DOI:
10.1039/D5SC00449G
(Review Article)
Chem. Sci., 2025,
16, 10141-10158
Harnessing C–H acetoxylation: a gateway to oxygen-enriched organic frameworks†
Received
18th January 2025
, Accepted 6th May 2025
First published on 7th May 2025
Abstract
Transition metal catalyzed C–H functionalization has emerged as a robust tool in organic synthesis, as it utilizes the most abundant functional group of an organic compound, i.e. C–H bonds, omitting the need for pre-functionalization. Selectively functionalizing a particular C–H bond out of numerous C–H bonds present in the molecular skeleton is a fascinating and difficult task to perform. To differentiate between almost identical C–H bonds, various strategies have evolved. Directing group (DG) assistance, non-directed functionalizations, and non-covalent interactions have significantly contributed to addressing the challenge of regioselectivity in targeting distinct C–H bonds. However, further advancements are still required. Among various C–H functionalizations, C–H acetoxylation is a pivotal organic transformation which enables direct functionalization of otherwise inert C–H bonds into versatile acetoxy groups. In this review, various strategies for C–H acetoxylation, i.e. directed and non-directed C–H acetoxylation, electrochemical C–H acetoxylation, and photo-induced C–H acetoxylation, are covered. A comprehensive coverage provided by this review will be extremely useful to chemists in both academia and industry, who are striving to incorporate oxygen into organic molecular skeletons.
Introduction
Regioselective functionalization of C–H bonds in organic frameworks through transition metal catalysis is an efficient and attractive alternative to existing conventional synthetic routes. Since an organic molecule contains several C–H bonds with almost the same chemical reactivity, selective functionalization of a particular C–H bond is crucial. Over the years, various methods have been devised for site-selective C(sp3)–H or C(sp2)–H functionalizations. C(sp2)–H bonds are relatively easy to activate, attributed to the π-assistance of organic compounds.1 The role of directing groups in C–H activation is important as these groups coordinate with a metal centre and drive it into the proximity of a particular C–H bond. C–H bond cleavage takes place to form a metallacycle intermediate which in the presence of a suitable reagent can undergo standard organometallic reactions to form C–C or C–X bonds. Directing group assisted ortho-C–H functionalization reactions of arenes are well explored as the metallacycle formed is thermodynamically stable. Directing group assisted meta- and para-C–H activation reactions are relatively difficult because of the unfavoured formation of large 12-18-membered metallacycles.2 Regioselective C–H acetoxylation reactions of arenes have significant importance since the corresponding acetoxylated products have great synthetic utility in the chemical industry.3 Acetoxy groups (OAc) can be easily transformed into hydroxy groups (OH) revealing a broad range of applications. Compounds containing acetoxy functional groups are abundant in natural products, biologically active molecules and functional materials.4,5 Acetoxy groups are also able to serve as a functional handle for many other organic transformations. Coupling reactions are also an important synthetic strategy to provide access to acetoxylated derivatives of arenes. However, prefunctionalization of substrates and formation of other by-products is a serious shortcoming of these transformations.6 Another traditional route to prepare acetoxylated arenes is Friedel–Crafts acylation followed by Baeyer–Villiger oxidation. There is a scarcity of reports on non-transition metal enabled and enzymatic C–H acetoxylation. Non-transition metal enabled transformations generally involve the attack of acetoxy cations onto the aromatic ring. Therefore, it is mainly applicable to electron-rich substrates and suffers from regioselectivity issues as well. Enzymatic C–H acetoxylation offers milder reaction conditions and also works with atmospheric oxygen as the oxidant. However, it is limited by narrow substrate scope, the requirement for co-factors, and lower catalytic efficiencies compared to nonenzymatic catalysts. In view of all these issues, transition metal catalysis serves as the backbone to carry out C–H acetoxylation, working on a range of substrates with good to excellent regioselectivity.7,8 Over the years, various directing groups have been developed for aliphatic compounds. However, in aliphatic acids there is a recent upsurge in the number of protocols which do not require an exogenous directing group, as inherent functionality of the molecule itself acts as the directing group.9 C–H activation will preferentially occur via the formation of a five- or six-membered metallacycle. Subsequent functionalization can be realised by using a suitable reagent. Generally, phenyl iodo diacetate PhI(OAc)2 [PIDA] is used as a common reagent for transition metal catalyzed C–H acetoxylation.10 This domain of C–H acetoxylation has witnessed a long journey of scientific development, i.e. from proximal to distal positions, or the replacement of a superstoichiometric amount of chemical oxidants by electricity. Non-directed and TDG approaches have also been probed to execute this transformation.11 Base metals such as cobalt are also known to catalyze this transformation, which adds up to the greener and economic aspect of such a transformation.12 Apart from these transformative strategies, looking at all these advances, it can easily be concluded that C–H acetoxylation stands as an efficient methodology in the quest to synthesize C–O bonds. This review aims to explore the state of the art in C–H acetoxylation, highlighting the strategic advances made to overcome the associated difficulties.
Proximal C(sp2)–H acetoxylation
Directed palladium-catalyzed C–H acetoxylation was first executed by Sanford and co-workers in 2004. In this protocol they enabled C–H acetoxylation of benzoquinoline. This compound is well documented in the domain of C–H activation, to provide facile cyclometallation under ambient conditions. Phenyl iodo diacetate (PIDA) acted as the OAc source and the oxidant to selectively provide the desired product in good yield. The reaction even worked with 8-methylquinoline derivatives, incorporating the OAc group at the benzylic position with excellent yields and selectivity (Scheme 1).13 A catalytic cycle was also proposed in which the first step involves the coordination of palladium to nitrogen to form a palladacyclic intermediate. The next step involves the oxidation of Pd(II) to Pd(IV).
 |
| | Scheme 1 Oxidative functionalization of proximal C–H bonds. | |
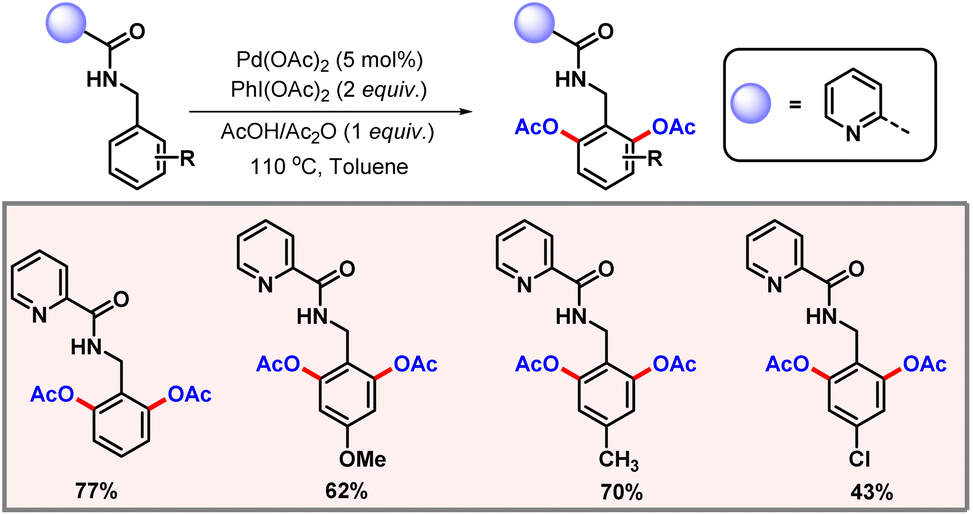
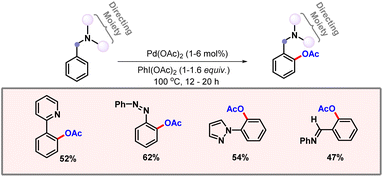
To terminate the catalytic cycle, reductive elimination generates the corresponding acetoxylated product. Later in 2009, Liang and co-workers introduced a bidentate directing group for proximal C–H acetoxylation (Scheme 2).14 Selective diacetoxylation of N-benzylpicolinamide took place in the presence of Pd(OAc)2 as a catalyst and PhI(OAc)2 as an oxidant. In the absence of Pd(OAc)2, no product formation was observed. Both pyridine and 8-aminoquinoline derivatives were successfully modified by this methodology to generate the corresponding mono- and diacetoxylated products.
 |
| | Scheme 2 Bidentate directing group guided proximal C–H acetoxylation. | |
However, substrates such as N-butylpicolinamide and N-(quinoline-8-yl) butyramide failed to promote the desired reaction, indicating that the methodology is ineffective for C(sp3)–H functionalization. Based upon the experimental evidence, a catalytic cycle was also proposed which highlights the necessary key steps of the transformation (Scheme 3).
 |
| | Scheme 3 Proposed catalytic cycle for bidentate directing group guided C–H acetoxylation. | |
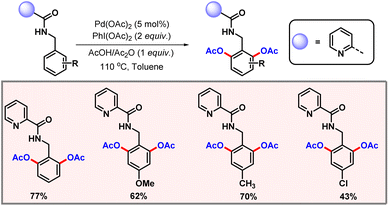
In 2008, the Wang group came up with a palladium-catalyzed ortho-C–H acetoxylation of anilides via C(sp2)–H oxidative activation (Scheme 4).15 The amide group itself acted as the directing group and acetic acid was utilized as the OAc source. Various anilide substrates were tested to evaluate the scope of this protocol. Notably, N-methyl acetanilide performed poorly under the optimized conditions, furnishing the product in lower yields. The most probable reason might be a steric effect of the methyl group inhibiting the coordination of the palladium with the amide group.
 |
| | Scheme 4
ortho-C–H acetoxylation of acetanilides. | |
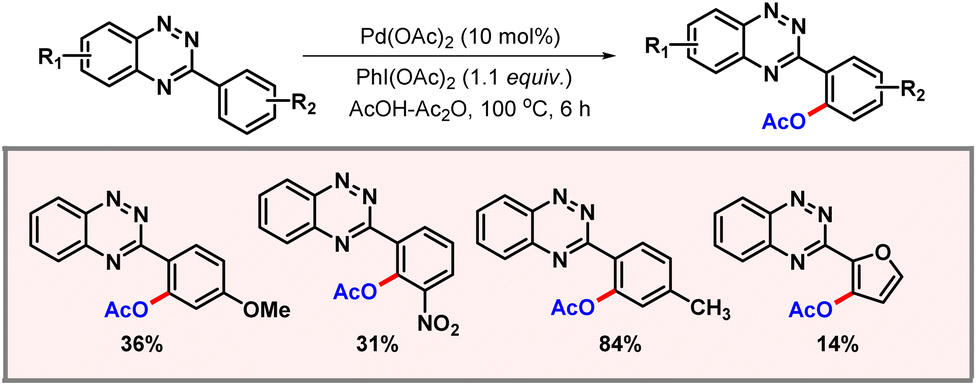
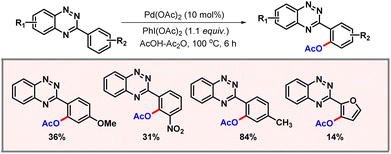
Following this report, in 2013 Huang and co-workers demonstrated ortho-C–H acetoxylation of arylbenzotriazines (Scheme 5).16 3-Arylbenzotriazines are important heterocycles, owing to their prevalence in aza-nucleosides, aza-nucleotides and herbicides. Therefore, synthetic derivatization of benzotriazines is of utmost importance. A variety of substrates containing both electron-donating and electron-withdrawing functional groups worked well to provide the desired products. It is worth noting that on doubling the amount of PhI(OAc)2, the diacetoxylated product was formed in significant amounts. Heterocycles containing benzotriazinyl residues were also transformed using the protocol, albeit in poorer yields.
 |
| | Scheme 5
ortho-C–H acetoxylation of 3-arylbenzotriazines. | |
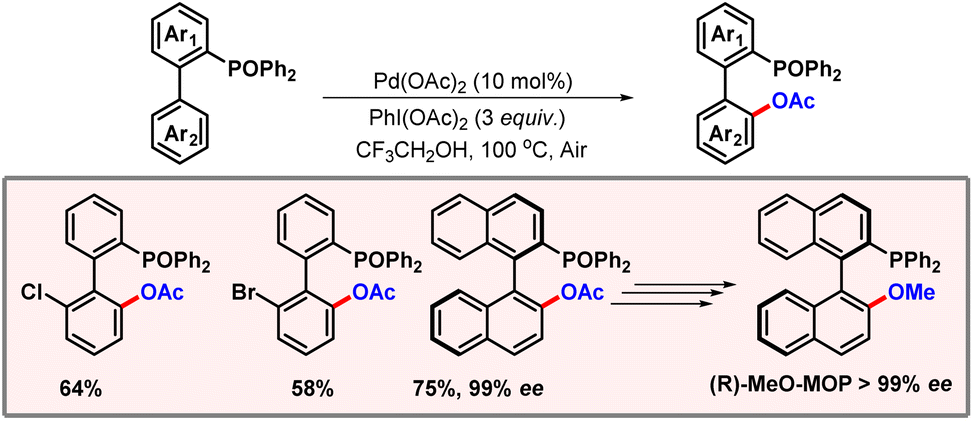
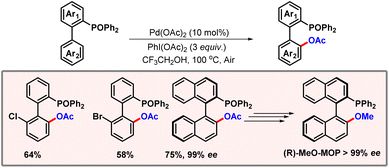
Later in 2014, the Yang group put forward a protocol for diphenylphosphine oxide directed ortho-C–H acetoxylation (Scheme 6).17 Phosphorus containing compounds are widely used as ligands in organic chemistry demonstrating the importance of their synthesis and further derivatization. This phosphorus oxide binds to palladium, forming a seven-membered metallacycle which leads to activation of the proximal C–H bond. Various other phosphorus oxides such as di-isopropyl or di-tert-butyl were also tested. However, diphenyl was found to be the most effective among them. Various kinds of functional groups were well tolerated showing the robustness of this protocol, albeit lower yields were observed in the case of electron-withdrawing substituents. When optically pure 2-diphenylphosphine oxide-1,10-binaphthyl was subjected to the reaction conditions, product formation was observed with almost complete stereo-retention. The obtained product was easily converted to an optically pure chiral ligand (R)-MeO-MOP with 40% cumulative yield, showcasing the applicability of this methodology.
