
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced polyelectrolyte complexation for the formation of stable films with reversible crosslinking†
Krisada
Auepattana-Aumrung
a,
Lauren M.
Bishop
b,
Kaden C.
Stevens
b,
Kevin A.
Stewart
b,
Daniel
Crespy
 *a and
Brent S.
Sumerlin
*b
*a and
Brent S.
Sumerlin
*b
aDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand. E-mail: daniel.crespy@vistec.ac.th
bGeorge & Josephine Butler Polymer Research Laboratory, Center for Macromolecular Science & Engineering, Department of Chemistry, University of Florida, Gainesville, Florida 32611, USA. E-mail: sumerlin@chem.ufl.edu
First published on 7th March 2025
Abstract
Thin films formed by complexation of oppositely charged polyelectrolytes have significant potential in applications such as separation membranes, biocompatible or anticorrosion coatings, and drug delivery systems. While layer-by-layer deposition is a reliable method for producing conformal films, this multi-step process limits scalability. In this study, we functionalize polymers with photoactive protecting and crosslinking groups, allowing a one-step approach for preparing polyelectrolyte complex (PEC) films. To achieve this goal, we introduced o-nitrobenzyl and coumarin groups into a polyanion. The o-nitrobenzyl protecting groups can be selectively deprotected upon exposure to 365 nm light, revealing charged pendent groups that initiate polyelectrolyte complexation. Simultaneously, the coumarin units in the copolymers undergo dimerization, enhancing the solvochemical stability of the PEC films. Notably, short-wave UV irradiation (254 nm) enables retrocyclization of the coumarin dimers, returning the PEC film to its uncrosslinked state. These UV-driven deprotection, crosslinking, and de-crosslinking processes provide a versatile and tunable platform for fabricating reversibly crosslinked films. By integrating photoresponsive polymers and reversible covalent linkages, this approach offers new opportunities for designing PEC materials with tunable dynamic properties for advanced applications.
Introduction
Polyelectrolytes are polymers containing ionizable groups that can bear positive or negative charges, classifying them as polycations or polyanions, respectively.1 When solutions of oppositely charged polyelectrolytes are combined, polyelectrolyte complexes (PECs) spontaneously emerge as either viscous liquids or solid films, depending on environmental conditions and polymer composition.2–6 The primary driving forces behind this self-assembly are the entropically favorable counterion release and enthalpic contributions from specific ion pairing between ionic sites on the polymer backbone or side chains.7–10 The functionality of PECs has generated significant interest in the development of PECs as membranes,11–13 coatings/films,14–16 hydrogels,17–19 electrospun fibers,20–22 and encapsulation technologies.23–25More recently, PEC thin films have emerged as an important class of materials with potential applications across engineering and technology. Layer-by-layer (LbL) assembly, which generates films by depositing alternating layers of polyanions and polycations onto a substrate, is widely used for preparing PEC films due to precise control over film thickness and material composition.26,27 However, the LbL approach requires a separate deposition for each layer, making it time-consuming and challenging to scale for industrial applications. A possibility of accelerating the formation of materials from polyelectrolyte complexes is to process them in the presence of large amounts of salts (“saloplastics”), which need to be removed after shaping the material.28 The formation of PEC coacervates via liquid–liquid phase separation in solutions of oppositely charged polymers offers a straightforward and more efficient method of preparing polyelectrolyte coatings. However, processing PECs into well-defined thin films or coatings remains a significant challenge. In 2021, Li et al. demonstrated a single-step method for the fabrication of PEC films, utilizing a volatile base as a trigger for complexation (Fig. 1A).29 Ammonia was used to tune the pH of poly(acrylic acid) (PAA), thereby preventing premature complexation when mixed with poly(ethylene imine) (PEI). This approach allows the formation of a homogeneous solution, with complexation between the two oppositely charged polyelectrolytes only occurring upon the evaporation of ammonia. However, this method required the addition of a volatile base (ammonia), which could affect the overall film quality. We propose a novel approach to address these limitations using stimuli-responsive polymers containing photolabile groups. These photolabile groups deprotect upon UV irradiation, revealing ionizable groups that facilitate the spontaneous complexation of oppositely charged polyelectrolytes.
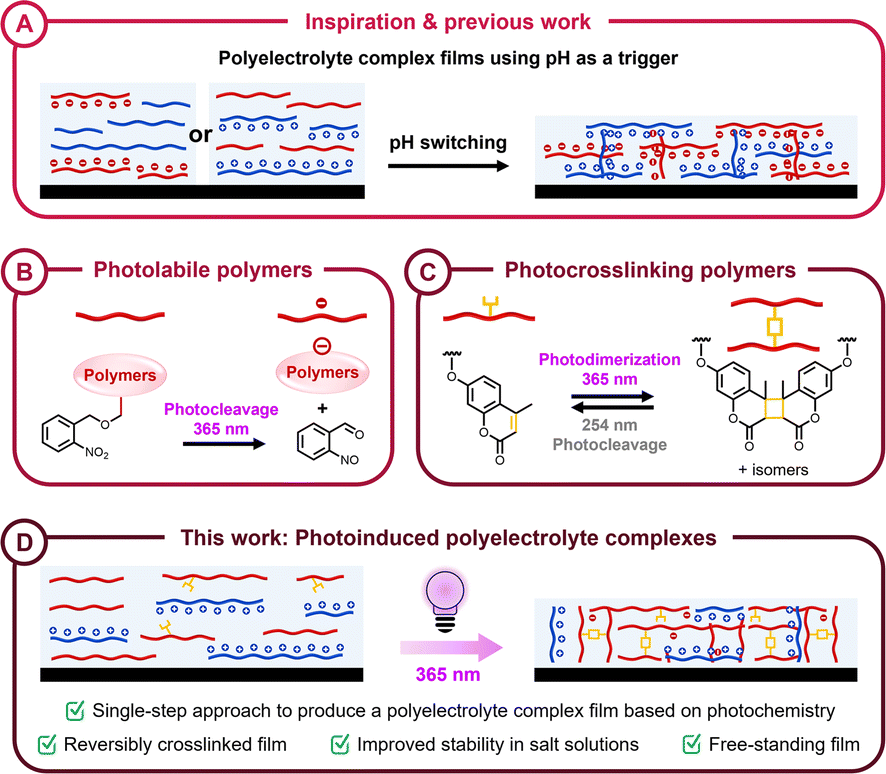 | ||
| Fig. 1 (A) Previous work demonstrating the preparation of polyelectrolyte complex films using pH as a trigger. Focus on concepts of (B) photolabile polymers containing o-nitrobenzyl groups and (C) photocrosslinking polymers containing coumarin groups. (D) This work, wherein polyelectrolyte complexes with reversible crosslinking is achieved using light as a trigger. | ||
