
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PVAylation: precision end-functionalized poly(vinyl alcohol) for site-selective bioconjugation†
Douglas E.
Soutar
 a,
Ho Fung
Mack
cd,
Melissa
Ligorio
a,
Akalabya
Bissoyi
cd,
Alexander N.
Baker
a and
Matthew I.
Gibson
*abcd
a,
Ho Fung
Mack
cd,
Melissa
Ligorio
a,
Akalabya
Bissoyi
cd,
Alexander N.
Baker
a and
Matthew I.
Gibson
*abcd
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: Matt.gibson@manchester.ac.uk
bWarwick Medical School, University of Warwick, Coventry, CV4 7AL, UK
cDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
First published on 24th April 2025
Abstract
The (bio)conjugation of polymers onto proteins enhances their pharmacokinetics and stability, most commonly using PEG (polyethylene glycol), but there is a need for alternatives. Poly(vinyl alcohol), PVA, is a water-soluble, biocompatible and environmentally degradable polymer, which also has the unique function of ice recrystallisation inhibition (IRI) which can aid the cryopreservation of biologics. Site-specific PVA bioconjugation (“PVAylation”) is underexplored due to the challenge of obtaining homogenous mono end-functional PVA. Here we show that following deprotection of the acetate (from the precursor poly(vinyl acetate)), the concurrent xanthate end-group reduction leads to a diversity of ambiguous end-groups which prevented precision conjugation. This is overcome by using a photo-catalyzed reduction of the omega-terminal xanthates to C–H, which is orthogonal to active-ester bioconjugation functionality at the alpha-chain terminus, demonstrated by MALDI-TOF mass spectrometry. This strategy enabled the preparation of well-defined mono-functional PVA displaying alkyne, biotin and O6-benzylguanine chain-end functionalities, which are each then used for covalent or non-covalent site-specific modification of three model proteins, introduce ice-binding function. These results will enable the synthesis of new bioconjugates containing PVA and be of particular benefit for low-temperature applications.
Introduction
Poly(vinyl alcohol) (PVA) is a water-soluble polymer with a unique combination of properties for a vinyl-derived polymer. It is degradable in the environment by action of microorganisms,1 and has low in vivo toxicity.2,3 PVA is also the most readily accessible, and most active, polymeric ice-recrystallization-inhibitor (IRI).4–7 Ice recrystallization is a significant problem during the storage and transport of biologics (or food stuffs), where ice growth can cause mechanical and osmotic damage.8–10 PVA has been non-specifically conjugated to lactate dehydrogenase and green fluorescent protein providing stability during several freeze/thaw cycles,11 by preventing irreversible aggregation. The addition of PVA into the solution also aided the cryopreservation of several proteins, but required a large molar excess relative to the protein.5 Alternatives, such as trehalose-side chain polymers have also been developed for protein stabilization during lyophilization, allowing aggregation-prone biologics such as insulin to be stabilized.12 Whilst trehalose-functional polymers are potent and have been proven to be safe in murine models,13 they are new-to-human polymers so require extensive testing.There are no examples, to the best of our knowledge, of the site-specific bioconjugation of well-defined PVA onto proteins. PVA-protein conjugates have, however, been shown to extend blood circulation time in vivo.14 Site-specific bioconjugation is essential to ensure the conjugation occurs away from active sites (for example, in enzymes) and to enable accurate characterization and hence reproducibility.15,16 PEGylation has been widely used but there is a desire to find alternatives to mitigate against anti-PEG antibodies,17,18 and several polymers such as polysarcosine19 and poly(oxazolines),20 as selected examples, have been explored.21 These polymers do not bring ice-binding or other advanced cryoprotective properties as bioconjugates and hence require additional excipients to enable storage and transport.
PVA has many synthetic barriers to its bioconjugation which has prevented its wider use despite its appealing properties. Firstly, it has hydroxyl groups along its backbone which can compete with nucleophile/electrophile conjugations. Secondly, PVA is produced by hydrolysis of poly(vinyl esters) such as poly(vinyl acetate) (PVAc), which necessitates any conjugation functionality to be tolerant to the deprotection conditions. Thirdly, VAc is a less-activated monomer with a tendency for chain transfer and head-to-head addition, which makes it challenging to polymerize.22 Head-to-head addition has complex effects on ‘living’ polymerization methods, as the stability of the adducts of the two possible chain ends with mediators is not equal.23 In both RAFT and ATRP, the dormant form of a growing chain is more stable in the head-to-head variant. In RAFT this leads to accumulation of head-to-head terminated chains, and at low degrees of polymerization, the polymer consists of a mixture of head-to-head and head-to-tail terminated chains. Cobalt mediated polymerization does not have the head-to-head problem, but offers less opportunities for precise end-group functionalization than RAFT.24 Advances in RAFT/MADIX polymerization allow facile access to well-defined PVAc.25 Particularly promising are photo-RAFT processes which bring advantages in terms of oxygen tolerance and spatio-temporal control over polymerization.26–28
The R or Z groups in RAFT derived polymers can be used to enable bioconjugation. For example, amine functional PVA synthesized with a phthalimide R group is deprotected on hydrolysis, but the fate of the Z group is not defined.29 Similar strategies have introduced azide30 and alkyne31 functional R groups. The thiocarbonyl functionality at the ⍵-terminus of RAFT-derived polymers can be reduced to a thiol for disfulfide or thio-Michael additions.32 This approach has been used to functionalize oligonucleotides and peptides,33 in addition to proteins, when using monomers which result in stable terminal thiols (unlike PVA).34 During deprotection of PVAc precursor to obtain PVA, the ⍵-terminal xanthate is cleaved, which to a first approximation would result in a useful thiol-end group. In the case of PVA, however, there is an alcohol on the same carbon as the thiol which is unlikely to be stable, similar to geminal diols. This ambiguity poses significant challenges for subsequent bioconjugation, because the end-group is not ‘pure’ for direct deployment as a conjugation reagent and may interfere with reactivity at the other chain end. With a sufficiently high degree of polymerization, the accumulation of head-to-head chain ends can make the thiol produced upon hydrolysis usable,35 however for short PVA lengths the thiol ambiguity problem remains.