 |
| | Scheme 6 Phosphine oxide directed ortho-C–H acetoxylation. | |
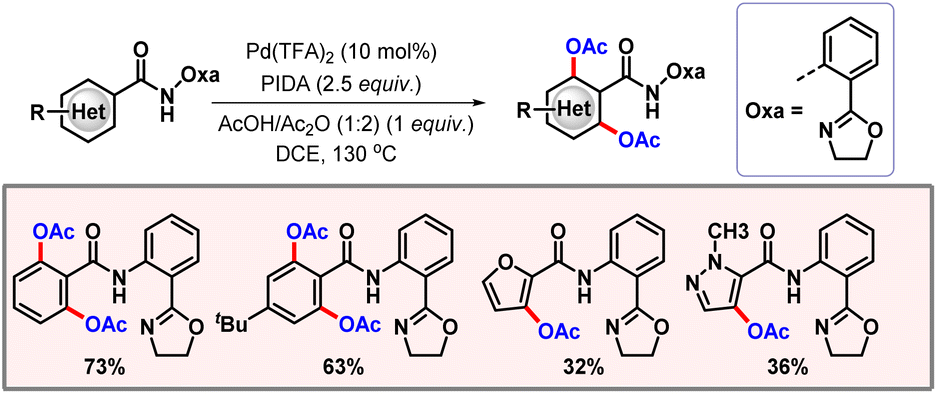
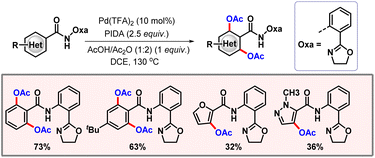
Along the line, Li and co-workers executed di-acetoxylation of arenes, enabled by an amide-tethered oxazoline based directing group (Scheme 7).18 A stringent optimization was performed to improve the catalytic efficacy of the process. Various heterocycles such as furan, quinoline, and thiophene, containing an amide group, were successfully transformed using this protocol. Kinetic isotope experiments (KIEs) revealed a kH/kD value of 1.1. Thus, C–H activation is most likely not the rate-limiting step of the transformation. Deuterium incorporation studies also aligned with the same fact, predicting C–H activation to be reversible. Utilization of azacycles such as pyridines and pyrimidines as directing groups is well established in the domain of C–H functionalization. However, it often causes problems, i.e. the formation of a strongly bound metallacycle sometimes leads to catalytic inactivity.19 To circumvent these issues, weakly coordinating DGs emerged as other efficient alternatives to promote the transformation. Functional groups such as carbonyl, ester, and amide fall under this category due to the low basicity of oxygen. In 2015, the Yu group disclosed the acetoxylation of Weinreb amides relying upon this weak coordination of the native functionality (Scheme 8).20
 |
| | Scheme 7
ortho-C–H acetoxylation of oxazoline tethered benzamides. | |
 |
| | Scheme 8
ortho-C–H acetoxylation of Weinreb amides. | |
PhI(OAc)2 was utilized as an oxidant to promote the transformation. Weinreb amides are good functional handles for the synthesis of aldehydes and ketones. Highly active benzylic positions also remained unaffected which showcases the ability of that directing group. Various substrates containing both electron-donating and electron-withdrawing groups were successfully transformed into the corresponding acetoxylated products with good yields and selectivities which demonstrates the generality of the protocol.
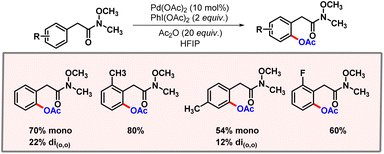
In 2016, Jiao and co-workers came up with a protocol for proximal C–H acetoxylation using isoxazolinyl as the directing group.21 A stringent optimization was performed which concluded Pd(OAc)2 (10 mol%) and PIDA (3 equiv.) to be optimal. Various substituents were tolerated on the isoxazoline ring, without affecting the outcome of the reaction to a significant extent. This observation clearly reveals the strong coordinating ability of the directing group to promote the desired reaction.
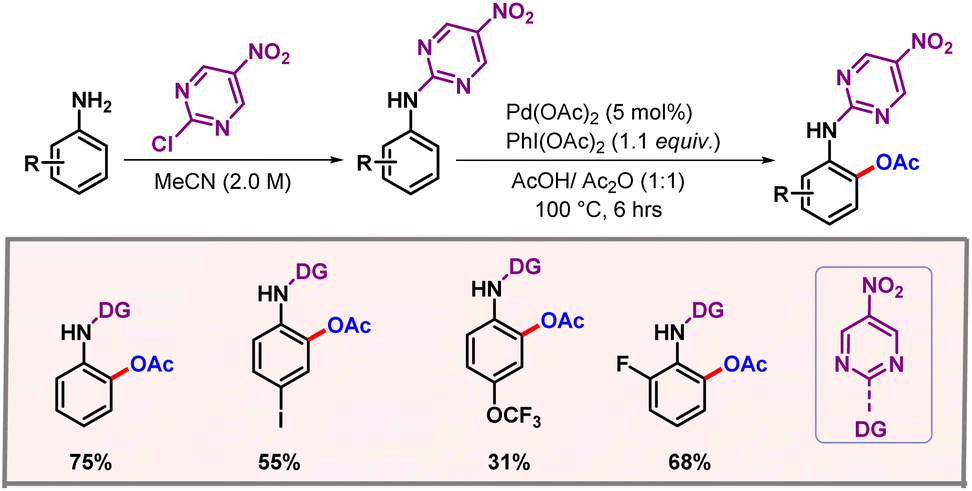
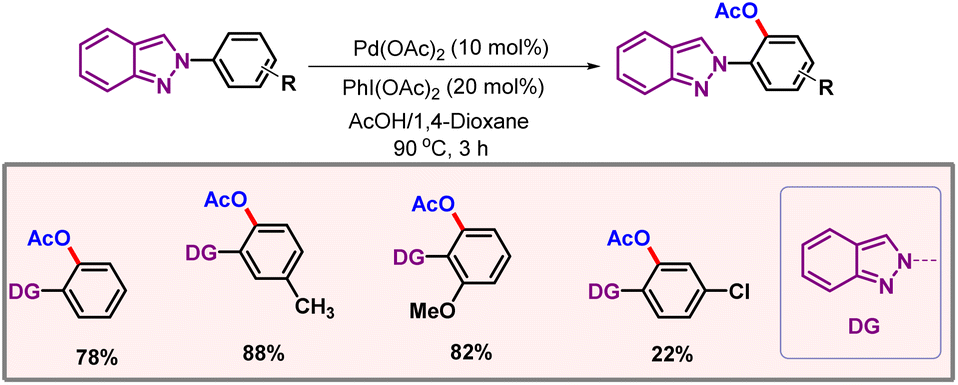
Later on, in 2019, Feng and co-workers showcased a protocol for ortho-C–H acetoxylation of anilines, utilizing pyrimidine as a directing group (Scheme 9).22 These DGs have been well documented for functionalization of phenols.23,24 However, anilines remained underdeveloped as anilines are generally not tolerated under oxidising conditions.25 The reaction utilized Pd(OAc)2 as the catalyst and a dual solvent combination of AcOH-Ac2O as the optimal solvent. Removal of the DG and OAc group could be easily performed, furnishing 2-aminophenols as the final product with good yield and selectivity. In 2021, Zhu, Song and co-workers extended the chelation assisted C–H activation method to ortho-C–H acetoxylation of 2-arylindazole derivatives, where an indazole moiety directed the metal to provide excellent regioselectivities (Scheme 10).26
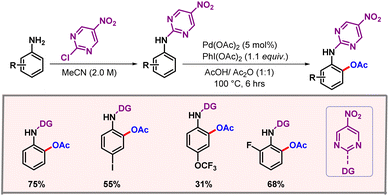 |
| | Scheme 9 Pyrimidine enabled ortho-C–H acetoxylation of anilines. | |
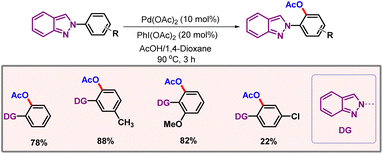 |
| | Scheme 10 Acetoxylation of 2-arylindazoles. | |
The substrate scope was evaluated, albeit yields obtained in the case of electron-deficient arenes were significantly poorer as compared to their electron-rich counterparts. The decrease in the yields was attributed to the less facile palladacycle formation in the case of electron-deficient arenes. Imidazo[1,2-a]pyridine is an abundant chromophore in medicinal and pharmaceutical chemistry. In 2024, Bakthadoss, Familoni and co-workers developed a protocol for acetoxylation of 2-aryloxyquinoline-3-carbaldehyde via palladium catalysis (Scheme 11).27 The aldehyde group present on the substrate remained unaffected during the transformation. The formation of mono- and bisfunctionalised products could be tuned selectively just by changing the amount of oxidant PIDA. Control experiments revealed that the aldehyde is not involved in chelation, whereas the nitrogen of the quinoline drives the reaction to furnish ortho-products. Pyrrolo[2,3-d]pyrimidine derivatives constitute an important class of heterocyclic compounds, which has a significant role in medicinal chemistry and drug discovery. In 2021, Zhang and co-workers came up with a palladium catalyzed C–H acetoxylation of pyrrolo[2,3-d]pyrimidine derivatives.28 The authors briefly described the role of sodium iodide in controlling the regioselectivity of the product. When 1.2 equivalents of NaI are used, the diacetoxylated product is predominantly formed. By increasing the amount of NaI, the monoacetoxylated product becomes the major product of the transformation.
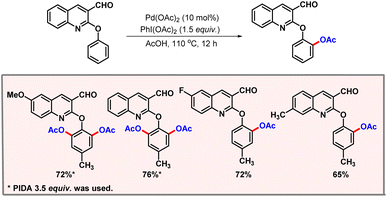 |
| | Scheme 11 Mono- and di-acetoxylation of 2-aryloxyquinoline-3-carbaldehydes. | |
Later, Correa and co-workers showcased a palladium catalyzed C(sp2)–H acetoxylation of peptides using the phenolic moiety of tyrosine as the directing group.29 Application of an easily removable 2-pyridyloxy group as the DG extends its applicability in late-stage functionalization. Along the line, Movassaghi and co-workers developed an efficient protocol for selective C-7 acetoxylation of indoline derivatives with the aid of an N1-acyl group (Scheme 12).30 The authors briefly studied the consequences of variation of electronic effects on the arene system. The absence of substitution at the C2 position led to a reduction in the reaction efficiency. An evaluation of the substrate scope reveals that increasing HOMO energy leads to a preference for the C-7 acetoxylated product. In 2022, Li and co-workers demonstrated a robust method for ortho-C(sp2)–H acetoxylation of phenylacetic acids and hydrocinnamic acids (Scheme 13).31 No external directing group was required in this protocol. KOAc is used as a base which is essential for promoting the necessary κ1-coordination leading to C–H bond cleavage, being the rate-limiting step of the transformation. Di-methyl substituted substrates and substituents at both ortho- and meta-positions were well tolerated under the optimized reaction conditions. Another highlight of the reaction was inherent inhibition of lactonization of acid, which generally leads to side products. In the same year, Volla and co-workers came up with a palladium catalyzed acetoxylation of indoles (Scheme 14).32 This protocol employed glycine as the transient directing group, which is involved in imine formation with indole-3-carbaldehyde.
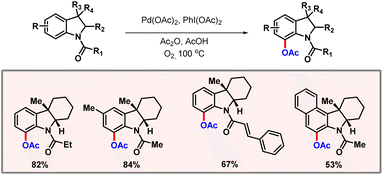 |
| | Scheme 12 Palladium catalyzed acetoxylation of indolines. | |
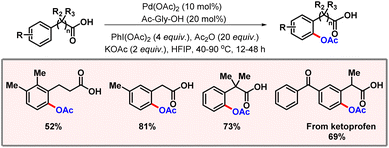 |
| | Scheme 13 Palladium catalyzed ortho-C(sp2)–H acetoxylation of phenyl acetic acids. | |
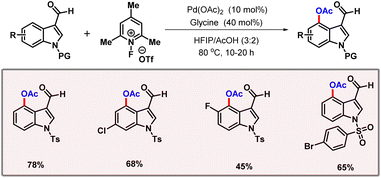 |
| | Scheme 14 Palladium catalyzed C-4 acetoxylation of indole-3-carbaldehydes. | |
The reaction proceeded well through formation of a six-membered palladacycle, bypassing the more stable five-membered metallacycle. After a stringent optimization, glycine as the TDG and 1-fluoro-2,4,6-trimethyl pyridinium triflate as the oxidant were found to be optimal. The reaction showcased a high tolerance for various protecting groups on nitrogen.
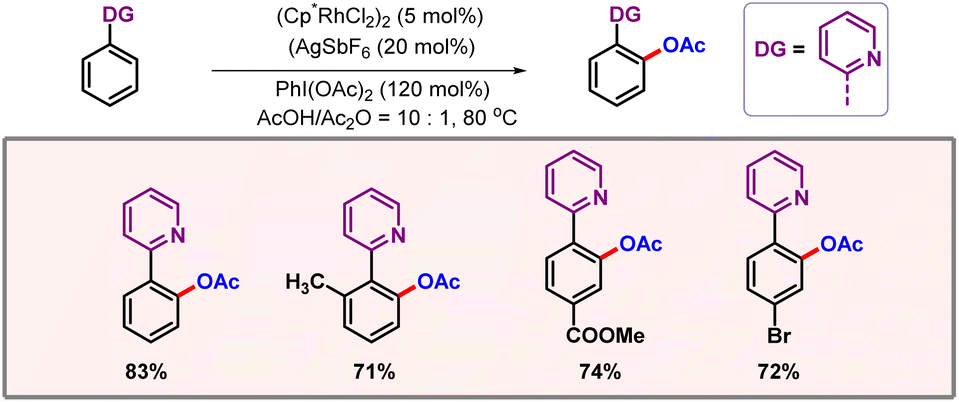
Progress in the domain of C–H activation is mainly centred on palladium. Exploration of other metals is necessary to further expand the boundaries of C–H activation. In 2017, Zhou and co-workers came up with a rhodium catalyzed ortho-acetoxylation of 2-phenyl pyridine derivatives (Scheme 15).33 The OAc group could be simply converted into OH, just by adding water to the reaction mixture. Generality of the protocol is clearly demonstrated by the diverse substrate scope which contained various arenes and heteroarenes.
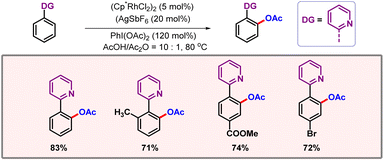 |
| | Scheme 15 ortho-C–H acetoxylation of phenyl pyridines. | |
Based upon experimental evidence, a catalytic cycle was also proposed. This involved cyclometallation followed by oxidation of Rh(III) to Rh(V) by PIDA. Subsequent reductive elimination to regenerate Rh(III) released the desired acetoxylated product (Scheme 16). Implementation of base metals in the domain of C–H activation is highly desired to make these transformations eco-friendlier and sustainable.
