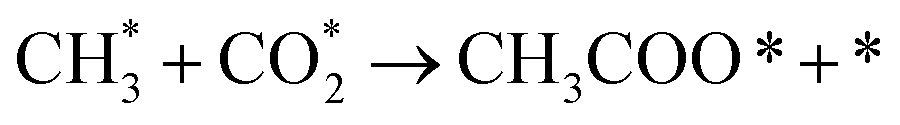 | (11) |
| CH3COȮ* → CH3COOH* + * | (12) |
| CH3COOH* → CH3COOH + * | (13) |
Tu et al. adopted a new atmospheric pressure reactor to achieve the one-step synthesis of liquid fuels and chemicals at room temperature by direct reforming of CO2 and CH4 (Fig. 3c). The overall selectivity for liquid chemicals is about 50–60%, and the main product is acetic acid. The molar ratio of CH4/CO2 and the type of catalyst can control the formation of different oxygenates.66
Dimethyl carbonate (DMC) is known as the “new base block” of organic synthesis in the 21st century, which can replace toxic phosgene, dimethyl sulfate, chloromethane and other reagents for methylation, carbonylation, methoxylation and other reactions, and is used to synthesize a variety of important fine chemicals. 77 From a green chemistry point of view, direct synthesis of DMC from CO2 and MeOH is a better route because the raw material (MeOH) is relatively cheap and the main by-product is water (H2O). 78 The reaction formula is as follows:
| 2CH3OH + CO2 → CO(OCH3)2 + H2O14 |
At present, although there are many studies on the catalytic mechanism of CH3OH and CO2 synthesis of DMC, it can be basically summarized into two types: the reaction mechanism of direct activation of CO2 and the reaction mechanism of first activation of CH3OH and then activation of CO2.
Catalytic mechanism of direct activation of CO2: the catalyst provides electrons to the π orbital of CO2 molecules, resulting in the bending of its molecular structure, and with the extension of C–O bonds, the formation of [CO2]−, thus realizing the activation of CO2, and further coupling with CH3OH to produce DMC under the synergistic action of the catalyst. The catalytic mechanism of the first activation of CH3OH and then activation of CO2 is that the catalyst causes CH3OH to lose protons to form [MeO]−, and then combines with CO2 to form [MeOCOO]−, and then the methyl group is transferred to form DMC. Li et al. synthesized a series of ZR-doped CeO2 nanorods by hydrothermal method (Fig. 3d), and studied in detail the effect of the doping content of Zr on the lattice structure, microstructure, especially the amount of oxygen vacancy as well as the catalytic activity of DMC synthesis from CO2 and CH3OH were studied in detail.67
2. Catalyst design for solar-driven catalytic C2+ product synthesis
2.1 Surface defect engineering
The generation of surface defects such as oxygen vacancies (OVs), interstitial defects, or cation states has been widely reported to enhance photo-driven catalytic activity.79–82 Oxygen vacancy engineering significantly enhances CO2 reduction performance by optimizing electronic structures and charge separation. Ye and co-works achieved self-doped SrTiO3–δ photocatalysts exhibit improved visible light absorption and charge mobility due to oxygen vacancies.79 Similarly, ultrathin W18O49 nanowires with abundant vacancies enhance CO2 activation and photochemical reduction efficiency.80 Furthermore, cation vacancies in ZnS enhance CO2 reduction by providing stable active sites that facilitate charge separation and CO2 adsorption. Unlike anion vacancies, they resist oxygen refilling, maintaining activity and promoting intermediates formation for improved CO2 photoreduction efficiency.81,82High temperature treatment in hydrogen atmosphere has been regarded as a powerful method for surface defect preparation, especially for oxygen vacancies. After the pioneering method was developed. Mao's team, it has been successfully used for oxygen vacancy introduction in a variety of metal oxide systems.83 As shown in the (Fig. 4a), during the high-temperature treatment process, hydrogen reduces TiO2 to other chemical substances, and a partially disordered structure can be found on the surface, and Ti4+ will become Ti3+ and form oxygen vacancy. This hydrogenation process will make the color of TiO2 change from white to black (Fig. 4b), greatly improving the light absorption range and enhancing the catalytic activity.
 | ||
| Fig. 4 (a) Schematic illustration of the concept to make black TiO2. (b) Pictures of white and black TiO2 nanomaterials.83 Copyright 2011, American Association for the Advancement of Science. (c) Pt-sensitized graphene-wrapped RBT.82 Copyright 2018, Royal Society of Chemistry. (d) Possible main reaction paths for acetic acid generation on ultrathin WO3·0.33H2O nanotubes.84 Copyright 2018, American Chemical Society. (e) Schematic illustration for the formation of VO-rich/poor atomically thin In2O3 porous sheets at different atmosphere.85 Copyright 2014, American Chemical Society. | ||
According to this method, Durrant et al. reported CH4 and ethane production using Pt-sensitized graphene-wrapped defect-induced TiO2 photocatalysts under sun-simulated light irradiation (Fig. 4c). In this example, TiO2 NPs were reduced with sodium borohydride at 350 °C in an argon atmosphere, creating defect-induced TiO2 with midgap states (Ti3+). The defect-induced TiO2 was then mixed with GO and annealed at 230 °C in a vacuum oven. Finally, Pt NPs were photo-deposited from H2PtCl6 aqueous solution with MeOH as a sacrificial agent. The authors proposed that the synergy between surface-Ti3+ sites and wrapped graphene enhanced ethane formation.82
Tatsumi Ishihara et al. reported the first synthesis of ultrathin WO3 0.33H2O nanotubes with a large amount of exposed surface Vo sites, achieving excellent and stable CO2 photoreduction to CH3COOH in pure water under solar light (Fig. 4d). The selectivity for acetic acid is up to 85%, with an average productivity of about 9.4 μmol g−1 h−1. Importantly, Vo sites in the catalyst remained stable, with their concentration unchanged even after 60 hours of reaction. Quantum chemical calculations and in situ DRIFT studies revealed the main reaction pathway: CO2 → ˙COOH → (COOH)2 → CH3COOH.84
Xie et al.85 have developed a rapid heating phase transition method to create defects, or oxygen vacancies, on the surface of two-dimensional materials. Using CeCO3OH, In(OH)3, WO3·xH2O, and cobaltous oxide ultra-thin nanosheets as precursors, the researchers prepared cerium oxide, In2O3, tungsten trioxide, and Co3O4 ultra-thin nanosheets through the phase transformation process (Fig. 4e). This method endows these materials with a narrowed band gap and higher carrier concentration, thereby enhancing visible light harvesting and improving carrier separation efficiency.85–88
Chemical reduction is an effective method for introducing surface defects into semiconductor materials. This can be achieved using reducing agents such as NaBH4, calcium hydride, and hydrazine, or reducing solvents like glycol and glycerol.89–91 When metal oxides are prepared using reducing solvents, the material typically loses oxygen atoms, creating oxygen vacancies. For example, in BiOCl, ethylene glycol reacts with oxygen atoms on the BiOCl (001) surface at 160 °C, forming oxygen vacancies. These vacancies extend the photo-absorption edge of the catalyst to visible light, enhance its ability to trap photogenerated electrons, and enable it to activate oxygen molecules to produce free radicals.92
Surface defect engineering provides a flexible and effective method to improve catalytic performance by selectively introducing or optimizing defect structures on the catalyst surface. This provides a new way to design more efficient and selective catalysts, and plays an important role in the research and application of catalytic CO2.
2.2 Bifunctional active sites design
The photo-driven hydrogenation of CO2 to produce C1 products like CO and CH4 has been extensively studied. However, methods for CO2 reduction to C2+ products show low activity and selectivity. In photothermal-catalytic systems, enhancing C2+ product generation while suppressing C1 formation remains challenging. This is because forming C2+ products requires C–C coupling reactions between intermediates, which face higher reaction barriers than C–H bond formation. Typically, the selectivity for C2–C4 hydrocarbons and C5+ hydrocarbons is limited to 58% and 48%,93 respectively, due to the Anderson–Schulz–Flory (ASF) distribution, significantly limiting C2+ production.Efficiently producing C2+ products requires high-selectivity C–C coupling in the CO2 reduction reaction. Catalysts with bifunctional active sites can overcome this limitation. Depending on the reaction pathway, such as the modified FTS pathway or the CH3OH pathway, the bifunctional catalyst components play different roles in producing value-added products, thus breaking the ASF distribution.94 Therefore, designing and regulating catalytic active sites for C–C coupling provides insights into creating catalysts that produce C2+ products other than CO. Bifunctional catalysts are classified based on the source and configuration of their active sites: heteronuclear multi-active sites, homonuclear multi-active sites, and combined integration of two kinds of active sites (Fig. 5).95 The first two types refer to catalysts composed of two adjacent active sites, differing in whether the active sites are the same. The third type involves randomly distributed homonuclear dual active sites.
 | ||
| Fig. 5 Representative categories of photo-driven CO2 reduction multi-active sites.95 Copyright 2014, Springer. | ||
Homonuclear multi-active site catalysts refer to identical and adjacent active sites embedded in a support structure, where the two active sites can interact by modulating the local electronic structure. This interaction results in different or similar structural behaviors. Ye et al. utilized a Co0 and Coδ+ dual active center catalyst supported on MgAl2O4 (Co-CoOx/MAO) for the efficient photothermal catalytic conversion of CO2 to C2–4 olefins.9 In this system, Co0 sites effectively adsorb and activate CO2 to produce C1 intermediates, while the electron-deficient Coδ+ sites reduce the energy barrier for the critical CHCH* intermediate, promoting the formation of C2–4 olefins. Compared to the Co/MAO catalyst, the Co–CoOx/MAO catalyst with dual active sites produces a substantial amount of multi-carbon products in the CO2 hydrogenation products (Fig. 6a). The activity for C2–4 hydrocarbons reached as high as 1303 μmol g−1 h−1. Additionally, using the same synthesis procedure, Co species were loaded onto TiO2 and Al2O3. The cobalt species on different supports exhibited similar product distributions in CO2 hydrogenation under identical reaction conditions (Fig. 6b), indicating the general applicability of the Co0–Coδ+ site design concept.
