DOI:
10.1039/D5SC00892A
(Edge Article)
Chem. Sci., 2025,
16, 7751-7758
A non-isothermal water formation cell for electrochemical heat recovery†
Received
4th February 2025
, Accepted 2nd April 2025
First published on 3rd April 2025
Abstract
Low-grade heat harvesting has emerged as a promising strategy to recover waste heat into usable energy. However, most of the thermo-electrochemical approaches are limited to redox reactions involving metal ion complexes and halide species, which often exhibit low heat-to-electricity conversion efficiencies. We demonstrate a heat harvesting approach based on a non-redox reaction; water formation driven by a net-zero hydrogen redox process. Under standard conditions, its positive entropy change enables the interconversion of nearly 30% of surrounding heat into electrical energy, resulting in a thermodynamic efficiency greater than unity. This water formation-based galvanic–thermogalvanic device demonstrated a temperature-insensitive maximum power density as high as ∼33.55 mW m−2 K−2. Notably, this figure of merit is ∼70 times higher than the state-of-the-art ferrocyanide–ferricyanide-based thermogalvanic devices, thereby extending the scope of electrochemical heat harvesting beyond conventional redox processes.
Introduction
The growing global energy requirement, accelerated by population and industrial expansion, has made the need for sustainable and efficient energy solutions inevitable.1–6 Traditional energy sources, such as fossil fuels, not only have limited reserves but also contribute significantly to environmental challenges like greenhouse gas emissions and climate change.7–11 Not only limited energy stock but also the wastage of energy in the form of heat drives this energy imbalance to a different level.12 Due to these factors, low-grade heat harvesting has emerged as a promising approach to utilize or store waste heat for energy recovery and conversion.13,14 Low-grade heat harvesting deals with converting dissipated heat from industrial power plants, natural heat gradients, and daily energy dissipation into usable energy forms.15–17 Electrochemical heat harvesting capitalizes on the thermodynamic properties of electrochemical processes to convert dissipated thermal energy directly into electrical energy in a thermogalvanic cell.18–20 Based on the Seebeck effect, the thermogalvanic cell mainly focuses on the generation of additional overall cell potential due to a temperature gradient (ΔT) between two temperature-dependent redox half cells.21–23 When one side of the thermogalvanic cell is raised to a higher temperature than the other half, the temperature gradient drives a difference in the reaction potentials of the two electrodes, which causes the redox couple to generate a voltage.24 The potential (ΔE) generated, the temperature difference (ΔT) and the Seebeck coefficient (α) are related to the equation ΔE = αΔT.22–24 Solid-state thermoelectric devices can show durability at high temperatures but have high costs and limitations of suitable materials.25 Non-aqueous Li-ion-based thermogalvanic systems have good conversion efficiency, but electrolyte degradation at high temperatures and flammability of the non-aqueous solvent are a barrier for their implementation.26,27 Organic redox electrolytes with high Seebeck effects are also utilized to improve the efficiency of thermogalvanic systems, but they lack conductivity, mass transport, and non-toxic traits.28–30 Due to these reasons, aqueous thermogalvanic systems have emerged as a potential electrochemical heat harvesting device due to their low cost, safe, non-toxic electrolytes, superior ionic conductivity, high mass transport, and availability of a variety of redox systems.20,31,32 However, electrochemical heat harvesting through aqueous thermogalvanic systems faces several challenges, including low heat-to-power conversion efficiency, limited partial molar entropy change, reduced solubility of redox couples, lower potential output, the low boiling point of electrolytes, and temperature-induced electrolyte degradation.19,33–35
In this study, we present an approach to heat harvesting using a non-redox reaction, specifically the water formation reaction resulting from the recombination of H+ and OH− ions, which can extract heat from the surroundings and convert it into an electrochemical driving force due to its positive entropy change and positive Seebeck coefficient, Calculation S1, ESI.† However, the non-redox nature of the water formation reaction poses a critical challenge for its harvesting as an electrical driving force. We have recently shown that the introduction of a hydrogen redox can aid the direct harvesting of water formation energy as an electrical driving force without a net redox.36–39 Our thermodynamic analysis (Calculation S1, ESI†) indicates that the positive entropy change associated with the water formation reaction allows for the harvesting of approximately 30% of the energy from the surroundings during its interconversion. We demonstrate that, for electrochemical water formation driven by hydrogen redox reactions, there is an inherent asymmetry in the temperature dependence of the electromotive force for the corresponding half-cell reactions. This asymmetry presents opportunities to harness heat gradients as an electromotive force in a non-isothermal electrochemical water formation cell. The incorporation of temperature gradient in a water formation cell maximizes the overall electrical energy output of the thermogalvanic device with a temperature-insensitive maximum power density as high as ∼33.55 mW m−2 K−2, which is around 70 times higher than the state-of-the-art ferrocyanide–ferricyanide-based thermogalvanic devices. This method overcomes several challenges associated with previously reported thermogalvanic devices, including low power and voltage outputs, reduced heat-to-electricity conversion efficiency, solubility issues with the redox couples, and an extremely low temperature-insensitive maximum power density.
Results and discussion
The general configuration of the non-isothermal water formation reaction-based thermogalvanic device consists of a cold anodic alkaline half-cell and a hot cathodic acidic half-cell. These half-cells are ionically separated by a Nafion-117 cation-conducting membrane, Scheme 1. In the anodic half-cell, Pt supported on carbon (Pt/C) is the anodic electrocatalyst, while in the cathodic half-cell, platinum electrodeposited on a titanium mesh (using a constant current of 4 mA cm−2 for 5 minutes) is used as the electrocatalyst. Due to the non-redox nature of the water formation reaction, we employed a hydrogen redox reaction within an acid–alkali decoupled architecture to harness water formation energy as an electrical driving force, all without a net redox process. In an alkaline medium, hydrogen redox reactions exhibit a relatively negative potential, whereas they show a relatively positive potential in an acidic medium (Fig. S1, ESI†). Based on the pH dependence of these reactions, we utilized the alkaline side as the anodic half-cell and the acidic side as the cathodic half-cell for the direct extraction of energy from water formation. This setup indicates that when hydrogen oxidation occurs in the alkaline half-cell (eqn (1)), the released electrons flow through the external circuit to power the load, while a corresponding hydrogen evolution reaction (HER) takes place in the cathodic half-cell (eqn (2)). The overall reaction (eqn (3)) does not consume hydrogen; instead, it results in the formation of water molecules. Consequently, the energy associated with water formation acts as the driving force for the operation of this electrochemical device.| | | Anodic half-cell reaction: H2 + 2OH− ⇄ 2H2O + 2e− | (1) |
| | | Cathodic half-cell reaction: 2H+ + 2e− ⇄ H2 | (2) |
| | | Overall cell reaction: 2H+ + 2OH− ⇄ 2H2O | (3) |
 |
| | Scheme 1 Schematic diagram of electrochemical water formation assisted galvanic–thermogalvanic device for waste heat recovery. | |
The electrochemical device designed to harvest the water formation energy via a hydrogen redox exhibits a peak power density of approximately 43 mW cm−2 at a peak current density of ∼107 mA cm−2 under room temperature and pressure, as shown in Fig. 1a (Blue trace). The polarization plot (orange trace in Fig. 1a and S2, ESI†) reveals that, without an H2 supply at the anodic half-cell of the device, no significant power or current density is generated. The galvanostatic polarization data, at a constant current density of 40 mA cm−2 (Fig. 1b) shows promising voltage output for a long duration. During galvanostatic discharge, the concentrations of H+ in the catholyte and OH− in the anolyte were monitored over time, and the post-operation concentrations of the anolyte and the catholyte were measured via acid–base titration (Fig. 1c). As the current is drawn from the device, the H+ and the OH− are consumed in accordance with the amount of charge passed during the discharge, Fig. 1d. Consequently, the cell potential decreases with time, as it is directly related to ΔpH between the two half-cells of the water formation energy harvesting device, Calculation S2, ESI.†Fig. 1b and c show that the electromotive force diminishes to nearly zero after ∼12.5 hours of operation at a constant current density, and the pH gradient converges to nearly zero. Notably, under open-circuit conditions, the concentration changes of H+/OH− ions were negligible over 48 hours (Fig. S3, ESI†), suggesting the self-discharge via the chemical recombination between the two half-cells is negligible. During the chronopotentiometry for the device (Fig. S4, ESI†), the in situ electrochemical mass spectrometry of the device's cathodic half-cells revealed H2 gas evolution at the cathodic exhaust, orange trace in Fig. 1e. When the cell is ON, the partial pressure of the H2 gas rises in the cathodic half-cell of the water formation energy harvesting device, Fig. 1e. Commensurately, when the cell is ON, the anodic half-cell demonstrates a decrease in the partial pressure of hydrogen, blue trace in Fig. 1e. These attest to the fact that there is no net hydrogen consumption during the operation of the device. Further, the quantity of generated hydrogen gas during the galvanostatic polarization (Fig. 1b) is ∼331 mL (13.5 millimoles) with ∼96.2% faradaic efficiency, Fig. 1f (Calculation S3, ESI†). Overall, these behaviours underscore the inevitability of H+/OH− gradients and the H2 redox in the operation of this water formation device.
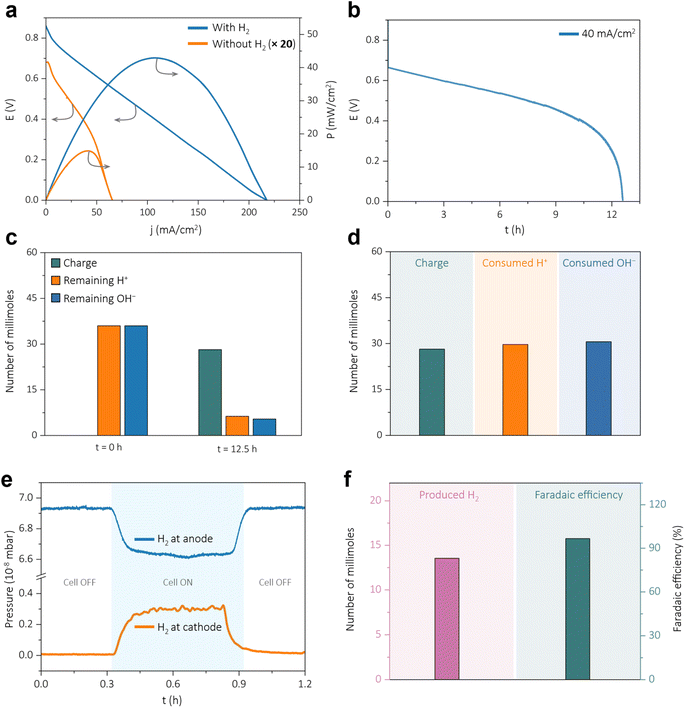 |
| | Fig. 1 (a) Polarization curve for water formation energy harvesting device with and without H2 as anodic feed gas stream (10 mL min−1). (b) Galvanostatic polarization at a constant current density of 40 mA cm−2 for water formation energy harvesting device with H2 as anodic feed gas stream (10 mL min−1) (c) correlation between the charge passed and the remaining amount of H+/OH− dual ion concentrations in the specific half cells after the long-term operation. (d) Consumption of H+ (catholyte) and OH− (anolyte) with respect to the amount of charge passed during galvanostatic discharge at a constant current density of 40 mA cm−2. (e) In situ electrochemical mass spectrometry of the cathodic half and the anodic half of the water formation cell. (f) Quantification and faradaic efficiency of the produced hydrogen. | |
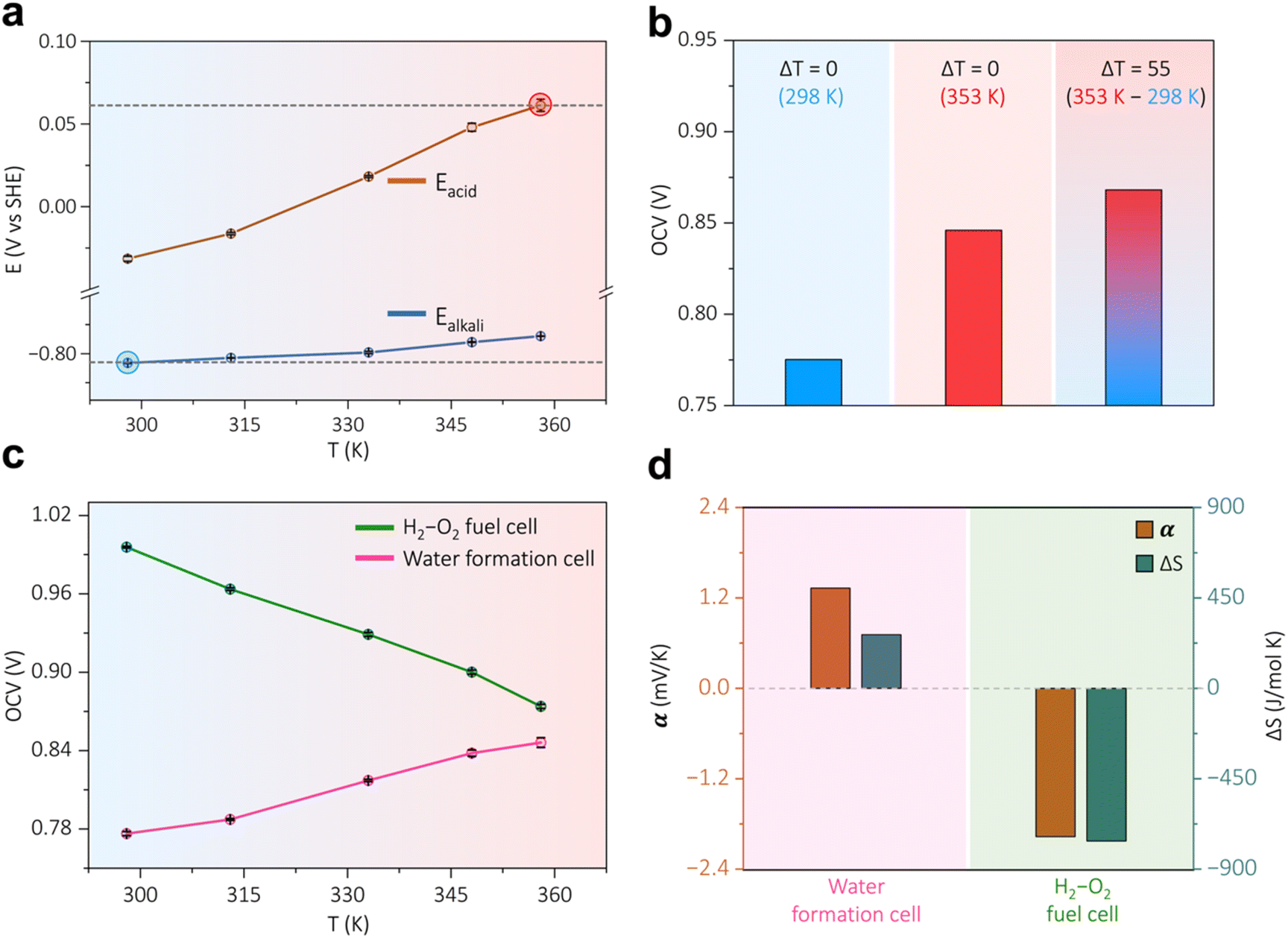
As already shown in Calculation S1, ESI,† the electrochemical water formation route has a positive entropy change; hence, this reaction can harvest heat from the surroundings directly as an electromotive force. A thermodynamic calculation shows that nearly 30% of the cell voltage or free energy is derived from the surroundings at room temperature and pressure, Calculation S1, ESI.† To investigate the influence of temperature on the water formation reaction, the temperature dependence of each half-cell reaction of the water formation cell is investigated. In the case of the acidic cathodic half-cell, it can be observed that the potential of hydrogen redox increased commensurately with the rise in temperature, (brown trace in Fig. 2a). When the surrounding temperature is increased from 298 K to 358 K, the potential of hydrogen redox in the acidic medium is commensurately increased by nearly 90 mV, Fig. S5, ESI.† This leads to a positive Seebeck coefficient of 1.62 ± 0.11 mV K−1 for the cathodic half-cell, which holds the potential to be utilized in a thermogalvanic cell. On the other hand, the alkaline hydrogen redox shows a moderate increment of potential with the rise in temperature (blue trace in Fig. 2a and S6, ESI†), resulting in a Seebeck coefficient of 0.42 ± 0.06 mV K−1. This indicates an inherent asymmetry in the temperature dependence of half-cell reaction voltages, likely due to the significantly higher mobility of protons compared to hydroxide ions.40,41 Conversely, the viscosity of the acidic solution decreases more significantly with rising temperature compared to that of the alkaline solution, which is expected to further enhance the ionic mobility and the voltage output. This increased proton mobility may contribute to a rise in entropy, subsequently enhancing the Seebeck coefficient. This ultimately provides us the opportunity to fabricate a water formation reaction-based galvanic–thermogalvanic device if the alkaline anode and acidic cathode are kept in cold and hot environments respectively (Fig. 2a). The open circuit voltage for the water formation device with a temperature gradient (ΔT = 55 K) is superior to the open circuit voltages under isothermal conditions, Fig. 2b. Although the open circuit voltage of the water formation cell increases with the surrounding temperature (pink trace in Fig. 2c), providing the ability to draw energy from the environment—unlike traditional H2–O2 fuel cells (green trace Fig. 2c and S7, ESI†), which lose energy as heat during operation. The inherent asymmetry in the temperature dependence of the half-cell reactions suggests that the voltage output of the device can be maximized in a non-isothermal configuration. This can be achieved by maintaining a temperature gradient, as indicated in Fig. 2a and b, rather than in an isothermal configuration.
 |
| | Fig. 2 (a) The variation of the potential of hydrogen redox in acidic (brown trace) and alkaline half-cells (blue trace) with respect to the surrounding temperature. (b) The net cell voltage of the device at non-isothermal conditions (ΔT = 55 K) and isothermal conditions (ΔT = 0 K). (c) The temperature dependency of open circuit voltage (OCV) for the water formation cell and an H2–O2 fuel cell. (d) The comparison of the Seebeck coefficient and partial molar entropy change for the water formation cell (WC) and the H2–O2 fuel cell (FC). | |
The water formation cell has a positive Seebeck coefficient value of 1.30 ± 0.12 mV K−1 along with partial molar entropy change of around 251 J mol−1 K−1 (Fig. 2d, Calculation S4, ESI†), which consolidates the fact of a thermodynamic efficiency value greater than 1, Calculation S1, ESI.† On the other hand, the H2–O2 fuel cell possesses a negative Seebeck coefficient with a magnitude of −1.96 ± 0.06 mV K−1 with a negative entropy change of – 756.43 J mol−1 K−1 (Calculation S4, ESI†), Fig. 2d. This attests to the fact that electrical energy output from a water-formation cell will increase with an increment in the temperature, which is not the case for the H2–O2 fuel cell. The whole cell OCV (voltage) vs. T (temperature) plot (Fig. 2c) is derived for both the devices by subtracting each potential value of the half cells (Fig. 2a, S6, and S7†) at respective temperatures. Nevertheless, as discussed before, in a non-isothermal configuration, the voltage output can be further enhanced (Fig. 2a and b). In order to experimentally validate this in the device mode, the open circuit voltage of the water formation cell was measured with, and without a temperature gradient between the two half cells, Fig. 3a. The temperature gradient (ΔT = 55 K) in the water formation cell leads to a rise in the open circuit voltage of the device with a magnitude of 73 mV, Fig. 3a. The polarization curve with a temperature gradient of 55 (orange trace in Fig. 3b) for this hybrid galvanic–thermogalvanic device based on water formation reaction exhibits superiority over the isothermal water formation cell, (blue trace in Fig. 3b). The peak power density has risen from 43 mW cm−2 to 53 mW cm−2 with the inclusion of a temperature gradient of 55, Fig. 3b. The overall energy output from this galvanic–thermogalvanic device, which relies on the water formation reaction, comprises both thermal and galvanic contributions. As illustrated in Calculation S5, ESI,† these contributions can be decoupled, revealing approximately 20% from thermal sources and 80% from galvanic sources, as shown in Fig. 3c (Calculation S5, ESI†). This suggests that a water formation-based hybrid galvanic–thermogalvanic device can boost the power density by nearly 24% with the inclusion of a temperature gradient (ΔT = 55 K), Calculation S5, ESI.† The galvanostatic polarization at a constant current density of 40 mA cm−2 also reflects higher performance metrics when there is a temperature gradient, Fig. 3d. The cathodic exhaust of the thermogalvanic cell has been investigated by in situ electrochemical mass spectrometry at a constant current of 40 mA cm−2, and in both cases, there is an evolution of pure H2 in the cathodic exhaust, Fig. 3e. It should be noted that an almost similar amount of hydrogen gas has evolved, however, with a higher potential output for the non-isothermal device, Fig. 3e and f.
 |
| | Fig. 3 (a) Open circuit voltages for the water formation-based thermogalvanic devices when kept at non-isothermal conditions (TA = 298 K, TC = 353 K) and isothermal conditions (TA and TC = 298 K). (b) Polarisation curves, (c) thermal and galvanic power output contributions, (d) chronopotentiometry at 40 mA cm−2, and (e) in situ electrochemical mass spectra of the cathodic half-cell of the water formation-based thermogalvanic device under non-isothermal conditions (TA = 298 K, TC = 353 K) and isothermal conditions (TA = TC = 298 K). (f) Hydrogen gas quantification and charge passed during the operation of the water formation-based device under non-isothermal conditions (ΔT = 55 K) and isothermal conditions (ΔT = 0 K). | |
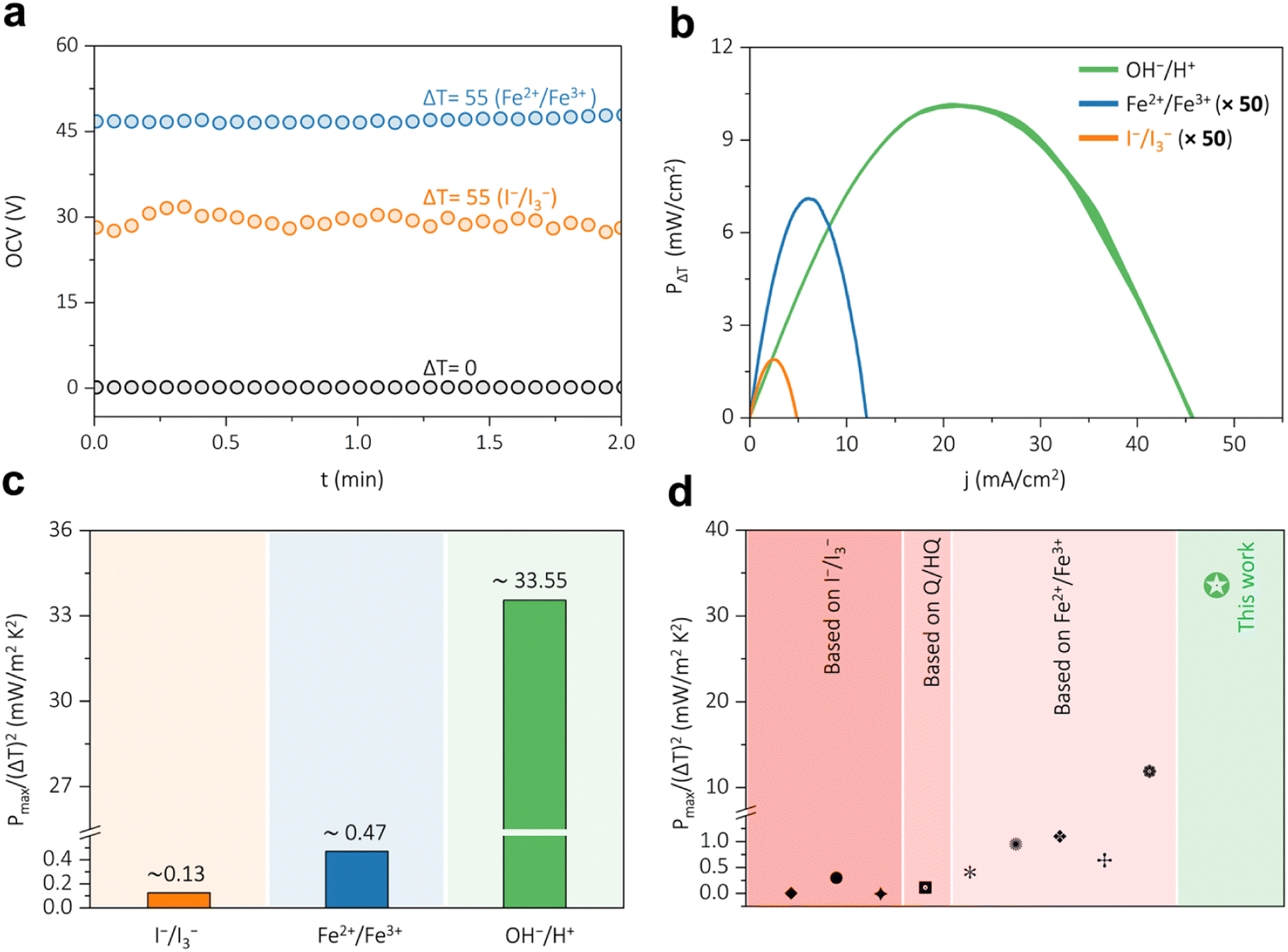
To demonstrate where this water formation reaction-based thermogalvanic cell stands, it is compared first with a state-of-the-art aqueous thermogalvanic device based on the ferrocyanide/ferricyanide (Fe2+/Fe3+). Initially, the temperature dependence was investigated with the equimolar ferrocyanide/ferricyanide (0.4 M Fe2+/0.4 M Fe3+) system, which shows a drop in the electromotive force with respect to a rise in temperature, Fig. S8, ESI.† The Seebeck coefficient of this system turns out to be negative in nature with a magnitude of −1.49 ± 0.13 mV K−1, which is opposite in nature with respect to the water formation reaction. The device consisting of a ferrocyanide/ferricyanide (Fe2+/Fe3+) system shows no appreciable open circuit voltage (OCV) under an isothermal state (ΔT = 0 K), black trace in Fig. 4a. Introduction of a temperature gradient of 55 K, in this ferrocyanide/ferricyanide redox system, led to the development of an open circuit voltage of nearly 47 mV for the (blue trace in Fig. 4b). The performance of the water formation cell is also compared with iodide/triiodide (I−/I3−) redox system and under a temperature gradient of 55 K, it demonstrated an OCV of nearly 30 mV (orange trace in Fig. 4b). The polarization curve for ferrocyanide/ferricyanide-based (Fe2+/Fe3+) thermogalvanic system provides a peak power density of ∼0.15 mW cm−2 at a peak current density of 6 mA cm−2, blue trace in Fig. 4b and S9, ESI.† Similarly, the thermogalvanic device based on iodide/triiodide (I−/I3−) redox system delivers a peak power density of ∼0.04 mW cm−2 at a peak current density of 2.43 mA cm−2, orange trace in Fig. 4c and S10, ESI.† A comparison of the thermal contributions of the studied thermogalvanic devices shows that the water formation-based galvanic–thermogalvanic device has an upper hand in interconverting heat to electricity, Fig. 4b. It should be noted that the performance metrics of the thermogalvanic devices based on the ferrocyanide/ferricyanide (Fe2+/Fe3+) and the iodide/triiodide (I−/I3−) redox system are displayed with a multiplication of 50, Fig. 4b and c, for comparative purposes. Furthermore, the thermal contribution of the water formation device is obtained from Fig. 3b by subtracting the current density and power density under non-isothermal conditions (ΔT = 55 K) and isothermal conditions (ΔT = 0). The temperature-insensitive maximum power density,18,22 which reflects the true performance metrics of a thermogalvanic device, is determined for the studied thermogalvanic systems, Fig. 4c (Calculation S6, ESI†). For the water formation cell, the temperature-insensitive maximum power density was calculated by subtracting the polarization curve under non-isothermal conditions (ΔT = 55 K) and isothermal conditions (ΔT = 0 K), which corresponds to the area of the shaded portion in Fig. 3b. This is expected to provide performance only due to the temperature gradients. Comparing the systems, the thermogalvanic device based on the water formation reaction outperforms the ferrocyanide/ferricyanide and the iodide-triiodide-based systems, Fig. 4c. The water formation-based thermogalvanic device exhibits a temperature-insensitive maximum power density of ∼33.55 mW m−2 K−2, which is nearly 70 times greater than the better-performing state-of-the-art ferrocyanide–ferricyanide based thermogalvanic system, Fig. 4c. The performance of this water formation reaction-based thermogalvanic device is also compared with various thermogalvanic devices reported in the literature,22,28,30,42–44 which demonstrate significance performance enhancement for heat harvesting through the water formation reaction, Fig, 4d and Table S1, ESI.† This enhancement in temperature-insensitive maximum power density and performance metrics is majorly attributed to the significantly higher solubility of H+ and OH− dual ions, their anomalously high ionic conductivities, and the higher entropy change values associated with the water formation reaction. Taken together; the water formation reaction-based thermogalvanic system holds promise for efficiently interconverting low-grade heat to electrical driving force via a pH gradient.
 |
| | Fig. 4 (a) The temperature dependency of the potential for the ferrocyanide/ferricyanide (0.4 M Fe2+/0.4 M Fe3+) and iodide/triiodide (0.8 M I−/I3−) redox systems and their corresponding open circuit voltages under isothermal and non-isothermal conditions. (b) Polarisation curves demonstrating the thermal contributions of the ferrocyanide/ferricyanide system, iodide/triiodide redox system, and water formation-based thermogalvanic device. The thermal contribution of the water formation device is obtained from Fig. 3b by subtracting the current density and power density under non isothermal conditions (ΔT = 55 K) and isothermal conditions (ΔT = 0 K). The power contributions of ferrocyanide/ferricyanide and iodide/triiodide systems are multiplied by 50 for comparative purposes. (c) Comparison of the temperature-insensitive maximum power density for the non-isothermal water formation cell, iodide/triiodide redox system, and ferrocyanide/ferricyanide redox system-based thermogalvanic devices. For the water formation cell, the temperature-insensitive maximum power density was calculated by subtracting the peak power density under non-isothermal conditions (ΔT = 55 K) and isothermal conditions (ΔT = 0 K). (d) Thermogalvanic device performance comparison of water formation cell with state-of-the-art systems reported in the literature, including the iodide/triiodide (I−/I3−), quinone/hydroquinone (Q/HQ), and the ferrocyanide/ferricyanide (Fe2+/Fe3+) redox systems (ref. 22, 28, 30 and 42–44). | |
Conclusions
Electrochemical heat harvesting through thermogalvanic cells exploits temperature gradients (ΔT) to generate electrical energy based on the Seebeck effect. The water formation reaction in an electrochemical cell has a positive Seebeck coefficient of nearly 1.30 mV K−1, which creates an opportunity for heat harvesting by decoupling the direct acid–alkali chemistry. The pH-dependent hydrogen redox has an inherent asymmetry in their temperature dependent response that in turn provide amble opportunities for waste heat recovery. An alkaline hydrogen redox at cold temperatures and an acidic hydrogen redox at hot temperatures noticeably boost voltage and electrical energy output in a non-isothermal water formation cell. This thermogalvanic device has a high temperature-insensitive maximum power output of ∼33.55 mW m−2 K−2, which is nearly 70 times higher than state-of-the-art ferricyanide-based heat harvesting devices, thereby holding promise for enhanced waste heat recovery. Since the proposed system is an aqueous thermogalvanic device, sustaining a temperature gradient, in the long run, is challenging due to heat dissipation at the membrane junction. In this context, the development of gel-based electrolytes may serve as a promising approach for limiting heat dissipation.32
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
R. M and S. S. Y. performed the experiments and analyzed the data with B. N. and H. P. M. O. T. supervised the work and wrote the manuscript with input from all the authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. O. T. is indebted to DST/TMD-HFC/2K18/58, DST-SERB (CRG/2020/002549), and DST-WTI (DST/TMD-EWO/WTI/2K19/EWFH/2019/272) for financial support. R. M. and S. S. Y acknowledges IISER Pune, India for fellowship and instrumental facilities.
References
- A. R. Kottaichamy, J. Tzadikov, A. Pedersen, J. Barrio, G. Mark, I. Liberman, A. Upcher, M. Volokh, I. Hod, S. Barzilai, M. Noked and M. Shalom, Adv. Energy Mater., 2024, 14, 2403817 CAS.
- A. V. Desai, R. Ettlinger, H. S. Seleghini, M. G. Stanzione, J. M. Cabañero, S. E. Ashbrook, R. E. Morris and A. R. Armstrong, J. Mater. Chem. A, 2024, 12, 12119–12125 CAS.
- S. Jerez, A. Pedersen, M. Ventura, L. Mazzoli, M. I. Pariente, M. Titirici, J. A. Melero and J. Barrio, Electrochim. Acta, 2024, 483, 144045 CAS.
- R. B. Jethwa, S. Mondal, B. Pant and S. A. Freunberger, Angew. Chem., Int. Ed., 2024, 63, e202316476 CrossRef CAS.
- Z. Li, K. Mali, P. Hapiot, S. De Feyter, A. J. Attias and S. F. L. Mertens, Adv. Funct. Mater., 2024, 2315861, 4–13 Search PubMed.
- E. Mourad, L. Coustan, P. Lannelongue, D. Zigah, A. Mehdi, A. Vioux, S. A. Freunberger, F. Favier and O. Fontaine, Nat. Mater., 2017, 16, 446–454 CrossRef CAS.
- A. H. Faqeeh and M. D. Symes, Electrochim. Acta, 2024, 493, 144345 CrossRef CAS.
- X. Wang, C. Zhang, M. Sawczyk, J. Sun, Q. Yuan, F. Chen, T. C. Mendes, P. C. Howlett, C. Fu, Y. Wang, X. Tan, D. J. Searles, P. Král, C. J. Hawker, A. K. Whittaker and M. Forsyth, Nat. Mater., 2022, 21, 1057–1065 CAS.
- E. H. Balaguera and J. Bisquert, Small, 2024, 1–8 Search PubMed.
- P. Prakash, B. Fall, J. Aguirre, L. A. Sonnenberg, P. R. Chinnam, S. Chereddy, D. A. Dikin, A. Venkatnathan, S. L. Wunder and M. J. Zdilla, Nat. Mater., 2023, 22, 627–635 CAS.
- L. Schulte, W. White, L. A. Renna and S. Ardo, Joule, 2021, 5, 2380–2394 CAS.
- Ø. Hodnebrog, G. Myhre, C. Jouan, T. Andrews, P. M. Forster, H. Jia, N. G. Loeb, D. J. L. Olivié, D. Paynter, J. Quaas, S. P. Raghuraman and M. Schulz, Commun. Earth Environ., 2024, 5, 1–9 Search PubMed.
- A. G. Olabi, K. Elsaid, E. T. Sayed, M. S. Mahmoud, T. Wilberforce, R. J. Hassiba and M. A. Abdelkareem, Nano Energy, 2021, 84, 105871 CAS.
- P. Simeoni, G. Ciotti, M. Cottes and A. Meneghetti, Energy, 2019, 175, 941–951 Search PubMed.
- R. Agathokleous, G. Bianchi, G. Panayiotou, L. Aresti, M. C. Argyrou, G. S. Georgiou, S. A. Tassou, H. Jouhara, S. A. Kalogirou, G. A. Florides and P. Christodoulides, Energy Procedia, 2019, 161, 489–496 CAS.
- C. Gao, S. W. Lee and Y. Yang, ACS Energy Lett., 2017, 2, 2326–2334 CAS.
- S. Pu, Y. Liao, K. Chen, J. Fu, S. Zhang, L. Ge, G. Conta, S. Bouzarif, T. Cheng, X. Hu, K. Liu and J. Chen, Nano Lett., 2020, 20, 3791–3797 Search PubMed.
- J. Duan, B. Yu, L. Huang, B. Hu, M. Xu, G. Feng and J. Zhou, Joule, 2021, 5, 768–779 Search PubMed.
- T. I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CAS.
- M. A. Buckingham, K. Laws, H. Li, Y. Kuang and L. Aldous, Cell Rep. Phys. Sci., 2021, 2, 100510 Search PubMed.
- H. Zhou, T. Yamada and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 10502–10507 CAS.
- J. Duan, G. Feng, B. Yu, J. Li, M. Chen, P. Yang, J. Feng, K. Liu and J. Zhou, Nat. Commun., 2018, 9, 1–8 Search PubMed.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2013, 6, 2639–2645 RSC.
- S. W. Lee, Y. Yang, H. W. Lee, H. Ghasemi, D. Kraemer, G. Chen and Y. Cui, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- L. Yang, H. Sun, S. Wang, L. Jiang and G. Sun, Int. J. Hydrogen Energy, 2017, 42, 25877–25881 CrossRef CAS.
- N. S. Hudak and G. G. Amatucci, J. Electrochem. Soc., 2011, 158, A572 CrossRef CAS.
- L. B. Hubrechtsen, L. L. De Taeye and P. M. Vereecken, Chem. Mater., 2023, 35, 5612–5630 Search PubMed.
- W. Fang, H. Luo, I. Mwamburi Mwakitawa, F. Yuan, X. Lin, Y. Wang, H. Yang, T. Shumilova, L. Hu, Y. Zheng, C. Li, J. Ouyang and K. Sun, ChemSusChem, 2024, e202401749 Search PubMed.
- H. Chen, H. Zou, F. Zhong, M. Qu, S. Zhao, X. Wei, D. Hong, Y. Song and Z. Liu, Nano Energy, 2024, 129, 109992 CrossRef CAS.
- T. Kim, J. S. Lee, G. Lee, H. Yoon, J. Yoon, T. J. Kang and Y. H. Kim, Nano Energy, 2017, 31, 160–167 CrossRef CAS.
- C. G. Han, Y. Bin Zhu, L. Yang, J. Chen, S. Liu, H. Wang, Y. Ma, D. Han and L. Niu, Energy Environ. Sci., 2023, 17, 1559–1569 Search PubMed.
- X. He, H. Sun, Z. Li, X. Chen, Z. Wang, Y. Niu, J. Jiang and C. Wang, J. Mater. Chem. A, 2022, 10, 20730–20755 Search PubMed.
- S. Jia, W. Qian, P. Yu, K. Li, M. Li, J. Lan, Y. H. Lin and X. Yang, Mater. Today Phys., 2024, 42, 101375 CrossRef CAS.
- J. Zhang, X. Li, J. Zheng, Z. Hu, Y. Xiao, W. Yang, S. Zhang, Z. Zhou and S. Han, Sol. Energy, 2024, 282, 112939 CrossRef CAS.
- W. Li, J. Ma, J. Qiu and S. Wang, Mater. Today Energy, 2022, 27, 101032 CrossRef CAS.
- R. Mondal, R. Thimmappa, B. Nayak, A. Dewan, M. C. Devendrachari, Q. Chen, Z. Wen and M. O. Thotiyl, Energy Environ. Sci., 2023, 16, 3860–3872 Search PubMed.
- Z. M. Bhat, D. Pandit, S. Ardo, R. Thimmappa, A. R. Kottaichamy, N. Christudas Dargily, M. C. Devendrachari and M. Ottakam Thotiyl, Joule, 2020, 4, 1730–1742 CrossRef CAS.
- R. Mondal, B. Nayak and M. Ottakam Thotiyl, J. Phys. Chem. Lett., 2024, 15, 6866–6871 CrossRef CAS.
- Z. Manzoor Bhat, R. Thimmappa, M. C. Devendrachari, A. R. Kottaichamy, S. P. Shafi, S. Varhade, M. Gautam and M. O. Thotiyl, J. Phys. Chem. Lett., 2018, 9, 388–392 CrossRef CAS.
- D. Marx, A. Chandra and M. E. Tuckerman, Chem. Rev., 2010, 110, 2174–2216 CAS.
- N. Agmon, H. J. Bakker, R. K. Campen, R. H. Henchman, P. Pohl, S. Roke, M. Thämer and A. Hassanali, Chem. Rev., 2016, 116, 7642–7672 CAS.
- H. Zhou, T. Yamada and N. Kimizuka, Sustainable Energy Fuels, 2018, 2, 472–478 CAS.
- H. Wang, X. Zhuang, W. Xie, H. Jin, R. Liu, B. Yu, J. Duan, L. Huang and J. Zhou, Cell Rep. Phys. Sci., 2022, 3, 100737 CAS.
- J. Wang, Y. Song, F. Yu, Y. Zeng, C. Wu, X. Qin, L. Peng, Y. Li, Y. Zhou, R. Tao, H. Liu, H. Zhu, M. Sun, W. Xu, C. Zhang and Z. Wang, Nat. Commun., 2024, 15, 1–9 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*