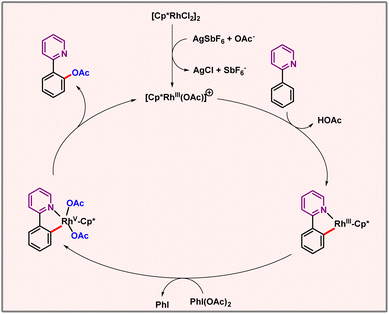 |
| | Scheme 16 Plausible mechanism of rhodium catalyzed acetoxylation. | |
Among various base metals, specifically cobalt catalyzed acetoxylation was put forward by the groups of Chatani and Deb.34,35 To further expand the spectrum of base metal catalysis, Guo and co-workers developed cobalt catalyzed ortho-acetoxylation of phenol derivatives (Scheme 17).36 The formed product could easily be hydrolysed to provide pyrocatechol, which is prevalent in many natural products and agrochemicals. Various DGs such as triazine and 2-methylpyridine were screened.
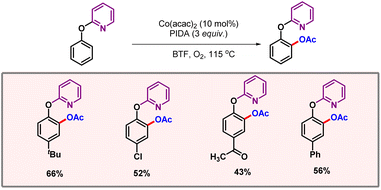 |
| | Scheme 17 Cobalt catalyzed C–H acetoxylation of phenoxy pyridines. | |
However, pyridine was found to be optimal. Notably, putting a linker between the phenol and pyridine moieties shuts down the reaction. A catalytic cycle was proposed, where cobalt first binds with the nitrogen of the pyridine DG. This intermediate was oxidised by oxidants such as O2 or PhI(OAc)2; subsequently, a metallacycle was formed. This cyclic intermediate underwent oxidation to provide Co(IV), followed by reductive elimination to release the acetoxylated product (Scheme 18). KIE studies revealed a kH/kD value of 2.25, which hints towards C–H cleavage being the rate-limiting step of the transformation.
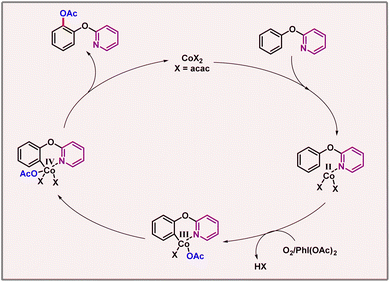 |
| | Scheme 18 Mechanism of cobalt catalyzed acetoxylation of phenoxypyridines. | |
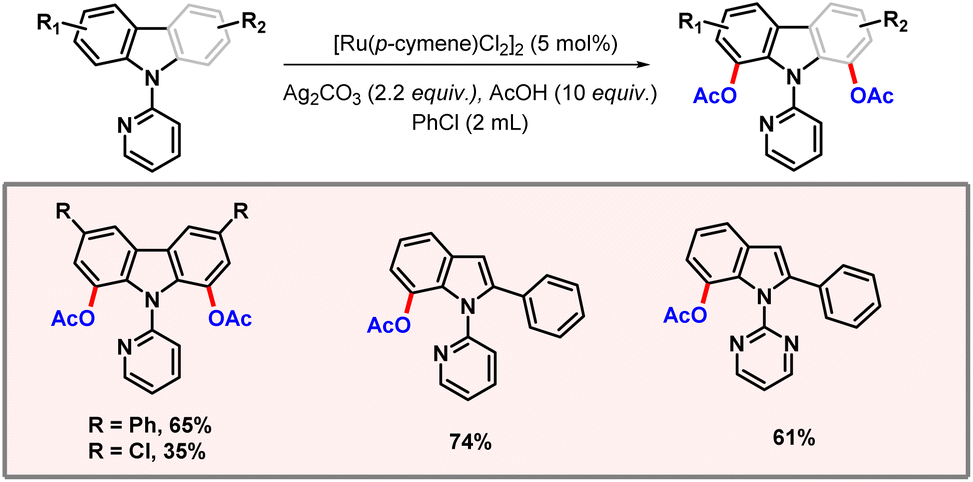
Another precious metal which is prevalent in the domain of C–H activation is ruthenium, relying upon [Ru(p-cymene)Cl2]2 as a catalyst. Miura and co-workers developed a protocol for acetoxylation of carbazole and indole substrates (Scheme 19).37,38 Regioselective acetoxylation at positions C1 and C7 of carbazole and indole was realised with the help of pyridine/pyrimidine directing groups. Acetic acid was used as an additive that might be the source of acetate ions. Ample substrate scope was achieved showcasing the robustness of the protocol. Notably, the presence of an aryl group at position C2 of indole was essential for the feasibility of the reaction. Electron-rich substrates were found to give better yields than electron-deficient ones.
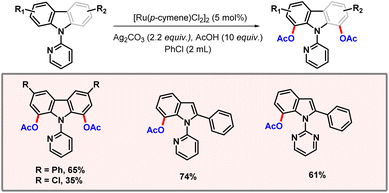 |
| | Scheme 19 C–H acetoxylation of indoles and carbazoles. | |
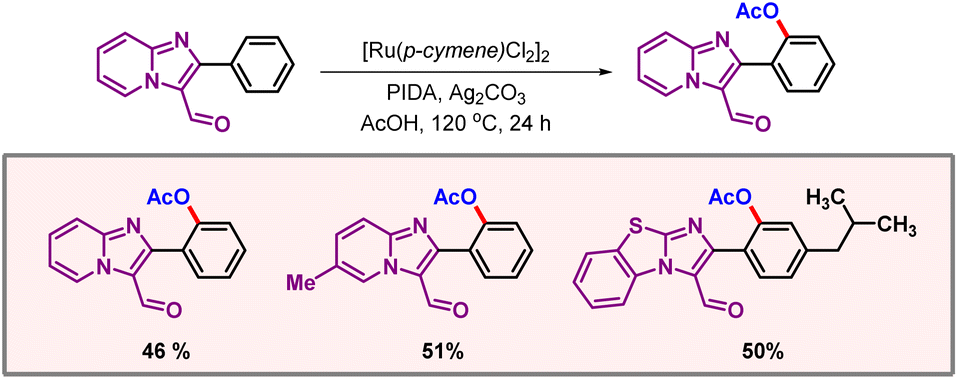
In 2023, Shankar and co-workers came up with a ruthenium catalyzed acetoxylation of imido[1,2-a]pyridine derivatives, which can later be transformed into corresponding hydroxyl derivatives (Scheme 20).39 RuCl2[p-cymene]2 was used as the catalyst and PIDA as an OAc source. Imidazole nitrogen binds to ruthenium to activate ortho-C–H bonds, leading to the desired product. Azacycles such as pyridine and quinoline are always difficult to functionalize through C–H activation, as it often leads to catalytic poisoning.
 |
| | Scheme 20 Acetoxylation of phenylimidazo[1,2-a]pyridine derivatives. | |
Distal C(sp2)–H acetoxylation
Regioselectivity is an important aspect of C–H functionalization which decides the quality of the reaction. Talking about arene systems, where three sites are available for C–H activation, there is ortho-, meta-, and para-selectivity. The directing group approach has played a crucial role in promoting regioselective C–H functionalization, and particular DGs have been developed to target a specific C–H bond out of numerous others. ortho-C–H activation is well explored since it proceeds via easily formed conformationally rigid five- or six-membered metallacycles.40 However, for meta- and para-C–H bonds, eleven-to twelve-membered and 17-18-membered metallacycles are required, respectively. Therefore, such processes become very difficult, owing to the thermodynamic instability of such large metallacycles. Thus, it is essential to design suitable directing groups that compensate for this high entropy demand of forming such a cyclophane-like pre-transition state that is able to selectively functionalize the meta-/para-C–H bonds. For the first time, in 2014 the Yu group showcased a removable cyano-based template that enabled the acetoxylation and olefination of distal meta-C–H bonds in anilines and benzylamine derivatives (Scheme 21).41 This reaction proceeds through a tricyclic cyclophane-like strained intermediate, involving a Pd(II)/Pd(IV) catalytic pathway.
 |
| | Scheme 21
meta-C–H acetoxylation of anilines. | |
In the case of distal C–H functionalization, conformation of the template becomes particularly important, as a marginal change in the conformation may lead to a significant decrease in product yield. It was also noted that placing a fluorine substituent in the template brought palladium closer in the vicinity of the meta-C-7 position, thus enhancing the efficiency of the reaction. A stringent optimization concluded PhI(OAc)2 to be the best reagent for acetoxylation. With the optimal conditions in hand, ample substrate scope was achieved which reflected the generality of the protocol. Later, the Yu group again reported another protocol for acetoxylation of indolines using a similar type of U-shaped template (Scheme 22).42 Indolines are more susceptible to ortho-, para-functionalization. However, using this sulfonyl linker reduced the electron density inside the ring and paved the way for regioselective meta-C–H functionalization. Bulky tert-butyl groups present in the substrate also helped in the formation of the tricyclic intermediate via the Thorpe–Ingold effect.
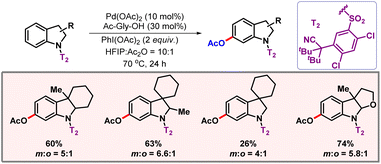 |
| | Scheme 22
meta-C–H acetoxylation of indolines. | |
Following this work, Li and co-workers described a protocol for meta-C–H acetoxylation of aniline derivatives assisted by a carbamate linker and nitrile as a directing group (Scheme 23).43 The majority of the carbamate assisted C–H functionalization reactions of anilines are known to provide ortho-selectivity. The carbamate template was prepared by incorporation of CO2 in aniline and subsequent directing group attachment through nucleophilic attack of the carboxylate group. After completion of the reaction, the DG could be easily removed under basic conditions to furnish meta-acetoxy-aniline as the final product. The same group later demonstrated meta-C–H acetoxylation and olefination of benzoic acid derivatives (Scheme 24).44 The same template-based strategy was applied to selectively activate the meta-C–H bond.
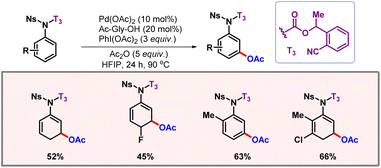 |
| | Scheme 23
meta-C–H acetoxylation of anilines. | |
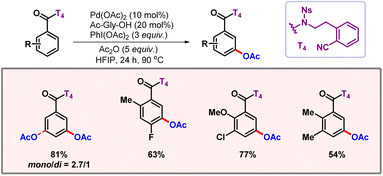 |
| | Scheme 24
meta-C–H acetoxylation of benzoic acids. | |
The carboxylic acid was converted to a nosyl-protected amide and the CN group in the template directed the palladium to the vicinity of the meta-C–H bond. The removal of the directing group was successfully achieved under mild conditions, to provide meta-functionalized benzoic acids as final products.
Along the line, our group also devised a protocol for meta-C–H acetoxylation, utilizing ultrasonics as the alternate source of energy (Scheme 25).45 Ultrasonic irradiation rocketed the regioselectivity and yield of the corresponding meta-acetoxylated product.
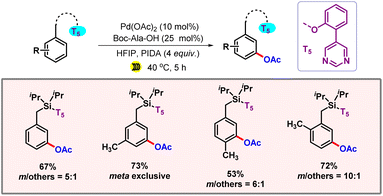 |
| | Scheme 25 Ultrasonic facilitated meta-C–H acetoxylation. | |
In 2016, our group again showcased template assisted meta-C–H hydroxylation and acetoxylation (Scheme 26).46 Notably, the change in the oxidant from PhI(TFA)2 to PhI(OAc)2 enabled the product formation with good yield and selectivity. HFIP as the solvent and N-acetylated amino acid as a ligand were crucial for the feasibility of the reaction. Kinetic studies concluded a zero-order with respect to the oxidant and first-order with respect to palladium. KIE studies revealed a (kH/kD) value of 3.02, which suggested that C–H activation is the rate-determining step. DFT studies concluded that HFIP plays a crucial role in the catalytic cycle by lowering the energy of a crucial intermediate and the corresponding transition states. A plausible mechanistic cycle was proposed which follows a Pd(II)/Pd(IV) pathway like usual acetoxylation reactions. It was also observed that with increasing linker length yield selectivity of the reaction decreased significantly which was attributed to more unfavorable transition states.
 |
| | Scheme 26
meta-C–H acetoxylation promoted by a sulfonyl tethered cyano directing group. | |
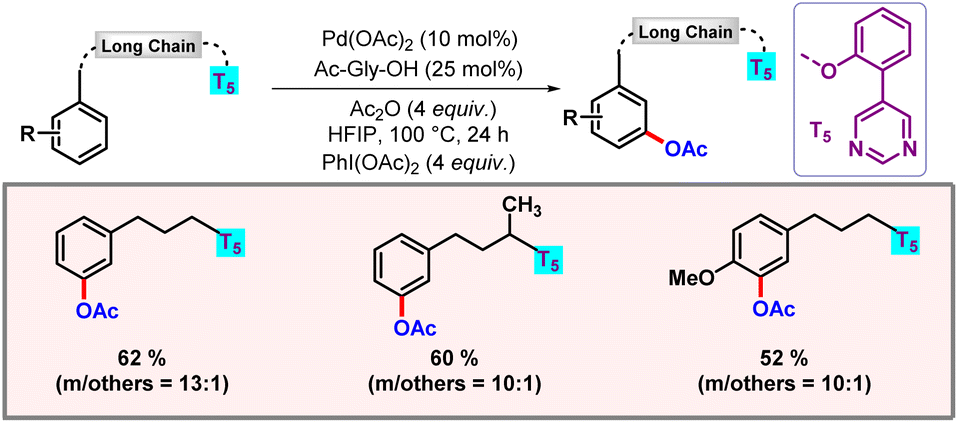
Following this work, our group disclosed a protocol for meta-functionalization of long chain flexible aryl alcohols, enabled by a pyrimidine directing group (Scheme 27).47 Excellent meta-selectivity was observed, ranging up to twenty atoms from the desired C–H bond. Various functionalizations such as acetoxylation, alkylation, alkenylation, and cyanation were realised using an ether tethered pyrimidine based directing group (DG). The hydrogen bonding between HFIP and the pyrimidine nitrogen of the directing group played a crucial role by enhancing the π-acidity of the palladium centre along with stabilization of the macrocyclic transition state.48
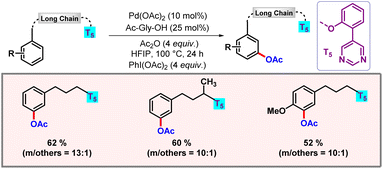 |
| | Scheme 27
meta-C–H functionalization of long chain aryl alcohols. | |
Later, Yu and co-workers in 2020 demonstrated regioselective meta-C–H acetoxylation and alkenylation of biaryl nitrile compounds (Scheme 28).49 The nitrile group is a versatile functional handle which can be converted to amines, acids, and other heterocycles.50 Control experiments suggested that the reaction proceeds via a concerted metalation–deprotonation (CMD) pathway in the presence of a pyridone ligand. Notably, in the presence of any other external oxidants such as AgOAc, a significant decrease in the yield was observed. This observation concludes that formation of a Pd–Ag heterobimetallic cluster is not required to promote the reaction. Therefore, this reaction proceeds through making use of bimetallic palladium as the active species. The pyridone ligand was found to have a crucial role in promoting meta-selectivity as compared to other ortho- and para-C–H bonds.
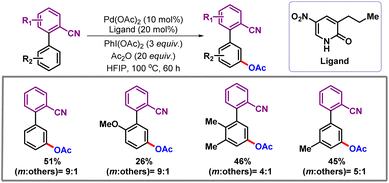 |
| | Scheme 28
meta-C–H acetoxylation of biaryl nitriles. | |
The reaction involved a CMD pathway where pyridone acted as the internal base. Regioselective para-C–H activation is a cumbersome transformation, as it requires the formation of a 17- to 18-membered metallacycle in the transition state, which imposes significant entropy constraints. Template guided C–H activation has emerged as an efficient strategy to address this task. However, para-C–H activation via templates presents several challenges, including the formation of large metallacycles, precise tuning of linker length, and the optimal orientation of donor atoms toward the para-C–H bond. Overcoming all these challenges, our group developed a D-shaped silyl tethered biphenyl nitrile-based template that selectively activates distal para-C–H bonds (Scheme 29).51 Utilizing this nitrile-based template, we successfully realised para-C–H acetoxylation under standard reaction conditions with Pd(OAc)2 as the catalyst and PhI(OAc)2 as the oxidant.
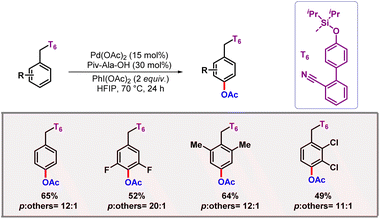 |
| | Scheme 29
para-C–H acetoxylation promoted by a silyl tethered cyano directing group. | |
Later, the Yu group came up with a protocol for para-C–H acetoxylation of benzoic acids (Scheme 30).52para-C–H activation of benzoic acids is strongly opposed by electronics of the molecule.53 Herein, an amide tethered biaryl nitrile directing group was employed, which enabled the activation of the distal para-C–H bond, overriding the electronic bias of the molecule. Conformational rigidity of biaryl also helped in achieving good para-selectivity. Notably, in situ generated PhI(OAc)2 by the reaction between PhIO and Ac2O was able to provide better yields than directly using the PhI(OAc)2. The yield further improved in an O2 atmosphere, owing to aerial oxidation of Pd(II) to Pd(IV).
 |
| | Scheme 30
para-C–H acetoxylation of benzoic acids. | |
Directed C(sp3)–H acetoxylation
Regioselective functionalization of inactive C(sp3)–H bonds is much more difficult in comparison with that of C(sp2)–H bonds.54 The inertness of C(sp3)–H bonds can be attributed to the high bond energy (∼98 kcal mol−1) and the relatively low acidity of C(sp3)–H bonds.55 In the absence of π-assistance, selective activation of C–H bonds becomes further more complex.56 Chelation assisted oxidation/acetoxylation of the C(sp3)–H bond was first disclosed by the Sanford group in 2004 (Scheme 31).57 Both oxime and pyrimidine substrates worked well in the presence of a palladium catalyst and PIDA as an oxidant. The protocol showcased high β-selectivity that was rationalized by formation of a stable five-membered metallacycle as an intermediate.58
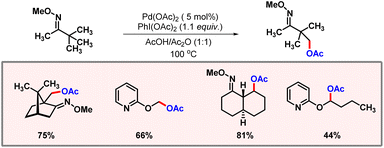 |
| | Scheme 31 Palladium catalyzed oxygenation of inactive C(sp3)–H bonds. | |
In 2006, Corey and co-workers also demonstrated β-C–H acetoxylation of N-phthaloyl-α-amino acids utilizing 8-aminoquinoline as the directing group.59 Later, Sahoo and co-workers executed primary β-C(sp3)–H acetoxylation of N-pivolyl-S-methyl-S-2-pyridylsulphoximines, enabled by a palladium catalyst and PIDA as an oxidant (Scheme 32).60 One of the highlights of the protocol was the selective formation of both mono- and disubstituted products by making some minor changes in the reaction conditions. Notably, PhI(OAc)2 acted only as the oxidising agent, while AcOH served as the OAc source.
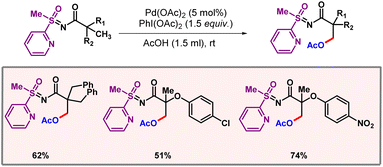 |
| | Scheme 32 Palladium catalyzed primary β-C(sp3)–H acetoxylation. | |
In the same year, Dong and co-workers executed oxime directed β-acetoxylation of alcohols (Scheme 33).61 Manifesting a transformation at the β-position of an alcohol is highly challenging, owing to the lack of electron density. Different kinds of substrates were well tolerated under optimized reaction conditions. KIE studies concluded C–H activation to be the rate-determining step of the reaction. Post-synthetic modifications further increased the versatility of this protocol.
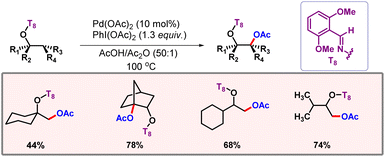 |
| | Scheme 33 β-C(sp3)–H acetoxylation of alcohols. | |
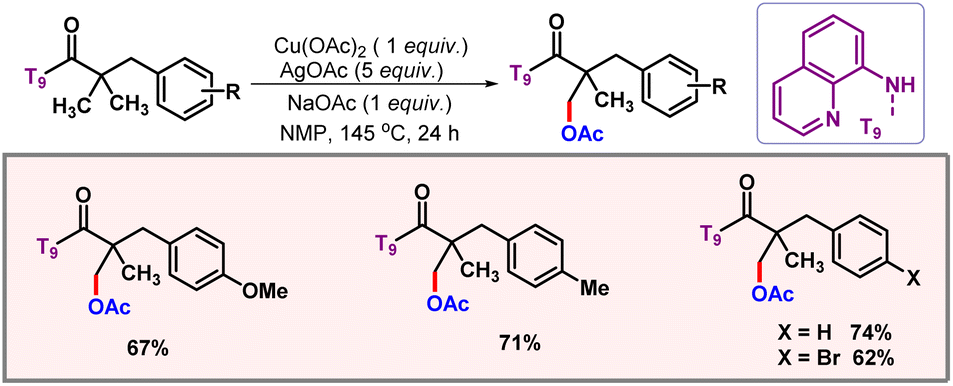
Along the line, the Kanai group put forward a bidentate directing group assisted acetoxylation of C(sp3)–H bonds by the combination of Cu(OAc)2 and AgOAc (Scheme 34).62 This protocol marked the entry of 3d metals into the domain of C(sp3)–H acetoxylation. A range of functional groups were well tolerated to provide the desired product. One of the shortcomings of this methodology was that in many substrates both mono- and disubstituted products were observed.
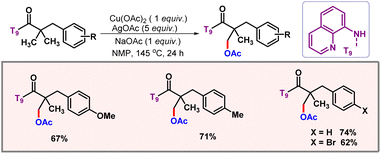 |
| | Scheme 34 Copper mediated C(sp3)–H acetoxylation. | |
In 2015, Zhao and co-workers developed a protocol for γ-C(sp3)–H acetoxylation of oxalyl amide protected aliphatic amines.63 Notably, implementation of acetic acid as an additive was crucial for spontaneity of the reaction. In the same year, the Zhang group also demonstrated an α-selective C(sp3)–H acetoxylation of amides, utilizing 1-aminoanthraquinone as the directing group (Scheme 35).64 It was observed that the reaction completely shuts down in the presence of a monodentate directing group. Various amides worked well with this protocol furnishing the desired products in good yields and selectivities. When an α-blocked substrate is subjected to the reaction, functionalization occurs at the β-position.
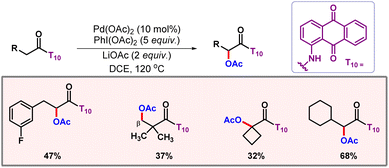 |
| | Scheme 35 Aminoanthraquinone directed C(sp3)–H acetoxylation of amides. | |
Later in 2016, Liu and co-workers disclosed another example of C(sp3)–H acetoxylation. 5-Methyl-isoxazole-3-carboxamide was used as the directing group to promote the desired reaction.65 Along the line, Mhaske and co-workers came up with C(sp3)–H acetoxylation of quinazolinone derivatives (Scheme 36).66 The reaction utilized Pd(OAc)2 as a catalyst, PIDA as an oxidant, and AcOH as solvent to enable product formation. The presence of a base was crucial to obtain the mono-acetoxylated product selectively.
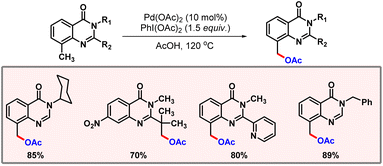 |
| | Scheme 36 C(sp3)–H acetoxylation of quinazolinone derivatives. | |
In the same year, Shi and co-workers developed a protocol for γ-C(sp3)–H acetoxylation of aliphatic primary amines (Scheme 37).67 To prohibit catalytic poisoning and to prevent the oxidation of amines, protonation of amines was a prerequisite. Functionalization at the γ-position was realised through a five-membered metallacycle, where the amine group acted as the directing group. Due to decomposition of amines under oxidising conditions somewhat lower yields were observed in many of the substrates. Protection of amines with an acetyl group led to a reduced reactivity.
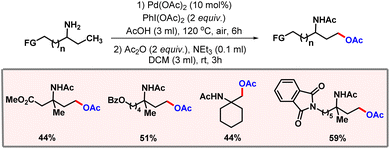 |
| | Scheme 37 Aliphatic C–H acetoxylation of functionalized primary amines. | |
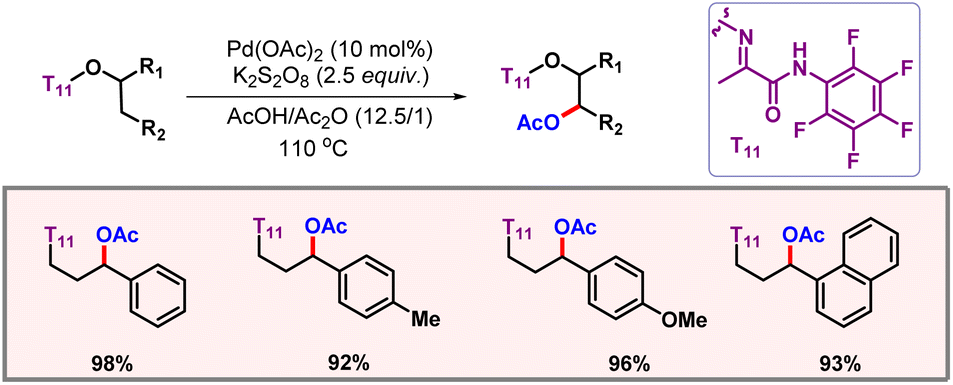
In 2019, the Xu group showcased a palladium catalyzed protocol for selective C(sp3)–H acetoxylation of masked alcohol derivatives using a bidentate auxiliary as the directing group (Scheme 38).68 The reaction worked well with various methyl, methylene, and benzylic C(sp3)–H bonds with readily available K2S2O8 as an oxidant. Mechanistic studies concluded C–H activation to be the rate-determining step. Removal of the directing group could also be realised to provide functionalized alcohols as the final products.
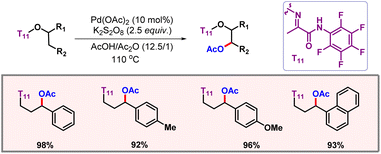 |
| | Scheme 38 Aliphatic C–H acetoxylation of masked alcohols. | |
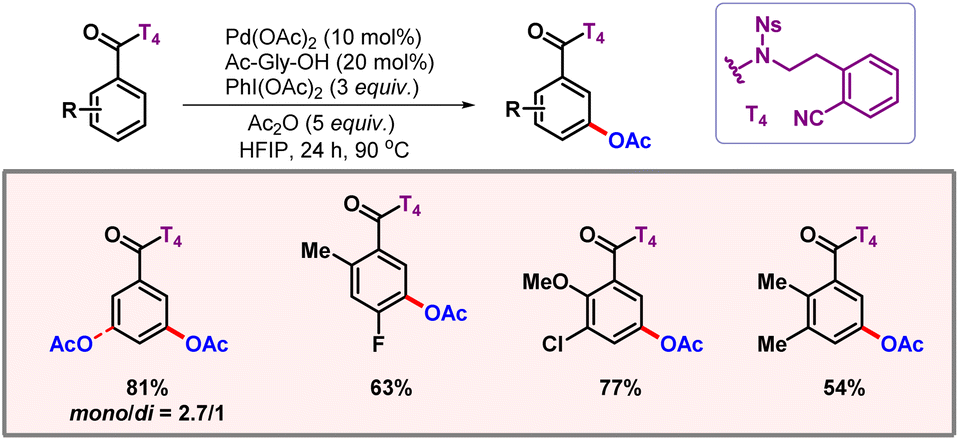
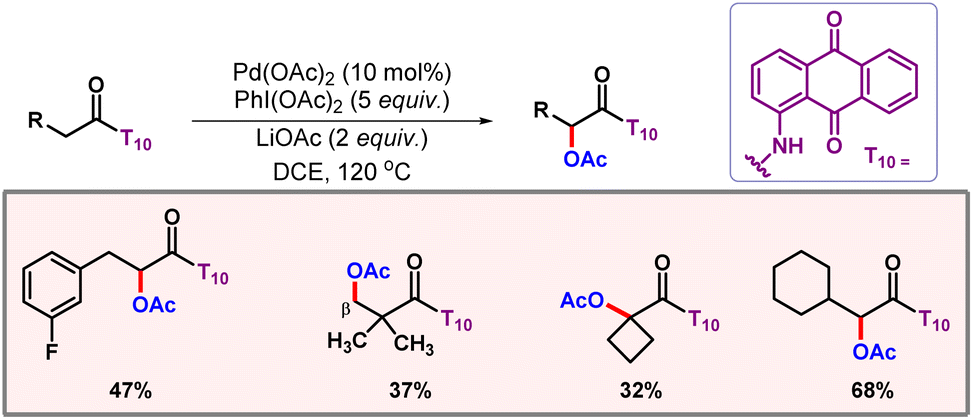
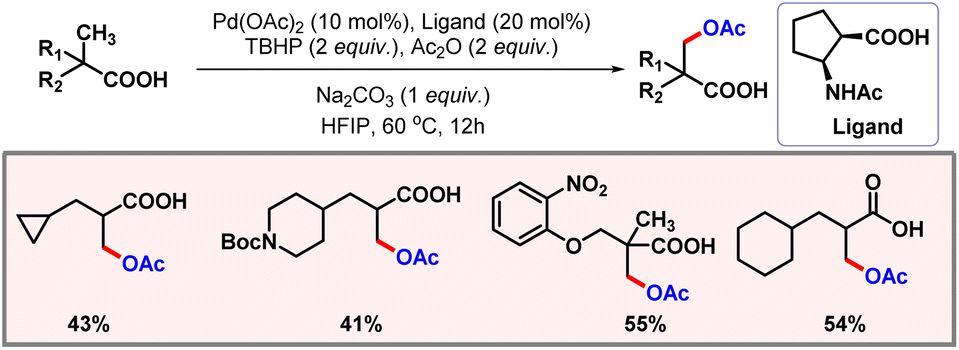
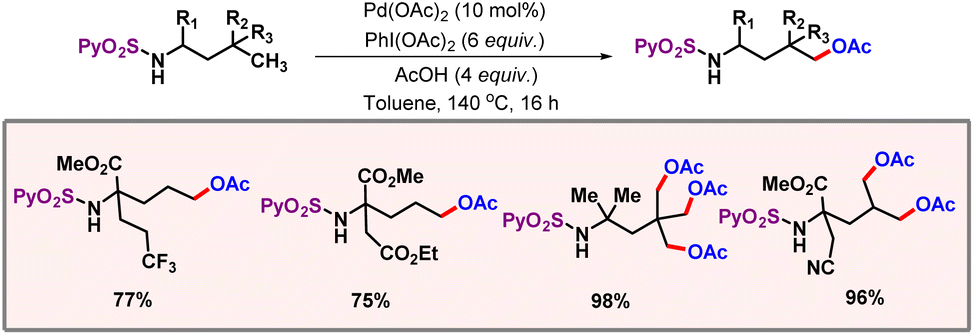
After this work, van Gemmeren and co-workers came up with a protocol for β-C(sp3)–H acetoxylation of aliphatic carboxylic acids (Scheme 39).69 Carboxylate itself served as the directing group as no other exogenous directing group was required. Optimization studies concluded that the use of an external base was necessary for this reaction, as it facilitates a κ1 coordination mode in which C–H activation becomes feasible.70 The external oxidant PIDA was used as the limiting reagent. A vast range of acid substrates were well tolerated under optimized reaction conditions to provide the desired products in good yields and selectivities.
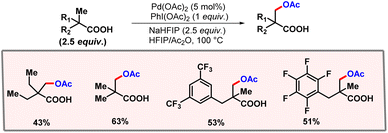 |
| | Scheme 39 β-C(sp3)–H acetoxylation of aliphatic carboxylic acids. | |
Along the line, Yu and co-workers demonstrated a transient directing group (TDG) enabled protocol to selectively functionalize the γ-position of alkyl amines (Scheme 40).71 The majority of the transformations of amines are based upon covalent directing group strategies which offer poor atom and step economy. Thus, this work emerged as an efficient alternative to those existing methodologies. 2-Hydroxynicotinaldehyde was employed as the TDG that transiently binds with the substrate through condensation, followed by product formation and subsequent removal. The reaction proceeded through the formation of a [6,5]-fused palladacycle intermediate. N-Fluoro-2,4,6-trimethyl tetrafluoroborate was found to be the optimal oxidant, providing the best results in terms of yield and selectivity. Various other oxidants such as PIDA, TBHP, and K2S2O8 were also screened. However, they were found to be inferior. Acetic acid was used as a co-solvent along with HFIP, which also acted as the acetate source. A vast range of amines containing various substitutions at different positions were well tolerated to provide the desired product. To make the isolation facile, final products were isolated in Bz-protected form.
 |
| | Scheme 40 TDG enabled γ-C(sp3)–H acetoxylation of aliphatic amines. | |
In the same year, Lu and co-workers developed a rhodium catalyzed protocol for selective C(sp3)–H acetoxylation of quinoline based substrates (Scheme 41).72 This reaction marked the first protocol for C(sp3)–H acetoxylation using rhodium catalysis. PIDA was used as the oxidant and implementation of Ac2O was found to improve the yield of the reaction.
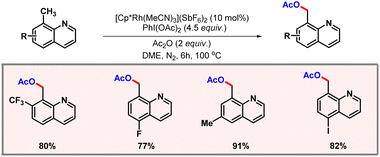 |
| | Scheme 41 C(sp3)–H acetoxylation of quinoline derivatives. | |
The substrate scope was evaluated to showcase the generality of the protocol. Mechanistic studies suggested C–H activation to be the rate-determining step of the transformation. In the same year, Yu and co-workers disclosed another protocol for β-C(sp3)–H acetoxylation of carboxylic acids (Scheme 42).73N-protected β-amino acid was used as a ligand, while TBHP acted as the oxidant. The presence of Ac2O was essential to promote formation of the desired product. In the absence of Ac2O, lactonization preferentially occurred in place of acetoxylation.
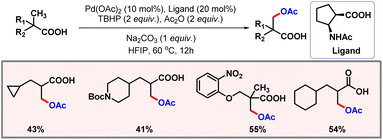 |
| | Scheme 42 β-C(sp3)–H acetoxylation of aliphatic carboxylic acids. | |
Later on, Hartwig and co-workers also demonstrated β-C(sp3)–H acetoxylation of alkylamine derivatives (Scheme 43).74 The reaction involved the formation of an uncommon four-membered metallacycle intermediate which is difficult to form. A number of directing groups were screened and salicylaldehyde was found to be the optimal. In the absence of salicylaldehyde the reaction remained completely silent.
 |
| | Scheme 43 β-C(sp3)–H acetoxylation of aliphatic amines. | |
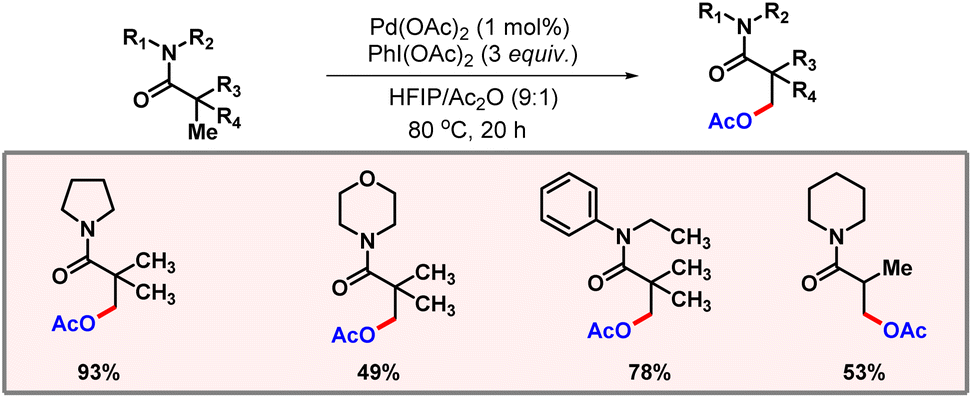
In non-polar solvents such as benzene, a mixture of both mono- and di-acetoxylated products was observed. A wide range of substrates containing different substituents were well tolerated, reflecting the generality of the protocol. In 2021, Punji and co-workers reported C(sp3)–H acetoxylation of tertiary amides (Scheme 44).75 The reaction was supposed to proceed via weak coordination of palladium to the carbonyl group. Low catalyst loading was one of the major highlights of this methodology along with broad substrate scope.
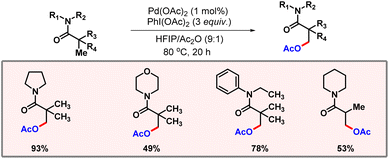 |
| | Scheme 44 β-C(sp3)–H acetoxylation of aliphatic amides. | |
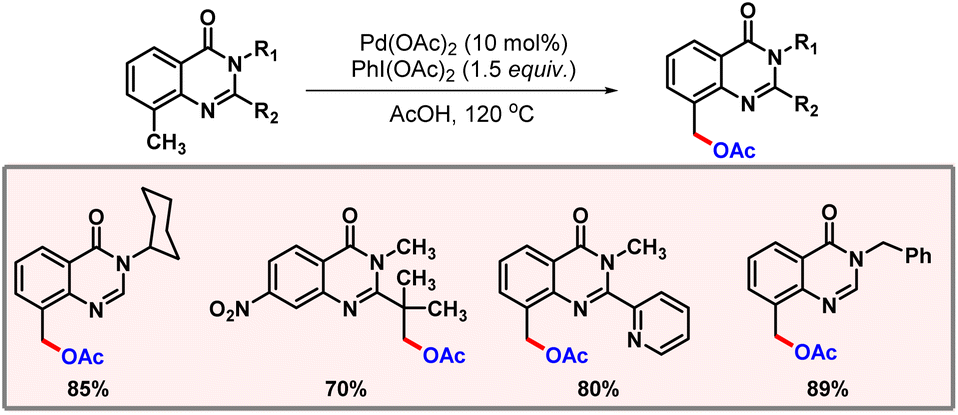
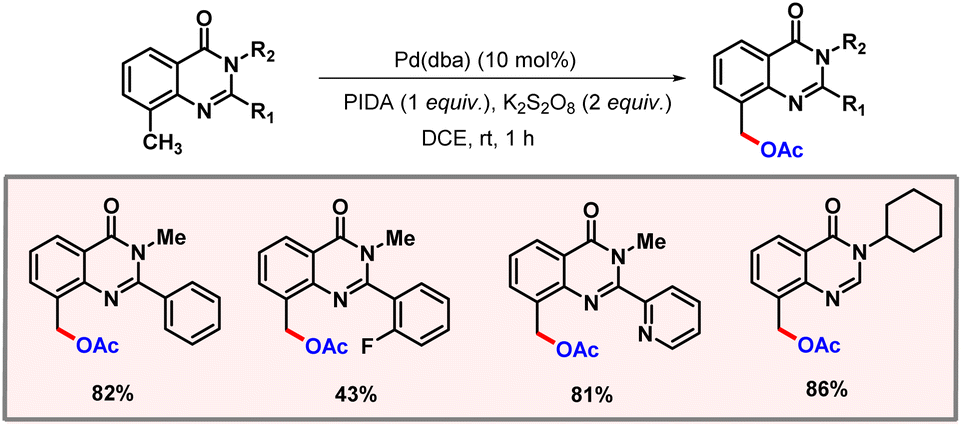
Quinazolinone-directed C(sp3)–H acetoxylation was first introduced by Mhaske and co-workers in 2017. Later in 2023, Jadhao and co-workers extended this protocol to ambient reaction conditions (Scheme 45).76 The reaction was carried out under mild conditions with Pd2(dba)3 as a catalyst, PIDA as an acetate source, and K2S2O8 as an additive for one hour. The nature of substituents at the C2 position was found to have a significant effect on the outcome of the reaction.
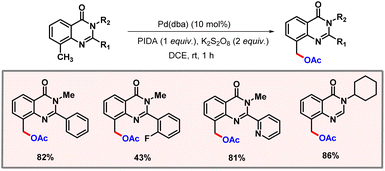 |
| | Scheme 45 Palladium catalyzed benzylic acetoxylation of quinazolinone moieties. | |
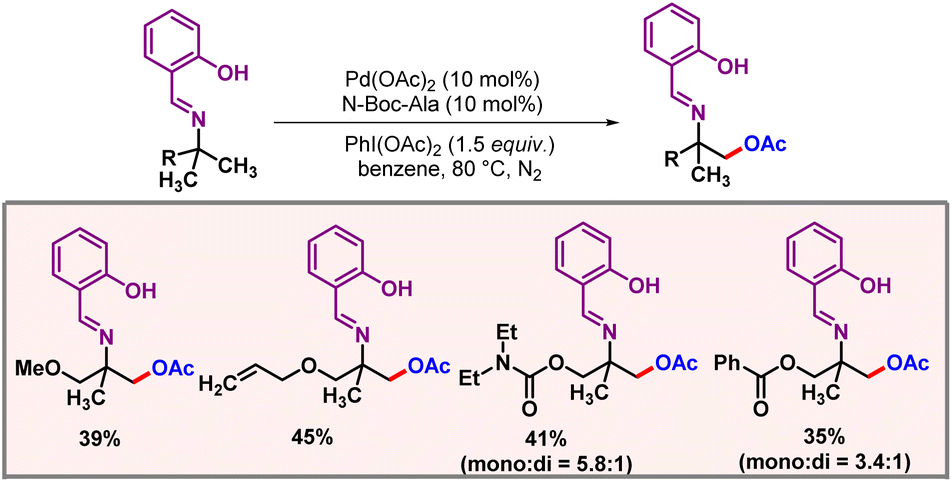
Realising δ-C(sp3)–H functionalization of amines is a challenging task as five-membered palladacycle formation is very facile which drives the reaction to the γ-position. In 2022, the Carretero group demonstrated an efficient protocol for δ-C(sp3)–H acetoxylation of N-protected amino acids using palladium catalysis (Scheme 46).77 The role of the N-(2-pyridyl) sulfonyl group as a directing template was crucial in attaining the desirable site selectivity and chemo-selectivity, i.e. to promote C–O bond formation over intramolecular C–N bond formation. PIDA was used as both an oxidant and an acetoxy source. A mechanism was also proposed where N-protected amine first coordinated to Pd(II), followed by oxidation to Pd(IV). Then, the OAc group acting as an inner sphere base promotes the C–H activation step. The generated AcOH during the C–H activation step later expedites intramolecular protonation of N-sulfonamide. Reductive elimination of this intermediate yields the desired product.
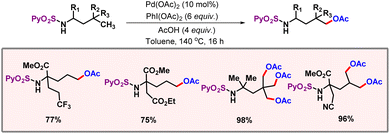 |
| | Scheme 46 Palladium catalyzed δ-C(sp3)–H acetoxylation of amines. | |
In the same year, Meng, Zhang, He and co-workers demonstrated palladium catalyzed γ-acetoxylation of alkylamides (Scheme 47).78 Herein, they focused on the structural influence of directing templates that is how variations of the directing group influenced the desired γ-C(sp3)–O bond formation instead of γ-C(sp3)–N bond formation. The 2-methoxyiminoacyl (MIA) group was found to be the optimal directing group and DFT studies also indicated that the energy barrier for the C–N elimination is higher than for the C–O elimination in the presence of MIA as the DG. Electron richness in MIA favours stable five-membered palladacycle formation.
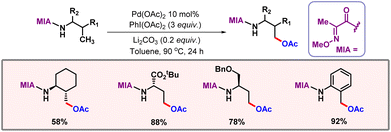 |
| | Scheme 47 Palladium catalyzed γ-acetoxylation of alkylamides. | |
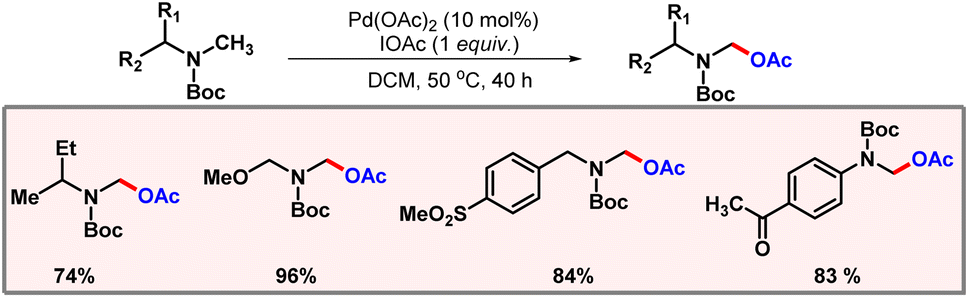
In 2006, the Yu group came up with a palladium catalyzed α-acetoxylation of Boc-protected N-methylamines (Scheme 48).79 IOAc was implemented as an oxidant which was supposed to oxidize Pd(II) to Pd(IV) in the catalytic cycle. Acetoxylation selectively occurred at the methyl position α to the nitrogen. A variety of Boc-protected amines were well tolerated under optimized reaction conditions.
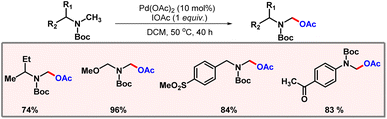 |
| | Scheme 48 Palladium catalyzed α-acetoxylation of amines. | |
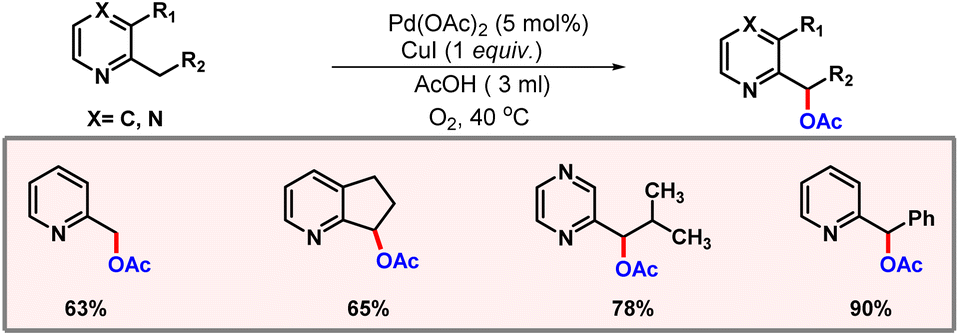
Later, Liang and co-workers demonstrated a transition metal free di-acetoxylation of amines at α- and β-positions using PhI(OAc)2.80 Following this, Jiang and co-workers developed a protocol for acetoxylation of C(sp3)–H bonds of 2-alkyl pyridine and pyrazine derivatives using oxygen as an oxidant (Scheme 49).81 Optimization studies concluded that both O2 and CuI are essential for the feasibility of the reaction. Acetic acid was used as the solvent, which itself acted as the acetate source.
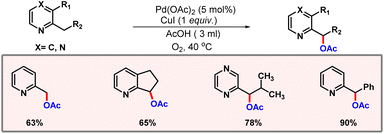 |
| | Scheme 49 C(sp3)–H acetoxylation of alkyl pyridines. | |
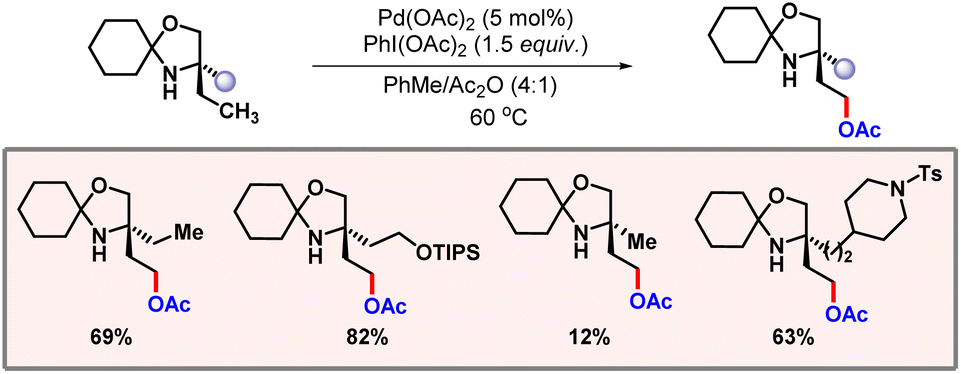
In 2015, Gaunt and co-workers demonstrated C(sp3)–H acetoxylation of amino alcohols by palladium catalysis (Scheme 50).82 Amino alcohols constitute an important class of organic compounds. Therefore, their diversification is a valuable transformation. Manifesting C–H activation in a substrate having an amine moiety is never easy, as it often leads to catalytic poisoning. This problem is somewhat resolved by attaching a directing auxiliary to the amine which inhibits this coordination and also directs the palladium into the proximity of the targeted C–H bond. However, this approach is not at all step economical, as attachment and removal of the DG adds two extra steps to the methodology. In the work of Gaunt this amino alcohol is first treated with cyclohexanone to form a hindered N,O-ketal moiety. This N,O-ketal type secondary amine system was found to undergo selective γ-acetoxylation under standard reaction conditions.
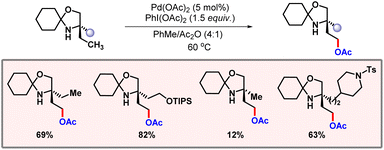 |
| | Scheme 50 γ-C(sp3)–H acetoxylation of amino alcohols. | |
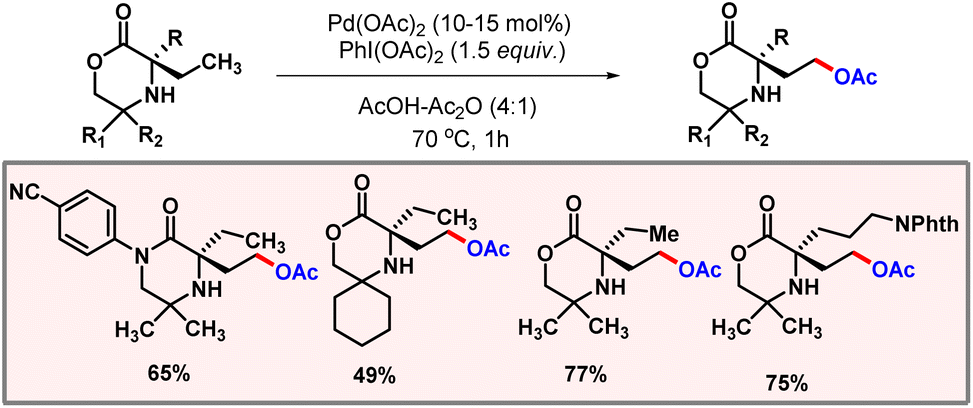
In 2019, the same group demonstrated γ-selective C(sp3)–H acetoxylation of morpholinone derivatives (Scheme 51).83 A broad range of morpholinone substrates underwent selective acetoxylation under optimized conditions. Kinetic studies revealed a kH/kD value of 2.8, suggesting that C–H bond cleavage is part of the turn-over limiting step. Detailed DFT calculations, along with kinetic studies, suggested that following the oxidation of the five-membered cyclopalladation complex, a dissociative ionization or SN2-type reductive elimination sequence yields the acetoxylated product. The preferential formation of the C–O bond (acetoxylated product) over the C–N bond (azetidine) was attributed to the significantly higher energy barrier for C–N reductive elimination. Given the rate-limiting nature of C–H bond cleavage, non-racemic binol-phosphoric acid ligands were assessed to induce enantioselectivity. Asymmetric acetoxylation (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 e.r.) was achieved using (R)-TRIP as a chiral ligand and I2/AgOAc as the oxidant system.
:15 e.r.) was achieved using (R)-TRIP as a chiral ligand and I2/AgOAc as the oxidant system.
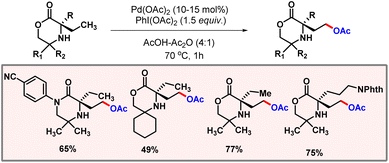 |
| | Scheme 51 C(sp3)–H acetoxylation of morpholinone derivatives. | |
Electrochemical C–H acetoxylation
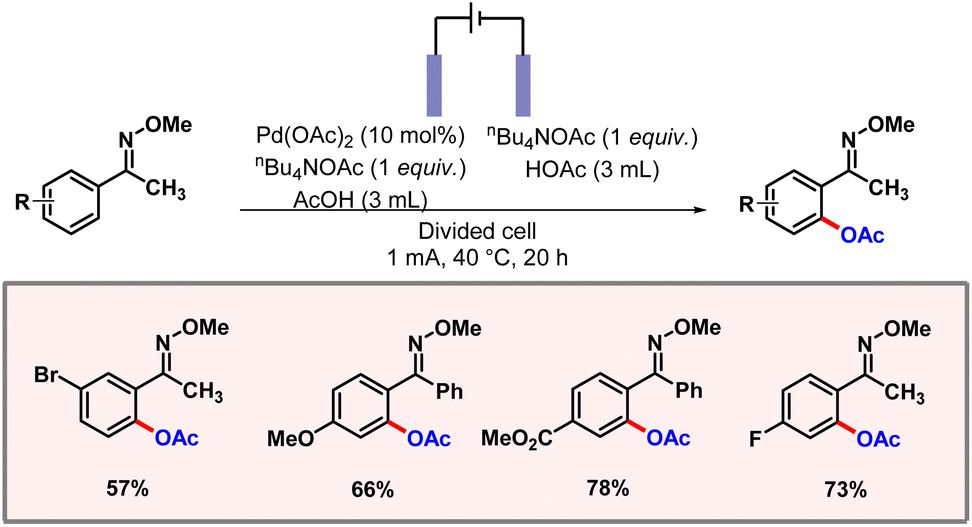
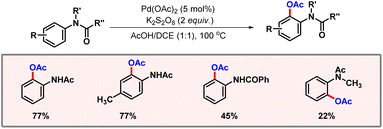
To a major extent, traditional C–H acetoxylation methods rely on transition metal catalysts and strong oxidants, which generate toxic waste and often cause severe functional group intolerance. Therefore, electrochemistry offers a cleaner and more sustainable approach by using electricity as the oxidant, reducing the need for hazardous reagents. Precise adjustment of the electrical voltage often allows the selectivity of a functionalization to be controlled. In this regard, the Zhang group demonstrated electrochemical C–H acetoxylation of aromatic oxime derivatives using palladium catalysis (Scheme 52).84 A constant current of 1 mA was flowing through the reaction mixture. Tetrabutylammonium acetate was found to be the optimal electrolyte in this reaction. The palladacycle undergoes anodic oxidation to release the product. Various aromatic oxime substrates worked well to produce the desired products in good yields and selectivities. KIE studies revealed the C–H activation step to be the rate-limiting step of the transformation.
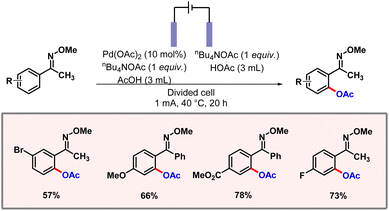 |
| | Scheme 52 Electrochemical ortho-acetoxylation of aromatic oximes. | |
In 2017, Mei and co-workers came up with another oxime directed C(sp3)–H electrochemical acetoxylation (Scheme 53).85 Sodium acetate was applied as an electrolyte. It was observed that the presence of both palladium acetate and electric current was essential for the generation of products. Various oxygen nucleophiles such as tosylate and alkoxide could also be incorporated into the molecular skeleton using this protocol. Cyclic voltammetry studies of the cyclopalladated species confirmed that the oxidation of the Pd(II) complex is followed by C–O reductive elimination.
 |
| | Scheme 53 Oxime directed C(sp3)–H acetoxylation. | |
Along the line, the Sanford group also came up with C(sp3)–H acetoxylation of 8-methylquinoline, enabled through electrochemistry (Scheme 54).86 Tetramethylammonium acetate played dual roles of electrolyte and an acetate source. Cyclovoltammetric studies were performed to investigate the mechanism. A divided cell provided better yields in comparison to an undivided cell. Another crucial observation was the diffusion of H2 from the cathode to the anode which leads to reduction of palladium from Pd(II) to Pd(0). To circumvent this issue, benzoquinone was used as an alternative chemical oxidant. The authors concluded that both the oxidants (chemical and electrochemical) played a crucial role in the fate of the reaction.