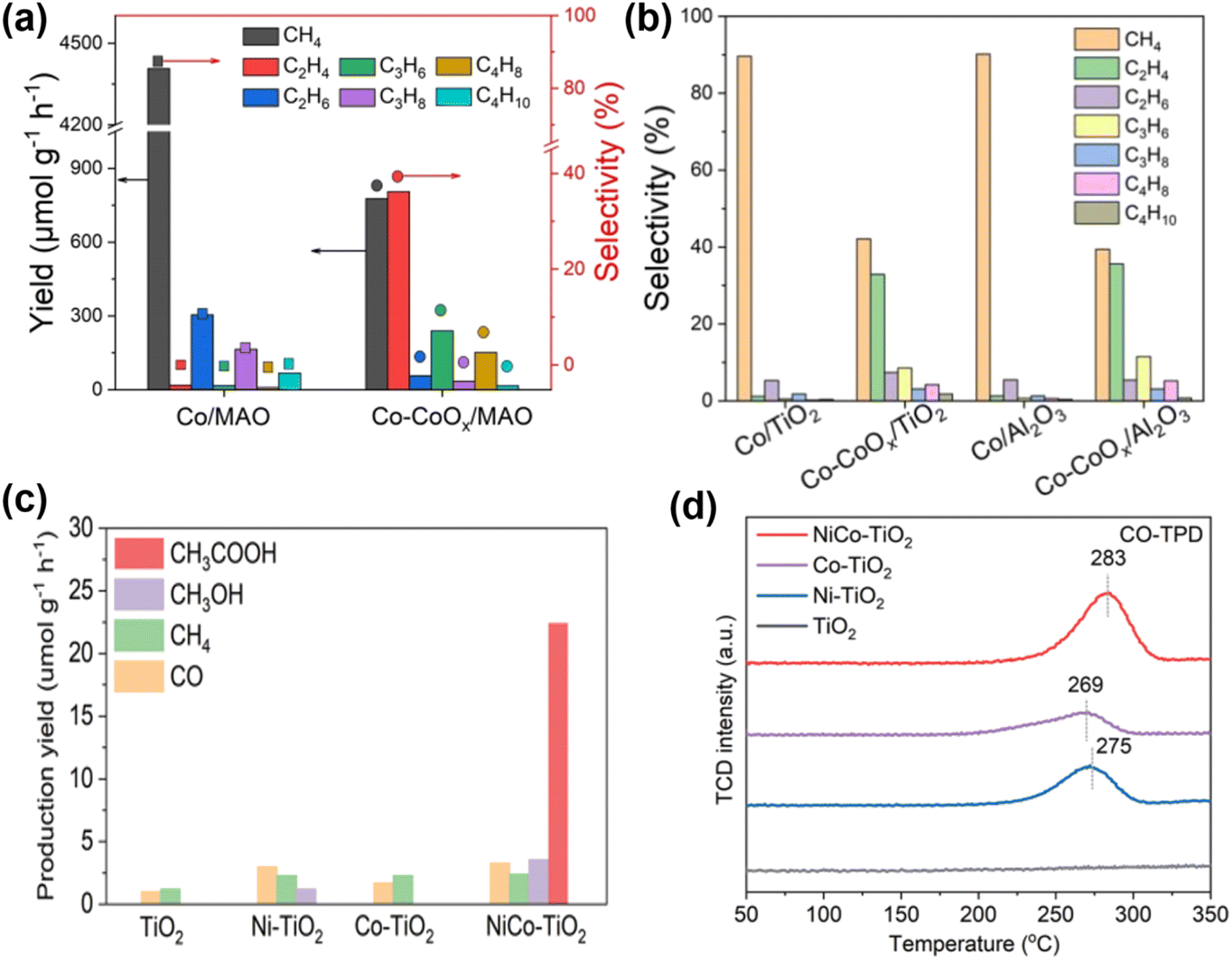 | ||
| Fig. 6 (a) Product yield and selectivity of CO2 hydrogenation in a closed gas circulation system. (b) Product selectivity relative to other catalysts.9 Copyright 2023, Wiley. (c) Yield of CH3COOH and other products under UV-visible light irradiation. (d) CO-TPD spectra of NiCo–TiO2, Ni–TiO2, Co–TiO2 and TiO2.96 Copyright 2022, Wiley-VCH Verlag. | ||
In contrast to homonuclear multi-active sites, heteronuclear multi-active sites have distinct active sites with varying electronic structures, making them potentially effective photocatalysts for CO2 reduction reaction (CO2RR). The electronic structure of such catalysts can be suitably tuned through synergistic effects, resulting in enhanced performance. Jia et al.96 synthesized a Ni and Co co-loaded TiO2 photocatalyst (NiCo–TiO2). The dual Ni and Co sites replace adjacent Ti atoms in the TiO2 lattice, forming an asymmetric dual-atom site photocatalyst. The asymmetric local electric field generated by the dual-atom sites creates asymmetric charge centers, promoting the separation of photogenerated carriers and facilitating multi-electron processes. This accelerates the CO2RR reaction, leading to high selectivity and activity for CH3COOH production. Photocatalytic CO2 reduction results show that CH3COOH is a major product for NiCo–TiO2, while the original TiO2, Ni–TiO2, and Co–TiO2 predominantly produce C1 species (Fig. 6c). Compared with Ni–TiO2 and Co–TiO2, the CO-TPD signal of NiCo–TiO2 shifts to higher temperatures, indicating superior chemisorption capacity in the dual-site state, mainly due to the electronic interaction of the bimetal, which promotes the formation of C2+ products post-coupling (Fig. 6d).
Two separate active sites are randomly distributed on the support, with some interconnected, offering unique advantages in CO2RR due to synergistic effects. Integrating different single atoms into the support enables synergistic catalysis in CO2 activation. For example, Wang et al.97 introduced P and Cu onto carbon nitride nanosheets, forming P–N and Cu–N4 dual active sites (P/Cu SAs@CN), achieving efficient carrier separation for photocatalytic CO2RR to produce C2H6. The dual active sites significantly enhanced CO2 reduction performance, with a C2H6 yield of 616.6 μmol g−1 h−1, 26 times higher than Cu SAs@CN, highlighting the crucial role of P in C2H6 production (Fig. 7a and b). Quasi-in situ XPS showed a positive shift of P2p after light irradiation on P/Cu SAs@CN, indicating P atoms' role in hole capture. P atoms capture photo-generated holes, aiding electron accumulation at Cu sites and enabling multi-electron transfer in CO2RR (Fig. 7c). Theoretical analysis shows electron-enriched Cu sites have a lower energy barrier for C–C coupling under light, favoring CO2RR and C2H6 formation. CO2 adsorbed onto Cu sites reduces to CO*, and then desorbed, while the rest forms CH4 and C2H6 through electron transfer and hydrogenation steps, with *OCCO to *OC–COH as key steps (Fig. 7d).
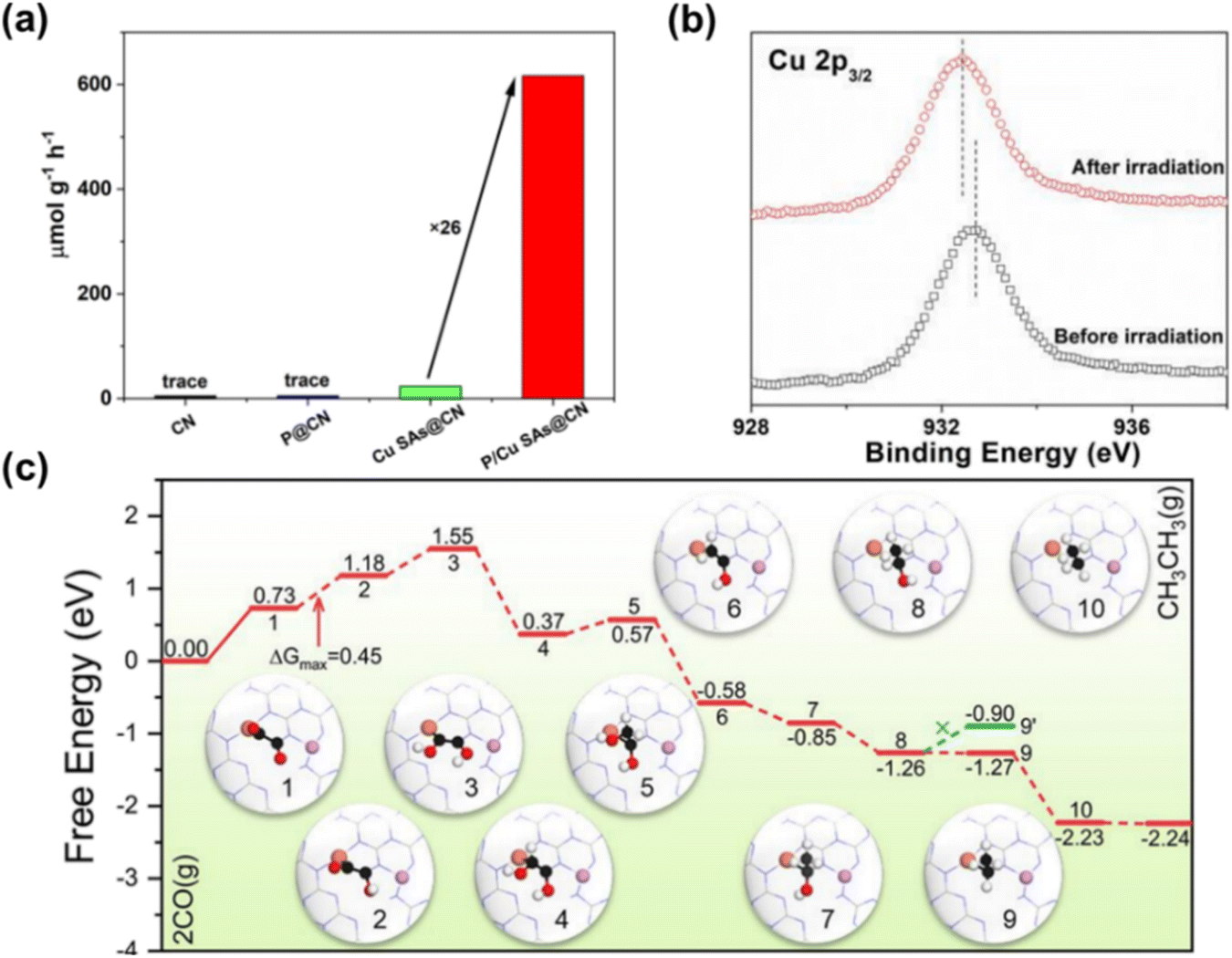 | ||
| Fig. 7 (a) Photo-driven CO2RR generates C2H6 on CN, P@CN, CuSAs@CN and P/Cu SAs@CN. (b) Cu 2p3/2 over P/Cu SAs@CN before and after light irradiation. (c) Free energy diagram of CO coupling reduction to C2 products on P/Cu SAs@CN photocatalyst and atomic structure of reaction intermediates.97 Copyright 2022, Wiley. | ||
2.3 Coupling catalytic effect of co-catalyst
Compared to typical surface catalytic reactions, the transfer of photogenerated carriers to the surface occurs much more rapidly, leading to the accumulation of interfacial charges.98 Reduced charge separation rates and increased reaction overpotentials result in poorer overall photocatalytic performance. Loading suitable co-catalysts can lower the overpotential of redox reactions, enhance electron–hole pair (EHP) separation at the interface, inhibit photodegradation, and ultimately improve photostability.99,100In semiconductor-cocatalyst composite systems, the electron dynamics during CO2 photoreduction can be divided into four processes (Fig. 8): (1) excitation of the semiconductor. (2) Transfer of photogenerated electrons from the semiconductor through the junction to the co-catalyst and coupling its hot electrons. (3) Transfer of photogenerated/hot electrons to the active sites of the co-catalyst. (4) Surface redox reactions at the active sites.101 Efficient electrons migration from the catalyst surface to the co-catalyst addresses issues such as significant carrier recombination, low solar energy utilization, and photocatalyst photodegradation.
 | ||
| Fig. 8 Typical process of semiconductor-cocatalyst composite system in CO2 photoreduction process.101 Copyright 2019, American Chemical Society. | ||
Co-catalysts enhance charge separation in photo-driven catalytic reactions by acting as sinks for photogenerated electrons (e−) or holes (h+), and coupling photothermal conversion at the composite sites. Li et al. modified ultrathin porous g-C3N4 nanosheets with AuCu alloy nanoparticles.102 These AuCu NPs facilitate rapid charge separation and partially activate CO2 molecules, aiding CO2 reduction. The local surface plasmon resonance (LSPR) of Au enhances light absorption and light-to-heat conversion, and promotes C–C coupling during ethanol formation. Transient photocurrent responses show that Cu/g-C3N4, Au/g-C3N4, and AuCu/g-C3N4 electrodes produce higher photocurrents than g-C3N4 alone, with AuCu/g-C3N4 showing the highest, six times greater than g-C3N4 (Fig. 9a), suggesting that AuCu nanoparticles significantly improve charge separation efficiency. Photothermal catalytic tests show increased activity of metal-loaded catalysts at 120 °C, particularly boosting ethanol yield on AuCu/g-C3N4 (Fig. 9b). The proposed mechanism for ethanol production involves photogenerated electrons and holes in g-C3N4 transferring to AuCu NPs under light. Positive charges on Au enhance CO2 adsorption, while charge transfer from Au to Cu enriches Cu with negative charges, promoting CO2˙− and *CO formation on Cu. Elevated temperatures enhance molecular motion, creating a synergistic effect between photocatalysis and thermocatalysis, promoting *CO dimerization and increasing CH3CH2OH yield (Fig. 9c).
 | ||
| Fig. 9 (a) Photocurrent response of g-C3N4 nanosheets, Cu/g-C3N4, Au/g-C3N4 and AuCu/g-C3N4 nanocomposites. (b) Photothermal catalytic reduction of CO2 performance (PTCR: 120 °C). (c) Schematic diagram of PTCR conversion of CO2 into ethanol via AuCu/g-C3N4 nanocomposite catalyst.102 Copyright 2020, Elsevier. | ||
The accumulation of photogenerated/hot electrons and holes on co-catalyst nanoparticles (NPs) leads to C2+ products formation through semiconductor interactions. Ji et al. developed Au–CeO2 nanocomposites with Au–O–Ce sites, enabling selective photocatalytic CO2 conversion to ethane.103 The Au–CeO2 interface ensures multi-electron reduction of CO*, enhancing CO2 conversion. Pristine CeO2 shows a CO yield of 2.02 μmol g−1 h−1 with no C2H6 over 4 hours. Increasing Au component shifts the primary product from CO to C2H6, peaking at 3 wt% Au with yields of 5.88 and 11.07 μmol g−1 h−1 for CO and C2H6, respectively (Fig. 10a). In situ FTIR reveals crucial C2+ intermediates like COCO* and COCOH* (Fig. 10b), absent in pristine CeO2, indicating the importance of the Au–CeO2 interface. Gibbs free energy (Fig. 10c) calculations show that the Au–CeO2 interface lowers the COOH* formation barrier from 1.09 eV to 0.50 eV, favoring COCO* formation over CO desorption. This reduction in energy barriers is key to producing C2H6, confirming the critical role of the Au–O–Ce interface in CO2 reduction to C2+ products.