Photolabile chemistries have become a powerful tool in polymer material design, introducing functional linkers or pendent groups that provide control over key material properties such as surface modifications, structural integrity, matrix degradation, and mechanical strength.30,31 The use of protecting groups in material synthesis involves temporarily masking a chemical functionality to enable on-demand chemical transformations, followed by selective deprotection to reveal the original functionality.32,33 Photochemical deprotection methods are orthogonal to most chemical functionalities, providing precise spatiotemporal control and selectively targeting photocleavable groups without interfering with chemically sensitive backbones or pendant groups.34,35 Among the many photolabile groups that have been studied, o-nitrobenzyl alcohol derivatives have particularly gained attention due to their high photocleavage efficiency under near-UV irradiation.36–38 Upon exposure to UV light, these groups undergo a photoisomerization process, converting into o-nitrosobenzaldehyde and simultaneously releasing a free carboxylic acid (Fig. 1B).33,39,40 This light-driven deprotection and subsequent revealing of an ionizable functional group provide a mild and straightforward stimulus to initiate complexation between oppositely charged polyelectrolytes.
The mechanical strength, stability, and tunability of polyelectrolyte complex (PEC) materials are critical for their practical application in various fields.19,41 However, achieving both robust physical properties and dynamic adaptability remains a challenge. Traditional PEC materials require exposure to aqueous solutions of variable salinity or pH to adjust or reverse their structures, which limits their utility in aqueous environments where the pH and salinity of the complex are outside a controlled setting.42 Such longstanding issues can, in principle, be overcome by introducing reversible/dynamic linkages within PECs, thereby enabling enhanced stability under harsh environments and a functional tool for fine-tuning the overall material properties.
Reversible covalent linkages have emerged as a powerful tool in the design of dynamic and adaptable materials, offering the stability of covalent bonds combined with the processability or reversibility typically associated with noncovalent interactions.43,44 These dynamic linkages allow for controlled assembly and disassembly of polymer networks, enabling the modulation of physical and chemical properties post-synthesis. A variety of reversible covalent chemistries have been explored for material fabrication, including photoresponsive cycloadducts,45–48 Diels–Alder adducts,49–53 disulfides,54–56 oximes,57,58 boronic esters,59,60 and Schiff bases,61–63 which impart self-healing and reprocessability to the resulting materials. Photoresponsive systems are particularly attractive due to the inherent spatiotemporal control and mild conditions associated with photochemical dynamicity. By carefully selecting the wavelength and intensity of light, specific covalent bonds can be formed or cleaved, allowing for precise tuning of material properties.64,65 This approach has been applied to the development of photoinduced polymer networks, where functional groups such as coumarins,66–69 anthracene derivatives,70–72 and pyrimidine or thymine bases73–75 undergo reversible dimerization and cleavage reactions under UV irradiation. Coumarin and its derivatives have been extensively studied due to their efficient dimerization and cleavage under UV irradiation, which occur without the need for photosensitizers or catalysts.76,77 Upon exposure to long-wave UV light (365 nm), coumarin undergoes a [2 + 2] cycloaddition, forming cyclobutane-linked dimers, while short-wave UV light (254 nm) induces a cycloreversion reaction, restoring their monomeric state (Fig. 1C).78–80 This photoreversible crosslinking behavior provides a versatile platform for controlling material properties, making coumarin-modified polymers highly suitable for the design of stimuli-responsive and reconfigurable materials.
Herein, we report a novel one-step method for fabricating stable polyelectrolyte complex films (Fig. 1D). This is achieved by introducing photolabile protecting groups to inhibit premature complexation between oppositely charged polyelectrolytes, thereby preventing undesired precipitation prior to film formation. Following film casting, long-wave UV irradiation (365 nm) induces the deprotection of ionizable groups, facilitating complexation between the polyelectrolytes. Simultaneously, coumarin units within the copolymers undergo photodimerization, enhancing the stability and overall functionality of the PEC films. Finally, we demonstrate how the crosslinked PEC films can be reverted to uncrosslinked PEC film upon short-wave UV irradiation (254 nm) due to the dynamicity of the coumarin linkages. This photochemical approach of on-demand, concomitant complexation and dynamic covalent crosslinking offers a versatile and accessible method for fabricating PEC films.