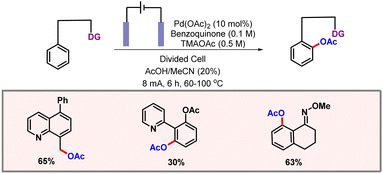 |
| | Scheme 54 Palladium catalyzed C–H acetoxylation via electrochemical oxidation. | |
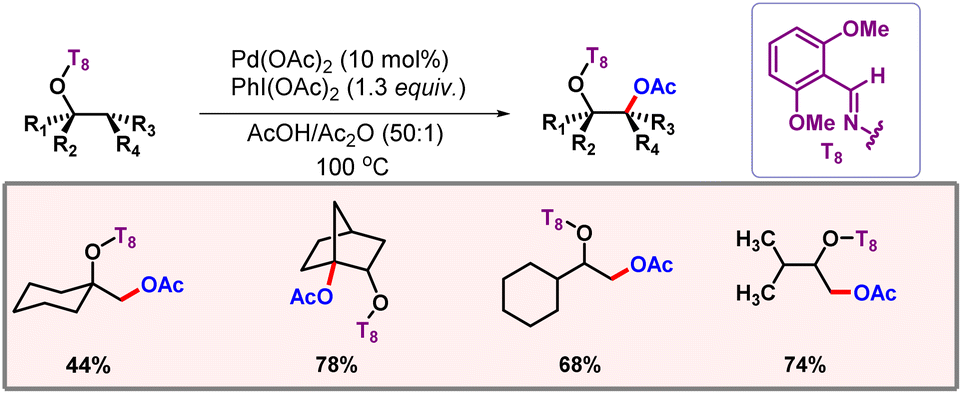
Very recently, Kakiuchi and co-workers disclosed electrochemical acetoxylation of alcohol derivatives using oxime ethers as the directing group.87 One of the key applications of this work included formation of 1,2-diols as the potential product. The reaction involved a reductive elimination from a Pd(IV) center to produce the desired product.
Photoinduced C–H acetoxylation
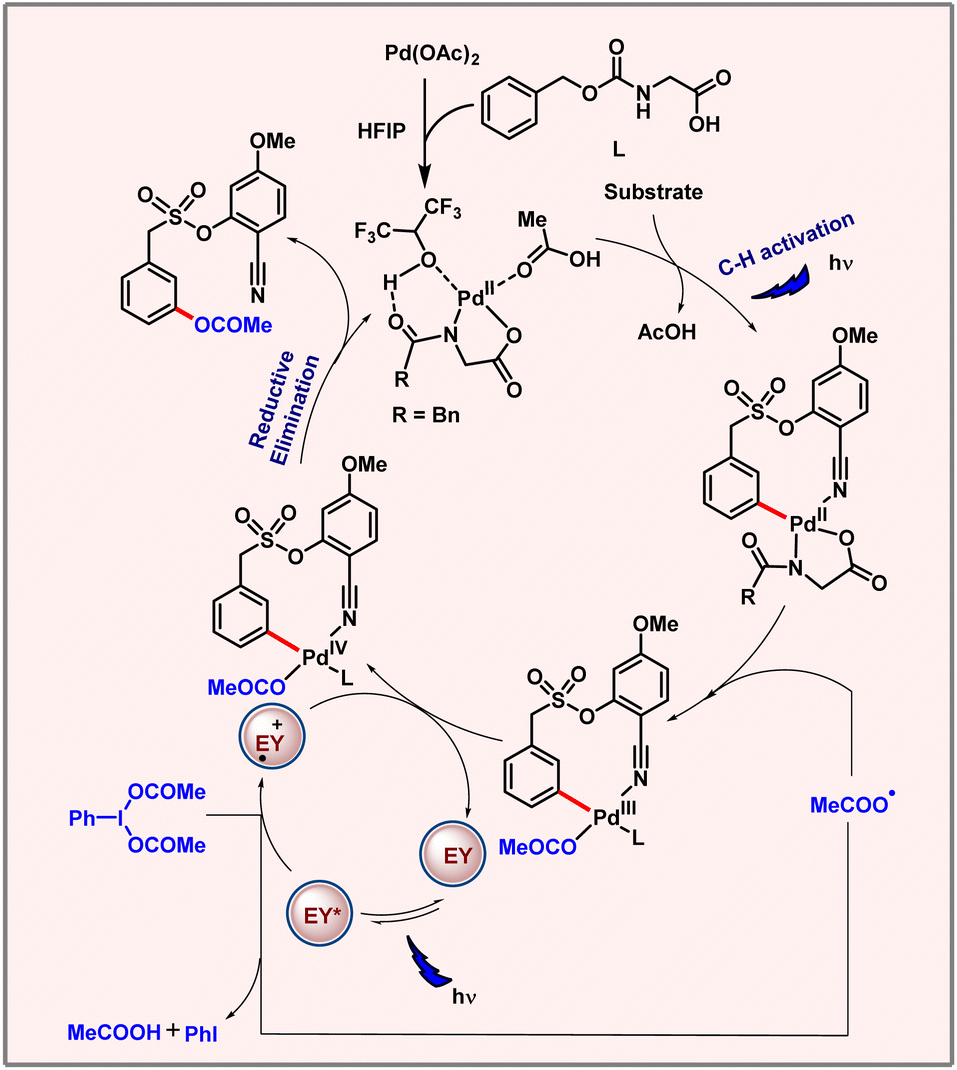
In 2023, our group came up with a palladium catalyzed meta-C–H oxygenation of arenes via a metallaphotocatalytic pathway (Scheme 55).88 This protocol unveils C–H acetoxylation at the meta-position of phenylacetic acids, biaryl alcohols and biaryl acids. The reaction proceeds through a radical pathway. Notably, only a weak coordinating nitrile based directing template was able to promote the desired product formation. Usage of PhI(TFA)2 as an OAc source instead of PhI(OAc)2 led to a meta-hydroxylated product. The proposed mechanism involves the formation of Pd(II)–Pd(III)–Pd(IV) intermediates unlike thermal conditions which follow the Pd(II)–Pd(IV) catalytic pathway. The reaction involved a palladacycle intermediate formation via C–H activation through photoexcitation. This Pd(II) intermediate binds with the OAc radical to form the Pd(III) intermediate. Oxidation of this intermediate by Eosin Y generates Pd(IV), followed by reductive elimination to release the desired product (Scheme 56). The C–H activation step is found to be rate-determining as suggested by a high KIE value of 2.5.
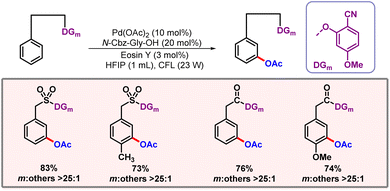 |
| | Scheme 55 Photoinduced meta-C–H acetoxylation of arenes. | |
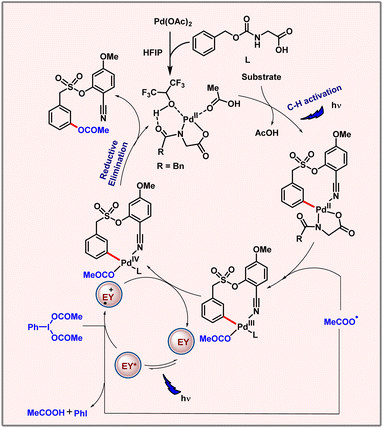 |
| | Scheme 56 Plausible catalytic cycle for photoinduced C–H acetoxylation. | |
Non-directed C–H acetoxylation
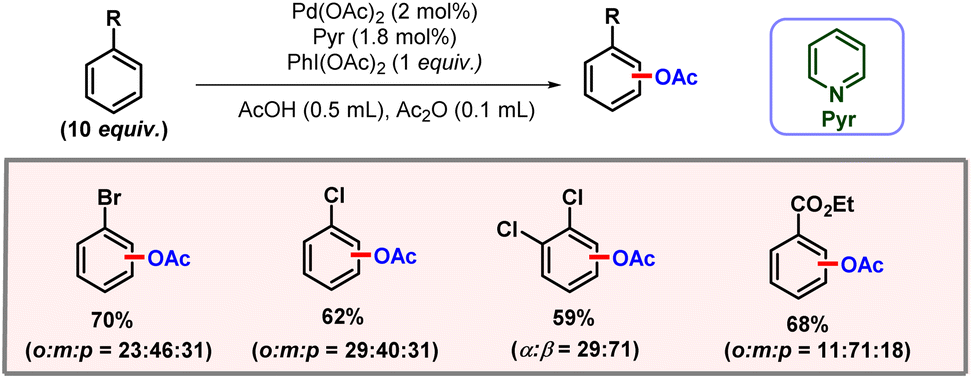
Non-directed C–H activation is advantageous as compared to directed C–H activation as it does not pose the requirement of a pre-installed directing group. It offers significant implications for efficiency, sustainability, and flexibility in molecular design. By bypassing the requirement of a directing group, non-directed C–H activation offers a reduction in the number of steps required for a transformation, thus leading to better step economy. Site selectivity in non-directed mode is generally governed by the inherent reactivity of C–H bonds, which depends upon several factors such as electronics, sterics, and the strength of the specific C–H bond. The concept of non-directed C–H activation was introduced by Fujiwara and Moritani in 1996. However, to induce reactivity, the substrate was required to be used in solvent amounts, diminishing its practicality.89 To address this limitation, in 2017, the Yu group developed a protocol for non-directed C–H olefination, enabled by pyridone ligands which allowed for the use of the substrate in stoichiometric amounts without compromising the reaction's efficiency.90 Further advances in this domain were made by the van Gemmeren and Maiti group, who introduced dual ligand strategies to improve both the yield and selectivity of the reaction.91,92 Talking about non-directed C–H acetoxylation, in 1996 Crabtree and co-workers showcased acetoxylation of naphthalene using a palladium catalyst and PIDA as an oxidant.93 In 2009, Suna and coworkers demonstrated selective C-3 acetoxylation of indole derivatives via an electrophilic palladation mechanism, using Pd(OAc)2 as the catalyst.94 Later in 2011, the Lei group also demonstrated a C-3 acetoxylation of indoles.95 The reaction showcased a high yield of the desired product under milder reaction conditions. Notably, weak bases were inferior as compared to strong bases such as KOH. It was also observed that electron-rich indoles were more active towards the reaction as compared to electron-deficient ones. In the absence of palladium, dimer formation of indole took place. However, the reaction was sluggish as compared to C–H acetoxylation in the presence of palladium. In the same year, Kwong and co-workers also came up with oxidative C-3 acetoxylation of indoles. Application of a mild base and mild reaction conditions were major highlights of the protocol.96 Later, the Sanford group conducted an in-depth mechanistic study on palladium catalyzed C–H acetoxylation of benzene derivatives (Scheme 57).97 Their research initially focused on the acetoxylation of benzene using PIDA as an oxidant, with pyridine serving as an assisting ligand. The study observed that maintaining a fixed metal–ligand combination while varying the oxidant led to notable shifts in the selectivity of product formation. Various pyridine derivatives were evaluated as ligands, revealing that sterically hindered pyridines resulted in reduced yield. They also proposed that nearly a 1:1 ratio of pyridine to Pd(OAc)2 allows for an open coordination model to the palladium centre, which is essential for facilitating C–H activation. Kinetic studies revealed that the C–H activation step was rate-determining with a value of kH/kD > 2.
 |
| | Scheme 57 Non-directed C–H acetoxylation of arenes. | |
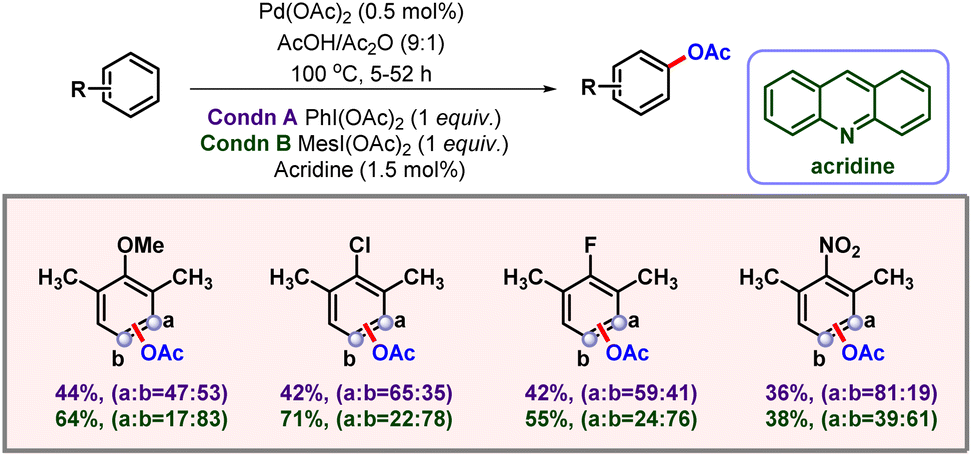
In 2013, the Sanford group again showcased ligand controlled C–H acetoxylation of simple arenes via palladium catalysis (Scheme 58).98 During their initial ligand screening it was observed that by changing the steric bulk around pyridine ligands, a considerable change could be induced into the selectivity of the product. After a stringent optimization, the acridine ligand was found to be the optimal ligand for meta-acetoxylation. Catalyst loading also significantly affected the yield and selectivity of the product formation. In this protocol under optimized conditions, the authors were not able to distinguish between meta- and para-C–H bonds, as 1:1 mixtures were observed. Various mono-, di- and trisubstituted arene substrates containing both electron-donating and electron-withdrawing groups furnished the desired products in good to moderate yields and selectivities.
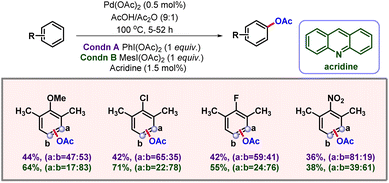 |
| | Scheme 58 Palladium catalyzed non-directed C–H acetoxylation of arenes. | |
In 2015 the same group came up with a full mechanistic investigation of palladium catalyzed non-directed C–H acetoxylation of benzene.99 Detailed kinetics analysis was performed; reaction orders with respect to the catalyst, substrate and oxidant were determined. The order with respect to all of these, combined with NMR experiments and the Hammett plot, provided a brief idea about the catalytic cycle, i.e. initial formation of a palladium acetate-pyridine dimer complex and then dissociation into its monomer. Followed by C–H activation, 2e− oxidation by PIDA takes place and subsequent reductive elimination occurs to form the desired product. Notably, attaching an electron-withdrawing group in the pyridine ligand accelerates the C–H activation step which is also the rate-determining step of this reaction. Later in 2016, the Fernández-Ibáñez group came up with a C–H acetoxylation of simple arenes using 6-fluoropicolinic acid as the ligand.100 This protocol opened a new avenue for formation of phenol from benzene with good catalytic efficacy. The same transformation can also be realized by using oxidizing agents such as K2Cr2O7. However, the TONs are comparatively low and biphenyl is observed as a potential side product. The reaction also furnished good to moderate selectivities with substituted arenes. Notably, picolinic acids containing an electron-withdrawing group at the sixth position worked better as compared to their electron-donating counterparts.