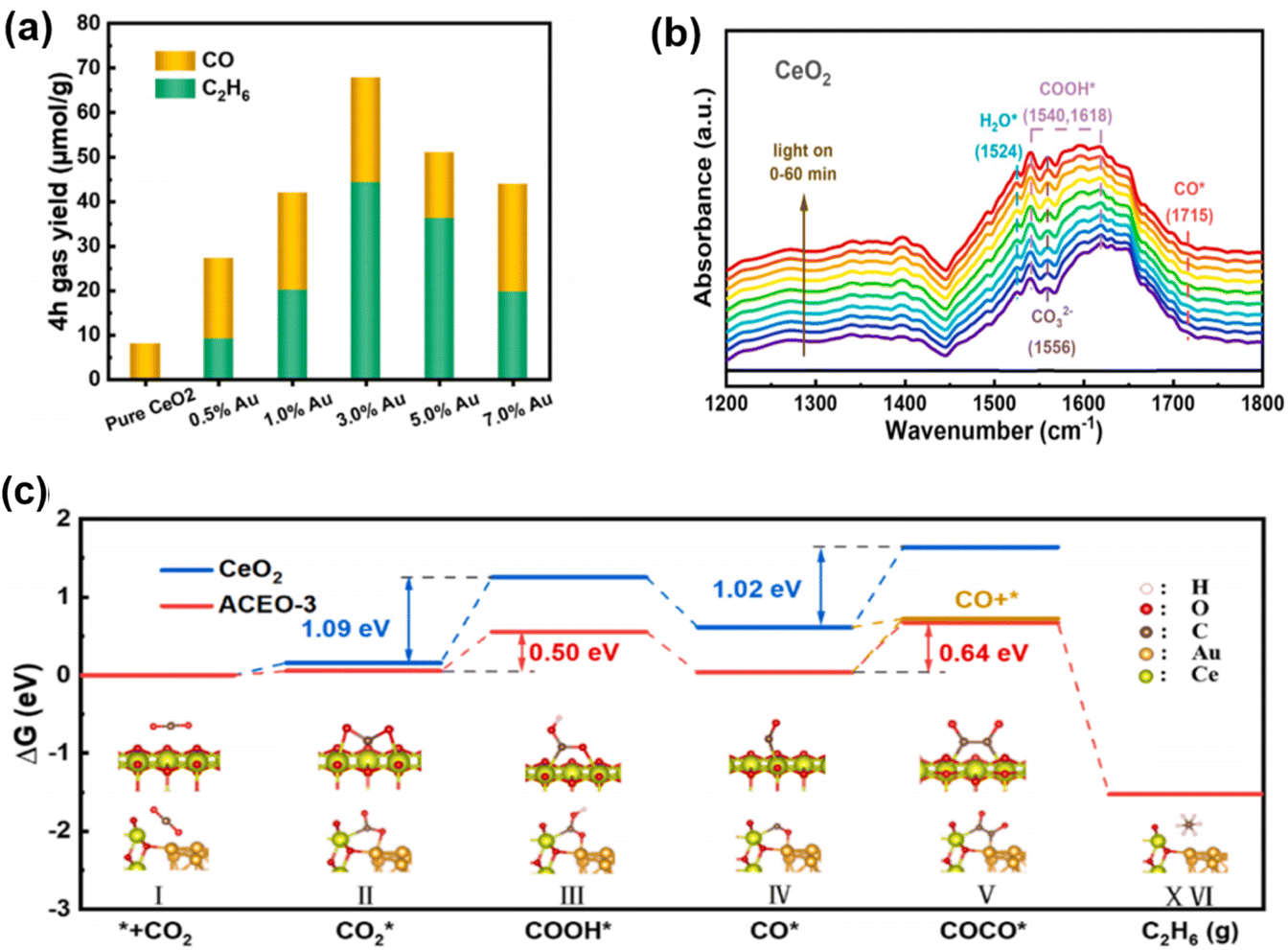 | ||
| Fig. 10 (a) CO and C2H6 generation rates as a function of Au content; in situ FTIR spectra of CO2 and H2O vapor mixtures co-adsorbed on (b) CeO2. (c) Reaction pathway for Gibbs free energy calculation of photocatalytic CO2 reduction.103 Copyright 2023, Elsevier. | ||
3. Solar-energy mediated CO2 reduction reactions
The catalytic conversion of CO2 into value-added C1 products such as CO, CH4, and CH3OH has emerged as a promising approach for mitigating carbon emissions.104–106 This strategy is closely aligned with the principles of green circular economy and environmental sustainability. Despite its potential, the primary challenge remains in enhancing the efficiency and selectivity of CO2 conversion processes to produce high-value products on an industrial scale. This review highlights recent advancements in photothermal catalytic systems for CO2 conversion, with a focus on not only the production of C1 compounds but also the synthesis of more complex and economically significant C2+ products. These developments provide valuable insights into sustainable carbon utilization and the design of advanced catalytic materials for high-performance applications.3.1 Photothermal catalytic reduction of CO2 to C1 products
H2 and CH4 are primary reductants used in the reduction of CO2, which correspond to hydrogenation and reforming.107 The following will provide an overview of the main reactions of catalytic reduction of CO2 to C1 products.However, RWGS is an endothermic reaction that requires high temperature conditions to proceed favorably, and the process consumes a lot of energy.108,109 RWGS reactions are thermodynamically favorable at high temperature, but can lead to carbon formation and unwanted byproducts. At the same time, too high temperature also faces the risk of metal sintering and coke deposition leading to catalyst deactivation. 110 At lower temperatures (below 500 °C), the reaction is kinetically unfavorable, necessitating extensive catalyst use. Therefore, current research focuses on developing different catalytic systems in photothermal catalytic processes and exploring milder catalytic conditions.
Research on metal catalysts is primarily focused on those based on Ni, Co, Cu, Fe, and Mo nanoparticles (NPs). Liu et al.111 developed a catalyst consisting of Co NPs with N-doped carbon, created through a two-step pyrolysis of a ZIF-67 precursor (Fig. 11a). Their Co@NC-700 catalyst demonstrated high activity and selectivity for the photocatalytic hydrogenation of CO2 to CO, achieving a CO generation rate of 0.75 mol gcat−1 h−1 and a CO selectivity of 92.6% under full-spectrum light. The exceptional performance was due to the synergistic effects of the well-coordinated Co NPs, carbon layer thickness, and N-doping species. The catalyst's durability is enhanced by carbon encapsulation, providing a simple and effective approach to producing highly active and long-lasting non-noble metal catalysts.
 | ||
| Fig. 11 (a) Schematic illustration for the preparation of N-doped carbon-coated Co NPs.111 Copyright 2023, Wiley. (b) High-resolution XPS spectra of O 1s for BiOBr and BC6.112 Copyright 2022, American Chemical Society. (c) Time course of CO and CH4 yield within 30 min and product selectivity for CO2 hydrogenation over Co and Co@CoN&C under irradiation.113 Copyright 2020, American Chemical Society. | ||
Yan and co-workers discovered that nanostructured BiOBr semiconductor catalyst, with its staggered layered structure, was a promising photocatalyst.112 They found frustrated Lewis pairs on the surface of BiOBr, serving as effective catalytic sites for CO2 reduction. Meanwhile, introducing Ce4+ and oxygen vacancies (Fig. 11b) optimized the electronic state and FLP properties, forming frustrated Lewis acid-base pairs that significantly enhance the CO2 reduction to CO. This work offers a strategy for constructing efficient photocatalysts and understanding the atomic-level mechanisms of photocatalysis.
The use of noble metals entails a significant cost. In order to reduce the price of the catalysts, non-noble transition metals have also been studied as part of catalysts in photothermal-catalytic reactions as a more economical option. Ning et al. synthesized a hybrid carbon-based catalyst with embedded Co NPs together with dispersed Co–N species, denoted as Co@CoN&C.113 The reactions were carried out at 55 kPa, using an equal amount of CO2 and H2 and a Xe lamp in a batch reactor. Initially, the photoinduced thermal-effect was demonstrated as evidenced by the increase of temperature of the catalyst surface during the reaction, which reached 518 °C, while the plain Co NP system increased only up to 300 °C. After 30 min of irradiation, the optimized showed 41% CO2 conversion and a CO selectivity of 91% (132 mmol CO g−1 h−1), meanwhile the unsupported Co NPs showed 43% CO selectivity (Fig. 11c). The high reaction rate of the hybrid catalyst was attributed to the significant photo-thermal effect, and the CO selectivity was improved by the special characteristics provided by the Co–N shell in comparison with plain Co NPs.
| CO2 + CH4 ⇔ 2CO + 2H2 ΔH298K = 247.3 kJ mol−1 | (14) |
It has been reported that integrating photothermal and photoelectric catalytic processes could achieve a redox cycle that promotes metal-to-metal charge transfer induced by light, enabling low-temperature DRM. Ma et al.116 demonstrated that the redox cycle photocatalyst Rh/CexWO3 (Fig. 12a–c) ensured the stability and anti-coking performance of light-driven DRM without external heating. The photothermal and photoelectric effects play a key role in light-driven DRM, surpassing thermodynamic limitations. Calculations indicated that the strong CO2 absorption of CexWO3, its high oxygen mobility, and the oxygen bridge reduce the initiation temperature of DRM. This advancement paves the way for developing low-temperature catalysts for DRM.
 | ||
| Fig. 12 (a) Schematic diagram for light-driven DRM reaction over Rh/CexWO3 catalyst. (b) In situ DRIFT spectraof Rh/CexWO3 measured in CH4/CO2 mixture under different temperature. (c) ISI-XPS spectra of W 4f of Rh/CexWO3 with and without illumination.116 Copyright 2022, Wiley. (d) Schematic diagram of the photothermal DRM reaction over the Ru/SrTiO3 catalyst. Adapted with permission from ref. 117 Copyright 2023, Elsevier. | ||
Recently, Tang and colleagues reported a photothermal catalyst Ru/SrTiO3 under 600 °C and 300 W xenon lamp irradiation, the yields of CO and H2 were 1.4–1.5 times higher than those achieved with the thermal catalytic method.117 In the Ru/SrTiO3 catalyst (Fig. 12d), light induced the transfer of electrons from SrTiO3 to Ru, which participated in CH4 dehydrogenation and H2 production, thereby reducing the reaction energy barrier. This study elucidated the mechanism of photothermal DRM, providing valuable guidance for the future solar photothermal conversion of greenhouse gases.
Among various CH4 activation reactions, methane coupling is a promising potential pathway for directly obtaining valuable C2+ hydrocarbons. Long et al.23 proposed an innovative structure, the single-atom nest, to enhance the performance of photochemical non-oxidative coupling of methane (NOCM). Pt single-atom nests were fabricated as proof-of-concept single-atom nest photocatalysts. The results showed that Pt single-atom nests led to a remarkable 3.2-fold enhancement in the photocatalytic C2H6 yield under simulated sunlight, compared to Pt single-atoms. This work presents a pioneering concept for improving the efficiency of coupling reactions.
Metal–organic frameworks (MOFs) are considered to be ideal carriers for active metal NPs. MOF-encapsulated metal NPs, due to the porous nature of MOFs, prevent sintering and aggregation of the metal NPs and facilitate the smooth diffusion of reactants and products within the nanochannels. Wang et al.118 successfully encapsulated Ir NPs of approximately 1.5 nm into UiO-66 (Fig. 13a), forming Ir@UiO-66. The Ir NPs effectively promoted the separation and transfer of photo-induced charge carriers in the excited UiO-66, thereby aiding the cleavage of H2 and the activation of CO2. Under light irradiation, Ir@UiO-66-2 showed an excellent CH4 yield of 19.9 mmol gcat−1 h−1 and high selectivity (95%) at 250 °C. This study offers a new strategy for designing CO2 methanation catalysts under mild conditions.
 | ||
| Fig. 13 (a) Proposed reaction mechanism for the thermal and photo-thermal catalytic Sabatier reaction process over Ir@UiO-66.118 Copyright 2022, Royal Society of Chemistry. (b) Feasible routes of the CO2 hydrogenation reaction on the Ru/TiO2 model surface under dark and light conditions.119 Copyright 2022, Elsevier. (c) In situ DRIFT spectra obtained from the surface species formed during the reaction for 3.5% Cd/TiO2.120 Copyright 2022, Elesiver. | ||
Solar-driven reactions are increasingly being considered to address the poor thermal catalytic performance of low-temperature methanation. These reactions are expected to reduce the excessive energy consumption of traditional thermal catalytic methods while extending the catalyst's lifespan. Zhou et al.119 designed a TiO2 photocatalyst loaded with Ru NPs (Fig. 13b), significantly improving the conversion efficiency of CO2 to CH4 under light irradiation. The photo-induced electrons transfer effectively promoted the dissociation of H2 and the activation of CO2 on Ru atoms. This work provides new theoretical insights into the activation and methanation of CO2 under light irradiation.