Here we introduce a practical and versatile method for the site-specific protein-PVA bioconjugation by precision modification of both chain ends to prevent side reactions. We first demonstrate the ambiguity of PVA end-groups during standard xanthate removal conditions, which prevent straightforward thio-based conjugations. To obtain precision PVAs, pentafluorophenyl (PFP) ester functional PVAc is first prepared by photo-RAFT polymerization using a bismuth oxide catalyst. The xanthate is then selectively reduced to a hydride using a phosphine and UV-light. The PFP ester functionality is not affected by this treatment, allowing it to be displaced by a biotin, alkyne, or O6-benzylguanine functional amine to produce ⍺-functional PVAc. Each step is monitored by MALDI-TOF mass spectrometry to assign the end-groups. Following acetate removal, the resulting functional PVAs are deployed for site-specific bioconjugation to model proteins using click, non-covalent, and biocatalytic bioconjugations to demonstrate broad scope. This synthetic approach effectively overcomes the obstacles to PVA being incorporated into protein therapeutics or biocatalysts and will enable downstream applications and wider use of PVAylation.
Experimental section
Materials
All chemicals were used as supplied unless otherwise stated. Water used throughout this work was 16 MΩ deionized water from an ELGA chorus 3 water purifier. Reagent grade solvents were used as supplied for reactions and purification. Tetrahydrofuran (THF) 99.5%, dichloromethane (DCM) 99%, ethyl acetate 99%, dimethyl sulfoxide (DMSO) 99.9%, pentane 99.5% and hexane (fraction from petroleum) were purchased from Fisher Scientific. Ethanol absolute was purchased from VWR chemicals. Methanol 99.8%, isopropyl alcohol 99.8%, UPHLC grade acetonitrile (for MALDI) and 99.9% anhydrous inhibitor free THF (for MALDI) were purchased from Sigma Aldrich.Vinyl acetate >99%, (3–20 ppm hydroquinone as inhibitor) was purchased from Sigma Aldrich and passed over a short plug of neutral alumina before polymerization to remove inhibitor. Potassium ethyl xanthogenate, 2-bromo-2-methyl propionic acid 98%, bismuth(III) oxide powder 99.999% trace metals basis, 1-ethylpiperidine hypophosphite, hydrazine hydrate 50–60% reagent grade, 5.4 M sodium methoxide in methanol, dibenzocyclooctyne-amine, DCTB 98%, 2,5-DHB 99%, sodium trifluoroacetate 99%, and potassium trifluoroacetate 99% were purchased from Sigma Aldrich. 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) was purchased from Carbosynth. Pentafluorophenol 99% and trifluoroacetic acid 99% were purchased from ABCR. Sulfuric acid 20%, magnesium sulfate were purchased from Fisher Scientific. Benzylamine 99% was purchased from Acros Organics. EZ-Link™ pentylamine-biotin was purchased from Thermo Scientific. 6-((4-(aminomethyl)benzyl)oxy)-7H-purin-2-amine (BG-NH2) was purchased from Fluorochem.
CDCl3, 0.03% TMS, was purchased from Apollo Scientific. CDCl3 (no TMS), DMSO-d6, and D2O were purchased from Sigma Aldrich. Octet® Streptavidin (SA) biosensors were purchased from Sartorius.
Physical and analytical methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, before spotting 0.25 μl of this mixture onto a pre-dried 0.25 μl DCTB spot.
:5, before spotting 0.25 μl of this mixture onto a pre-dried 0.25 μl DCTB spot.
For PVA samples, a ‘TA30’ solution was prepared by mixing 7 parts TFA solution (0.01% trifluoracetic acid in water) with 3 parts acetonitrile. PVA samples were dissolved in TA30 at concentration 2 mg ml−1. A saturated solution of 2,5-DHB in TA30 was prepared. A 2 mg ml−1 solution of KTFA in TA30 was prepared. The sample, KTFA and 2,5-DHB solutions were mixed at a ratio of 1:1:10 respectively and 1 μl of this solution was spotted onto the plate. The plate was dried under an extractor hood at room temperature. KTFA was used as a cationization agent because this reliably produced a single distribution rather than various H+, Na+, K+ adducts.
Splat, biolayer interferometry and other associated techniques are detailed in the ESI.†
Synthetic procedures
Xanthate and polymer synthesis methods are detailed in the ESI† as well as NMR spectra, SEC data, and methods for MALDI-TOF MS, BLI, SDS-PAGE and IRI assay.Results and discussion