Conclusion
C–H acetoxylation has emerged as a transformative strategy in modern organic synthesis, providing a streamlined pathway for incorporating oxygen functionalities into complex molecular frameworks. By leveraging this direct functionalization method, chemists can bypass traditional multistep protocols, thereby enhancing both efficiency and atom economy in synthetic routes. Many strategies have evolved such as the use of covalently tethered directing groups, transient directed groups and non-directed C–H functionalization to boost the applicability of C–H acetoxylation. Along with these traditional methods, more sustainable approaches such as photochemical and electrochemical ones have also been explored to execute this transformation. Additionally, non-noble metals have also been exploited in the domain of C–H acetoxylation, promoting the sustainability of these transformations. Base metals have also entered this territory. However, they are still somewhat underexplored. Looking ahead, the continued exploration of new catalysts, reaction conditions, and substrate classes will undoubtedly expand the applicability of C–H acetoxylation. As this domain of C–H activation will flourish further, C–H acetoxylation is surely going to be a powerful weapon in chemists’ munitions to incorporate oxygen into molecular organic frameworks.
Data availability
This review article does not involve the generation or analysis of new data. All data referenced and discussed in this review are available from the original published studies, which are cited in the text. Therefore, a data availability statement is not applicable.
Author contributions
D. M. and J. G. proposed the topic and developed the concept of this review. J. G., B. D., D. G., P. K. S., and S. M. co-wrote the manuscript with thoughtful input from D. B. W. and D. M.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank SERB India (CRG/2022/004197) for financial support. We also thank Sunita Sanghi Centre of Aging and Neurodegenerative diseases (SCAN) [DO/2023-SSAN002-005] for the financial support. The Prime Minister Research Fellowship, Ministry of Education, India (JG) and (BD), is greatly acknowledged.
Notes and references
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed.
- W. Ali, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2024, 53, 9904–9953 RSC.
- C. J. Mulligan, S. M. Bagale, O. J. Newton, J. S. Parker and K. K. M. Hii, ACS Sustainable Chem. Eng., 2019, 7, 1611–1615 CrossRef CAS.
- R. Sheng, X. Lin, J. Li, Y. Jiang, Z. Shang and Y. Hu, Bioorg. Med. Chem. Lett., 2005, 15, 3834–3837 CrossRef CAS PubMed.
- G. Martelli, M. Cirillo, V. Giraldi and D. Giacomini, Bioorg. Chem., 2022, 120, 105580 CrossRef CAS PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- A. Goswami, P. P. Saikia, N. C. Barua, M. Bordoloi, A. Yadav, T. C. Bora, B. K. Gogoi, A. K. Saxena, N. Suri and M. Sharma, Bioorg. Med. Chem. Lett., 2010, 20, 359–361 CrossRef CAS PubMed.
- T. A. Hong Nguyen and D.-R. Hou, Org. Lett., 2021, 23, 8127–8131 CrossRef CAS.
- J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887–10909 RSC.
- S. Vittal, M. Mujahid Alam, M. Hussien, M. Amanullah, P. M. Pisal and V. Ravi, ChemistrySelect, 2023, 8, e202204240 CrossRef CAS.
- J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed.
- J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef CAS.
- F.-R. Gou, X.-C. Wang, P.-F. Huo, H.-P. Bi, Z.-H. Guan and Y.-M. Liang, Org. Lett., 2009, 11, 5726–5729 CrossRef CAS.
- G.-W. Wang, T.-T. Yuan and X.-L. Wu, J. Org. Chem., 2008, 73, 4717–4720 CrossRef CAS.
- X. Ren, J. Liu, H. Yan, X. Shi, S. Yang, J. Li and G. Huang, Synlett, 2013, 24, 1395–1398 CrossRef CAS.
- H. Zhang, R.-B. Hu, X.-Y. Zhang, S.-X. Li and S.-D. Yang, Chem. Commun., 2014, 50, 4686–4689 RSC.
- B. Liu, X. Huang, X. Wang, Z. Ge and R. Li, Org. Chem. Front., 2015, 2, 860 RSC.
- Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Nature, 2014, 515, 389–393 CrossRef CAS PubMed.
- G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS.
- C. Geng, M. Jiang, L. Feng and P. Jiao, RSC Adv., 2016, 6, 56971–56976 RSC.
- J. Zhao, Y. Huang, G. Ma, L. Lin and P. Feng, Organometallics, 2019, 38, 2084–2091 CrossRef CAS.
- J. Chen, Q. Pang, Y. Sun and X. Li, J. Org. Chem., 2011, 76, 3523–3526 CrossRef PubMed.
- S. Gu, C. Chen and W. Chen, J. Org. Chem., 2009, 74, 7203–7206 CrossRef CAS.
- G. Qian, B. Liu, Q. Tan, S. Zhang and B. Xu, Eur. J. Org Chem., 2014, 4837–4843 CrossRef CAS.
- X.-Y. Wang, Y. Li, L. Shi, X. Zhu, X.-Q. Hao and M.-P. Song, Tetrahedron, 2021, 93, 132277 CrossRef CAS.
- M. Bakthadoss, O. S. Aina, T. T. Reddy, J. U. Izunobi and O. B. Familoni, RSC Adv., 2024, 14, 13306–13310 RSC.
- Z. Mao, Y. Jiang, M. Liu and X. Zhang, Tetrahedron Lett., 2021, 84, 153435 CrossRef CAS.
- I. Urruzuno, P. Andrade-Sampedro and A. Correa, Eur. J. Org Chem., 2023, 26, e202201489 CrossRef CAS.
- K. M. Flynn, K. L. White and M. Movassaghi, J. Org. Chem., 2022, 87, 2975–2984 CrossRef CAS PubMed.
- X. Wang, H. Wang, C. Zhou, L. Yang, L. Fu and G. Li, Chem. Commun., 2022, 58, 4993–4996 RSC.
- A. Singh, A. Dey, K. Pal, O. P. Dash and C. M. R. Volla, Org. Lett., 2022, 24, 1941–1946 CrossRef CAS PubMed.
- Y. Wu and B. Zhou, Org. Lett., 2017, 19, 3532–3535 CrossRef CAS PubMed.
- R. Ueno, S. Natsui and N. Chatani, Org. Lett., 2018, 20, 1062–1065 CrossRef CAS PubMed.
- W. Sarkar, A. Bhowmik, A. Mishra, T. K. Vats and I. Deb, Adv. Synth. Catal., 2018, 360, 3228–3232 CrossRef CAS.
- Q. Gou, X. Tan, M. Zhang, M. Ran, T. Yuan, S. He, L. Zhou, T. Cao and F. Luo, Org. Lett., 2020, 22, 1966–1971 CrossRef CAS PubMed.
- T. Okada, K. Nobushige, T. Satoh and M. Miura, Org. Lett., 2016, 18, 1150–1153 CrossRef CAS.
- N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed.
- J. A. Tali and R. Shankar, Org. Lett., 2023, 25, 3200–3205 CrossRef CAS PubMed.
- O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed.
- G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed.
- L. Yang, L. Fu and G. Li, Adv. Synth. Catal., 2017, 359, 2235–2240 CrossRef CAS.
- S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed.
- R. Jayarajan, H. B. Chandrashekar, A. K. Dalvi and D. Maiti, Chem.–Eur. J., 2020, 26, 11426–11430 CrossRef CAS PubMed.
- A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147–3153 RSC.
- R. Jayarajan, J. Das, S. Bag, R. Chowdhury and D. Maiti, Angew. Chem., Int. Ed., 2018, 57, 7659–7663 CrossRef CAS PubMed.
- Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 3191–3195 CrossRef CAS PubMed.
- Z. Fan, K. L. Bay, X. Chen, Z. Zhuang, H. S. Park, K.-S. Yeung, K. N. Houk and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 4770–4777 CrossRef CAS.
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS.
- M. Li, M. Shang, H. Xu, X. Wang, H.-X. Dai and J.-Q. Yu, Org. Lett., 2019, 21, 540–544 CrossRef CAS PubMed.
- Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed.
- J. Grover, A. T. Sebastian, S. Maiti, A. C. Bissember and D. Maiti, Chem. Soc. Rev., 2025, 54, 2006–2053 RSC.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- B. D. Dangel, J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2001, 123, 8149–8150 CrossRef CAS.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS.
- R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724–3727 CrossRef CAS PubMed.
- Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991–16994 CrossRef CAS PubMed.
- Z. Wang, Y. Kuninobu and M. Kanai, Org. Lett., 2014, 16, 4790–4793 CrossRef CAS PubMed.
- P. Liu, J. Han, C. P. Chen, D. Q. Shi and Y. S. Zhao, RSC Adv., 2015, 5, 28430–28434 RSC.
- M. Wang, Y. Yang, Z. Fan, Z. Cheng, W. Zhu and A. Zhang, Chem. Commun., 2015, 51, 3219–3222 RSC.
- K. K. Pasunooti, R. Yang, B. Banerjee, T. Yap and C.-F. Liu, Org. Lett., 2016, 18, 2696–2699 CrossRef CAS.
- D. N. Garad and S. B. Mhaske, J. Org. Chem., 2017, 82, 10470–10478 CrossRef CAS.
- K. Chen, D. Wang, Z.-W. Li, Z. Liu, F. Pan, Y.-F. Zhang and Z.-J. Shi, Org. Chem. Front., 2017, 4, 2097–2101 RSC.
- B.-X. Wang, Y.-J. Mao, H.-Y. Hao, Q.-Z. Wu, K. Zhou, S.-J. Lou and D.-Q. Xu, Chem. Commun., 2019, 55, 7049–7052 RSC.
- K. K. Ghosh, A. Uttry, A. Koldemir, M. Ong and M. van Gemmeren, Org. Lett., 2019, 21, 7154–7157 CrossRef CAS PubMed.
- A. Uttry and M. Van Gemmeren, Synlett, 2018, 29, 1937–1943 CrossRef CAS.
- Y.-Q. Chen, Y. Wu, Z. Wang, J. X. Qiao and J.-Q. Yu, ACS Catal., 2020, 10, 5657–5662 CrossRef CAS PubMed.
- Y.-K. Chen, Y.-S. Kang, H.-J. Xu, P. Zhang, J. Zhao, T. Li, W.-Y. Sun and Y. Lu, Org. Lett., 2020, 22, 5390–5395 CrossRef CAS PubMed.
- Z. Zhuang, A. N. Herron, Z. Fan and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 6769–6776 CrossRef CAS PubMed.
- B. Su, A. Bunescu, Y. Qiu, S. J. Zuend, M. Ernst and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 7912–7919 CrossRef CAS.
- M. Vijaykumar and B. Punji, J. Org. Chem., 2021, 86, 8172–8181 CrossRef CAS.
- S. S. Gaikwad, S. C. Zyate, S. B. Waghmode and A. R. Jadhao, Tetrahedron, 2023, 138, 133405 CrossRef CAS.
- M. Martínez-Mingo, A. García-Viada, D. S. Prendes, I. Alonso, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2022, 61, e202209865 CrossRef.
- P.-Y. Liu, S.-C. Zhao, M.-Y. Zhang, L. Song, C. Wang, F. Yu, Q. Meng, Z. Zhang and Y.-P. He, J. Org. Chem., 2022, 87, 6378–6386 CrossRef CAS.
- D.-H. Wang, X.-S. Hao, D.-F. Wu and J.-Q. Yu, Org. Lett., 2006, 8, 3387–3390 CrossRef CAS PubMed.
- X.-Z. Shu, X.-F. Xia, Y.-F. Yang, K.-G. Ji, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2009, 74, 7464–7469 CrossRef CAS.
- H. Jiang, H. Chen, A. Wang and X. Liu, Chem. Commun., 2010, 46, 7259–7261 RSC.
- J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009–1016 CrossRef CAS.
- C. S. Buettner, D. Willcox, B. G. N. Chappell and M. J. Gaunt, Chem. Sci., 2018, 10, 83–89 RSC.
- Y.-Q. Li, Q.-L. Yang, P. Fang, T.-S. Mei and D. Zhang, Org. Lett., 2017, 19, 2905–2908 CrossRef CAS.
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed.
- A. Shrestha, M. Lee, A. L. Dunn and M. S. Sanford, Org. Lett., 2018, 20, 204–207 CrossRef CAS PubMed.
- D. Ogawa, A. Sasaki, T. Kochi and F. Kakiuchi, Org. Biomol. Chem., 2024, 22, 7696–7701 RSC.
- W. Ali, A. Saha, H. Ge and D. Maiti, JACS Au, 2023, 3, 1790–1799 CrossRef CAS.
- Y. Fujiwara, I. Moritani, S. Danno, R. Asano and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166–7169 CrossRef CAS PubMed.
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- P. Wedi, M. Farizyan, K. Bergander, C. Mück-Lichtenfeld and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 15641–15649 CrossRef CAS PubMed.
- S. K. Sinha, S. Panja, J. Grover, P. S. Hazra, S. Pandit, Y. Bairagi, X. Zhang and D. Maiti, J. Am. Chem. Soc., 2022, 144, 12032–12042 CrossRef CAS PubMed.
- T. Yoneyama and R. H. Crabtree, J. Mol. Catal. A: Chem., 1996, 108, 35–40 CrossRef CAS.
- I. Mutule, E. Suna, K. Olofsson and B. Pelcman, J. Org. Chem., 2009, 74, 7195–7198 CrossRef CAS PubMed.
- Q. Liu, G. Li, H. Yi, P. Wu, J. Liu and A. Lei, Chem.–Eur. J., 2011, 17, 2353–2357 CrossRef CAS PubMed.
- P. Y. Choy, C. P. Lau and F. Y. Kwong, J. Org. Chem., 2011, 76, 80–84 CrossRef CAS PubMed.
- M. H. Emmert, A. K. Cook, Y. J. Xie and M. S. Sanford, Angew. Chem., Int. Ed., 2011, 50, 9409–9412 CrossRef CAS.
- A. K. Cook, M. H. Emmert and M. S. Sanford, Org. Lett., 2013, 15, 5428–5431 CrossRef CAS.
- A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109–3118 CrossRef CAS PubMed.
- C. Valderas, K. Naksomboon and M. Á. Fernández-Ibáñez, ChemCatChem, 2016, 8, 3213–3217 CrossRef CAS.
Footnote |
| † Dedicated to Prof. Dr Reinhard Brückner on the occasion of his 70th birthday |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Siddhartha
Maiti
c,
Daniel B.
Werz
b,
Siddhartha
Maiti
c,
Daniel B.
Werz