It has been reported that the combination of two component catalysts is an effective way to improve catalytic activity: a metal oxide for CO2 activation and a metal or reducible metal oxide for H2 activation. Li et al.120 have developed a novel Cd/TiO2 catalyst (Fig. 13c), which exhibited a CH3OH selectivity of 81%, a CO2 conversion rate of 15.8%, and a CH4 selectivity of less than 0.7%. Experimental and computational studies indicated that the unique electronic properties of Cd clusters supported by the TiO2 matrix were responsible for the high selectivity of hydrogenating CO2 to CH3OH via the HCOO* pathway at the interfacial catalytic sites.
This discovery underscores the significance of the synergistic action between the two components of the catalyst: the metal oxide facilitates CO2 activation and transformation, while the metal or reducible metal oxide promotes H2 activation and dissociation. This finding highlights the importance of material design and understanding the underlying mechanisms in catalysis.
3.2 C2–4 hydrocarbons
In addition to producing CO, CH4, CH3OH, and other C1 products, CO2 reduction can also yield multi-carbon hydrocarbons (C2–4) with broader applications. The production of C2–4 compounds can be achieved through methods such as petroleum refining, direct or indirect conversion of synthesis gas, or CO2 hydrogenation. This process addresses environmental issues like the “greenhouse effect” resulting from rising CO2 concentrations. Therefore, the photocatalytic or photo-thermal catalytic conversion of CO2 into C2–4 products is an effective approach for mitigating energy shortages. The conversion from C1 products to multi-carbon products requires a C–C coupling reaction. Currently, there are two main methods to achieve this: (1) the Fischer–Tropsch process, where CO2 is first converted into CO through the reverse water-gas shift reaction and then into C2–4via the Fischer–Tropsch process;121 (2) the CH3OH process, where CO2 is hydrogenated to produce CH3OH, which is then converted into C2–4 through the methanol-to-hydrocarbons process. These methods represent important steps in developing sustainable and efficient technologies for CO2 conversion.122 | ||
| Fig. 14 (a) The proposed photocatalytic CO2 hydrogenation mechanism on NiFe-x alloys catalysts.123 Copyright 2022, Elsevier BV. (b) Schematic diagram of the multiple channels for electrons and holes transfer between In2O3, In2S3 and rGO.124 Copyright 2023, Elsevier. (c) Product selectivity and CO2 conversion at different reaction temperatures over NiFe-300 catalyst. 123 Copyright 2022, Elsevier BV. (d and e) The atomic structure configuration (colour codes: light blue (Ti), blue (Cu), and red (O)). (f) Photocatalytic product evolution as a function of light irradiation times on Cu-Ti-VO/Ti0.91O2-SL.125 Copyright 2023, Springer Nature. | ||
Zhang and co-workers 126 successfully prepared a series of new Fe-based catalysts with different chemical compositions using FeMgAl-LDH catalysts through hydrogen reduction at temperatures from 300 °C to 700 °C, of which catalyst obtained by hydrogen reduction at 500 °C (Fe-500) demonstrated excellent CO2 hydrogenation capabilities (Fig. 15a). These high-performance Fe-based catalysts were used for photothermal CO2 hydrogenation to C2+ hydrocarbons at atmospheric pressure. The FeO nanoparticles and FeOx loaded on MgO–Al2O3 showed a CO2 conversion rate of 50.1%, C2+ selectivity of 52.9%, and excellent stability over 22 hours of continuous testing (Fig. 15b). Zhang et al.52 also developed novel CoFe-based catalysts, using layered-double-hydroxide (LDH) nanosheets for hydrogen reduction to generate C2+ hydrocarbons. The CoFe-650 catalyst demonstrated remarkable selectivity toward hydrocarbons, with 60% CH4 and 35% C2+ (Fig. 15c). Additionally, Zhang et al.127 reported a hydrophobic core–shell Fe-based FTS catalyst, Fe/Fe3C-c, prepared by a three-step process including hydrothermal synthesis, thermal decomposition, and hydrophobic modification(Fig. 15d). The optimized Fe/Fe3C-c catalyst, driven by light irradiation, delivered attractive light-olefins selectivities of 48.0% with an O/P ratio of 10.1 for batch-type reactions and 49.0% with an O/P ratio of 12.2 for flow-type reactions (Fig. 15e).
 | ||
| Fig. 15 (a) HRTEM image of Fe-500. (b) The hydrocarbon product distribution obtained over Fe-500 under UV-vis irradiation.126 Copyright 2021, Wiley. (c) Illustration of the different CoFe-x catalysts formed by hydrogen reduction of a CoFeAl-LDH nanosheet precursor at different temperatures. The CO2 hydrogenation selectivity of each CoFe-x catalyst is indicated.52 Copyright 2018, Wiley. (d) Raman spectra of PB, Fe/Fe3C and Fe/Fe3C-c catalysts. (e) The FTS performance of the Fe/Fe3C-c catalyst in a flow system (1 ML min−1).127 Copyright 2024, Wiley-VCH Verlag. | ||
Yan and colleagues 128 synthesized graphdiyne/In2O3 (GDY-IO) nanocomposites for gas-phase photocatalytic CO2 reduction to C2+ hydrocarbons. By adjusting the composite ratio of GDY and In2O3, the 0.4% GDY-IO catalyst exhibited optimal C2+ hydrocarbons production activity and a high selectivity of 14% under simulated solar light irradiation and atmospheric pressure (Fig. 16a, b). Ozin et al.129 reported that cobalt ferrite (CoFe2O4) spinel produced C2–4 hydrocarbons in a single-step, ambient-pressure photocatalytic hydrogenation of CO2, with a rate of 1.1 mmol g−1 h−1, a selectivity of 29.8%, and a conversion yield of 12.9%. The CoFe2O4 reconstructed into a CoFe–CoFe2O4 alloy-spinel nanocomposite, facilitating the light-assisted transformation of CO2 to CO and subsequent hydrogenation to C2–4 hydrocarbons (Fig. 16c and d).
 | ||
| Fig. 16 (a) The charge carrier separation pathways and photocatalytic mechanism for CO2 hydrogenation over graphdiyne-modified In2O3 composite. (b) Products of photocatalytic CO2 reduction on various catalysts.128 Copyright 2024, Elsevier. (c) Possible reaction pathway of FC2 for CO2 hydrogenation. (d) The corresponding selectivity of CO, CH4, and C2–4 hydrocarbons for sample FC2 with and without light irradiation in the temperature range of 200–300 °C and atmospheric press.129 Copyright 2023, Wiley. | ||
Corma et al.130 investigated the CO2 hydrogenation into hydrocarbons promoted by sunlight on Co nanoparticles covered by thin carbon layers. They achieved nearly 100% selectivity to hydrocarbons, with increased selectivity towards C2+ hydrocarbons and alcohols (mainly ethanol) using Na-promoted Co nanoparticles (Na–Co@C) (Fig. 17a and b). Furthermore, Corma et al.131 prepared nickel–iron oxide nanoparticles by decomposing a mixed oxalate precursor and used them as catalysts for sunlight-promoted CO2 hydrogenation (Fig. 17c and d). The intimate contact between crystalline NiO and γ-Fe2O3 (and possibly amorphous iron oxide) domains in NiFeOx, NiFe3Ox, and NiFe9Ox proved highly efficient for generating light alkanes, which can be used as synthetic fuels.
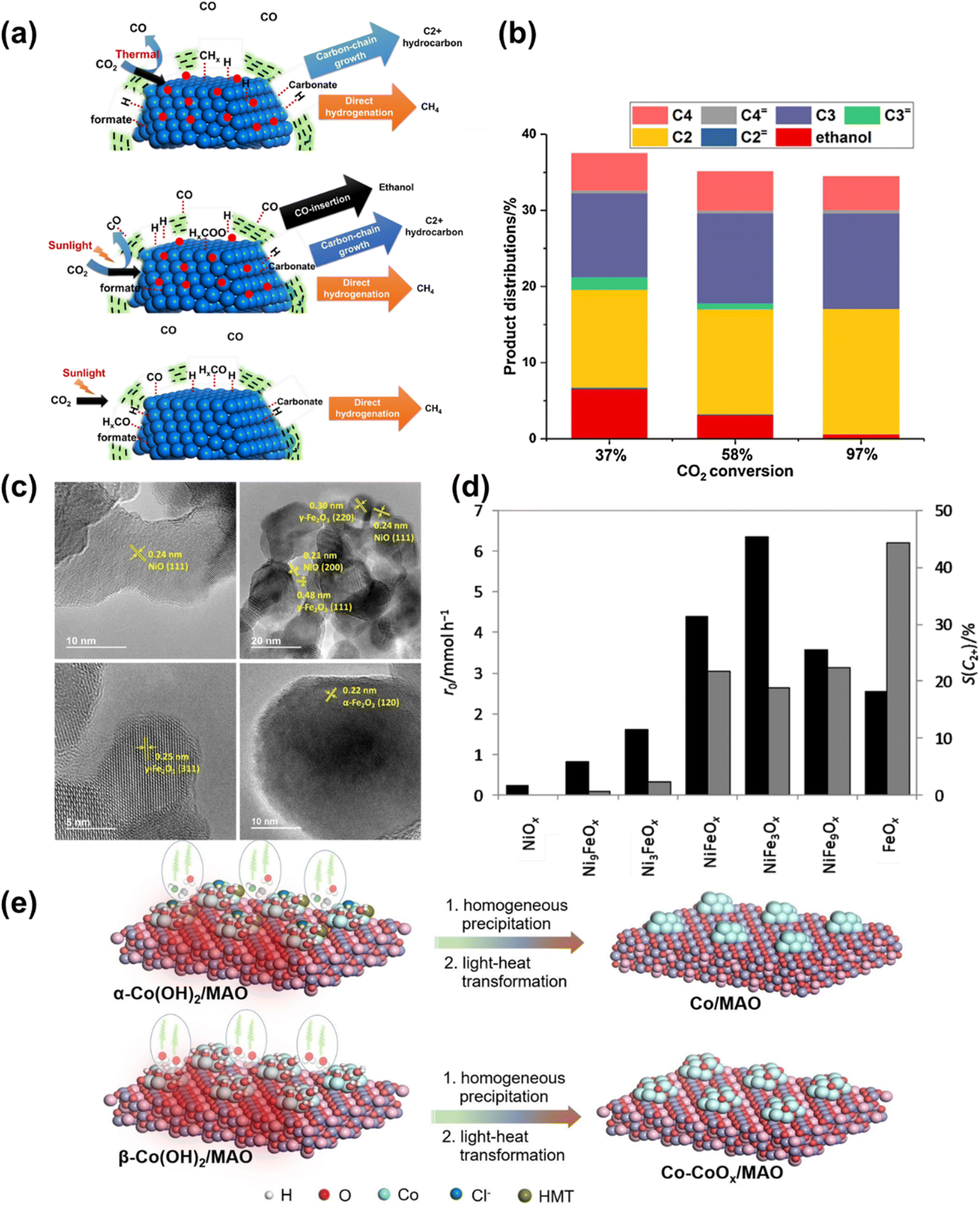 | ||
| Fig. 17 (a) Schematic illustration of the CO2 hydrogenation on Na–Co@C and Co@C catalysts under thermal and photothermal conditions. Sodium is shown with red circles. (b) Selectivity to C2+ hydrocarbons and ethanol under photothermal conditions at different CO2 conversion.130 Copyright 2018, Elsevier. (c) HRTEM micrographs (200 kV) of NiyFe1−yOx materials: Ni9FeOx, NiFe3Ox, NiFe9Ox, and FeOx. (d) Initial CO2 conversion rate (r0, black bars) and hydrocarbon selectivity towards C2+ products [S(C2+), excluding CO, grey bars] obtained from CO2 hydrogenation experiments on the series of NiyFe1−yOx materials.131 Copyright 2018, Springer. (e) A schematic illustration of the two-step growth of α- and β-Co(OH)2 nanosheets on MAO and light-to-heat reduction.9 Copyright 2023, Wiley. | ||
Amin et al.132 prepared Ag-loaded TiO2 nanoparticles with varying Ag concentrations using an improved sol–gel method and fixed them on a monolithic support. The Ag/TiO2 catalyst demonstrated significant photocatalytic activity for the dynamic reduction of CO2 to CO via the RWGS reaction in both continuous cell and integrated photoreactors. Compared to battery-type photoreactors, integrated photoreactors effectively converted CO2 into CO as the main product through Ag/TiO2, while also producing multi-carbon products like C2–4.