Results and discussion
The synthesis of the photoresponsive functional monomers o-nitrobenzyl acrylate (NBA) and 7-(2-acryloyloxyethoxy)-4-methylcoumarin (CoumAc) were adapted from previous literature procedures (Fig. 2A).81–84 NBA was synthesized by the reaction of acryloyl chloride with o-nitrobenzyl alcohol in the presence of triethylamine. A polymerizable acrylate derivative of coumarin was synthesized via a reaction between 4-methylumbelliferone and 2-chloroethanol, followed by a reaction of acryloyl chloride in the presence of triethylamine to yield CoumAc. Both monomers were thoroughly purified via vacuum distillation and silica gel column chromatography. The chemical structures of the intermediates and monomers were confirmed by 1H NMR spectroscopy (Fig. S1–S3†). | ||
| Fig. 2 Reaction schemes for (A) the preparation of the monomer o-nitrobenzyl acrylate (NBA) and 7-(2-acryloyloxyethoxy)-4-methylcoumarin (CoumAc) and (B) the conventional radical polymerization of CoumAc, N-hydroxyethyl acrylamide (HEAA), and NBA to obtain photolabile polymers P(CoumAcx-co-HEAAy-co-NBAz). | ||
The photoactive copolymers were designed using photolabile o-nitrobenzyl and photoresponsive coumarin monomers (NBA and CoumAc, respectively), along with a comonomer, N-hydroxyethyl acrylamide (HEAA) [P(CoumAcx-co-HEAAy-co-NBAz)]. HEAA was selected as a comonomer for polyelectrolyte complex (PEC) films due to its hydrophilic and non-ionic nature. P(CoumAcx-co-HEAAy-co-NBAz) copolymers were synthesized via free radical polymerization at 70 °C in dimethyl sulfoxide (DMSO), employing azobis(isobutyronitrile) (AIBN) as the initiator (Fig. 2B). The photolabile anionic copolymers were synthesized with varying molar ratios of CoumAc to investigate the influence of coumarin crosslink density on the formation of polyelectrolyte complexes (PECs) (Fig. S4–S6†). The molar ratio of CoumAc was determined by integrating the signals associated with the aromatic protons of CoumAc relative to the –CH2– protons of NBA via1H NMR spectroscopy. For all synthesized polymers, NMR analysis confirmed that the incorporation of coumarin units in the purified polymers closely matched the initial monomer feed ratio (Table S1†). The apparent number-average molecular weight (Mn) ranged from 30 to 33 kg mol−1.
The photoreactivity of the P(CoumAcx-co-HEAAy-co-NBAz) copolymers was first examined in solution. Under UV irradiation at 365 nm, photocleavage of the o-nitrobenzyl units and photodimerization of the coumarin units occurred simultaneously (Fig. 3A). The release of the o-nitrobenzyl group was investigated by 1H NMR spectroscopy in solution at different irradiation time intervals. Both P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) and P(HEAA0.90-co-NBA0.10) copolymers were dissolved in DMSO-d6 and irradiated with UV light at 365 nm. For the o-nitrobenzyl deprotection study, DMSO-d6 was selected as solvent for minimizing issues associated with proton exchange. Because the deprotection reaction generates carboxylic acid groups, using water could lead to pH-dependent chemical shift variations and broadened signals due to exchange of labile protons, hence complicating quantitative analysis. A representative temporal evolution of 1H NMR spectra of the copolymers is shown in Fig. 3B and S7.† The cleavage of o-nitrobenzyl groups from the copolymers was monitored by comparing the signals of the –CH2– protons from the o-nitrobenzyl group (HA) with the proton of the newly formed –CHO group (HB).85 The signal for the –CH2– protons of the o-nitrobenzyl group at 5.4 ppm gradually decreased with increasing irradiation time, while a new peak corresponding to the –CHO group at 12.1 ppm increased proportionally. After 5 h of UV irradiation, the cumulative release of o-nitrobenzyl groups from P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) copolymer reached approximately 93% (Fig. 3C). Similarly, the cumulative release from the copolymer without coumarin, P(HEAA0.90-co-NBA0.10), was approximately 92% (Fig. S8†), indicating that the incorporation of coumarin unit into the copolymers did not adversely affect the photoreactivity of the o-nitrobenzyl group.
 | ||
| Fig. 3 (A) Reaction scheme for the photoreactions of the copolymers after irradiating with UV light at 365 nm. (B) Temporal evolution of 1H NMR spectra and (C) release profiles of the o-nitrobenzyl group from P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) in DMSO-d6. UV-vis study of the reversible crosslinking reaction of P(CoumAc0.03-co-HEAA0.87-co-AA0.10) in water. (D) UV-vis spectra of photodimerization and (E) percent coumarin dimerization as a function of irradiation time at 365 nm. (F) UV-vis spectra of photocleavage and (G) percent coumarin cleavage as a function of irradiation time at 254 nm. | ||
To investigate the reversible photodimerization of the coumarin groups, a copolymer without NBA, P(CoumAc0.03-co-HEAA0.87-co-AA0.10), was prepared (Fig. S9†) as the released o-nitrobenzyl group exhibited strong absorption in the same wavelength region as the coumarin compound.86 The solution dimerization and cleavage behavior of the copolymers were monitored using UV-vis spectroscopy. The characteristic absorbance of coumarin at 320 nm corresponds to the vinyl group of the α,β-unsaturated ester.84 A dilute aqueous solution of P(CoumAc0.03-co-HEAA0.87-co-AA0.10) (0.1 wt%) was irradiated with 365 nm UV light, and the absorbance at 320 nm was recorded as a function of irradiation time. For the isomerization reactions of coumarin derivatives, water was used to match the conditions under which the polyelectrolyte complexes were formed and studied. While solvent polarity and hydrogen bonding can influence photochemical reactions, coumarin dimerization and cycloreversion reactions are primarily governed by intramolecular interactions, making them relatively insensitive to solvent quality in this case. However, in highly hydrogen-bonding solvents like water, subtle differences in reaction kinetics may occur due to changes in coumarin solubility and local microenvironment effects. The local polymer environment, particularly in polyelectrolyte complexes, can influence reaction efficiency by altering the spatial arrangement and accessibility of reactive groups, as demonstrated in studies on single-chain nanoparticle formation via solvent-induced polymer collapse. Notably, previous studies have shown that methanol enhances coumarin dimerization due to its polarity, which increases the quantum yield of the reaction.87 This suggests that solvent-mediated compaction or solvophobic interactions, as seen in polymer systems, may play a role in tuning reaction kinetics, particularly in aqueous or hydrogen-bonding environments.