The primary aim of this study was to develop an accessible yet precise synthetic methodology for site-selective protein bioconjugation of poly(vinyl alcohol) (PVA) at a single chain end. This would enable its wider exploration as an alternative to other water-soluble polymers (such as PEG) and enable PVA's advanced functions such as ice-binding to be integrated into biohybrid conjugates. First, to synthesise poly(vinyl acetate) (PVAc) with high end-group fidelity, PET-RAFT/MADIX was employed, using 2-((ethoxycarbonothioyl)thio)-2-methylpropanoic acid, (ECTTMPA) and pentafluorophenyl 2-((ethoxycarbonothioyl)thio)-2-methylpropanoate (PFP-ECTTMPA) (Fig. 1). VAc is a less activated monomer with a tendency for side reactions including chain transfer, which makes it difficult to achieve high conversion without higher initiator concentrations when conventional RAFT polymerization is used. Photoiniferter and PET-RAFT polymerizations can produce polymers with higher end-group fidelity than those produced using thermal initiators, due to the absence initiator-derived chains.36 Several photocatalysts such as tris(2-phenylpyridine)iridium have been reported,37 but bismuth oxide was chosen here, due to ease of removal (heterogeneous catalyst), which combined with the lack of need for deoxygenation27,38 makes this method accessible. The mechanism of this method involves reduction of molecular oxygen by the photocatalyst such that it no longer inhibits propagation, but trace oxygen species also play a role in the catalytic cycle.39 Using this polymerization system, PVAc was prepared using two chain transfer agents (Fig. 1), reaching at least 75% conversion overnight, with dispersity below 1.2 (Table 1). Relatively low DPs were targeted to enable full characterization throughout.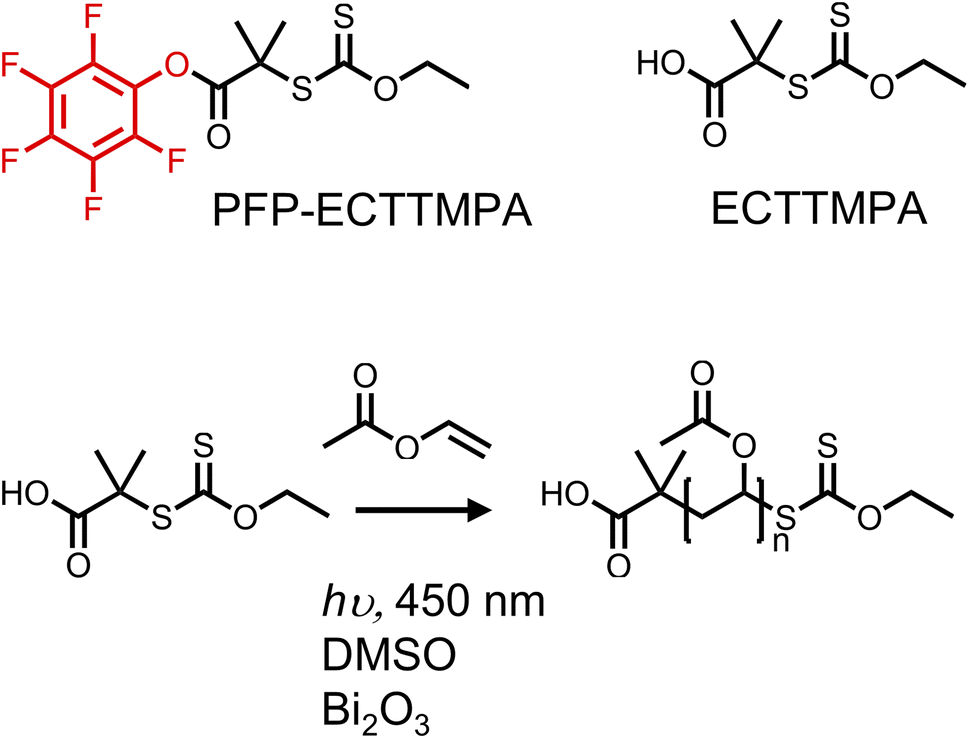 | ||
| Fig. 1 PET-RAFT of vinyl acetate with bismuth oxide as catalyst, under 450 nm (blue) light irradiation, using either ECTTMPA or PFP-ECTTMPA as the CTA. | ||
| Polymer | [M]:[CTA] | CTA | Conversion | Time | M n, Đ (SEC)a | DP, (NMR)b | DP (theoretical)c |
|---|---|---|---|---|---|---|---|
| a SEC was performed in DMF verses poly(methyl methacrylate) standards. b Calculated using the 1H NMR integration of the xanthate O-ethyl CH2 compared to the PVAc backbone integrations. c Conversion multiplied by [M]:[CTA]. | |||||||
| PVAc19-Xan | 25 | ECTTMPA | 76% | 18h | 3800, 1.17 | 19 | 19 |
| PFP-PVAc23-Xan | 25 | PFP-ECTTMPA | 85% | 16h | 3900, 1.19 | 23 | 21 |
We first attempted to use ECTTMPA derived PVAc to produce ω-thiol-terminated polymers suitable for thiol-ene reactions with maleimides. However, we observed that the conversion to the succinimide product was low and irreproducible. MALDI-MS evidence revealed that a thioaldehyde (or a –CHCH–SH functionality) is the main ω-end-group present after deacetylation of PVAc19-Xan (Fig. S18†), suggesting water is eliminated from the ω-chain end following deacetylation (Fig. 2). There is likely a complex mixture of products and the equilibrium will shift with the preparation and analysis conditions, and hence results in barriers to precision bioconjugation. We also attempted to capture the thiol in situ during a ‘one-pot’ thiol-ene reaction of PVAc19-Xan with benzyl acrylamide, which did not reliably produce the thiol-ene product (Fig. S21†), supporting the hypothesis that the thiol adjacent to an acetate may also be unstable or engaging in side-reactions. This highlights a key barrier to the deployment of RAFT-derived PVA as a bioconjugation reagent, as the terminal sulfur containing species may interfere with any conjugation chemistry at the other chain end, while not being ‘pure’ enough itself to be used. The aim of this study was not to quantify these equilibrium products but to develop the tools to prevent them being a problem, making RAFT-derived PVAc a viable route to end-functional poly(vinyl alcohol).
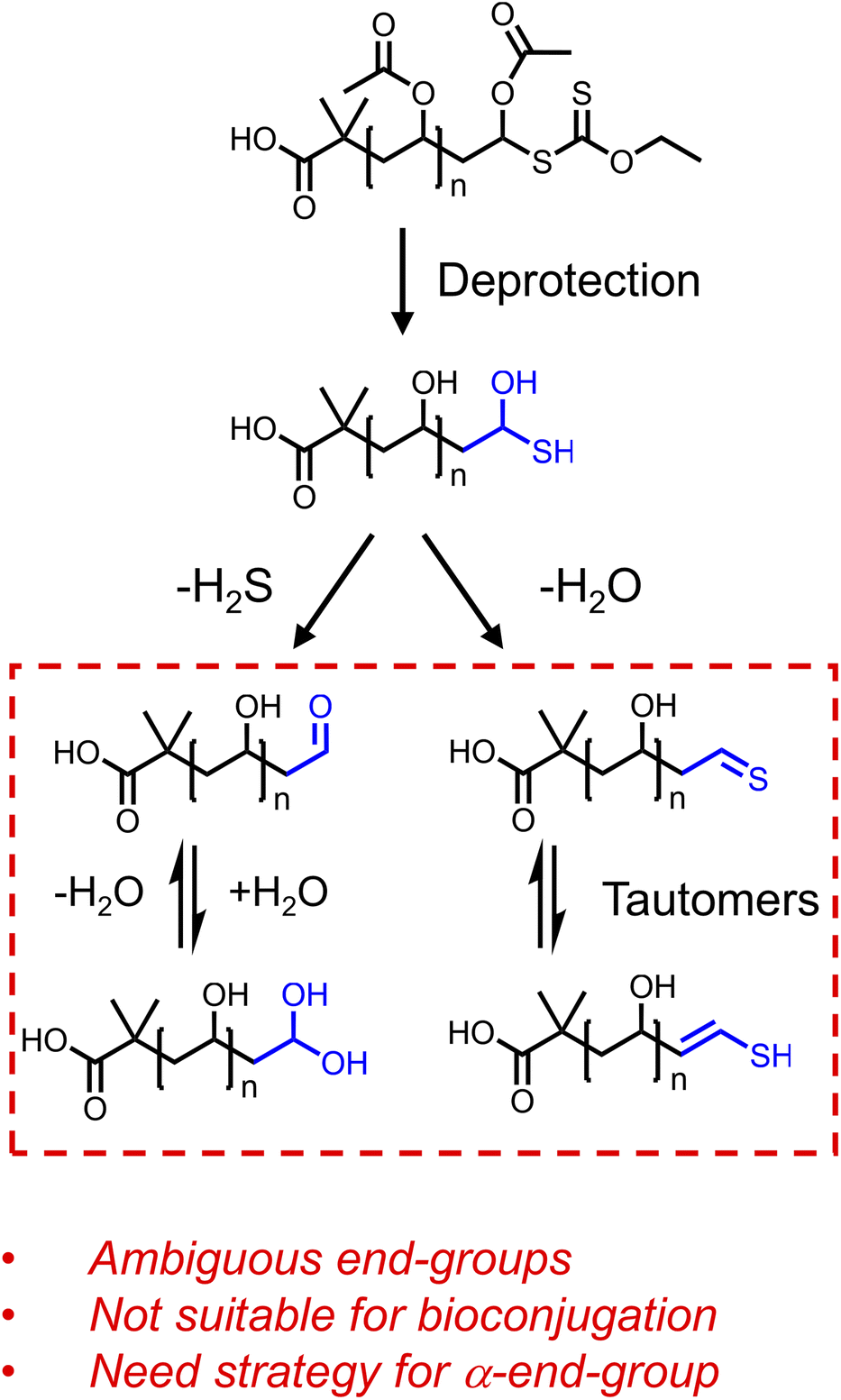 | ||
| Fig. 2 End-group ambiguity following removal of the acetate groups from PVAc. The possible pathways are shown based on H2O or H2S loss supported by MALDI-MS evidence (ESI†). | ||
To address the above problem, our proposed synthetic strategy was to remove the xanthate ω-end-group before removal of the acetate groups, to prevent the ⍵-terminal ambiguity. We could then exploit a pentafluorophenol (PFP) ester group at the α-terminus of the PVAc to install diverse bioconjugation functionalities. A key consideration here, is that any methodology must retain the PFP ester at the ⍺-terminus, to subsequently install biorthogonal functional groups which themselves would not be tolerant to radical polymerization conditions.
Treatment with UV light and N-ethylpiperidine hypophosphite was identified as a suitable method for reduction of a xanthate to terminal hydride while avoiding high temperatures which could degrade the activated ester (Fig. 3A).40 Photoexcitation of the xanthate ester leads to cleavage at the C–S bond, forming a thiyl radical and a polymer radical. The EPHP then acts as a hydrogen atom donor, capping the polymer. The thiyl radical likely forms ethyl xanthic acid as a side product. We first tested the method on PVAc-Xan19 (Table 1), with an average degree of polymerization of 19 which allows end-groups to be resolved in 1H NMR. Following 3 hours irradiation at 380 nm in the presence of approximately 30-fold molar excess of EPHP, quantitative removal of the xanthate CH2 peak at 4.64 ppm in 1H NMR was observed. The peak at 6.62 ppm corresponding to the terminal CH(OAc)Xan was also removed. A new peak attributable to C(OAc)H2 appeared at 4.05 ppm (Fig. 3B). MALDI-TOF MS revealed a clear shift in the observed molecular weight, corresponding to replacement of the xanthate with hydride across the entire distribution (Fig. 3C and D).
 | ||
| Fig. 3 Photocatalytic reduction of xanthate to C–H. (A) Reaction scheme. (B) Region of 1H NMR showing disappearance of CH2 of xanthate at 4.64 ppm and appearance of terminal C(OAc)H2 at 4.08 ppm (Fig. S9† for full spectra). MALDI-MS mass spectra for (C) PVAc19-Xan and (D) PVAc19-H. Peaks correspond to K+ adducts of the polymers. | ||
The end-group cleavage method was then applied to PFP-PVAc23-Xan, with the aim of retaining the PFP group. PFP-PVAc23-Xan was reacted with 17 equivalents EPHP for 6 hours. MALDI-MS confirmed xanthate removal (Fig. 4B and C) with complete retention of the PFP end group, supported by 19F NMR evidence showing no new fluorine environments (Fig. 4E and F). Comparison of molecular weight distributions from SEC revealed a minimal amount of higher molecular weight material after EPHP treatment (Fig. S24†), which was not observable by MALDI-TOF MS.
 | ||
| Fig. 4 H(A) Reaction scheme for transformation of PFP-PVAc-Xan to Bzl-PVAc: MALDI-TOF MS spectra showing K+ adducts of polymers for; (B) PFP-PVAc23-Xan; (C) PFP-PVAc23-H; (D) Bzl-PVAc23-H 19F NMR of the polymers (E) PFP-PVAc23-Xan; (F) PFP-PVAc23-H; (G) Bzl-PVAc23-H. | ||
To confirm the availability of the ⍺-terminal PFP for substitution, the isolated PFP-PVAc23-H was reacted with an excess of benzylamine (to provide a handle easily quantified by 1H NMR). After 2 hours, 19F NMR of the reaction mixture showed complete removal of the PFP-ester to release pentafluorophenol (Fig. 4G). The purified polymer was analysed by 1H NMR (Fig. S14†) and by MALDI-TOF MS (Fig. 4D) showing the expected masses consistent with installation of the benzyl group.
Benzylamide-PVAc23-H was dissolved in methanol and treated with aqueous hydrazine hydrate solution to remove the acetate groups to produce benzylamide modified PVA, Bzl-PVA23, which was purified by precipitation. The structure for Bzl-PVA23 was confirmed using 1H NMR (Fig. S15†) and MALDI-TOF MS (Fig. 5) demonstrating homogeneous PVA with precisely defined end-groups was obtained by this two-step functionalisation protocol.
 | ||
| Fig. 5 Homogeneous Bzl-PVA23 produced by hydrolysis of Bzl-PVAc23 (A) 1H NMR spectra in D2O showing installation of the benzyl group into PVA; (B) structure and exact mass for K+ ion of Bzl-PVA23. (C) MALDI-TOF mass spectrum of Bzl-PVA23. | ||
Having determined a route to α-functional PVA, we wanted to explore the installation of diverse functionalities suitable for bioconjugation. We chose biotin, dibenzocyclooctyne (DBCO), and O6-benzylguanine. For each substituent, we used their respective amine to substitute the PFP-PVAc-H in a similar manner to benzylamine. The routes to PVA differed in the deprotection method, where for DBCO and O6-benzylguanine methanolic NaOMe was used to avoid side reactions with hydrazine (Fig. 6). Synthesis of each substituted PVA was confirmed by MALDI-TOF MS (Fig. 7). Substitution of biotin-PVAc23 and retention of the biotin after deacetylation was also confirmed by 1H NMR (Fig. S16 and S17†).
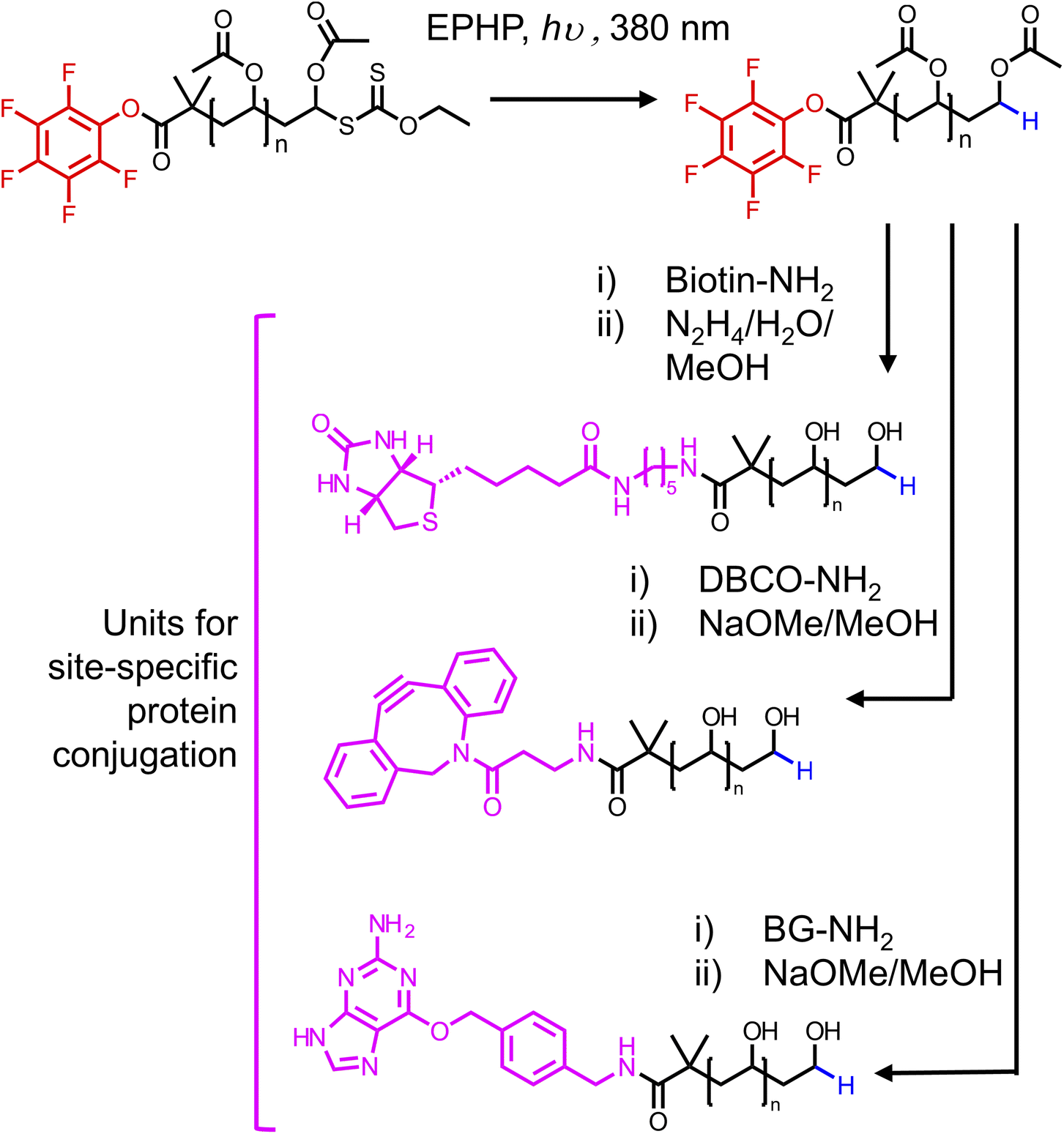 | ||
| Fig. 6 Synthetic routes to obtain ⍺-functional PVA suitable for bioorthogonal conjugation to proteins. | ||
 | ||
| Fig. 7 Structures and MALDI-TOF mass spectra for (A) biotin-PVA, (B) DBCO-PVA and (C) BG-PVA. | ||
Biotin engages in strong non-covalent interactions with streptavidin.41 Biolayer interferometry (BLI) was used to demonstrate the selective interaction between the biotinylated PVA and streptavidin. BLI is a surface-sensitive technique, with an increase in signal output corresponding to mass captured. Commercial streptavidin-coated BLI sensors were exposed to biotin alone, biotin-PVA23, or PVA23 at the same molar concentration. The biotin-PVA gave a large irreversible signal confirming capture, while the biotin alone gave a small signal (Fig. 8). Unmodified PVA and buffer controls showed no signal, confirming the selective bioconjugation of biotin-PVA.
 | ||
| Fig. 8 BLI (biolayer interferometry) to assess capture of biotin-PVA23 to streptavidin-coated sensors. (A) Schematic of the interaction and capture of biotin-PVA23 onto the sensor. (B) BLI output showing specific capture of the biotin-PVA (magenta), verses unmodified PVA (blue), biotin alone (green) and buffer alone (black). | ||
Encouraged by the above, an azide-alkyne cycloaddition (SPAAC) ‘click’ reaction was used to conjugate DBCO-PVA23 with mono-azido bovine serum albumin (BSA). An excess (>100 molar equivalents) of DBCO-PVA23 was reacted at room temperature overnight in phosphate buffer with BSA-azide at a 0.25 mg.ml−1. The conjugation was confirmed by a shift in the molecular weight visible by both FPLC (Fig. 9A) and SDS-PAGE (Fig. S26†). By both methods, the shift in retention volume/retention factor corresponded to approximately 2 kDa, which is consistent with successful bioconjugation. Using the ‘splat’ assay,4,42 the PVA23-BSA conjugate was shown to retain ice recrystallisation inhibition (when corrected to total PVA concentration) demonstrating that bioconjugation does not negatively impact PVA's additional functionality (Fig. S25†).
 | ||
| Fig. 9 Bioconjugations of DBCO-PVA23 and BG-PVA23. (A) Extrapolated molecular weight distributions from FPLC to confirm the conjugation of DBCO-PVA23-H to BSA-azide. (B) MALDI-MS spectra of the 1+ charge state of snap protein compared to a mixture of snap protein and BG-PVA23. | ||
Next, the conjugation of O6-benzylguanine-PVA to SNAP-tag protein43 was confirmed by MALDI-TOF MS (Fig. 9B). After reaction of 4 equivalents PVA with snap tag at 37 °C for 30 minutes in PBS with DTT, the mass spectrum shows a new species at 21.1 kDa, adjacent to snap protein at 19.7 kDa. The new peak was also visible (with higher resolution) in the 2+ and 3+ charge states (Fig. S27†). While the conjugation was not quantitative, it demonstrates the single addition. We suspect a different DTT concentration or use of an alternative reducing agent in the buffer may improve the extent of conjugation since the remaining snap protein is mainly the form reduced by DTT, but we did not optimize further as the aim was to demonstrate the installation and bioavailability of the chain-end functionality for bioconjugation.
To demonstrate the versatility of this method, SNAP-tag conjugation with a longer PVA (BG-PVA110) was undertaken. It was observed that the EPHP treatment step required longer (24 hours) UV exposure time to remove the xanthate fully, which in methanol resulted in partial hydrolysis of the PFP-ester. Changing the solvent to 50:50 mixture of THF:IPA allowed complete xanthate removal and retention of the PFP-ester (Fig. S30†). Following addition of O6-benzylguanine and hydrolysis, this polymer was successful conjugated to the SNAP-tag protein, shown by MALDI-TOF MS (Fig. 10).
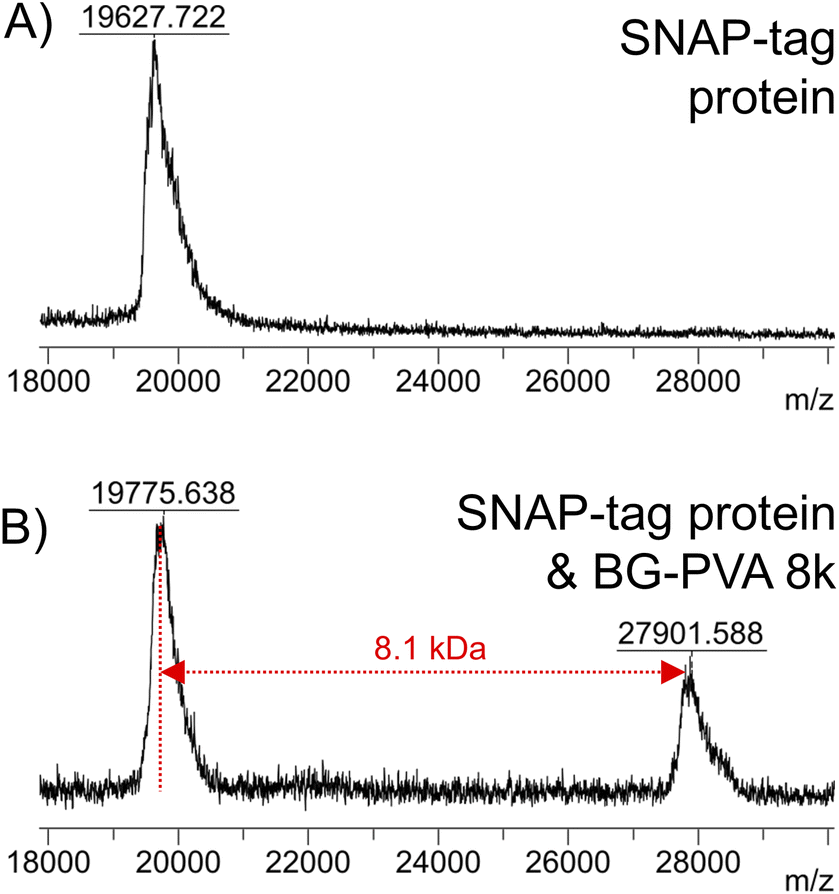 | ||
| Fig. 10 MALDI-TOF mass spectra of (A) SNAP-tag protein (A) and reaction mixture of snap tag protein + BG-PVA110 (B). | ||
Conclusions
In conclusion, we have demonstrated a synthetic route for the site-specific bioconjugation of poly(vinyl alcohol), PVA, onto proteins to enable this versatile, but challenging, polymer to be integrated into bioconjugates. Firstly, a photochemical route was used to reduce the RAFT/MADIX-derived xanthate end-group to a C–H bond of the PVA precursor, PVAc, using N-ethylpiperidine hypophosphite. This step prevents the generation of ambiguous (sulfur containing) ω end-groups at the chain-end of PVA would otherwise interfere with conjugations, and are themselves unsuitable for precision conjugation. The photoredox strategy was shown to be compatible with pentafluorophenyl functional ⍺ chain ends, which were subsequently available to install the required bioorthogonally reactive units for protein conjugation, which themselves are not always tolerant to controlled radical polymerization conditions. Following complete hydrolysis of acetates of the modified PVAc, precision PVA was obtained bearing alkyne, biotin and benzylguanine end-groups, for strain promoted click, streptavidin capture or ‘SNAP’ tag bioconjugations, respectively. Bioconjugation was demonstrated using a range of methods including MALDI-TOF MS, fast protein liquid chromatography (FPLC), gel electrophoresis and biolayer interferometry. The PVA-BSA conjugate retained the ice-binding and ice-recrystallisation inhibition activity of PVA, which is not introduced by PEG or other water-soluble biocompatible polymers. PVA bioconjugation is appealing as an alternative to the standard PEGylation, bringing advanced functionality including (environmental) biodegradability and ice binding/recrystallisation inhibition functionality, not found in most synthetic polymers. Future work will explore bioconjugates of PVA, particularly for intrinsically cryo-stable bioconjugates with resistance to protein degradation.Data availability
All data is within the supporting information.Author contributions
DES, ML, ANB, HFM conducted experiments. DES and MIG devised experiments and analyzed the data. MIG directed the research. MIG and ANB supervised DES. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Lijiang Song (University of Warwick) is thanked for mass spectrometry support. The Polymer RTP at the University of Warwick are thanked for technical support for collecting SEC data. MIG thanks the ERC for a Consolidator Grant (866056), the Royal Society for an Industry Fellowship (191037) joint with Cytiva and the University of Manchester for funding. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) license to any author-accepted manuscript version arising from this submission.References
- S. Matsumura, H. Kurita and H. Shimokobe, Anaerobic Biodegradability of Polyvinyl Alcohol, Biotechnol. Lett., 1993, 15(7), 749–754, DOI:10.1007/BF01080150.
- G. Rivera-Hernández, M. Antunes-Ricardo, P. Martínez-Morales and M. L. Sánchez, Polyvinyl Alcohol Based-Drug Delivery Systems for Cancer Treatment, Int. J. Pharm., 2021, 600, 120478, DOI:10.1016/j.ijpharm.2021.120478.
- C. C. DeMerlis and D. R. Schoneker, Review of the Oral Toxicity of Polyvinyl Alcohol (PVA), Food Chem. Toxicol., 2003, 41(3), 319–326, DOI:10.1016/S0278-6915(02)00258-2.
- C. I. Biggs, C. Stubbs, B. Graham, A. E. R. Fayter, M. Hasan and M. I. Gibson, Mimicking the Ice Recrystallization Activity of Biological Antifreezes. When Is a New Polymer “Active”, Macromol. Biosci., 2019, 19(7), 1900082, DOI:10.1002/mabi.201900082.
- D. E. Mitchell, A. E. R. Fayter, R. C. Deller, M. Hasan, J. Gutierrez-Marcos and M. I. Gibson, Ice-Recrystallization Inhibiting Polymers Protect Proteins against Freeze-Stress and Enable Glycerol-Free Cryostorage, Mater. Horiz., 2019, 6(2), 364–368, 10.1039/C8MH00727F.
- M. Hasan, A. E. R. Fayter and M. I. Gibson, Ice Recrystallization Inhibiting Polymers Enable Glycerol-Free Cryopreservation of Microorganisms, Biomacromolecules, 2018, 19(8), 3371–3376, DOI:10.1021/acs.biomac.8b00660.
- F. Bachtiger, T. R. Congdon, C. Stubbs, M. I. Gibson and G. C. Sosso, The Atomistic Details of the Ice Recrystallisation Inhibition Activity of PVA, Nat. Commun., 2021, 12(1), 1323, DOI:10.1038/s41467-021-21717-z.
- Y. B. Yu, K. T. Briggs, M. B. Taraban, R. G. Brinson and J. P. Marino, Grand Challenges in Pharmaceutical Research Series: Ridding the Cold Chain for Biologics, Pharm. Res., 2021, 38(1), 3–7, DOI:10.1007/s11095-021-03008-w.
- R. Fang, P. Sane, I. Borges Sebastião and B. Bhatnagar, Stresses, Stabilization, and Recent Insights in Freezing of Biologics, in Principles and Practices of Lyophilization in Product Development and Manufacturing, Jameel, F., ed. Springer International Publishing, Cham, 2023, pp. 189–197, DOI:10.1007/978-3-031-12634-5_11.
- B. S. Bhatnagar, R. H. Bogner and M. J. Pikal, Protein Stability During Freezing: Separation of Stresses and Mechanisms of Protein Stabilization, Pharm. Dev. Technol., 2007, 12(5), 505–523, DOI:10.1080/10837450701481157.
- A. E. R. Fayter, M. Hasan, T. R. Congdon, I. Kontopoulou and M. I. Gibson, Ice Recrystallisation Inhibiting Polymers Prevent Irreversible Protein Aggregation during Solvent-Free Cryopreservation as Additives and as Covalent Polymer-Protein Conjugates, Eur. Polym. J., 2020, 140, 110036, DOI:10.1016/j.eurpolymj.2020.110036.
- J. Lee, E.-W. Lin, U. Y. Lau, J. L. Hedrick, E. Bat and H. D. Maynard, Trehalose Glycopolymers as Excipients for Protein Stabilization, Biomacromolecules, 2013, 14(8), 2561–2569, DOI:10.1021/bm4003046.
- M. B. Gelb, K. M. M. Messina, D. Vinciguerra, J. H. Ko, J. Collins, M. Tamboline, S. Xu, F. J. Ibarrondo and H. D. Maynard, Poly(Trehalose Methacrylate) as an Excipient for Insulin Stabilization: Mechanism and Safety, ACS Appl. Mater. Interfaces, 2022, 14(33), 37410–37423, DOI:10.1021/acsami.2c09301.
- Y. Kojima and H. Maeda, Evaluation of Poly(Vinyl Alcohol) for Protein Tailoring: Improvements in Pharmacokinetic Properties of Superoxide Dismutase, J. Bioact. Compat. Polym., 1993, 8(2), 115–131, DOI:10.1177/088391159300800202.
- J. H. Ko and H. D. Maynard, A Guide to Maximizing the Therapeutic Potential of Protein–Polymer Conjugates by Rational Design, Chem. Soc. Rev., 2018, 47(24), 8998–9014, 10.1039/C8CS00606G.
- X. Liu and W. Gao, Precision Conjugation: An Emerging Tool for Generating Protein–Polymer Conjugates, Angew. Chem., Int. Ed., 2021, 60(20), 11024–11035, DOI:10.1002/anie.202003708.
- B.-M. Chen, T.-L. Cheng and S. R. Roffler, Polyethylene Glycol Immunogenicity: Theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies, ACS Nano, 2021, 15(9), 14022–14048, DOI:10.1021/acsnano.1c05922.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Poly(Ethylene Glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives, Angew. Chem., Int. Ed., 2010, 49(36), 6288–6308, DOI:10.1002/anie.200902672.
- Y. Hu, Y. Hou, H. Wang and H. Lu, Polysarcosine as an Alternative to PEG for Therapeutic Protein Conjugation, Bioconjug. Chem., 2018, 29(7), 2232–2238, DOI:10.1021/acs.bioconjchem.8b00237.
- A. Mero, Z. Fang, G. Pasut, F. M. Veronese and T. X. Viegas, Selective Conjugation of Poly(2-Ethyl 2-Oxazoline) to Granulocyte Colony Stimulating Factor, J. Controlled Release, 2012, 159(3), 353–361, DOI:10.1016/j.jconrel.2012.02.025.
- M. A. Gauthier and H.-A. Klok, Peptide/Protein–Polymer Conjugates: Synthetic Strategies and Design Concepts, Chem. Commun., 2008,(23), 2591–2611, 10.1039/B719689J.
- P. J. Flory and F. S. Leutner, Occurrence of Head-to-Head Arrangements of Structural Units in Polyvinyl Alcohol, J. Polym. Sci., 1948, 3(6), 880–890, DOI:10.1002/pol.1948.120030608.
- A. Morin, C. Detrembleur, C. Jérôme, P. de Tullio, R. Poli and A. Debuigne, Effect of Head-to-Head Addition in Vinyl Acetate Controlled Radical Polymerization: Why Is Co(Acac) 2 -Mediated Polymerization so Much Better?, Macromolecules, 2013, 46(11), 4303–4312, DOI:10.1021/ma400651a.
- A. Debuigne, J.-R. Caille and R. Jérôme, Synthesis of End-Functional Poly(Vinyl Acetate) by Cobalt-Mediated Radical Polymerization, Macromolecules, 2005, 38(13), 5452–5458, DOI:10.1021/ma047726q.
- S. Harrisson, X. Liu, J.-N. Ollagnier, O. Coutelier, J.-D. Marty and M. Destarac, RAFT Polymerization of Vinyl Esters: Synthesis and Applications, Polymers, 2014, 6(5), 1437–1488, DOI:10.3390/polym6051437.
- M. A. S. N. Weerasinghe, N. D. A. Watuthanthrige and D. Konkolewicz, Exploring High Molecular Weight Vinyl Ester Polymers Made by PET-RAFT, Polym. Chem., 2024, 15(9), 868–877, 10.1039/D4PY00065J.
- K. Hakobyan, T. Gegenhuber, C. S. P. McErlean and M. Müllner, Visible-Light-Driven MADIX Polymerisation via a Reusable, Low-Cost, and Non-Toxic Bismuth Oxide Photocatalyst, Angew. Chem., Int. Ed., 2019, 58(6), 1828–1832, DOI:10.1002/anie.201811721.
- S. Shanmugam, J. Xu and C. Boyer, Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization of Vinyl Acetate and N-Vinylpyrrolidinone: Kinetic and Oxygen Tolerance Study, Macromolecules, 2014, 47(15), 4930–4942, DOI:10.1021/ma500842u.
- A. A. A. Smith, T. Hussmann, J. Elich, A. Postma, M.-H. Alves and A. N. Zelikin, Macromolecular Design of Poly(Vinyl Alcohol) by RAFT Polymerization, Polym. Chem., 2011, 3(1), 85–88, 10.1039/C1PY00429H.
- J. Muller, F. Marchandeau, B. Prelot, J. Zajac, J.-J. Robin and S. Monge, Self-Organization in Water of Well-Defined Amphiphilic Poly(Vinyl Acetate)-b-Poly(Vinyl Alcohol) Diblock Copolymers, Polym. Chem., 2015, 6(16), 3063–3073, 10.1039/C5PY00091B.
- J. R. Góis, A. V. Popov, T. Guliashvili, A. C. Serra and J. F. J. Coelho, Synthesis of Functionalized Poly(Vinyl Acetate) Mediated by Alkyne-Terminated RAFT Agents, RSC Adv., 2015, 5(111), 91225–91234, 10.1039/C5RA15580K.
- H. Willcock and R. K. O'Reilly, End Group Removal and Modification of RAFT Polymers, Polym. Chem., 2010, 1(2), 149–157, 10.1039/B9PY00340A.
- C. Boyer, V. Bulmus and T. P. Davis, Efficient Usage of Thiocarbonates for Both the Production and the Biofunctionalization of Polymers, Macromol. Rapid Commun., 2009, 30(7), 493–497, DOI:10.1002/marc.200800708.
- M. Li, P. De, H. Li and B. S. Sumerlin, Conjugation of RAFT-Generated Polymers to Proteins by Two Consecutive Thiol–Ene Reactions, Polym. Chem., 2010, 1(6), 854–859, 10.1039/C0PY00025F.
- G. Murakami, T. Kubo and K. Satoh, Precision Synthesis of End-Functionalized Star Poly(Vinyl Alcohol)s by RAFT Polymerization and Post-Polymerization Modification, Precis. Chem., 2023, 1(4), 256–263, DOI:10.1021/prechem.3c00051.
- M. A. Beres, C. Boyer, M. Hartlieb, D. Konkolewicz, G. G. Qiao, B. S. Sumerlin and S. Perrier, RAFT with Light: A User Guide to Using Thiocarbonylthio Compounds in Photopolymerizations, ACS Polym. Au, 2025 DOI:10.1021/acspolymersau.4c00101.
- S. Shanmugam, J. Xu and C. Boyer, Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization of Vinyl Acetate and N-Vinylpyrrolidinone: Kinetic and Oxygen Tolerance Study, Macromolecules, 2014, 47(15), 4930–4942, DOI:10.1021/ma500842u.
- I. Kontopoulou, T. R. Congdon, S. Bassett, B. Mair and M. I. Gibson, Synthesis of Poly(Vinyl Alcohol) by Blue Light Bismuth Oxide Photocatalysed RAFT. Evaluation of the Impact of Freeze/Thaw Cycling on Ice Recrystallisation Inhibition, Polym. Chem., 2022, 13(32), 4692–4700, 10.1039/D2PY00852A.
- L. Zhang, C. Wu, K. Jung, Y. H. Ng and C. Boyer, An Oxygen Paradox: Catalytic Use of Oxygen in Radical Photopolymerization, Angew. Chem., Int. Ed., 2019, 58(47), 16811–16814, DOI:10.1002/anie.201909014.
- R. N. Carmean, C. A. Figg, G. M. Scheutz, T. Kubo and B. S. Sumerlin, Catalyst-Free Photoinduced End-Group Removal of Thiocarbonylthio Functionality, ACS Macro Lett., 2017, 6(2), 185–189, DOI:10.1021/acsmacrolett.7b00038.
- N. M. Green, Avidin. 3. The Nature of the Biotin-Binding Site, Biochem. J., 1963, 89(3), 599–609, DOI:10.1042/bj0890599.
- C. A. Knight, D. Wen and R. A. Laursen, Nonequilibrium Antifreeze Peptides and the Recrystallization of Ice, Cryobiology, 1995, 32(1), 23–34, DOI:10.1006/cryo.1995.1002.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, A General Method for the Covalent Labeling of Fusion Proteins with Small Molecules in Vivo, Nat. Biotechnol., 2003, 21(1), 86–89, DOI:10.1038/nbt765.
Footnote |
| † Electronic supplementary information (ESI) available: Including additional methods, synthetic information, and underpinning data. See DOI: https://doi.org/10.1039/d5sc00772k |
| This journal is © The Royal Society of Chemistry 2025 |