Building on this progress, we constructed a C–C coupling center for C1 intermediates via the in situ formation of Co0–Coδ+ interface double sites on MgAl2O4 (Co–CoOx/MAO) (Fig. 17e).9 The Co–CoOx/MAO exhibited a high C2–4 hydrocarbons production rate of 1303 μmol g−1 h−1, with a total organic carbon selectivity of 62.5% for C2–4 hydrocarbons under light irradiation and a high olefin to paraffin ratio (≈11), demonstrating its efficiency and selectivity for multi-carbon hydrocarbon production. Future research could focus on optimizing catalyst compositions, enhancing light absorption, and improving reaction kinetics to further increase the yield and selectivity of C2+ products.
3.3 C4+ hydrocarbons
Despite successful reports on the hydrogenation of CO2 to produce value-added C2+ chemicals, synthesizing C4+ remains a significant challenge due to the high barrier of C–C coupling. C5+ hydrocarbons (liquid gasoline), which contain five or more carbon atoms, are important products in the petrochemical industry. In photocatalytic CO2 hydrogenation reactions, generating C5+ products faces higher energy barriers due to the C–C coupling reaction. There has not yet been a decisive study on the production of liquid gasoline from CO2 hydrogenation.Zhang et al.52 developed an Al2O3-supported CoFe alloy catalyst, adjusting its surface chemical composition by changing the reduction temperature of the CoFeAl-LDH nanosheet precursor, thereby optimizing the activity and product selectivity of CO2 hydrogenation (Fig. 15c). The CoFe-650 catalyst achieved efficient hydrogenation of CO2 to C4+ with a selectivity of 8.3% under photothermal conditions, demonstrating the potential for using solar energy to produce high-value chemicals. Subsequently, Zhang et al. prepared a series of novel Fe-based catalysts with varying chemical compositions by reducing MgFeAl layered double hydroxide nanosheets at temperatures ranging from 300 °C to 700 °C.126 Using a 300 W xenon lamp as the light source, the Fe-500 catalyst showed excellent stability for C2+ hydrocarbons under UV-visible light irradiation over 22 hours of continuous testing, with a C5+ selectivity of 6.3% (Fig. 18b), maintaining its activity and selectivity without significant deactivation. Ma and his research team 133 developed a K-promoted Ru/Fe3O4 catalyst, achieving the production of high-value chemicals and fuels through photocatalytic CO2 hydrogenation (Fig. 18a). When using the K–Ru/Fe3O4 catalyst for CO2 hydrogenation, the selectivity of C5+ reached 19.0%, indicating that the introduction of a potassium promoter can enhance C–C coupling, thereby improving the yield of C5+ products. Yan et al.134 incorporated magnetic cobalt ions into ultra-thin BiOCl sheets to achieve selective control of the photocatalytic CO2 reduction reaction. Co–BiOCl can selectively convert CO2 into CH4 and C2+ products, with a hydrocarbon selectivity of up to 76.9%, a performance not possessed by the original BiOCl (Fig. 18c). These results were significant for improving the efficiency of converting CO2 into high-value chemicals and demonstrates the potential of photocatalytic technology to convert CO2 into useful chemicals under mild conditions. Under light conditions, electron transfer on the catalyst surface can change the adsorption/desorption dynamics of reaction intermediates, affecting the reaction path and making the distribution of C5+ products significantly different from the thermal catalysis process, providing a new strategy for solar-driven CO2 conversion.
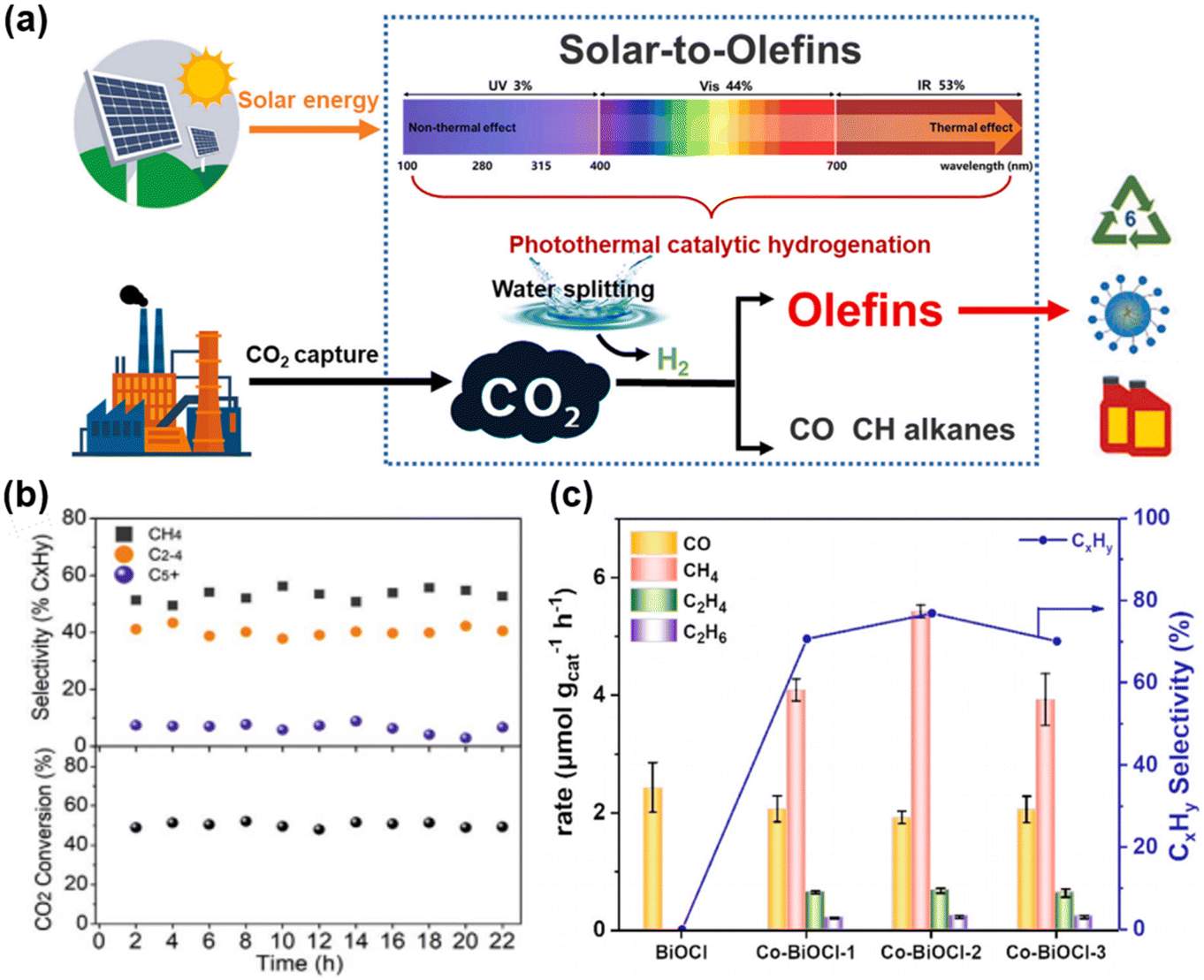 | ||
| Fig. 18 (a) Photodriven hydrogenation of CO2 to lower olefins, which can be efficiently performed in a photothermally catalytic way with the K–Ru/Fe3O4 catalyst presented in this work.133 Copyright 2024, Cell Press. (b) The CO2 conversion and product selectivities over Fe-500 under UV-vis irradiation using a flow system with a flow rate of 5 mL min−1.126 Copyright 2021, Wiley. (c) Photocatalytic CO2 hydrogenation activity and selectivity of CxHy on Co–BiOCl with different Co content after 4 h light irradiation of CxHy on Fe–BiOCl, Cu–BiOCl, Ni–BiOCl and Zn–BiOCl.134 Copyright 2024, Elsevier. | ||
Song et al.135 highlighted the dynamic evolution of active iron and carbon species in CO2 hydrogenation over Fe–Zr catalysts supported on different zirconia forms (monoclinic m-ZrO2 and tetragonal t-ZrO2). During CO pre-reduction, the FeO intermediate species on the Fe–K/m-ZrO2 catalyst indicated mild and sufficient reduction of Fe2O3 on m-ZrO2 (Fig. 19a), promoting the carbonization reaction to form smaller and more active iron species (Fe3O4 and χ-Fe5C2). When the Fe content reached 15 wt%, the Fe–K/m-ZrO2 catalyst showed a significant CO2 conversion rate (38.8%) and light olefins selectivity (42.8%). Jing et al.129 reported a photocatalyst, cobalt ferrite (CoFe2O4), which converts CO2 into C2–4 hydrocarbons through a photothermal catalytic process at ambient pressure and temperature (Fig. 19b). The active sites are formed by the transformation of CoFe2O4 into a nano-composite of CoFe alloy and CoFe2O4, synergistically promoting the conversion of CO2 to CO and subsequent CO hydrogenation, effectively producing C2–4 hydrocarbons. A 35 hours stability test under atmospheric pressure and light exposure showed this sample continuously producing C2–4 hydrocarbons, along with CO and CH4, achieving a C2–4 hydrocarbons selectivity of 29.8% and a high CO2 conversion efficiency (12.9%) (Fig. 19c). Mechanism studies showed CO2 was first adsorbed on the CoFe2O4 surface, then converted into formate intermediates under hydrogen action. These intermediates further hydrogenated to produce CO and water through the RWGS reaction, with CO further hydrogenated under CoFe alloy catalysis to produce C2–4 hydrocarbons via the FTS process. Zhang's team 136 reported a new composite catalyst, InZrOx-Beta, showing high selectivity in hydrogenating CO2 to butane-enriched C4 alkanes. Depositing SiO2 on the surface of InZrOx effectively inhibited metallic In migration, preventing rapid deactivation of the zeolite part (Fig. 19d). Under photothermal conditions of 315 °C and 3.0 MPa, the InZrOx-Beta composite catalyst achieved a butane selectivity of 53.4%, a CO2 conversion rate of 20.4%, and a CH4 selectivity of about 2% (Fig. 19e). Through the above analysis, the design of two-component catalysts offers a promising strategy to enhance both the activity and selectivity for the production of C4+ hydrocarbons. By combining complementary active sites, these catalysts facilitate complex reaction pathways, such as C–C coupling and hydrogenation, enabling more efficient synthesis of higher-order hydrocarbons.
 | ||
| Fig. 19 (a) Relationship between the physicochemical properties of different types of ZrO2 supports and the structure and activity of Fe–K/ZrO2 catalysts.135 Copyright 2021, AMER CHEMICAL SOC. (b). HRTEM image of spent CoFe2O4 (inset image is FFT pattern). (c) Stability of CoFe2O4 over 35 hours showing the solar-powered hydrogenation of CO2 production rate and selectivity towards CO, CH4, and C2–4, respectively.129 Copyright 2023, Wiley. (d) Reaction scheme of butane formation via the side-chain route of aromatic-based cycle. (e) CH3OH conversion and product distribution for MTH in H2 atmosphere over H-Beta.136 Copyright 2023, Springer Nature. | ||
Zhang and co-workers 137 reported an iron nitride (Fe2N) encapsulated in carbon nanoparticles (Fe2N@C) (Fig. 20a), which showed excellent performance in selectively hydrogenating CO2 to C2+ hydrocarbons. By encapsulating the catalyst with a carbon shell, Fe2N@C exhibited outstanding durability, achieving a C5+ selectivity of 7.52% under pressurized conditions of 1 MPa (Fig. 20b), enhancing stability and long-term application potential. Zeng et al.138 activated the CuFeO2 catalyst under atmospheric pressure to convert CO2 and hydrogen (H2) into long-chain olefins (C4+). Under these conditions, the activated CuFeO2 catalyst showed a CO2 conversion rate of 27.3% and a C4+ = selectivity of 66.9%. In a 50 hours stability test, the CO2 conversion rate remained stable, but the C4+ = selectivity decreased to 57.6% (Fig. 20c). At a pressure of 30 bar, the C4+ = selectivity improved to 66.3%. The experimental results showed that the catalyst's performance could be effectively controlled, and regeneration could be achieved when the catalyst deactivates by adjusting the reaction pressure. In atmospheric pressure CO2 hydrogenation, the CuFeO2 catalyst remained stable, though its selectivity decreased over time. Increasing the pressure restored catalyst performance, which is significant for industrial applications and catalyst design. Recently, we reported a Cu-promoted CoFe alloy catalyst, which still produced C5+ products under atmospheric pressure, though the C5+ selectivity was only 5.7%. The natural light-driven CO2 hydrogenation reaction showed excellent activity and selectivity at a weak solar irradiance intensity of 0.45 kW m−2 (0.45 suns) (Fig. 20d).56
 | ||
| Fig. 20 (a) Synthetic process of Fe2N@C nanoparticles and the in situ restructuring transformation of Fe2N@C into iron carbides during the selective hydrogenation of CO2 to C2+ hydrocarbons. (b) CO2 conversion rate and hydrocarbons selectivity over Fe2N@C and Fe@C.137 Copyright 2021, Wiley. (c) Stability test of activated CuFeO2.138 Copyright 2022, Springer Nature. (d) The CO2 hydrogenation performance of sunlightdriven Cu0.65Co0.35Fe2 catalyst.56 Copyright 2024, Wiley-VCH Verlag. | ||
These findings suggest promising directions for future research aimed at enhancing C5+ selectivity. Key strategies could include optimizing the metal composition and electronic structure of the active sites, as well as refining catalyst–support interactions to stabilize intermediate species during chain propagation. Additionally, tailoring reaction conditions, such as pressure and temperature, in conjunction with advanced photothermal reactor designs, may further improve catalytic performance.
