
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape sorting of two distinct amino acid residues at the multiple binding sites of a porous metal-macrocycle framework†
Shohei
Tashiro
 *a,
Kosuke
Nakata
a,
Ryunosuke
Hayashi
a and
Mitsuhiko
Shionoya
*ab
*a,
Kosuke
Nakata
a,
Ryunosuke
Hayashi
a and
Mitsuhiko
Shionoya
*ab
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tashiro@chem.s.u-tokyo.ac.jp
bResearch Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: shionoya@rs.tus.ac.jp
First published on 2nd June 2025
Abstract
The arrangement of amino acids within crystalline porous materials represents a unique approach to enhance their functionalities such as catalysis, separation and sensing. In particular, the simultaneous arrangement of distinct residues in crystalline frameworks, i.e., shape sorting, remains one of the most important challenges. Here, we demonstrate the shape sorting of two distinct amino acid residues, tryptophan and serine, within porous metal-macrocycle framework-1 via precise molecular recognition at multiple binding sites on the pore surface. Single-crystal X-ray diffraction analysis showed that the indole ring of an N-protected tryptophan molecule was effectively encapsulated within a binding pocket located at the bottom corners of the nanochannel, forming multipoint hydrogen bonds and CH–π interactions. In addition to tryptophan, N-protected serine was adsorbed onto the ceiling sites via multipoint hydrogen bonds. Moreover, modifying their protecting groups allowed us to control the relative positions of the two residues in the nanochannel. We further tested the co-adsorption of both residues in selective separation experiments. The results suggest that designing porous crystals with multiple binding sites is an effective strategy for precise heteroleptic arrangement of amino acid residues, resulting in enhanced functionalization of porous materials.
Introduction
Porous crystals, such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and hydrogen-bonded organic frameworks (HOFs), are promising candidates for new functional materials in modern chemistry,1 because nanometer-sized pores can be engineered at will by combining specific building blocks.2 Thus, the well-defined pores serve as excellent platforms for guest adsorption, enabling a wide variety of types of guest species to arrange themselves within the pores or on the surfaces.3,4 For example, the placement of gas molecules or structurally simple, small molecules, such as CO2,5 O2,6 H2,7 N2,8 H2O9 and hydrocarbons10–12 into porous materials has been extensively studied, primarily for storage, separation and transformation applications. In addition to simple molecules, the arrangement of biomolecules exhibiting complex structures is also an important and developing area of research: for example, nucleobases have been incorporated into MOFs via multipoint hydrogen bonds reminiscent of natural base-paring.13,14 Similarly, the structure determination of other natural compounds has also been studied by the crystalline sponge methods.15–19 Furthermore, although the enzyme encapsulation has enabled the control of enzyme activity and selectivity,20,21 determination of the enzyme inclusion structures via crystal structure analysis remains a major challenge.Amino acids, with more than 20 distinct residues, provide another attractive category as guest species to arrange within porous crystals because their arranged structures exhibit several unique functions that mimic enzymatic behavior. Conventional methods for arranging amino acids within porous crystals typically involve post-synthetic modification of the pore surfaces through the formation of covalent bonds, and these approaches have been successfully applied to catalysis, separation and storage.22–26 More recently, amino acids have also been arranged within porous frameworks via non-covalent or coordination bonds in addition to covalent arrangements. For example, Li et al. demonstrated the non-covalent arrangement of tryptophan at well-defined binding sites within MOFs, and their structures were determined by single-crystal X-ray diffraction (ScXRD).27 Furthermore, Liu et al. achieved post-synthetic modification of MOFs with two distinct amino acids, primarily via coordination bonds. The resulting supramolecular structure was characterized using spectroscopic and theoretical methods and then applied to a unique sensing system.28 However, the precise arrangement of multiple amino acid residues remains a major challenge (Fig. 1a) due to the difficulty in determining the crystal structure, even though it is essential for further functionalization of the porous materials.
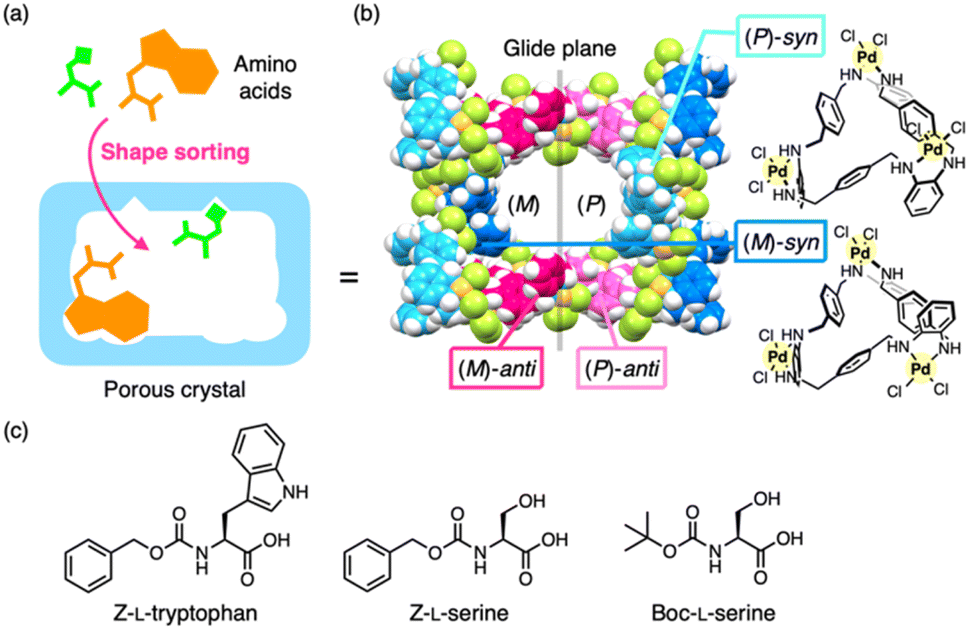 | ||
| Fig. 1 Outline of this study. (a) Shape sorting of two amino acid residues in a nanospace with different binding sites. (b) Porous metal-macrocycle framework-1 (MMF-1), which is composed of four isomeric metal-macrocycles. (c) Chemical structures of N-protected L-tryptophan and L-serine. Z = benzyloxycarbonyl, Boc = tert-butoxycarbonyl. | ||
Here, we present the shape sorting of two distinct amino acid residues within a crystalline nanochannel and the crystal structure determination of their co-located structures. Our group previously developed porous metal-macrocycle frameworks (MMFs) via the self-assembly of helically-twisted trinuclear PdII macrocycles.29 Among the MMF series, MMF-1 is composed of four stereoisomers of PdII macrocycles and features nanochannels with multiple binding sites on the enantiomeric (P)- and (M)-pore surfaces (Fig. 1b).30 Shape sorting of multiple aromatic compounds has been demonstrated in previous studies,30–32 and more recently, site-selective arrangement of the amino acid residue, serine, has also been demonstrated.33 In this study, based on these previous findings for MMF-1, the simultaneous arrangement of serine and tryptophan residues on the pore surfaces was confirmed by ScXRD analysis (Fig. 1). These residues were effectively recognized at their complementary binding sites via multipoint non-covalent interactions, and the relative positions of the two residues in the nanochannel were controlled by altering the N-terminal protecting groups. Previously, pairwise recognition of two amino acids has been achieved in solution using discrete host molecules,34 which has also been applied to control protein–protein interactions.35,36 Recently, heteroleptic pairwise recognition of distinct residues has finally been accomplished within the cavity of cucurbit[8]uril.37 However, to the best of our knowledge, simultaneous recognition of two amino acids in crystalline hosts, specifically heteroleptic pairwise recognition and its crystal structure determination, has not been realized.
Results and discussion
We previously reported that serine residues are site-selectively adsorbed onto the ceiling sites of the MMF-1 nanochannel.33 To achieve shape sorting of multiple amino acid residues within a single nanochannel, we aimed to explore an additional distinct residue that could be adsorbed at the bottom portion of the nanochannel without competing with the serine residue already adsorbed on the ceiling. Because many types of aromatic molecules are typically adsorbed in the corner pockets at the bottom,29 this binding pocket was expected to effectively recognize aromatic amino acid residues. We then examined three N-protected aromatic amino acids, Z-L-phenylalanine, Z-L-tyrosine and Z-L-tryptophan (Z = benzyloxycarbonyl), as potential guests for the bottom-corner pockets (Fig. 1c). Protection of the amino groups was essential to prevent degradation of MMF-1 due to unprotected free amino groups. As a result, Z-L-histidine cannot be used as a guest due to the presence of the coordinating side chain. In a typical procedure, MMF-1 crystals were soaked in an acetonitrile solution of Z-L-phenylalanine, Z-L-tyrosine or Z-L-tryptophan at room temperature and then analyzed by ScXRD at −180 °C. As a result, only Z-L-tryptophan was adsorbed in the corner pockets (Fig. 2a and b), whereas Z-L-phenylalanine and Z-L-tyrosine were not observed in any pockets, indicating that the corner pockets of MMF selectively recognize tryptophan residues in a residue-specific manner. | ||
| Fig. 2 Arrangement structure of Z-L-tryptophan in MMF-1. (a) Crystal structure of the MMF-1 nanochannel adsorbing Z-L-tryptophan, represented by the CPK model (C: yellow). The percentage values indicate the chemical occupancies of the guests. (b) Electron density maps of the Z-L-tryptophan at the (M)- and (P)-sides, with the contour levels of 1.06σ and 0.97σ, respectively. (c) Adsorption structure of Z-L-tryptophan at both sides. MMF-1 and the guest molecule are represented as surface and stick models, respectively. Magenta dots indicate non-covalent interactions among the guests, framework and solvents. Magenta “D” and “A” indicate hydrogen bond donors and acceptors, respectively. Pd: pale yellow, Cl: pale green, N: blue, H: white, C (MMF-1): gray. (d) The crystal structure of Z-L-tryptophan at the (M)-side (red) superimposed with the structurally inverted Z-L-tryptophan at the (P)-side (blue). (e) Hirshfeld surfaces mapped with the dnorm, of Z-L-tryptophan at both sides and their 2D fingerprint plots, where the di and de values represent the closest internal and external distances, respectively, from given points on the Hirshfeld surface. | ||
To understand the binding mode specific to the tryptophan residue, the binding structure was further analyzed by focusing on the non-covalent interactions between the guests, the framework and solvent molecules. For example, the N–H group of the indole ring formed hydrogen bonds with one of the three PdCl2 moieties of the syn-isomers exposed on the pore surfaces. Furthermore, the electron-rich π-surface of the indole ring interacted with multiple C–H moieties of the framework and with acetonitrile molecules co-adsorbed in the pockets, forming CH–π interactions. Moreover, the water molecules trapped on the pore surface formed weak hydrogen bonds with the Ar–H moieties of the indole ring (Fig. 2c). These efficient multipoint non-covalent interactions may provide specific molecular recognition for the tryptophan residue in the corner pockets, while the solvent molecules may play a cooperative role in stabilizing the binding of the indole ring.
Notably, the occupancies of the guests in the enantiomerically paired pockets are significantly different (93.0% and 55.0% for the (M)- and (P)-pockets, respectively), which was also supported by the Flack parameter (0.16(4)). This difference may be due to the slightly different binding modes of the indole rings at the (M)- and (P)-sites. For example, the indole ring in the (M)-pocket was positioned close to the pore surface and formed effective CH–π interactions with the framework (Fig. 2d). The different binding modes at the (M)- and (P)-sites were verified by their distinct fingerprint plots from the Hirshfeld surface analysis (Fig. 2e).38 This analysis also suggested that weak non-covalent interactions, such as CH–π and van der Waals forces, act cooperatively in the molecular recognition. Furthermore, the main-chain structure of the amino acid was partially observed in the (M)-site but not in the (P)-site due to disorder. These differences are likely the result of steric repulsion around the chiral carbon center, suggesting that the enantiomeric binding pockets can recognize the chiral structure of tryptophan even when the chiral carbon is positioned far from the indole ring.
The uptake of Z-L-tryptophan into MMF-1 was further confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy of the guest@MMF-1 crystals after dissolution in DMSO-d6/DCl (Fig. S19†). The 1H NMR spectra showed that approximately three molecules of Z-L-tryptophan were encapsulated into the nanochannel per unit space. Similar experiments were conducted with Z-L-phenylalanine or Z-L-tyrosine, and these non-adsorbed amino acid derivatives were also encapsulated within the channel (approximately 3 molecules per unit space) (Fig. S20 and S21†). These results indicate that the uptake of amino acids was comparable among these residues, but the adsorption efficiency to the pore surface, especially the corner pockets, varied significantly due to the different interaction modes with the pore surface.
Based on this finding, we next investigated the shape-sorting of tryptophan and serine in a single nanochannel. MMF-1 crystals were soaked in a mixed solution containing Z-L-tryptophan (950 mM) and Z-L-serine (300 mM) in acetonitrile at room temperature for 5 days and then analyzed by ScXRD at −180 °C. As a result, both serine and tryptophan residues were concurrently observed at the ceiling and bottom corner sites, respectively (Fig. 3). Remarkably, the binding modes of both residues were nearly identical to those observed in the single-guest experiments. For example, the indole rings were identified at the bottom corner pockets via hydrogen bonds and CH–π interactions, while the main-chain structure was observed only on the (M)-side, similar to the single bonding case (Fig. 3b). Hirshfeld surface analysis also verified the binding modes similar to those observed in the single-guest experiments (Fig. S9–S11†). Moreover, the arrangement of serine residues was successfully observed at the ceiling via multipoint hydrogen bonds, which is similar to our previous study that showed a single binding of Z-L-serine at the ceiling (Fig. 3b).33
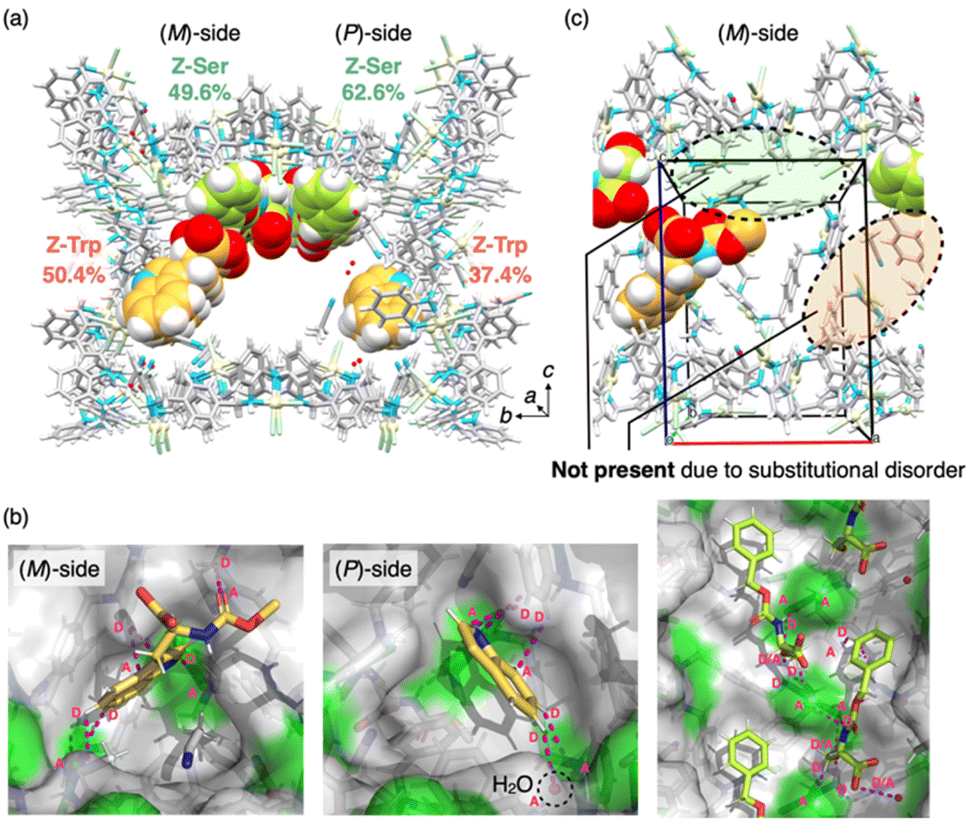 | ||
| Fig. 3 Simultaneous arrangement structure of Z-L-tryptophan and Z-L-serine in MMF-1. (a) Crystal structure of the MMF-1 nanochannel adsorbing Z-L-tryptophan (C: yellow) and Z-L-serine (C: green) represented by the CPK model. The percentage values indicate the chemical occupancy of the guest. (b) Adsorption structures of Z-L-tryptophan on both sides and Z-L-serine on the ceiling. MMF-1 and guest are represented as a surface and a stick model, respectively. Magenta dots indicate non-covalent interactions among the guests, framework and solvents. Magenta “D” and “A” indicate hydrogen bond donors and acceptors, respectively. (c) Enlarged view of the (M)-side surface of the unit cell. Pd: pale yellow, Cl: pale green, N: blue, H: white, C of MMF-1: gray. | ||
However, closer inspection of the co-adsorbed structure revealed that the Z-L-tryptophan on the (M)-side was substitutionally disordered for steric reasons with Z-L-serine (Fig. S12†), preventing them from coexisting in the same unit space (Fig. 3c). The occupancies of tryptophan and serine on the (M)-side (50.4% and 49.6%, respectively), optimized to prevent coexistence, indicated that all the (M)-sides were equally occupied by tryptophan and serine. Therefore, to co-adsorb two different amino acids in the same unit space while avoiding steric interference, we used another serine derivative with a different N-protecting group, Boc-L-serine, because Boc-protected serine is known to occupy only one site on the ceiling, whereas Z-protected serine occupies both the enantiomeric (M)- and (P)-sites on the ceiling.33
To test this hypothesis, MMF-1 crystals were soaked in a mixed solution containing Z-L-tryptophan and Boc-L-serine in acetonitrile at room temperature for 5 days and then analyzed by ScXRD at −180 °C. As expected, both amino acids were co-adsorbed on the pore surface (Fig. 4a), specifically, the indole rings of the tryptophan residues were observed in the both corner pockets via hydrogen bonds and CH–π interactions (Fig. 4b). As observed in single-guest experiments, their binding modes in both enantio-paired pockets were slightly different from each other (Fig. S18†), which was also verified by Hirshfeld surface analysis (Fig. 4d). Notably, the molecular structure of Z-L-tryptophan at the (M)-site was fully assigned without loss of structure due to disorder, in contrast to the partial structure observed in the single-guest experiments. In addition to tryptophan residues, Boc-L-serine was also observed at the ceiling. Essentially, similar to our previous study,33 only the (P)-ceiling part was diastereoselectively occupied by the L-serine residue via multipoint hydrogen bonds.
 | ||
| Fig. 4 Simultaneous arrangement structure of Z-L-tryptophan and Boc-L-serine in MMF-1. (a) Crystal structure of the MMF-1 nanochannel adsorbing Z-L-tryptophan (C: yellow) and Boc-L-serine (C: green), represented by a CPK model. The percentage values indicate the chemical occupancy of the guests. (b) Adsorption structures of Z-L-tryptophan at both sides and Boc-L-serine at the ceiling. MMF-1 and the guests are represented as surface and stick models, respectively. Magenta dots indicate non-covalent interactions among the guests, framework and solvents. Magenta “D” and “A” indicate hydrogen bond donors and acceptors, respectively. (c) Enlarged view of the (M)-side surface of the unit cell. Pd: pale yellow, Cl: pale green, N: blue, H: white, C of MMF-1: gray. (d) The Hirshfeld surfaces of Z-L-tryptophan on both sides, mapped with dnorm, and their 2D fingerprint plots. The di and de values represent the closest internal and external distances, respectively, from given points on the Hirshfeld surface. | ||
Therefore, we investigated the possibility that both residues could exist simultaneously in the same unit space by carefully examining the obtained structure. The steric collision between both residues was evaluated on the (M)-side, and the closest interatomic distance between them was 2.96 Å (Fig. 4c), indicating that no substitutional disorder occurred between them, unlike in the combination of Z-L-tryptophan and Z-L-serine. Moreover, the occupancies of tryptophan and serine residues were optimized to 68.1% and 59.6%, respectively, further demonstrating that both amino acids could exist in the same unit space. This cooperative arrangement is due to the fact that Boc-L-serine diastereoselectively adsorbs only at the (P)-site,33 creating enough space for Z-L-tryptophan to adsorb at the (M)-site. These results demonstrated that by appropriately selecting amino acids, different amino acid residues can be precisely arranged as predicted beforehand. In addition, the C-terminal carboxy group of each residue is asymmetrically located but relatively close to each other, making such a structure potentially useful for a variety of applications such as asymmetric catalysis and chiral separations.
Finally, the simultaneous binding of tryptophan and serine residues was tested in a preliminary selective separation experiment involving multiple amino acids. For example, MMF-1 crystals were soaked in a CD3CN solution containing equal concentrations of six different Z-protected residues (Z-L-serine, Z-L-tryptophan, Z-L-phenylalanine, Z-L-leucine, Z-L-valine and Z-L-alanine) at 20 °C for 1 day. The crystals were collected by filtration and then re-soaked in CD3CN for one more day to extract the encapsulated residues from the crystals (Fig. 5a). When the extract was analyzed by 1H NMR spectroscopy, it was observed that both serine and tryptophan were preferentially adsorbed to MMF-1, likely due to their simultaneous binding to their respective recognition sites, as discussed earlier (Fig. 5b). Therefore, this result supports the effective recognition of the two residues observed by ScXRD. Since the amount of amino acid uptake estimated by 1H NMR analysis is similar whether observed by ScXRD or not, the selective adsorption is likely due to the inhibition of the uptake of other residues by the two residues that preferentially occupy the nanospace of MMF-1.
 | ||
| Fig. 5 Selective separation experiment of Z-protected amino acids. (a) Schematic of the experiment. (b) Adsorption ratios of the six residues in MMF-1 estimated by 1H NMR analysis of the extract. | ||
Conclusions
Shape sorting of two distinct amino acids, tryptophan and serine, was demonstrated in a single nanochannel of porous MMF-1 crystals with multiple binding sites. X-ray crystallographic analysis confirmed the simultaneous arrangement of both structures within the nanochannel and elucidated the non-covalent interactions that stabilize the arrangement. In particular, the chirality of tryptophan residues was recognized in the enantiomeric binding pockets, as verified by distinct diastereomeric binding modes. Furthermore, the relative positions of the tryptophan and serine residues could be controlled by modifying the N-protecting group of the serine residue, allowing the simultaneous arrangement of Z-L-tryptophan and Boc-L-serine in the same unit space. Moreover, this approach was successfully applied to a selective separation experiment involving a mixture of six different amino acids, although future applications as separation matrices require kinetic and thermodynamic investigations. To the best of our knowledge, this is the first demonstration of precise arrangement of two distinct amino acid residues within a crystalline host framework, achieved by selective molecular recognition. Furthermore, because single guest adsorption of dipeptides has already been demonstrated,33 this concept could potentially be extended to precise arrangements of oligopeptides with different sequences or chirality. Therefore, these findings provide important insights into the development of highly functional porous materials based on precise arrangement of amino acids and peptides, such as enzyme-mimetic catalysts, selective separation media and custom sensors.Data availability
Data supporting this study have been included in the ESI.† Crystallographic data has been deposited at the Cambridge Crystallographic Data Centre under CCDC no. 2453192 (Z-Trp@MMF), 2255578 (Z-Trp@MMF, disordered model), 2255579 (Z-Trp-Z-Ser@MMF) and 2255580 (Z-Trp-Boc-Ser@MMF) and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
All the authors designed the project. S.T., K.N. and R.H. analyzed the results. S.T. and M.S. wrote the manuscript. K.N. and R.H. performed the experiments.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by JSPS KAKENHI (Grant No. JP16H06509 (Coordination Asymmetry)) to M. S., JP15H05478 (Encouragement of Young Scientists (A)) and JP24H01174 (2.5D Materials) to S. T., and JST PRESTO, grant number JPMJPR22A8 to S. T. We would like to thank Editage (https://www.editage.jp) for English language editing.Notes and references
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS.
- S. Liu, L. Wei, T. Zeng, W. Jiang, Y. Qiu, X. Yao, Q. Wang, Y. Zhao and Y.-B. Zhang, J. Am. Chem. Soc., 2024, 146, 34053–34063 CrossRef CAS PubMed.
- Z. Lu, H. G. W. Godfrey, I. da Silva, Y. Cheng, M. Savage, F. Tuna, E. J. L. Mclnnes, S. J. Teat, K. J. Gagnon, M. D. Frogley, P. Manuel, S. Rudić, A. J. Ramirez-Cuesta, T. L. Easun, S. Yang and M. Schröder, Nat. Commun., 2017, 8, 14212 CrossRef CAS PubMed.
- R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M. Kobayashi, H.-C. Chang, T. C. Ozawa, M. Suzuki, M. Sakata and M. Takata, Science, 2002, 298, 2358–2361 CrossRef CAS PubMed.
- V. K. Peterson, Y. Liu, C. M. Brown and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 15578–15579 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS PubMed.
- N. Hanikel, X. Pei, S. Chheda, H. Lyu, W. Jeong, J. Sauer, L. Gagliardi and O. M. Yaghi, Science, 2021, 374, 454–459 CrossRef CAS PubMed.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- L. Li, R.-B. Lin, R. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Science, 2018, 362, 443–446 CrossRef CAS PubMed.
- H. Cai, M. Li, X.-R. Lin, W. Chen, G.-H. Chen, X.-C. Huang and D. Li, Angew. Chem., Int. Ed., 2015, 54, 10454–10459 CrossRef CAS PubMed.
- S. L. Anderson, P. G. Boyd, A. Gładysiak, T. N. Nguyen, R. G. Palgrave, D. Kubicki, L. Emsley, D. Bradshaw, M. J. Rosseinsky, B. Smit and K. C. Stylianou, Nat. Commun., 2019, 10, 1612 CrossRef PubMed.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed.
- Y. Matsuda, T. Mitsuhashi, S. Lee, M. Hoshino, T. Mori, M. Okada, H. Zhang, F. Hayashi, M. Fujita and I. Abe, Angew. Chem., Int. Ed., 2016, 55, 5785–5788 CrossRef CAS PubMed.
- S. Lee, E. A. Kapustin and O. M. Yaghi, Science, 2016, 353, 808–811 CrossRef CAS PubMed.
- J.-G. Song, J. Zheng, R.-J. Wei, Y.-L. Huang, J. Jiang, G.-H. Ning, Y. Wang, W. Lu, W.-C. Ye and D. Li, Chem, 2024, 10, 924–937 CAS.
- W. He, Y. Yu, K. Iizuka, H. Takezawa and M. Fujita, Nat. Chem., 2025, 17, 653–662 CrossRef CAS PubMed.
- V. Lykourinou, Y. Chen, X.-S. Wang, L. Meng, T. Hoang, L.-J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed.
- X. Wang, L. He, J. Sumner, S. Qian, Q. Zhang, H. O'Neill, Y. Mao, C. Chen, A. M. Al-Enizi, A. Nafady and S. Ma, Nat. Commun., 2023, 14, 973 CrossRef CAS PubMed.
- D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS PubMed.
- J. Bonnefoy, A. Legrand, E. A. Quadrelli, J. Canivet and D. Farrusseng, J. Am. Chem. Soc., 2015, 137, 9409–9416 CrossRef CAS PubMed.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef CAS PubMed.
- R. Newar, N. Akhtar, N. Antil, A. Kumar, S. Shukla, W. Begum and K. Manna, Angew. Chem., Int. Ed., 2021, 60, 10964–10970 CrossRef CAS PubMed.
- H. Lyu, O. I.-F. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 2387–2396 CrossRef CAS PubMed.
- H. Cai, Y.-X. Wu, Z. Lu, D. Luo, J.-X. Sun, G.-W. Wu, M. Li, Y.-B. Wei, L.-M. Zhong and D. Li, J. Am. Chem. Soc., 2022, 144, 9559–9563 CrossRef CAS PubMed.
- Z. Wang, Y. Huang, K. Xu, Y. Zhong, C. He, L. Jiang, J. Sun, Z. Rao, J. Zhu, J. Huang, F. Xiao, H. Liu and B. Y. Xia, Nat. Commun., 2023, 14, 69 CrossRef CAS PubMed.
- S. Tashiro and M. Shionoya, Acc. Chem. Res., 2020, 53, 632–643 CrossRef CAS PubMed.
- S. Tashiro, R. Kubota and M. Shionoya, J. Am. Chem. Soc., 2012, 134, 2461–2464 CrossRef CAS PubMed.
- S. Tashiro, T. Umeki, R. Kubota and M. Shionoya, Angew. Chem., Int. Ed., 2014, 53, 8310–8315 CrossRef CAS PubMed.
- S. Tashiro, T. Umeki, R. Kubota and M. Shionoya, Faraday Discuss., 2021, 225, 197–209 RSC.
- S. Tashiro, K. Nakata, R. Hayashi and M. Shionoya, Small, 2021, 17, 2005803 CrossRef CAS PubMed.
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS PubMed.
- P. J. de Vink, J. M. Briels, T. Schrader, L.-G. Milroy, L. Brunsveld and C. Ottmann, Angew. Chem., Int. Ed., 2017, 56, 8998–9002 CrossRef CAS PubMed.
- K. O. Ramberg, S. Engilberge, T. Skorek and P. B. Crowley, J. Am. Chem. Soc., 2021, 143, 1896–1907 CrossRef CAS PubMed.
- X. Chen, Z. Huang, R. L. Sala, A. M. McLean, G. Wu, K. Sokołowski, K. King, J. A. McCune and O. A. Scherman, J. Am. Chem. Soc., 2022, 144, 8474–8479 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic analysis, preparation and characterization of the crystals, and crystallographic data could be found in the ESI. CCDC 2255578–2255580, 2453192. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00795j |
| This journal is © The Royal Society of Chemistry 2025 |