As expected, the absorbance gradually decreased as the coumarin units dimerized (Fig. 3D). Approximately 84% of coumarin moieties formed dimers after 4 h of irradiation (Fig. 3E). Given that coumarin dimers can revert to their monomeric form under 254 nm UV light, we hypothesized that the original polymer could be regenerated. Upon irradiation with 254 nm UV light, the absorbance at 320 nm recovered (Fig. 3F), indicating that approximately 79% of the coumarin dimers were cleaved back into their constituent monomers within 4 min (Fig. 3G). However, complete cycloreversion was not achieved, which we attribute to an equilibrium between the forward and reverse reactions under short-wave UV exposure.84,88–90 Therefore, under storage conditions (e.g., in the dark), we believe a significant fraction of the coumarin moieties remains in the dimerized (crosslinked) state. The persistence of these coumarin crosslinks enhances the overall stability of the polymer network, as evidenced by the improved durability of the free-standing polyelectrolyte complex films in both deionized water and 1 M NaCl aqueous solutions. Although our study did not include long-term shelf-life testing under various storage conditions, the observed equilibrium suggests that the polymer is likely to retain its crosslinked structure over prolonged periods in the absence of short-wave UV light. Notably, when comparing the dimerization under 365 nm irradiation to the cleavage of the o-nitrobenzyl groups, the rates of these processes were found to be comparable (Fig. 3B–G).
We hypothesize that at high polymer concentrations, increased intermolecular interactions and solution viscosity may lead to slower reaction rates, as diffusion-controlled processes such as photoisomerization and proton transfer could be hindered. Additionally, self-screening effects may reduce the efficiency of UV-light absorption in highly concentrated solutions, potentially affecting photoconversion uniformity. For the o-nitrobenzyl deprotection, the carboxylic acid ratios should remain consistent under both dilute and concentrated conditions, provided that sufficient UV exposure is provided to complete the reaction. However, aggregation or altered local microenvironments in concentrated samples may slightly reduce the efficiency of the reaction. Coumarin dimerization may increase at higher concentrations due to enhanced monomer proximity, but photodimerization and cycloreversion efficiency could vary with light penetration depth, leading to potential variability in crosslink density in bulk samples compared to predicted efficiency in dilute solutions.
PECs were prepared by mixing solutions of our photoresponsive anionic copolymer with the established polycation (Fig. 4A). Specifically, to initiate the complexation process, stock solutions of P(CoumAcx-co-HEAAy-co-NBAz) and the polycation poly(N,N-diallyldimethylammonium chloride) (PDADMAC) were prepared in deionized water and transferred to a 1.5 mL Eppendorf tube (Table S2†), followed by immediate vortexing to ensure homogeneous mixing. The mixed solutions were irradiated with UV light at 365 nm for 4 h, inducing the release of the o-nitrobenzyl group from the P(CoumAcx-co-HEAAy-co-NBAz) copolymer, which generated acrylic acid units. At a pH above the pKa of the poly(acrylic acid) (pKa = ∼4.5),91,92 the carboxyl groups were deprotonated and revealed negatively charged residues on the copolymers, leading to the formation of complex coacervates with PDADMAC (Fig. 4B).
 | ||
| Fig. 4 (A) Schematic representation of the preparation of polyelectrolyte complexes. (B) Optical micrographs and (C) photographs of polyelectrolyte complexes of P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) or P(HEAA0.90-co-NBA0.10) with PDADMAC under different polyelectrolyte concentrations. | ||
To investigate the effects of polyelectrolyte (PE) concentration on the PEC formation, PECs with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 matching ratio between polyanion and polycation repeating units with total PE concentrations during complexation of 1, 5, and 10 wt% were prepared (Table S2†). At 1 and 5 wt% total PE concentrations, liquid-like coacervates were formed, indicating successful complexation (Fig. 4C). However, complexation was inhibited when total polymer concentrations exceeded 5 wt% due to self-suppression effects that hindered PEC formation.93,94 These effects result from enhanced polymer chain interactions and steric hindrance at higher concentrations, which reduce the availability of charged groups for effective complexation. The effect of charge ratios on the PEC formation was further explored by maintaining a total PE concentration at 5 wt% while altering the charge ratios between P(CoumAcx-co-HEAAy-co-NBAz) and PDADMAC to 0.5:1, 1:1, and 1.5:1 (Table S3†). The successful formation of PECs at a stoichiometric 1:1 charge ratio was evidenced by the observation of liquid-like coacervates, which indicate efficient complexation between the polyelectrolytes, as shown in Fig. S10.† PECs were formed most effectively at a 1:1 charge ratio, where entropic gain from complexation between polycations and polyanions is maximized, resulting in stable complexes. Deviations from this ratio disrupt charge neutralization, leading to reduced stability and incomplete complexation.95–97 Similar results were obtained using the photolabile anionic copolymer without coumarin units, P(HEAA0.90-co-NBA0.10), confirming the presence of coumarin units did not hinder PEC formation (Fig. S10†).
:1 matching ratio between polyanion and polycation repeating units with total PE concentrations during complexation of 1, 5, and 10 wt% were prepared (Table S2†). At 1 and 5 wt% total PE concentrations, liquid-like coacervates were formed, indicating successful complexation (Fig. 4C). However, complexation was inhibited when total polymer concentrations exceeded 5 wt% due to self-suppression effects that hindered PEC formation.93,94 These effects result from enhanced polymer chain interactions and steric hindrance at higher concentrations, which reduce the availability of charged groups for effective complexation. The effect of charge ratios on the PEC formation was further explored by maintaining a total PE concentration at 5 wt% while altering the charge ratios between P(CoumAcx-co-HEAAy-co-NBAz) and PDADMAC to 0.5:1, 1:1, and 1.5:1 (Table S3†). The successful formation of PECs at a stoichiometric 1:1 charge ratio was evidenced by the observation of liquid-like coacervates, which indicate efficient complexation between the polyelectrolytes, as shown in Fig. S10.† PECs were formed most effectively at a 1:1 charge ratio, where entropic gain from complexation between polycations and polyanions is maximized, resulting in stable complexes. Deviations from this ratio disrupt charge neutralization, leading to reduced stability and incomplete complexation.95–97 Similar results were obtained using the photolabile anionic copolymer without coumarin units, P(HEAA0.90-co-NBA0.10), confirming the presence of coumarin units did not hinder PEC formation (Fig. S10†).
With the optimal PE concentrations and charge ratios identified for stable PEC formation, we next sought to translate these conditions into the preparation of PEC coatings. By leveraging the charge-matched conditions, PE coatings were fabricated by drop-casting, and their structural and surface properties under UV irradiation were examined. A mixture containing photolabile anionic polymer, cationic polymer, and deionized water was drop-cast onto dried glass substrates. The total PE concentration was kept at 5 wt%. The coated substrates were irradiated with UV light at 365 nm for 4 h and dried at 25 °C for 24 h (Fig. 5A). The thickness measured by profilometry of irradiated PE coatings containing 0, 1, and 3 mol% CoumAc were 5.88 ± 0.12, 5.72 ± 0.06, and 6.05 ± 0.11 µm, respectively, whereas the thickness of the PE coatings without UV irradiation was slightly higher (Fig. S11†). UV-induced complexation significantly altered the surface properties, as indicated by the contact angle measurements. Indeed, the static water contact angle of P(CoumAc0.03-co-HEAA0.87-co-NBA0.10)/PDADMAC coatings increased from 72 ± 2° to 105 ± 2° after UV exposure (Fig. 5B). The increase in contact angle was attributed to UV-induced complexation between oppositely charged polyelectrolytes, which leads to the formation of a more compact structure that reduces the exposure of hydrophilic groups. Additionally, UV-induced crosslinking further rigidifies the network, restricting the mobility of the hydrophilic groups and reducing their availability for water interaction. These effects collectively result in a significant decrease in surface hydrophilicity, as evidenced by the increase in contact angle.
 | ||
| Fig. 5 (A) Schematic for the preparation of polyelectrolyte complex films. (B) Contact angle of P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) coatings without and with UV irradiation at 365 nm. Weight loss analysis of P(CoumAcx-co-HEAAy-co-NBAz) coatings (C) without and (D) with UV irradiation at 365 nm after immersing in deionized water. (E) Photocleavage reaction of P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) under UV irradiation at 254 nm. (F) Stability of P(CoumAc0.03-co-HEAA0.87-co-NBA0.10) films in a 1 M NaCl aqueous solution following UV irradiation at 254 nm. | ||
The stability of the PE coatings in water was evaluated gravimetrically by monitoring weight loss over immersion time. PE coatings containing 0, 1, and 3 mol% CoumAc were prepared on glass substrates with and without UV irradiation. The coatings were first immersed in deionized water, and the weight change was measured as a function of immersion time. Non-irradiated coatings were completely removed from the substrates within 5 min (Fig. 5C and S12A†), while UV-irradiated coatings remained on the substrates for up to 5 d (Fig. 5D and S12B†). Notably, incomplete complexation during film formation results in the presence of a fraction of water-soluble polymer chains or low-molecular-weight species that are not fully incorporated into the crosslinked network. Moreover, this initial mass loss is due to leaching of o-nitrosobenzaldehyde following photodeprotection. Upon immersion, these components rapidly diffuse out, leading to an initial decrease in mass. Afterward, the remaining crosslinked network reaches equilibrium, inducing a stabilization of the mass over time. Moreover, the remaining weight of the UV-irradiated coatings increased proportionally with CoumAc content. Indeed, the remaining weight of the PECs coatings containing 0, 1, and 3 mol% CoumAc were ∼4, 21, and 60% of the original weight, respectively. PECs exhibit higher water stability than individual PEs due to strong electrostatic interactions between oppositely charged polymers, which neutralize charges and reduce water solubility.98
To further confirm the role of coumarin crosslinking on the improved stability, free-standing PEC films were immersed in a 1 M NaCl aqueous solution (Fig. S13A†). The PECs films without coumarin rapidly broke into pieces and sank to the bottom of the vial after 3 min, whereas the PECs films containing 3 mol% CoumAc remained structurally intact and fully sank to the bottom of the vial after 30 d (Fig. S13B†). This result demonstrates the significance of the role coumarin-based, covalent crosslinking has in reinforcing PEC film stability. Although our results demonstrate that coumarin crosslinking prevents the films from dissolving, we acknowledge that high ionic strength could potentially weaken noncovalent electrostatic interactions that contribute to complex formation. However, we believe this underscores the utility of our orthogonal methodology in that while the polyelectrolyte complexes are initially formed through electrostatic interactions, the subsequent covalent crosslinking via coumarin dimerization locks in the network structure. This covalent reinforcement is expected to preserve the integrity of the complexes, even if some degree of ionic screening occurs in high-salt conditions. Moreover, our extended stability tests show that free-standing PEC films containing 3 mol% CoumAc remain intact for up to 30 d in a 1 M NaCl solution (Fig. S13B†), suggesting that the internal complexes do not undergo significant destabilization over time.
The reversible photocleavage of coumarin dimers under short-wave UV light (254 nm) is demonstrated in Fig. 5E. Upon exposure to 254 nm UV, cycloreversion reactions occurred, wherein crosslinked coumarin dimers were partially converted back to their de-crosslinked form.99 This photoreversible process enables the transition of films between a chemically crosslinked state, induced by long-wave UV irradiation (365 nm), and a de-crosslinked, soluble state under short-wave UV exposure (254 nm). To confirm this process also occurs in PECs, free-standing films of P(CoumAc0.03-co-HEAA0.87-co-NBA0.10)/PDADMAC, which had been previously crosslinked under long-wave UV light, were immersed in a 1 M NaCl aqueous solution and then irradiated with a 254 nm UV light (Fig. 5F). Within 15 min of irradiation, the films began to dissolve and sink to the bottom of vials, confirming that the coumarin crosslinks had been sufficiently cleaved. After an additional 90 min of short-wave UV exposure and overnight immersion in the salt solution, the film fully disintegrated, providing strong evidence of coumarin dimer reversion leading to soluble, uncrosslinked copolymers. Compared to the films without UV irradiation at 254 nm, the films remained stable throughout the experiment. Additionally, differential scanning calorimetry (DSC) of the PECs films after irradiating with short-wave UV light revealed a decrease in the glass transition temperature (Tg) compared to the original PECs films (Fig. S14†), further confirming the de-crosslinking reaction of coumarin units. Indeed, our copolymer, composed of o-nitrobenzyl acrylate, coumarin acrylate, and 2-hydroxyethyl acrylamide, exhibits a Tg of 53 °C when not exposed to UV light. This relatively high Tg is attributable to the rigid, aromatic nature of the o-nitrobenzyl acrylate segments and the acrylamide comonomer units. Upon irradiation with 365 nm UV light, photoinitiated coumarin dimerization increases the crosslink density, resulting in an increase of Tg to 57 °C due to enhanced network rigidity and restricted chain mobility. However, subsequent exposure to 254 nm UV light leads to a significant reduction in Tg to 38 °C. We believe indeed that o-nitrosobenzaldehyde acts as a plasticizer during the photolysis of o-nitrobenzyl groups, reducing intermolecular interactions and increasing the flexibility of the polymer network. Moreover, under 254 nm irradiation, the cleavage of coumarin dimers should further increase chain mobility. These effects collectively lead to the observed decrease in Tg for the film exposed to both 365 and 254 nm UV light.
The ability to reversibly modulate crosslinking in PEC films through light exposure provides a functional platform for designing materials with tunable mechanical and solubility properties. Such materials could be of interest for applications in smart coatings, self-healing materials, and responsive hydrogels, where environmental stimuli, such as light, can be used to trigger structural changes and control material performance. Additionally, precise control over crosslinking and de-crosslinking offers potential for recyclable or reprocessable materials, which is highly advantageous in reducing waste and improving the sustainability of polymer-based systems. These findings underscore the importance of coumarin-based photo-crosslinking mechanisms in developing advanced PEC films with reversible, light-responsive properties.
Conclusions
We have developed a photomediated approach for fabricating stable polyelectrolyte complex (PEC) coatings with reversible crosslinking. By copolymerizing vinyl monomers containing o-nitrobenzyl and coumarin units with a water-soluble comonomer, we synthesized copolymers capable of forming stable PECs with PDADMAC upon irradiation with long-wave UV light (365 nm). Crosslinking within the PECs was achieved via the dimerization of coumarin units, significantly enhancing the coatings' stability in both water and salt solutions. Importantly, the coumarin-based cyclobutane dimers enabled reversible crosslinking, as exposure to short-wave UV light (254 nm) restored the PECs to their soluble state, offering a viable pathway to both a robust and recyclable material system. This conceptual design can be further adapted to impart tailored functionalities by modifying the photolabile groups, stimuli-responsive triggers, or comonomer units, allowing for a broad range of practical applications. Moreover, this approach opens the possibility of incorporating active payloads within the films, which could be released upon deprotection of the photolabile groups, further broadening the potential utility of these materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. A.: conceptualization, methodology, visualization, formal analysis, investigation and writing – original draft. L. M. B.: formal analysis, validation and writing – review & editing. K. C. S.: conceptualization, methodology, validation and writing – review & editing. K. A. S.: validation and writing – review & editing. D. C.: conceptualization, funding acquisition, validation, supervision and writing – review & editing. B. S. S.: conceptualization, funding acquisition, validation, supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding support from the National Research Council of Thailand (NRCT, project code 4815847). This material is based on work supported by the National Science Foundation (DMR-2404144) and DoD through the ARO (W911NF2410050 and W911NF2310260).Notes and references
- A. V. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS.
- J. v. d. Gucht, E. Spruijt, M. Lemmers and M. A. Cohen Stuart, J. Colloid Interface Sci., 2011, 361, 407–422 CrossRef PubMed.
- A. Veis, Adv. Colloid Interface Sci., 2011, 167, 2–11 CrossRef CAS PubMed.
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS.
- A. E. Neitzel, Y. N. Fang, B. Yu, A. M. Rumyantsev, J. J. de Pablo and M. V. Tirrell, Macromolecules, 2021, 54, 6878–6890 CrossRef CAS PubMed.
- K. O. Margossian, M. U. Brown, T. Emrick and M. Muthukumar, Nat. Commun., 2022, 13, 2250 CrossRef CAS PubMed.
- Z. Ou and M. Muthukumar, J. Chem. Phys., 2006, 124, 154902 CrossRef PubMed.
- J. Fu and J. B. Schlenoff, J. Am. Chem. Soc., 2016, 138, 980–990 CrossRef CAS PubMed.
- S. Chen and Z.-G. Wang, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2209975119 CrossRef CAS PubMed.
- K. C. Stevens and M. V. Tirrell, ACS Macro Lett., 2024, 13, 688–694 CrossRef CAS PubMed.
- Q. Zhao, Q. F. An, Y. Ji, J. Qian and C. Gao, J. Membr. Sci., 2011, 379, 19–45 CrossRef CAS.
- X.-S. Wang, Y.-L. Ji, P.-Y. Zheng, Q.-F. An, Q. Zhao, K.-R. Lee, J.-W. Qian and C.-J. Gao, J. Mater. Chem. A, 2015, 3, 7296–7303 RSC.
- K. Sadman, D. E. Delgado, Y. Won, Q. Wang, K. A. Gray and K. R. Shull, ACS Appl. Mater. Interfaces, 2019, 11, 16018–16026 CrossRef CAS PubMed.
- K. D. Kelly and J. B. Schlenoff, ACS Appl. Mater. Interfaces, 2015, 7, 13980–13986 CrossRef CAS PubMed.
- J. D. Delgado and J. B. Schlenoff, Macromolecules, 2019, 52, 7812–7820 CrossRef CAS.
- J. Akintola, Y. Chen, Z. A. Digby and J. B. Schlenoff, ACS Appl. Mater. Interfaces, 2023, 15, 50058–50068 CrossRef CAS PubMed.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- S. Srivastava, A. E. Levi, D. J. Goldfeld and M. V. Tirrell, Macromolecules, 2020, 53, 5763–5774 CrossRef CAS.
- D. Li, T. Göckler, U. Schepers and S. Srivastava, Macromolecules, 2022, 55, 4481–4491 CrossRef CAS.
- X. Meng, S. L. Perry and J. D. Schiffman, ACS Macro Lett., 2017, 6, 505–511 CrossRef CAS PubMed.
- X. Meng, J. D. Schiffman and S. L. Perry, Macromolecules, 2018, 51, 8821–8832 CrossRef CAS.
- A. Baer, N. Horbelt, M. Nijemeisland, S. J. Garcia, P. Fratzl, S. Schmidt, G. Mayer and M. J. Harrington, ACS Nano, 2019, 13, 4992–5001 CrossRef CAS PubMed.
- M. A. J. Mazumder, F. Shen, N. A. D. Burke, M. A. Potter and H. D. H. Stöver, Biomacromolecules, 2008, 9, 2292–2300 CrossRef CAS PubMed.
- K. A. Black, D. Priftis, S. L. Perry, J. Yip, W. Y. Byun and M. Tirrell, ACS Macro Lett., 2014, 3, 1088–1091 CrossRef CAS PubMed.
- M. Zhao and N. S. Zacharia, J. Chem. Phys., 2018, 149, 163326 CrossRef PubMed.
- J. J. Richardson, M. Björnmalm and F. Caruso, Science, 2015, 348, aaa2491 CrossRef PubMed.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- P. Schaaf and J. B. Schlenoff, Adv. Mater., 2015, 27, 2420–2432 CrossRef CAS PubMed.
- J. Li, G. van Ewijk, D. J. van Dijken, J. van der Gucht and W. M. de Vos, ACS Appl. Mater. Interfaces, 2021, 13, 21844–21853 CrossRef CAS PubMed.
- H. Yu, J. Li, D. Wu, Z. Qiu and Y. Zhang, Chem. Soc. Rev., 2010, 39, 464–473 RSC.
- P. J. LeValley, R. Neelarapu, B. P. Sutherland, S. Dasgupta, C. J. Kloxin and A. M. Kloxin, J. Am. Chem. Soc., 2020, 142, 4671–4679 CrossRef CAS PubMed.
- F. Lind, K. Markelov and A. Studer, Chem. Sci., 2023, 14, 12615–12620 RSC.
- M. P. O'Hagan, Z. Duan, F. Huang, S. Laps, J. Dong, F. Xia and I. Willner, Chem. Rev., 2023, 123, 6839–6887 CrossRef PubMed.
- A. Blanc and C. G. Bochet, J. Org. Chem., 2002, 67, 5567–5577 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- J. D. Kahl and M. M. Greenberg, J. Am. Chem. Soc., 1999, 121, 597–604 CrossRef CAS.
- F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 2091–2158 CrossRef CAS PubMed.
- X. Bai, Z. Li, S. Jockusch, N. J. Turro and J. Ju, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 409–413 CrossRef CAS PubMed.
- J. E. T. Corrie, A. Barth, V. R. N. Munasinghe, D. R. Trentham and M. C. Hutter, J. Am. Chem. Soc., 2003, 125, 8546–8554 CrossRef CAS PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- T. Boudou, T. Crouzier, K. Ren, G. Blin and C. Picart, Adv. Mater., 2010, 22, 441–467 CrossRef CAS PubMed.
- C. H. Porcel and J. B. Schlenoff, Biomacromolecules, 2009, 10, 2968–2975 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- Y. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka, S. M. Pham and R. M. Leblanc, Macromolecules, 2002, 35, 5228–5234 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- L. A. Connal, R. Vestberg, C. J. Hawker and G. G. Qiao, Adv. Funct. Mater., 2008, 18, 3315–3322 CrossRef CAS.
- C. P. Kabb, C. S. O'Bryan, C. D. Morley, T. E. Angelini and B. S. Sumerlin, Chem. Sci., 2019, 10, 7702–7708 RSC.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- S. Schäfer and G. Kickelbick, Macromolecules, 2018, 51, 6099–6110 CrossRef.
- E. G. Fuller, H. Sun, R. D. Dhavalikar, M. Unni, G. M. Scheutz, B. S. Sumerlin and C. Rinaldi, ACS Appl. Polym. Mater., 2019, 1, 211–220 CrossRef CAS.
- Y. Jiang and N. Hadjichristidis, Angew. Chem., Int. Ed., 2021, 60, 331–337 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- Q. Zhang, D.-H. Qu, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2022, 144, 2022–2033 CrossRef CAS PubMed.
- B.-S. Wang, Q. Zhang, Z.-Q. Wang, C.-Y. Shi, X.-Q. Gong, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2023, 62, e202215329 CrossRef CAS PubMed.
- S. Mukherjee, A. P. Bapat, M. R. Hill and B. S. Sumerlin, Polym. Chem., 2014, 5, 6923–6931 RSC.
- S. Mukherjee, W. L. A. Brooks, Y. Dai and B. S. Sumerlin, Polym. Chem., 2016, 7, 1971–1978 RSC.
- J. N. Cambre, D. Roy and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3373–3382 CrossRef CAS.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 RSC.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678–8684 CrossRef CAS PubMed.
- S. Bhattacharya, R. S. Phatake, S. Nabha Barnea, N. Zerby, J.-J. Zhu, R. Shikler, N. G. Lemcoff and R. Jelinek, ACS Nano, 2019, 13, 1433–1442 CrossRef CAS PubMed.
- S. Li, N. Chen, X. Li, Y. Li, Z. Xie, Z. Ma, J. Zhao, X. Hou and X. Yuan, Adv. Funct. Mater., 2020, 30, 2000130 CrossRef CAS.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- G. Das, T. Prakasam, N. Alkhatib, R. G. AbdulHalim, F. Chandra, S. K. Sharma, B. Garai, S. Varghese, M. A. Addicoat, F. Ravaux, R. Pasricha, R. Jagannathan, N. i. Saleh, S. Kirmizialtin, M. A. Olson and A. Trabolsi, Nat. Commun., 2023, 14, 3765 CrossRef CAS PubMed.
- C. N. Zhu, C. Y. Li, H. Wang, W. Hong, F. Huang, Q. Zheng and Z. L. Wu, Adv. Mater., 2021, 33, 2008057 CrossRef CAS PubMed.
- M. B. Sims, B. Zhang, Z. M. Gdowski, T. P. Lodge and F. S. Bates, Macromolecules, 2022, 55, 3317–3324 CrossRef CAS.
- S. Inacker, J. Fanelli, S. I. Ivlev and N. A. Hampp, Macromolecules, 2022, 55, 8461–8471 CrossRef CAS.
- M. B. Sims, J. W. Goetze, G. D. Gorbea, Z. M. Gdowski, T. P. Lodge and F. S. Bates, ACS Appl. Mater. Interfaces, 2023, 15, 10044–10052 CrossRef CAS PubMed.
- J. Van Damme, O. van den Berg, J. Brancart, L. Vlaminck, C. Huyck, G. Van Assche, B. Van Mele and F. Du Prez, Macromolecules, 2017, 50, 1930–1938 CrossRef CAS.
- J. Van Damme and F. Du Prez, Prog. Polym. Sci., 2018, 82, 92–119 CrossRef CAS.
- T. Hughes, G. P. Simon and K. Saito, ACS Appl. Mater. Interfaces, 2019, 11, 19429–19443 CrossRef CAS PubMed.
- P. Johnston, C. Braybrook and K. Saito, Chem. Sci., 2012, 3, 2301–2306 RSC.
- H. Sun, H. Fan and X. Peng, J. Org. Chem., 2014, 79, 11359–11369 CrossRef CAS PubMed.
- B. Tang, M. Pauls, C. Bannwarth and S. Hecht, J. Am. Chem. Soc., 2024, 146, 45–50 CrossRef CAS PubMed.
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3078 CrossRef CAS PubMed.
- T. Hughes, G. P. Simon and K. Saito, Mater. Horiz., 2019, 6, 1762–1773 RSC.
- T. Defize, J.-M. Thomassin, H. Ottevaere, C. Malherbe, G. Eppe, R. Jellali, M. Alexandre, C. Jérôme and R. Riva, Macromolecules, 2019, 52, 444–456 CrossRef CAS.
- D. Lu, M. Zhu, S. Wu, Q. Lian, W. Wang, D. Adlam, J. A. Hoyland and B. R. Saunders, Adv. Funct. Mater., 2020, 30, 1909359 CrossRef CAS.
- H. Mardani, H. Roghani-Mamaqani, S. Shahi and M. Salami-Kalajahi, ACS Appl. Polym. Mater., 2022, 4, 1816–1825 CrossRef CAS.
- K. H. Shaughnessy, P. Kim and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 2123–2132 CrossRef CAS.
- X. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632–2643 CrossRef CAS.
- L. Ionov and S. Diez, J. Am. Chem. Soc., 2009, 131, 13315–13319 CrossRef CAS PubMed.
- C. P. Kabb, C. S. O'Bryan, C. C. Deng, T. E. Angelini and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2018, 10, 16793–16801 CrossRef CAS PubMed.
- R. Cheng, J. Chen, Z. Liu, Z. Liu and J. Jiang, Macromol. Rapid Commun., 2016, 37, 514–520 CrossRef CAS PubMed.
- Y. V. Il'ichev, M. A. Schwörer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef PubMed.
- G. M. Scheutz, J. Elgoyhen, K. C. Bentz, Y. Xia, H. Sun, J. Zhao, D. A. Savin and B. S. Sumerlin, Polym. Chem., 2021, 12, 4462–4466 RSC.
- Y. Chen and J.-D. Wu, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1867–1875 CrossRef CAS.
- Y. Chen and J.-L. Geh, Polymer, 1996, 37, 4481–4486 CrossRef CAS.
- S. R. Trenor, T. E. Long and B. J. Love, Macromol. Chem. Phys., 2004, 205, 715–723 CrossRef CAS.
- L. Liu, S.-Z. Luo, B. Wang and Z. Guo, Appl. Surf. Sci., 2015, 345, 116–121 CrossRef CAS.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- A. Veis, E. Bodor and S. Mussell, Biopolymers, 1967, 5, 37–59 CrossRef CAS PubMed.
- L. Li, S. Srivastava, M. Andreev, A. B. Marciel, J. J. de Pablo and M. V. Tirrell, Macromolecules, 2018, 51, 2988–2995 CrossRef CAS.
- R. Chollakup, W. Smitthipong, C. D. Eisenbach and M. Tirrell, Macromolecules, 2010, 43, 2518–2528 CrossRef CAS.
- E. Spruijt, A. H. Westphal, J. W. Borst, M. A. Cohen Stuart and J. van der Gucht, Macromolecules, 2010, 43, 6476–6484 CrossRef CAS.
- M. Lemmers, E. Spruijt, L. Beun, R. Fokkink, F. Leermakers, G. Portale, M. A. Cohen Stuart and J. van der Gucht, Soft Matter, 2012, 8, 104–117 RSC.
- S. Adhikari, M. A. Leaf and M. Muthukumar, J. Chem. Phys., 2018, 149, 163308 CrossRef PubMed.
- M. Jiang, N. Paul, N. Bieniek, T. Buckup, N. Hampp and M. Motzkus, Phys. Chem. Chem. Phys., 2017, 19, 4597–4606 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthetic protocols, methods, instrumentation, 1H NMR spectra of monomers and polymers, release profile of photolabile group, polyelectrolyte complex (PEC) formation, thickness of coatings, stability of coatings and free-standing films, DSC data, and composition of polymers and PECs. See DOI: https://doi.org/10.1039/d5sc00637f |
| This journal is © The Royal Society of Chemistry 2025 |