Looking forward, the integration of such catalysts with renewable energy sources, particularly under solar-driven conditions, holds significant potential for sustainable carbon utilization. The development of robust, scalable systems for selective C5+ hydrocarbon production could pave the way for new advancements in solar-to-chemical energy conversion and contribute to achieving carbon-neutral chemical manufacturing.
3.4 Ethanol, acetic acid and other products
Photothermal catalysis for the conversion of CO2 into high-value-added chemicals, such as fuels, is extremely promising as it provides a valuable source of these chemicals while also helping to reduce CO2 emissions that affect the global climate. 139 This process involves the coupling of photoelectrons produced by the photocatalyst and the dissolution of CO2 in a water medium, enabling the catalytic conversion of CO2 into liquid fuels such as ethanol and acetic acid.140Sira Srinives et al.141 utilized a one-step hydrothermal process for the synthesis of precious metal-doped TiO2/GO composites. Cu–TiO2/GO showed the best photocatalytic performance, which could be attributed to remarkable key parameters, including a high specific surface area, a narrowed band gap energy, and a big crystallite size (Fig. 21a).
 | ||
| Fig. 21 (a) Schematic diagram exhibiting proposed reaction mechanisms for the photoreduction of CO2 on Ag–TiO2/GO, Pd–TiO2/GO, and Cu–TiO2/GO.141 Copyright 2021, Royal Society of Chemistry. (b) C2 product yield rates, evolutions over time and catalytic stabilities of pristine meso-Co3O4, mr-meso-Co3O4, and meso-Co3O4@CoO. (c) Pathway on surfacereduced meso-Co3O4.142 Copyright 2023, American Chemical Society. (d) The proposed reaction mechanism for the photocatalytic CO2 reduction conversion to CH3CHO.143 Copyright 2022, Royal Society of Chemistry. | ||
Acetic acid is a valuable chemical suitable for industrial intermediates and fuel additives. Zhang et al.142 reported the controllable preparation of highly ordered mesoporous cobalt oxides with modulated surface states to achieve efficient photothermal water-steam reforming of CO2 to C2 products with high activity and tunable selectivity (Fig. 21b and c). Pristine mesoporous Co3O4 exhibited an acetic acid selectivity of 96% with a yield rate of 73.44 μmol g−1 h−1.
Acetaldehyde (CH3CHO) is an indispensable intermediate in the production of high value-added chemicals, and is widely used in medicine, pesticides and spices. 144 Photocatalytic conversion of CO2 to acetaldehyde has become a promising method for sustainable production of acetaldehyde. Wang et al.143 Reported the selective photocatalytic hydrogenation of CO2 to CH3CHO using a modified polymeric carbon nitride (PCN) under mild conditions (Fig. 21d). The locally crystallized PCN offers a photocatalytic activity of 1814.7 μmol h−1 g−1 with a high selectivity of 98.3% for CH3CHO production and a quantum efficiency of 22.4% at 385 nm, outperforming all the state-of-art CO2 photocatalysts. The promoted formation of the *OCCHO intermediate on the locally crystallized PCN is disclosed as the key factor leading to the highly selective CH3CHO generation.
3.5 Dimethyl carbonate, ethyl-carbonate and other carbonates
Compared to CO2 reduction into C1 chemicals like CH3OH or formate, non-redox CO2 chemical fixation offers a route to high-value long-chain products. DMC, with its methoxy and carbonyl groups, can replace toxic phosgene and dimethyl sulfate in methylation and carbonylation reactions and serves as an important electrolyte solvent for lithium-ion batteries due to its high dielectric constant.145–153 Thus, direct synthesis of DMC from CO2 has significant theoretical and practical value. Chen et al. reported light-driven CO2 activation into DMC over B2O3 atomic layers with oxygen vacancies, achieving nearly 100% selectivity under mild conditions.154 These vacancies provided unsaturated sites, enhancing CO2 and CH3OH adsorption. Xie et al.155 found that frustrated Lewis pairs (FLP) sites on CeO2 with rich FLP sites improved reaction activity, achieving a high yield of DMC up to 15.3 mmol g−1 (Fig. 22a and b). They used density functional theory to investigate the reaction mechanism on the FLP site, providing insights into CO2 chemical conversion to long-chain chemicals. They proposed an in-plane heterostructure strategy to construct Lewis acid-base sites for efficient CO2 activation. Using ultrathin in-plane Cu2O/Cu heterostructures as a prototype, they showed that Lewis acid-base sites on the heterointerface facilitate mixed C and O dual coordination on the surface, strengthening CO2 adsorption and activation. DRIFTS revealed that these sites could activate CO2 to CO2− species, a key intermediate for CO2 fixation (Fig. 22c–f). | ||
| Fig. 22 (a) Illustration of reaction route for the direct DMC synthesis from CO2 and CH3OH on CeO2. (b) TEM image of P–CeO2-NR, where the inset is corresponding HRTEM image. (c–f) In situ DRIFTS for the direct DMC synthesis from CO2 and CH3OH under simulated experimental condition. 155 Copyright 2022, Wiley. (g) Illustrations of reaction mechanism for the carbonylation of ethanol with CO2 on Cu2O–SrTiCuO3–x under UV/Visible light irradiation. (h) CO2 activation rate and formation rate of self-products from CO2. 156 Copyright 2023, Wiley. | ||
Despite significant progress in CO2 chemical fixation to DMC, major challenges and opportunities remain.157 The overall performance of DMC generation from CO2 still falls short of practical application requirements, necessitating the development of advanced catalysts. Therefore, exploring and constructing new catalyst systems with high efficiency for activating CO2 into DMC is crucial and presents many future opportunities.
The carbonylation of ethanol using CO2 as a carbonyl source to synthesize value-added esters holds significant potential but remains a considerable challenge due to the difficulty of activating inert CO2.158 Harsh reaction conditions often lead to undesired side reactions, including the activation of the α-C–H bond and even cleavage of the C–C bond in ethanol, which hinders the selective activation of the O–H bond.159 To address these challenges, we propose a photo-thermal cooperative strategy for the carbonylation of ethanol with CO2. This approach utilizes photo-catalysis to activate CO2 into reactive CO, assisted by *H generated from the thermally-catalyzed dissociation of the alcoholic O–H bond.
To realize this strategy, a Cu2O–SrTiCuO3–x catalyst has been designed, featuring abundant interfacial sites and oxygen vacancies to facilitate the reaction(Fig. 22g and h).156 Under photo-thermal conditions (3.29 W cm−2 UV/Visible light irradiation, 144 °C, and 0.2 MPa CO2), the catalyst achieved an ethyl formate production rate of 320 μmol g−1 h−1 with an impressive selectivity of 85.6% toward the targeted activation of the alcoholic O–H bond. This work demonstrates the potential of leveraging photo-thermal synergy and tailored catalytic materials for efficient CO2 utilization and selective carbonates production under mild conditions.
4. Conclusions and outlooks
Solar-driven catalytic conversion of CO2 into value-added C2+ chemicals/fuels has been a focus of research in the past decades due to its significance in the energy and environment fields. However, due to the slow reduction kinetics, the photocatalytic reduction of CO2 remains challenging. The state-of-the-art photocatalytic systems in such route suffer from low efficiency, which is still far from practical application. Therefore, it is of great interest to explore alternate catalytic reaction routes driven by solar energy for efficient CO2 reduction. Recent pioneering work by several groups, including ours, has shown that photocatalysis coupling of photothermal catalysis for CO2 reduction is a novel and promising strategy for CO2 catalytic conversion into C2+ products.This review has explored the current state of light-driven CO2 reduction, tracing its historical development, elucidating the underlying mechanisms, and examining the pivotal advancements in catalyst design and emphasizing its potential for converting CO2 into valuable C2+ products. Historically, photothermal CO2 reduction has progressed from basic studies to more advanced applications, utilizing the combined effects of light absorption and heat to drive catalytic reaction. Integrating photothermal effects with photocatalysis has significantly improved the efficiency and selectivity of CO2 reduction, especially for producing C2+ hydrocarbons. Understanding the mechanisms of photothermal CO2 conversion has revealed pathways for creating C2+ products, highlighting the crucial role of simultaneously activating CO2 and hydrogen sources under solar light.
Catalyst design is critical for advancing solar-driven synthesis of C2+ products. Surface defect engineering has proven effective in enhancing catalyst performance by creating active sites that improve CO2 adsorption and activation. Additionally, designing bifunctional active sites facilitates multi-electron transfer processes, boosting the production of long-chain hydrocarbons. These advancements in catalyst design are essential for addressing the challenges of selectively producing C2+ products.
Solar-driven CO2 reduction involves various pathways, from producing C1 products like CO, CH4 and CH3OH to high value-added C2–4 hydrocarbons. Photothermal catalysis shows great promise in increasing the efficiency of these reactions by providing the necessary heat to drive endothermic steps and stabilize intermediates. However, producing C5+ hydrocarbons remains challenging due to the complexity of the reaction mechanisms and the need for highly selective and stable catalysts.
Looking ahead, producing C5+ products is a promising but challenging goal that requires innovative catalyst designs and reaction engineering. Future research should focus on developing bifunctional catalysts with highly selective active sites to enable the formation of longer-chain hydrocarbons while minimizing side reactions. Develop reactor systems that maximize light absorption and distribution within the catalytic environment, ensuring efficient utilization of photonic energy for optimizing the intermediates selective coupling and accelerating the reaction rate of CO2 hydrogenation. Our group has developed advanced photothermal reactor devices that harness natural outdoor sunlight for CO2 conversion, producing valuable products such as CO, CH4, and multi-hydrocarbons.8,56 These systems offer a scalable, sustainable solution for efficient CO2 hydrogenation, providing a promising path for carbon capture and utilization. Additionally, integrating computational modeling and in situ characterization techniques will be crucial for understanding the detailed reaction pathways and optimizing conditions for C5+ synthesis.
The advancement of light-driven photothermal CO2 reduction goes beyond environmental benefits, providing a sustainable route for chemical synthesis and energy storage. Overcoming current limitations will enable scalable and economically viable CO2 conversion technologies. Producing higher-order hydrocarbons like ethanol, acetic acid, and various carbonates can significantly impact the chemical industry by offering renewable alternatives to fossil-based materials.
In summary, significant progress has been made in photothermal CO2 reduction, but developing effective strategies for synthesizing C2+ products remains an exciting challenge. Continued interdisciplinary efforts, combining advances in materials science, catalysis, and reaction engineering, are essential to fully utilize solar energy for the sustainable and selective conversion of CO2 into a wide range of valuable chemicals. The future of this field is promising, with the potential to greatly contribute to environmental sustainability and the growth of green chemistry.
Data availability
Data availability is not applicable to this review article as no new data were created or analyzed in this study.Author contributions
All of the authors contributed to the manuscript preparation. S. N. and J. Y. conceived the outline of the manuscript. X. W., S. Z., S. N., C. Y., L. L., L. T., R. L., X. Y., Y. Z., S. C. wrote the original draft of the manuscript. X. W., S. Z., S. N., J. W. and J. Y. discussed and helped revise the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22402046, U23A20139), the Hebei Natural Science Foundation (Grant nos. B2023201020, B2024201096, 2023HBQZYCXY001, E2023201019), the Hebei Education Department Foundation (Grant No. QN2025190, CXZX2025016), the China Postdoctoral Science Foundation (Grant No. 2023M743770), Industry-University-Research Cooperation Major Projects of Shijiazhuang City (241130477A) and Hebei Province Innovation Capability Enhancement Plan Project (22567620H).References
- S. Yang, D. Yang, W. Shi, C. Deng, C. Chen and S. Feng, Environ. Sci. Pollut. Res., 2023, 30, 81725–81744 CrossRef CAS PubMed.
- Z. Xiao, P. Li, H. Zhang, S. Zhang, Y. Zhao, J. Gu, Z. Lian, G. Li, J. J. Zou and D. Wang, J. Colloid Interface Sci., 2024, 672, 642–653 CrossRef CAS PubMed.
- P. Zhang, X. Sui, Y. Wang, Z. Wang, J. Zhao, Y. Weng and J. Long, J. Am. Chem. Soc., 2023, 145, 5769–5777 CrossRef CAS PubMed.
- Z. Lu, Y. F. Xu, Z. S. Zhang, J. C. Sun, X. Ding, W. Sun, W. G. Tu, Y. Zhou, Y. F. Yao, G. A. Ozin, L. Wang and Z. G. Zou, J. Am. Chem. Soc., 2023, 145, 26052–26060 CrossRef CAS PubMed.
- C. Qiu, J. Sun, M. Li, C. Mao, R. Song, Z. Zhang and L. Wang, J. Am. Chem. Soc., 2024, 146, 33997–34007 CrossRef CAS PubMed.
- Y. J. Li, H. M. Liu, L. Ma, J. X. Liu and D. H. He, Acta Phys.-Chim. Sin., 2024, 40, 2308005 CrossRef.
- H. H. Ou, S. B. Ning, P. Zhu, S. H. Chen, A. Han, Q. Kang, Z. F. Hu, J. H. Ye, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- C. Lv, X. Bai, S. Ning, C. X. Song, Q. Guan, B. Liu, Y. Li and J. Ye, ACS Nano, 2023, 17, 1725–1738 CrossRef CAS PubMed.
- S. Ning, H. Ou, Y. Li, C. Lv, S. Wang, D. Wang and J. Ye, Angew. Chem., Int. Ed., 2023, 62, e202302253 CrossRef CAS PubMed.
- J. Q. Zhao, R. Shi, G. I. N. Waterhouse and T. R. Zhang, Nano Energy, 2022, 102, 107650 CrossRef CAS.
- G. Y. Chen, H. Ågren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- L. Yuan, M. Y. Qi, Z. R. Tang and Y. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 21150–21172 CrossRef CAS PubMed.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, 1903915 CrossRef CAS PubMed.
- J. Xie, Y. H. Jiang, S. Y. Li, P. Xu, Q. Zheng, X. Y. Fan, H. L. Peng and Z. Y. Tang, Acta Phys.-Chim. Sin., 2023, 39, 2306037 Search PubMed.
- L. K. Zhao, Y. H. Qi, L. Z. Song, S. B. Ning, S. X. Ouyang, H. Xu and J. H. Ye, Angew. Chem., Int. Ed., 2019, 58, 7708–7712 CrossRef CAS PubMed.
- G. G. Liu, T. Wang, H. B. Zhang, X. G. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. H. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
- J. J. Wang, S. Kattel, C. J. Hawxhurst, J. H. Lee, B. M. Tackett, K. Chang, N. Rui, C. J. Liu and J. G. G. Chen, Angew. Chem., Int. Ed., 2019, 58, 6271–6275 CrossRef CAS PubMed.
- H. Song, X. G. Meng, Z. J. Wang, H. M. Liu and J. H. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- F. Jiao, J. J. Li, X. L. Pan, J. P. Xiao, H. B. Li, H. Ma, M. M. Wei, Y. Pan, Z. Y. Zhou, M. R. Li, S. Miao, J. Li, Y. F. Zhu, D. Xiao, T. He, J. H. Yang, F. Qi, Q. Fu and X. H. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- H. B. Zhang, J. Wei, J. C. Dong, G. G. Liu, L. Shi, P. F. An, G. X. Zhao, J. T. Kong, X. J. Wang, X. G. Meng, J. Zhang and J. H. Ye, Angew. Chem., Int. Ed., 2016, 55, 14308–14312 Search PubMed.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS PubMed.
- E. Pipelzadeh, V. Rudolph, G. Hanson, C. Noble and L. Z. Wang, Appl. Catal., B, 2017, 218, 672–678 CrossRef CAS.
- P. Zhang, J. Li, H. Huang, X. Sui, H. Zeng, H. Lu, Y. Wang, Y. Jia and J. Long, J. Am. Chem. Soc., 2024, 146, 24150–24157 CrossRef CAS PubMed.
- C. Chen, D. Liu, Y. J. Wu, W. B. Bi, X. K. Sun, X. Chen, W. Liu, L. Xu, H. W. Song and Q. L. Dai, Nano Energy, 2018, 53, 849–862 CrossRef CAS.
- P. Sabatier, Comptes Rendus, 1902, 134, 514–516 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Q. Zhang, A. Mirzaei, Y. Wang, G. Song, C. Wang, L. V. Besteiro, A. O. Govorov, M. Chaker and D. Ma, Appl. Catal., B, 2022, 317, 121792 CrossRef CAS.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- L. Buzzetti, G. E. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- M. Sayed, J. Yu, G. Liu and M. Jaroniec, Chem. Rev., 2022, 122, 10484–10537 CrossRef CAS PubMed.
- Z. Wei, W. Wang, W. Li, X. Bai, J. Zhao, E. C. Tse, D. L. Phillips and Y. Zhu, Angew. Chem., Int. Ed., 2021, 60, 8236–8242 CrossRef CAS PubMed.
- Q. Zhang, S. Huang, J. Deng, D. T. Gangadharan, F. Yang, Z. Xu, G. Giorgi, M. Palummo, M. Chaker and D. Ma, Adv. Funct. Mater., 2019, 29, 1902486 CrossRef.
- Y. Zhao, S. Zhang, R. Shi, G. I. Waterhouse, J. Tang and T. Zhang, Mater. Today, 2020, 34, 78–91 CrossRef CAS.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed.
- Q. Zhang, F. Yang, Z. Xu, M. Chaker and D. Ma, Nanoscale Horiz., 2019, 4, 579–591 RSC.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924–936 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- J. Kizlink, Collect. Czech. Chem. Commun., 1993, 58, 1399–1402 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Sci. Technol., 1984, 5, 1–117 Search PubMed.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505–516 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- A. Kumar and G. Pandey, Mater. Sci. Eng. Int. J., 2017, 1, 1–10 Search PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed.
- Z. Q. Wang, Z. Q. Yang, Z. C. Kadirova, M. N. Guo, R. M. Fang, J. He, Y. F. Yan and J. Y. Ran, Coord. Chem. Rev., 2022, 473, 37 CrossRef.
- Y. Li, R. Z. Li, Z. H. Li, W. Q. Wei, S. X. Ouyang, H. Yuan and T. R. Zhang, Chem. Res. Chin. Univ., 2020, 36, 1006–1012 CrossRef CAS.
- L. L. Zhu, M. M. Gao, C. K. N. Peh and G. W. Ho, Mater. Horiz., 2018, 5, 323–343 RSC.
- R. Ma, J. Sun, D. H. Li and J. J. Wei, Int. J. Hydrogen Energy, 2020, 45, 30288–30324 CrossRef CAS.
- S. W. Chen, X. Cao, N. Wang, L. Ma, H. R. Zhu, M. Willander, Y. Jie and Z. L. Wang, Adv. Energy Mater., 2017, 7, 1601255 CrossRef.
- W. Zhang, L. Wang, K. Wang, M. U. Khan, M. Wang, H. Li and J. Zeng, Small, 2017, 13, 1602583 CrossRef PubMed.
- Y. He, Q. Lei, C. Li, Y. Han, Z. Shi and S. Feng, Mater. Today, 2021, 50, 358–384 CrossRef CAS.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L. Wu, C. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed.
- Y. Zhou, Z. Wang, L. Huang, S. Zaman, K. Lei, T. Yue, Z. a. Li, B. You and B. Y. Xia, Adv. Energy Mater., 2021, 11, 2003159 CrossRef CAS.
- N. L. Reddy, V. N. Rao, M. Vijayakumar, R. Santhosh, S. Anandan, M. Karthik, M. Shankar, K. R. Reddy, N. P. Shetti and M. Nadagouda, Int. J. Hydrogen Energy, 2019, 44, 10453–10472 CrossRef CAS.
- S. Ning, Y. Sun, S. Ouyang, Y. Qi and J. Ye, Appl. Catal., B, 2022, 310, 121063 CrossRef CAS.
- S. Ning, J. Wang, X. Wu, L. Li, S. Zhang, S. Chen, X. Ren, L. Gao, Y. Hao, C. Lv, Y. Li and J. Ye, Adv. Funct. Mater., 2024, 34, 2400798 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- T. Numpilai, C. K. Cheng, J. Limtrakul and T. Witoon, Process Saf. Environ. Prot., 2021, 151, 401–427 CrossRef CAS.
- Q. Yang, H. Liu, Y. Lin, D. Su, Y. Tang and L. Chen, Adv. Mater., 2024, 36, 2310912 CrossRef CAS PubMed.
- X. Ding, W. Liu, J. Zhao, L. Wang and Z. Zou, Adv. Mater., 2025, 37, 2312093 CrossRef CAS PubMed.
- M. Xu, X. Qin, Y. Xu, M. Wang, X. Liu and D. Ma, Nat. Commun., 2022, 13, 6720 CrossRef CAS PubMed.
- R. C. Brady III and R. Pettit, J. Am. Chem. Soc., 1981, 103, 1287–1289 CrossRef.
- G. K. K. Gunasooriya, A. P. van Bavel, H. P. Kuipers and M. Saeys, ACS Catal., 2016, 6, 3660–3664 CrossRef CAS.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- C. Yang, R. Mu, G. Wang, J. Song, H. Tian, Z.-J. Zhao and J. Gong, Chem. Sci., 2019, 10, 3161–3167 RSC.
- L. Wang, Y. Yi, C. Wu, H. Guo and X. Tu, Angew. Chem. Int. Ed., 2017, 129, 13867–13871 CrossRef.
- B. Liu, C. Li, G. Zhang, X. Yao, S. S. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Energy, 1997, 22, 343–348 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Catal. Today, 1996, 28, 261–266 CrossRef CAS.
- A. W. Budiman, J. S. Nam, J. H. Park, R. I. Mukti, T. S. Chang, J. W. Bae and M. J. Choi, Catal. Surv. Asia, 2016, 20, 173–193 CrossRef CAS.
- W. Jian-Feng, Y. Si-Min, W. W. David, F. Yan-Xin, B. Shi, Z. Chuan-Wei, G. Qiang, H. Jun and W. Wei, J. Am. Chem. Soc., 2013, 135, 13567–13573 CrossRef PubMed.
- S. Wang, S. Guo, Y. Luo, Z. Qin, Y. Chen, M. Dong, J. Li, W. Fan and J. Wang, Catal. Sci. Technol., 2019, 9, 6613–6626 RSC.
- P. Zhang, X. Yang, X. Hou, J. Mi, Z. Yuan, J. Huang and C. Stampfl, Catal. Sci. Technol., 2019, 9, 6297–6307 RSC.
- Y. Zhao, C. Cui, J. Han, H. Wang, X. Zhu and Q. Ge, J. Am. Chem. Soc., 2016, 138, 10191–10198 CrossRef CAS PubMed.
- B. D. Montejo-Valencia, Y. J. Pagán-Torres, M. M. Martínez-Iñesta and M. C. Curet-Arana, ACS Catal., 2017, 7, 6719–6728 CrossRef CAS.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- S. Fang and K. Fujimoto, Appl. Catal., A, 1996, 142, L1–L3 Search PubMed.
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Energy Environ. Sci., 2011, 4, 4211–4219 RSC.
- G. Xi, S. Ouyang, P. Li, J. Ye, Q. Ma and N. Su, Angew. Chem., Int. Ed., 2012, 51, 2395–2399 CrossRef CAS PubMed.
- H. Pang, X. Meng, P. Li, K. Chang, W. Zhou, X. Wang, X. Zhang, W. Jevasuwan, N. Fukata, D. Wang and J. Ye, ACS Energy Lett., 2019, 4, 1387–1393 CrossRef CAS.
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S.-I. In, Energy Environ. Sci., 2018, 11, 3183–3193 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- S. Sun, M. Watanabe, J. Wu, Q. An and T. Ishihara, J. Am. Chem. Soc., 2018, 140, 6474–6482 CrossRef CAS PubMed.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS PubMed.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, 1903915 CrossRef CAS PubMed.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- Y. Sun, J. Chen, X. Du, J. Cui, X. Chen, C. Wu, X. Yang, L. Liu and J. Ye, Angew. Chem. Int. Ed., 2024, 63, e202410802 Search PubMed.
- L. Ye, X. Jin, Y. Leng, Y. Su, H. Xie and C. Liu, J. Power Sources, 2015, 293, 409–415 CrossRef CAS.
- C. Mao, F. Zuo, Y. Hou, X. Bu and P. Feng, Angew. Chem., Int. Ed., 2014, 126, 10653–10657 Search PubMed.
- W. Fang, M. Xing and J. Zhang, Appl. Catal., B, 2014, 160, 240–246 CrossRef.
- J. Jiang, L. Zhang, H. Li, W. He and J. J. Yin, Nanoscale, 2013, 5, 10573–10581 RSC.
- R. B. Anderson, Fischer-Tropsch synthesis, United Kingdom: N. p., 1984, p. 311 Search PubMed.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC.
- Z. Wang, G. Zou, J. H. Park and K. Zhang, Sci. China Mater., 2024, 67, 397–423 CrossRef CAS.
- G. Jia, M. Sun, Y. Wang, Y. Shi, L. Zhang, X. Cui, B. Huang and J. C. Yu, Adv. Funct. Mater., 2022, 32, 2206817 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS PubMed.
- M. O. Olagunju, X. Poole, P. Blackwelder, M. P. Thomas, B. S. Guiton, D. Shukla, J. L. Cohn, B. Surnar, S. Dhar and E. M. Zahran, ACS Appl. Nano Mater., 2020, 3, 4904–4912 CrossRef CAS.
- S. Cao, Y. Li, B. Zhu, M. Jaroniec and J. Yu, J. Catal., 2017, 349, 208–217 CrossRef CAS.
- D. Gao, W. Liu, Y. Xu, P. Wang, J. Fan and H. Yu, Appl. Catal., B, 2020, 260, 118190 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- P. Li, L. Liu, W. An, H. Wang, H. Guo, Y. Liang and W. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS.
- J. Ji, R. Li, H. Zhang, Y. Duan, Q. Liu, H. Wang and Z. Shen, Appl. Catal., B, 2023, 321, 122020 Search PubMed.
- R. Schlögl, Chem. Sustain. Energy Mater., 2010, 3, 209–222 Search PubMed.
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- C. Qiu, J. Sun, M. Li, C. Mao, R. Song, Z. Zhang, D. D. Perovic, J. Y. Howe, L. Wang and G. A. Ozin, J. Am. Chem. Soc., 2024, 146, 33997–34007 Search PubMed.
- M. Zhang, Y. Zhu, J. Yan, J. Xie, T. Yu, M. Peng, J. Zhao, Y. Xu, A. Li, C. Jia, L. He, M. Wang, W. Zhou, R. Wang, H. Jiang, C. Shi, J. Rodriguez and D. Ma, Angew. Chem., Int. Ed., 2024, e202418645 Search PubMed.
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li and M. Tong, Adv. Mater., 2021, 33, 2102576 CrossRef CAS PubMed.
- J. Ma, J. Yu, G. Chen, Y. Bai, S. Liu, Y. Hu, M. Al-Mamun, Y. Wang, W. Gong and D. Liu, Adv. Mater., 2023, 35, 2302537 CrossRef CAS PubMed.
- Q. Wang, Z. Miao, Y. Zhang, T. Yan, L. Meng and X. Wang, ACS Catal., 2022, 12, 4016–4025 Search PubMed.
- S. B. Ning, H. Xu, Y. H. Qi, L. Z. Song, Q. Q. Zhang, S. X. Ouyang and J. H. Ye, ACS Catal., 2020, 10, 4726–4736 Search PubMed.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou and M. Li, Science, 2016, 351, 1065–1068 Search PubMed.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi and Y. Dai, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- Y. Yang, Z. Chai, X. Qin, Z. Zhang, A. Muhetaer, C. Wang, H. Huang, C. Yang, D. Ma and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202200567 Search PubMed.
- Y. Tang, Y. Li, W. Bao, W. Yan, J. Zhang, Y. Huang, H. Li, Z. Wang, M. Liu and F. Yu, Appl. Catal., B, 2023, 338, 123054 CrossRef CAS.
- Y. Tang, Z. Yang, C. Guo, H. Han, Y. Jiang, Z. Wang, J. Liu, L. Wu and F. Wang, J. Mater. Chem. A, 2022, 10, 12157–12167 RSC.
- Y. Li, Z. Liu, Z. Rao, F. Yu, W. Bao, Y. Tang, H. Zhao, J. Zhang, Z. Wang, J. Li, Z. Huang, Y. Zhou, Y. Li and B. Dai, Appl. Catal., B, 2022, 319, 121903 CrossRef CAS.
- J. Wang, J. Meeprasert, Z. Han, H. Wang, Z. Feng, C. Tang, F. Sha, S. Tang, G. Li and E. A. Pidko, Chin. J. Catal., 2022, 43, 761–770 CrossRef CAS.
- D. Tian, Y. Men, S. Liu, J. Wang, Z. Li, K. Qin, T. Shi and W. An, Colloids Surf. A Physicochem. Eng. Asp., 2022, 653, 129945 CrossRef CAS.
- Q. Yang, V. A. Kondratenko, S. A. Petrov, D. E. Doronkin, E. Saraçi, H. Lund, A. Arinchtein, R. Kraehnert, A. S. Skrypnik and A. A. Matvienko, Angew. Chem., Int. Ed., 2022, 61, e202116517 CrossRef CAS PubMed.
- S. Zhu, N. Li, D. Zhang and T. Yan, J. CO2 Util., 2022, 64, 102177 CrossRef CAS.
- Y. Zhang, W. Li, F. Tian, N. Cai, Q. Guan, D. Zhang, W. Ran, N. Li and T. Yan, Chem. Eng. J., 2023, 477, 147129 CrossRef CAS.
- Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long, W. Zhang, Y. Yang, Y. Zhang, Y. Yao, H. Chi, J. Wang, Q. Shen, Y. Xiong, Z. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS PubMed.
- Z. Li, J. Liu, R. Shi, G. I. Waterhouse, X. D. Wen and T. Zhang, Adv. Energy Mater., 2021, 11, 2002783 CrossRef CAS.
- Y. Shi, Z. Li, Q. Hao, R. Li, Y. Li, L. Guo, S. Ouyang, H. Yuan and T. Zhang, Adv. Funct. Mater., 2024, 34, 2308670 CrossRef CAS.
- W. Li, Y. Zhang, Y. Wang, W. Ran, Q. Guan, W. Yi, L. Zhang, D. Zhang, N. Li and T. Yan, Appl. Catal., B, 2024, 340, 123267 CrossRef CAS.
- R. Song, Z. Li, J. Guo, P. N. Duchesne, C. Qiu, C. Mao, J. Jia, S. Tang, Y. F. Xu, W. Zhou, L. Wang, W. Sun, X. Yan, L. Guo, D. Jing and G. Ozin, Angew. Chem., Int. Ed., 2023, 135, e202304470 CrossRef.
- L. Liu, A. V. Puga, J. Cored, P. Concepción, V. Pérez-Dieste, H. García and A. Corma, Appl. Catal., B, 2018, 235, 186–196 CrossRef CAS.
- A. V. Puga and A. Corma, Top. Catal., 2018, 61, 1810–1819 CrossRef CAS.
- M. Tahir and N. A. S. Amin, Int. J. Hydrogen Energy, 2017, 42, 15507–15522 CrossRef CAS.
- C. Song, Z. Wang, J. Zhao, X. Qin, M. Peng, Z. Gao, M. Xu, Y. Xu, J. Yan and Y. Bi, Chem Catal., 2024, 4, 100960 CrossRef CAS.
- W. Li, Y. Zhang, W. Ran, Y. Wang, F. Tian, F. Zhang, M. Xu, D. Zhang, N. Li and T. Yan, Appl. Catal., B, 2024, 351, 123978 CrossRef CAS.
- J. Huang, S. Jiang, M. Wang, X. Wang, J. Gao and C. Song, ACS Sustain. Chem. Eng., 2021, 9, 7891–7903 CrossRef CAS.
- H. Wang, S. Fan, S. Guo, S. Wang, Z. Qin, M. Dong, H. Zhu, W. Fan and J. Wang, Nat. Commun., 2023, 14, 2627 CrossRef CAS PubMed.
- B. Zhao, M. Sun, F. Chen, Y. Shi, Y. Yu, X. Li and B. Zhang, Angew. Chem., Int. Ed., 2021, 133, 4546–4550 CrossRef.
- Z. Li, W. Wu, M. Wang, Y. Wang, X. Ma, L. Luo, Y. Chen, K. Fan, Y. Pan, H. Li and J. Zeng, Nat. Commun., 2022, 13, 2396 CrossRef CAS PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and M. Tawalbeh, Appl. Surf. Sci., 2019, 483, 363–372 CrossRef CAS.
- N. Lertthanaphol, N. Pienutsa, K. Chusri, T. Sornsuchat, P. Chanthara, P. Seeharaj, P. Kim-Lohsoontorn and S. Srinives, ACS Omega, 2021, 6, 35769–35779 CrossRef CAS PubMed.
- Y. Xiong, W. Zhao, D. Gu, Z. Tie, W. Zhang and Z. Jin, Nano Lett., 2023, 23, 4876–4884 CrossRef CAS PubMed.
- Q. Liu, H. Cheng, T. Chen, T. W. B. Lo, Z. Xiang and F. Wang, Energy Environ. Sci., 2022, 15, 225–233 RSC.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinberner and A. Westfechtel, Ullmann's encycl, Chem. Ind., 2006, 14, 74–116 Search PubMed.
- S. Subramanian, Y. Song, D. Kim and C. T. Yavuz, ACS Energy Lett., 2020, 5, 1689–1700 CrossRef CAS.
- P. Kumar, V. C. Srivastava, U. L. Štangar, B. Mušič, I. M. Mishra and Y. Meng, Catal. Rev., 2021, 63, 363–421 CrossRef CAS.
- S. Subramanian, J. Oppenheim, D. Kim, T. S. Nguyen, W. M. Silo, B. Kim, W. A. Goddard and C. T. Yavuz, Chem, 2019, 5, 3232–3242 CAS.
- N. Thonemann and M. Pizzol, Energy Environ. Sci., 2019, 12, 2253–2263 RSC.
- H. Ohno, M. Ikhlayel, M. Tamura, K. Nakao, K. Suzuki, K. Morita, Y. Kato, K. Tomishige and Y. Fukushima, Green Chem., 2021, 23, 457–469 RSC.
- M. Liu, M. Konstantinova, L. Negahdar and J. McGregor, Chem. Eng. Sci., 2021, 231, 116267 CrossRef CAS.
- M.-Y. Zhou, X.-Q. Ding, J.-F. Ding, L.-P. Hou, P. Shi, J. Xie, B.-Q. Li, J.-Q. Huang, X.-Q. Zhang and Q. Zhang, Joule, 2022, 6, 2122–2137 Search PubMed.
- Z. Chang, Y. Qiao, H. Yang, X. Cao, X. Zhu, P. He and H. Zhou, Angew. Chem., Int. Ed., 2021, 60, 15572–15581 CrossRef CAS PubMed.
- S. Zhang, G. Yang, S. Liu, X. Li, X. Wang, Z. Wang and L. Chen, Nano Energy, 2020, 70, 104486 CrossRef CAS.
- S. Chen, H. Wang, Z. Kang, S. Jin, X. Zhang, X. Zheng, Z. Qi, J. Zhu, B. Pan and Y. Xie, Nat. Commun., 2019, 10, 788 CrossRef CAS PubMed.
- L. Li, W. Liu, R. Chen, S. Shang, X. Zhang, H. Wang, H. Zhang, B. Ye and Y. Xie, Angew. Chem., Int. Ed., 2022, 134, e202214490 Search PubMed.
- J. Zhang, C. Shang, Z. An, Y. Zhu, H. Song, Z. Chai, X. Shu, L. Zheng and J. He, Angew. Chem., Int. Ed., 2023, 62, e202312068 CrossRef CAS PubMed.
- G. Hou, Q. Wang, D. Xu, H. Fan, K. Liu, Y. Li, X. K. Gu and M. Ding, Angew. Chem., Int. Ed., 2024, 63, e202402053 CrossRef CAS PubMed.
- Y. Cao, R. Guo, M. Ma, Z. Huang and Y. Zhou, Acta Phys.-Chim. Sin., 2024, 40, 2303029 CrossRef.
- Y. F. Liang, M. Bilal, L. Y. Tang, T. Z. Wang, Y. Q. Guan, Z. Cheng, M. Zhu, J. Wei and N. Jiao, Chem. Rev., 2023, 123, 12313–12370 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |