
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromotion or suppression of hydrogen evolution activity? The competition between sodium cations and quaternary ammonium ions at the metal/water interface†
Shilin
Bo
,
Yang
Xiang
 ,
Qiong
Xiang
,
Li
Li
,
Xun
Huang
* and
Zidong
Wei
*
,
Qiong
Xiang
,
Li
Li
,
Xun
Huang
* and
Zidong
Wei
*
Center of Advanced Electrochemical Energy, State Key Laboratory of Advanced Chemical Power Sources, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 40004, P. R. China. E-mail: huangxun@cqu.edu.cn; zdwei@cqu.edu.cn
First published on 11th April 2025
Abstract
Quaternary ammonium salts (QASs) are frequently utilized to modulate the structure of the cathodic electric double layer in processes such as water electrolysis and hydrogenation reactions. However, literature reports have shown that QASs can both suppress and promote hydrogen evolution activity, yet the underlying mechanisms remain incompletely understood. In this study, we experimentally observed that the presence of QASs alone accelerates hydrogen evolution compared to NaOH solutions. Conversely, when the QAS is combined with Na+ or H+, it inhibits hydrogen evolution. Ab initio molecular dynamics simulation and surface-enhanced infrared absorption spectroscopy results indicate that Na+ ions disrupt the hydrogen bond network at the interface, leading to a disorder in the water chain arrangement. In contrast, QASs enhance the hydrogen bond network, thereby facilitating the hydrogen evolution reaction. However, coexistence of Na+ and QASs leads to hydration competition, creating gaps in the hydrogen bond network near the surface and impeding hydrogen transport. These findings enhance our understanding of QASs in hydrogen evolution and guide future interface modulation strategies.
1. Introduction
Electrochemical processes, such as water electrolysis,1,2 and electrosynthesis, and devices such as fuel cells and batteries, have garnered substantial interest for their promise in addressing energy and environmental challenges.3–6 Understanding the microstructure, charge and mass transport mechanisms, and reaction kinetics is crucial to enhance their energy efficiency.7–11 Water, the ubiquitous solvent in electrochemical systems, hosts a hydrogen bond network at the electrode/electrolyte interface that profoundly influences charge and hydrogen transfer, ultimately dictating the rates of not just hydrogen evolution (HER) and oxidation (HOR) reactions,1,12–15 but also those involving CO2 and unsaturated organic hydrogenation.16–19Altering the electrolyte composition is a simple yet effective strategy to engineer the electrode/electrolyte interfacial structure and thereby tune HER activity. Extensive literature studies have demonstrated a clear cation-dependent HER activity trend (for example, Li+ > Na+ > K+ > Cs+) in alkaline electrolytes. This phenomenon is mechanistically attributed to two interrelated factors: (1) smaller cations (e.g., Li+) preferentially lower the water dissociation energy by destabilizing HO–H bonds at catalytic interfaces through strong non-covalent interactions;20 (2) compact cations like Li+ enhance the dynamic reorganization of interfacial water molecules, facilitating rapid proton transfer via optimized hydrogen-bond relays. In contrast, larger cations (e.g., Cs+) disrupt the continuity of hydrogen-bond networks, introducing kinetic bottlenecks.21,22 Recent advances further highlight the critical role of the outer Helmholtz plane (OHP) in governing HER kinetics. Li et al.23,24 proposed that the interfacial proton transfer efficiency in the OHP layer is more vital in determining the HER activity. Tang et al.25 also proved that multivalent Mg2+ with smaller ion radii outperform monovalent Na+ and larger Ca2+ in enhancing hydrogen-bond connectivity, likely due to their stronger polarizing power and optimized interfacial hydration structures.
In the area of electrosynthesis,18,26–29 water acts as a proton donor to produce chemicals and fuels, while its participation in the HER needs to be avoided.30 One well-known example is the electrochemical hydrodimerization of acrylonitrile, which is an industrial route developed by Monsanto to produce adiponitrile, the precursor of nylon-6.6.13,31,32 A small amount of quaternary ammonium salts (QASs) was added into the electrolyte, which greatly improves the selectivity and Faraday efficiency of adiponitrile.6,33–35 It has been widely accepted that QASs can assemble at the Cd cathode/water interface, generating a hydrophobic layer that inhibits the adsorption of hydrogen, thus preventing side reactions including the HER and direct protonation of acrylonitrile to form propionitrile. A similar effect of QASs can also be found in the hydrogenation of CO,36 CO2,37 aldehydes,38etc. For example, during the electrocatalytic reduction of CO, it was found the interaction between the interfacial water molecules and the CO adsorbed on the surface is critical for the formation of ethylene. Larger cations (e.g., propyl4N+ and butyl4N+) interrupt such interactions, and thus inhibit ethylene production.32,36 Meanwhile, Wang et al.37 studied how the interfacial water structure on the surface of Cu(111)@PDMS affects the selectivity of the CO2 reduction reaction. They found that the hydrophobic electrodes formed a strong hydrogen bond structure, which hinders the rearrangement of water molecules, increases the metal-hydrogen distance, and finally inhibits the kinetic process of hydrolysis dissociation and hydrogen evolution reactions.17,39
Recently, it was also reported that the addition of QASs can effectively establish a strengthened hydrogen bonding network.40–42 For example, the reaction rate of the HER in alkaline media is two orders of magnitude lower than that under acidic conditions,43 attributed to the disruption of the hydrogen bond network by alkali metal cations,21,44 Zhao et al. demonstrated that large organic cations such as tetraethylammonium (TEA) and tetrapropylammonium (TPA) can foster long-range ordering of interfacial water molecules, generating hydrogen-bonded water chains that bridge the electrode and bulk electrolyte, thereby leading to faster HER and HOR kinetics then observed for K+.33 Fernández-Vidal and Koper further revealed that the acceleration of tetrabutylammonium is more obvious at higher concentrations, attributed to its accumulation in the diffusion layer.45
The above discussion reveals that, in some cases organic cations can improve the HER by enhancing the hydrogen bond network, while in other cases organic cations inhibit the HER due to their hydrophobicity, and a full understanding of the role of QASs in the interfacial hydrogen bond network under different conditions is lacking. In this work, we investigated the impact of QASs on the HER in acidic and alkaline media across different cathodes (Pt and Cd). Kinetic experiments showed that HER activity follows the order of TPA > TEA > TMA > NaOH, implying an enhancement by QASs. However, in NaOH solution, the addition of QASs significantly decreases HER activity, and this suppression is more obvious with longer alkyl side chains. Machine learning accelerated ab initio molecular dynamics (ML-AIMD) analysis together with surface-enhanced infrared absorption spectroscopy (SEIRAS) revealed that alkali metal cations such as Na+ disrupt the interfacial hydrogen bond network, causing water chain disorder. Conversely, QASs enhance the hydrogen bond network, thereby promoting the HER. However, when Na+ and QASs coexist, the competition of hydration between QASs and Na+ leaves a gap in the hydrogen bond network near the surface, which hinders the transportation of protons through the hydrogen bond. These findings enhance our comprehension of the role of QASs in organic synthesis and steer interface modulation strategies.
2. Methods
2.1 Computational details
The Pt/water interface is simulated using explicit solvent model and a four-layer, 4 × 4 orthogonal Pt (111) slab, possessing a surface area of 1.23 nm2. Similarly, for the Cd/water interface, a four-layer, 2 × 4 orthogonal Cd (101) slab with a surface area of 1.51 nm2 is utilized. As shown in Fig. S1 and S2,† both metal/water interfaces are characterized by a water layer and a vacuum layer, each measuring 15 Å in thickness, contributing to an overall height of 35 Å along the z-axis. To account for the effect of long-range water interaction, an implicit solvation model is implemented within the vacuum layer. All the ab initio computational molecular dynamics (AIMD) simulations are performed using the Vienna Ab initio Simulation Package (VASP).34,46,47 To enhance computational efficiency, a machine learning approach implanted in the VASP is employed, leveraging existing data to rapidly predict energy and force (Fig. S3†).48–51 The exchange-related energies are calculated using the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) framework,52 while the electron core is treated by the projected augmented wave (PAW) method.53,54 A plane wave cut-off energy of 400 eV is utilized to extend the wave function, in conjunction with a first-order Methfessel-Paxton55 scheme with a smear value of 0. To minimize computational costs, non-spin polarization is adopted in all simulations. Grimme's semi-empirical zero-damping D3 method is implemented, incorporating dispersion correction in all calculations.10,56,57 The AIMD simulations are executed with a time step of 1 fs, and a NoseHoover thermostat maintains the regular ensemble condition (NVT) at a target temperature of 300 K.58,59 Gamma points of the Brillouin zone were used in these AIMD simulations without consideration of symmetry.2.2 Experimental methods
The metal pieces, Pb and Cd, each measuring 1 cm × 1 cm, were meticulously cleaned and polished to eliminate the surface oxide layer. These cleaned pieces were then utilized as the cathodes for the hydrogen evolution reaction (HER). For the electrochemical measurements, Hg/HgO served as the alkaline reference electrode, while Ag/AgCl was used as the acidic reference electrode. The experiments were conducted in an H-cell containing 50 mL of electrolyte. To determine the solution resistance of the system, electrochemical impedance spectroscopy (EIS) was employed, with measurements conducted over the frequency range of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz to 0.1 Hz at an amplitude of 10 mV. Additionally, linear sweep voltammetry (LSV) was utilized to assess the electrocatalytic activity at a stirring speed of 600 rpm. The pH values of the electrolytes used for LSV tests are quite stable in each group, measuring around 13.25 in 0.1 M MOH, 13.03 in 0.1 M NaOH + 0.1 M MOH and 1.35 in 0.1 M H2SO4 + 0.1 M MOH (M = TMA, TEA, TPA, Fig. S4†). In situ infrared spectra were acquired using a Fourier transform infrared spectroscopy (Bruker Vertex 70V) configured with ATR-SEIRAS.
000 Hz to 0.1 Hz at an amplitude of 10 mV. Additionally, linear sweep voltammetry (LSV) was utilized to assess the electrocatalytic activity at a stirring speed of 600 rpm. The pH values of the electrolytes used for LSV tests are quite stable in each group, measuring around 13.25 in 0.1 M MOH, 13.03 in 0.1 M NaOH + 0.1 M MOH and 1.35 in 0.1 M H2SO4 + 0.1 M MOH (M = TMA, TEA, TPA, Fig. S4†). In situ infrared spectra were acquired using a Fourier transform infrared spectroscopy (Bruker Vertex 70V) configured with ATR-SEIRAS.
Cadmium powder was uniformly deposited onto a Si prism coated with a thin Au film via the drop-casting technique. Prior to acquiring the infrared spectra, the working electrode underwent multi-potential chronoamperometry (CA) activation to ensure a pristine electrode surface. Each potential was maintained for a duration of 120 seconds, with infrared spectra being recorded at intervals of 20 seconds.
3. Results and discussion
3.1 Effect of QASs and Na+ on HER activity
Quaternary ammonium ions significantly impact the HER rate at metal electrodes. To compare their effects with those of traditional alkali metal cations, we conducted HER experiments using Pt, known for its high HER activity, and Cd, which exhibits low HER activity and has been applied in adiponitrile synthesis from acrylonitrile hydrodimerization. As shown in Fig. 1(a1), the HER current density trends on Cd follow the sequence TPAOH > TEAOH > TMAOH ≈ NaOH, implying that long-chain QASs can enhance HER activity, consistent with previous reports.33,45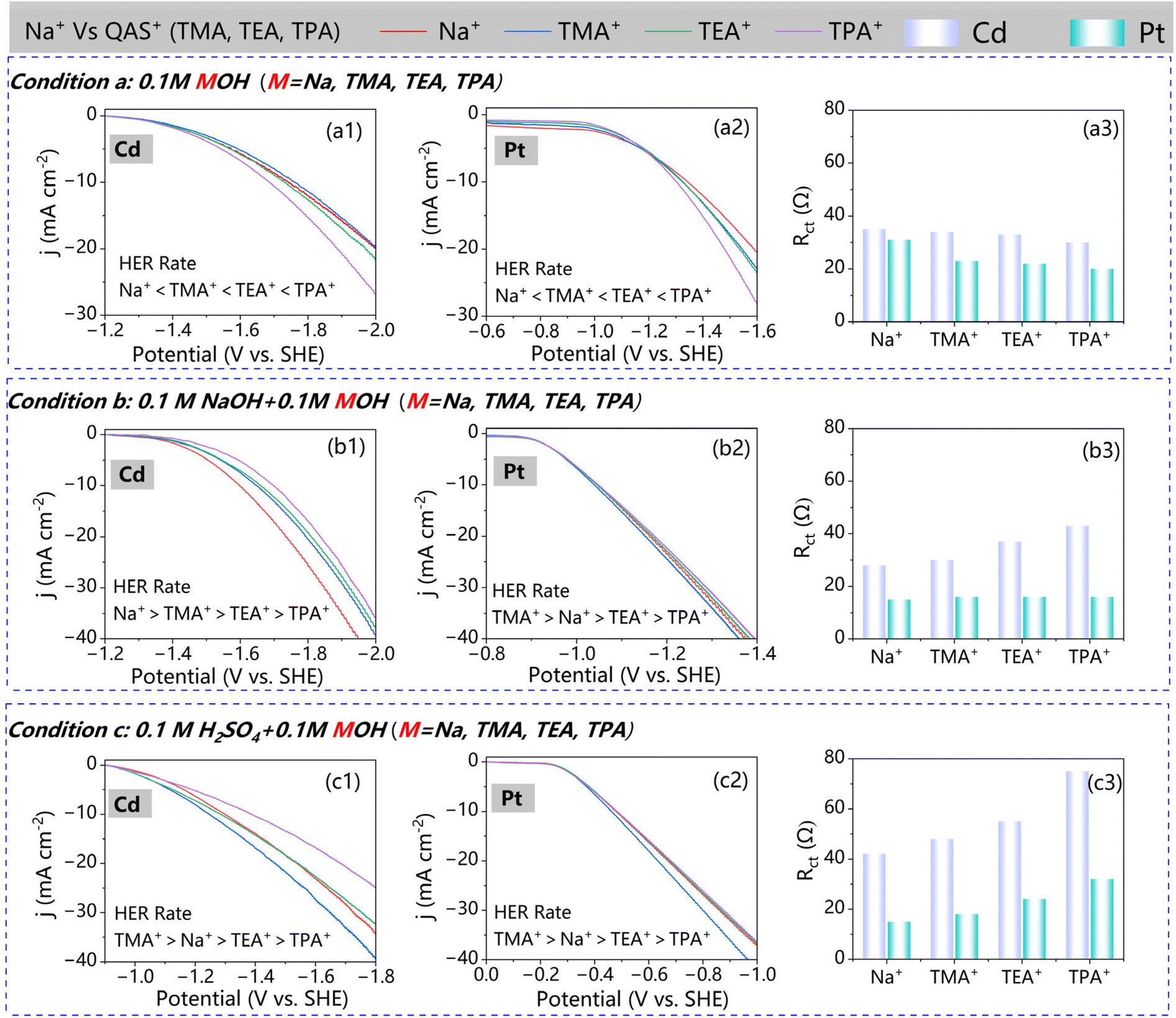 | ||
| Fig. 1 (a1–a3) LSV polarization curves and charge transfer resistances on Cd and Pt electrodes in different electrolytes of 0.1 M MOH. (b1–b3) LSV polarization curves and charge transfer resistances on Cd and Pt electrodes in different electrolytes in 0.1 M NaOH and 0.1 M MOH (M = Na, TMA, TEA, TPA). (c1–c3) LSV polarization curves and charge transfer resistances on Cd and Pt electrodes in different electrolytes in 0.1 M H+ and 0.1 M MOH (M = Na, TMA, TEA, TPA). | ||
However, Pt electrodes exhibited remarkable potential-dependent behaviour: in the low polarization regime (0.2–1.2 VSHE), current densities followed an inverse order (TPAOH < TEAOH < TMAOH < NaOH), while the conventional QAS enhancement re-emerged at higher overpotentials (>1.2 VSHE) with TPAOH > TEAOH > TMAOH > NaOH. This difference is more significant when the solution is not stirred (Fig. S5†). Due to the larger size of organic cations compared to Na+, QASs require higher potentials to reach the metal surface, and thus the enhancement of QASs is only effective in the high polarization zone. This polarity-dependent inversion highlights the critical role of cation diffusion resistance in modulating HER activity, particularly evident on highly catalytic Pt surfaces where mass transport limitations become significant under static conditions.
Additionally, the charge transfer resistance (Rct) at open current potential exhibits contrasting trends on Pt and Cd. As illustrated in Fig. 1(a3), with increasing branch chain length of the QAS, Rct on Cd decreases while it also decreases on Pt. The HER on both Pt and Cd in alkaline solution follows the Tafel–Volmer mechanism, with the Volmer step (H2O + e− ↔ Had + OH−) as the rate-determining step (RDS).60,61 Pt, as an active metal, easily transfers electrons to water molecules to generate Had. Organic cations enhance the interfacial hydrogen bond network, facilitating the Volmer step. Conversely, Cd, being an inactive metal with a higher work function, struggles to release electrons with large organic cations, which reduce the electric field intensity at the inner Helmholtz layer.
Nevertheless, in alkaline media containing Na+, a distinguished phenomenon can be noticed. Fig. 1(b1) shows that, in the presence of Na+, the addition of QASs consistently decreases HER current density on Cd cathodes, with this inhibitory effect intensified by longer-branched QASs. On Pt electrodes, the activity variation is much smaller than that on Cd. Only TMAOH accelerates the HER slightly, whereas other QASs resulted in reduced current density. Notably, the Rct measured on Pt remains stable across these four electrolytes, suggesting that the energy transfer barrier remains unaffected by QASs. Moreover, in acidic media, the HER current density is enhanced by TMA+ compared to Na+, albeit with a decrement as cation size increases. TMA+ also promotes HER activity on Pt, albeit to a lesser extent compared to under other conditions. Overall, the performance of QASs in acidic media with abundant H+ is similar to that in bases containing Na+.
We also obtained the Tafel slopes on Pt in the presence of Na+ or H+. As shown in Fig. S6,† the Tafel slopes indicate the same HER activity order. Besides, after IR compensation, the LSV curves retain the same tendency (Fig. S7 and S8†). These experimental results suggest that the effect of QASs on the HER depends not only on the properties of the electrode surface, but also on the microenvironment at the interface, especially the nature of cations involved.
3.2 Effect of cations on the interfacial hydrogen bond structure
The characteristics of the hydrogen bond network in both the presence and absence of TEA were assessed using in situ surface-enhanced infrared absorption spectroscopy (SEIRAS) in the attenuated total reflectance (ATR) configuration. As shown in Fig. 2, ATR-SEIRAS experiments revealed that, at cathodic currents below 60 μA in NaOH alkaline solutions, the hydrogen bond network remains intact, exhibiting solely the vibration peak of hydrogen-bonded water ν(O–H). Conversely, currents exceeding 80 μA disrupt the hydrogen bond network, evident from the emergence of the negative vibration peak of ν(O–H). In TEAOH solution, the hydrogen bond network remains intact at all cathodic currents. The increasing ν(O–H) peak area along with the reduced peak wavenumber suggests the stabilizing effect on the hydrogen bond network of TEA. By comparing the peak area at lower current, it can be found that Na+ leads to more hydrogen bonds with less intensity than TEA, which again underscores that the enhancement due to TEA of hydrogen bond strength and HER activity becomes more pronounced at a more negative potential. However, in combined NaOH and TEAOH solutions, the negative vibration peaks appear at the beginning of the current scan, and the area of this negative peak increases with applied current, implying that the coexistence of Na+ and TEA significantly destroys the hydrogen bond network. These observations of hydrogen bond variation are in accordance with the experimental HER activity in different solutions (Fig. 1a1–a3). | ||
| Fig. 2 In situ ATR-SEIRAS spectra at currents from 0 μA to −100 μA in Cd electrolytes: (a) in Ar saturated solutions of 0.1 M NaOH; (b) in Ar saturated solutions of 0.1 M TEAOH; (c) in Ar saturated solutions of 0.05 M NaOH and 0.05 M TEAOH. | ||
To provide a deeper understanding of our experimental results, we performed AIMD simulations on EDLs featuring various cation species. As depicted in Fig. 3(a), despite starting slightly distant from the metal surface prior to the AIMD simulation, Na+ exhibit a tendency to permeate the inner Helmholtz layer, disrupting the structural integrity of the first water layer. Conversely, TEA ions are more inclined to remain at a distance from the metal surface, allowing a fully intact first water layer to persist at the interface. This observation elucidates the superior HER activity exhibited by TEA compared to Na+, despite the interface experiencing a reduced water density owing to the hydrophobic nature of TEA. In scenarios where TEA and Na+ coexist, the smaller Na+ ions still manage to traverse the water layer, whereas TEA maintains a distance of one water layer from the surface. Both the hydrophobic properies of TEA and the disruption of the surface water layer contribute to the decline in HER activity.
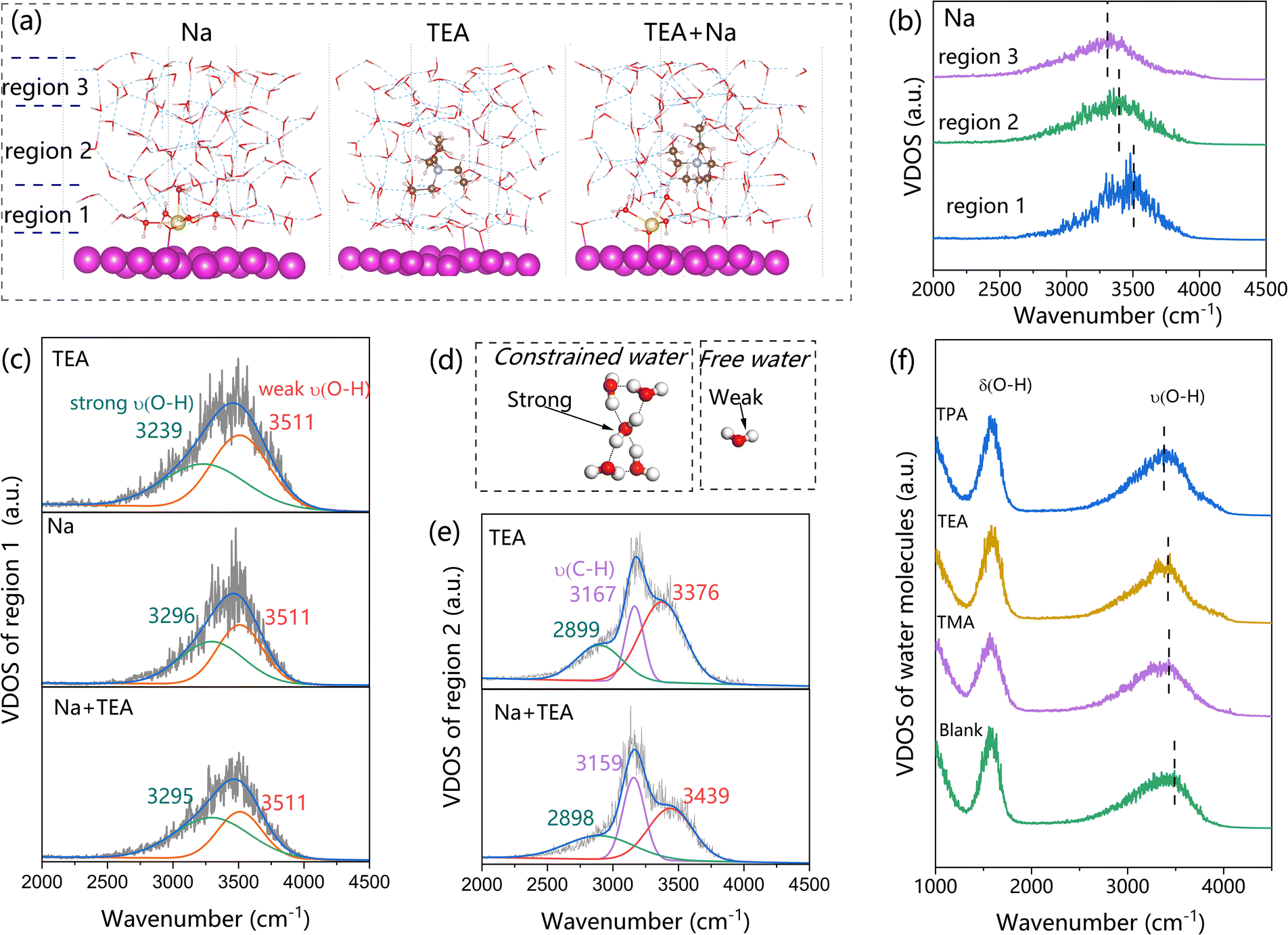 | ||
| Fig. 3 VDOS spectra of Cd(101)/water interfaces with various cations. (a) Distribution of interfacial water molecules after a 10 ps AIMD simulation; (b) VDOS spectra of water molecules stratified into different layers in the presence of Na+; (c) deconvolution of VDOS peaks for the first water layer with differing cations; (d) schematic illustration distinguishing between weak and strong H2O vibrations; (e) VDOS spectra of the second water layer in the presence of TEA or TEA combined with Na; (f) influence of QAS size on the VDOS spectra. | ||
The vibrational density of states (VDOS) for interfacial water molecules, derived from molecular dynamics trajectories, was analyzed in detail. The peaks observed at approximately 3300 cm−1 and 1550 cm−1 closely correspond to experimental data, validating the accuracy of our VDOS calculations. Typically, an increase in wavenumber indicates a weakening of hydrogen bonding, and conversely, a decrease suggests strengthening. Based on the proximity to the Cd(101) surface, interfacial water was categorized into three regions: Region 1, adjacent to the metal surface; Region 2, surrounding TEA molecules; and Region 3, above TEA. As illustrated in Fig. 3(b), in the presence of only Na+, the wavenumber of ν(O–H) VDOS peaks follows the trend Region 1 > Region 2 > Region 3, suggesting that Na+ weakens hydrogen bonds near the metal surface, consistent with previous reports.19
To assess the impact of cation type on hydrogen bonding, the ν(O–H) peaks are further differentiated into two subpeaks: one representing stronger ν(O–H) in highly hydrogen-bonded, constrained water molecules, and the other weaker ν(O–H) in less ordered water (Fig. 3(c and d)). The weak ν(O–H) subpeak remains at 3511 cm−1, while the stronger subpeak shifts from 3239 cm−1 to 3296 cm−1 upon replacing TEA with Na+, indicating more weakened hydrogen bonding in the first water layer. Notably, the ν(O–H) peak resolution in the presence of both Na+ and TEA is nearly identical to that with only Na+, suggesting that adding TEA to NaOH solution does not enhance the hydrogen bond strength of the first water layer.
As illustrated in Fig. 3(e), the VDOS peak within the 2000 cm−1 to 4000 cm−1 range in region 2 can be resolved into three subpeaks at ∼2899 cm−1, ∼3167 cm−1, and ∼3376 cm−1, corresponding to strong ν(O–H), ν(C–H), and weak ν(O–H), respectively. Notably, the wavenumber of the strong ν(O–H) peak remains consistent regardless of the presence of Na+. However, the addition of Na+ shifted the peak of weak ν(O–H) from 3376 cm−1 to 3439 cm−1, probably due to more free water. Since the hydrogen bond network in region 2 is essential for faster hydrogen transfer, the less ordered structure caused by Na+ would inhibit the HER process.
Fig. 3(f) further compares VDOS for various QASs, showing that incorporating organic QASs with longer alkyl chain lengths significantly enhances interactions among interfacial H2O molecules, leading to a denser hydrogen bonding network that facilitates proton activation and transport during the HER. An additional advantage of employing larger QASs lies in the elevated proportion of water molecules where the hydrogen atom is oriented towards the metal surface, thereby favorably influencing the HER process (as depicted in Fig. S9†). Furthermore, the incorporation of QASs enhances the overall hydrogen bonding network within the NaOH solution (Fig. S10a†), suggesting that the inhibitory effect of QASs on the HER in the presence of Na+ ions primarily stems from the weakening of hydrogen bonds within the inner water layer. Notably, as illustrated in Fig. S10b,† the enhancing effect of QASs is more pronounced under acidic conditions (where H+ serves as the cation) compared to alkaline conditions (where Na+ is the cation). This observation elucidates the enhancement of HER activity observed in H2SO4 solutions upon the addition of smaller QAS molecules (Fig. 1c1).
3.3 Hydration competition between Na+ and QASs
Na+ and QASs primarily exist in the electric double layer as hydrated cations, yet their distinct sizes and structures lead to varied hydration capabilities. The distribution of water molecules surrounding the QAS can be quantified by analyzing the radial distribution of N–O. Fig. 4(a) illustrates that the N–O distance between TMA and water exhibits three peaks at 4.35 Å, 6.75 Å, and 8.85 Å, indicating the presence of three well-defined water layers around TMA ions. For TEA and TPA, which possess longer side chains, water molecules have the ability to penetrate the spaces between these chains, leading to a less structured arrangement near the QAS ions. The good hydration ability of TMA explains its promotion effect in NaOH (Fig. 1b2) and H2SO4 solutions (Fig. 1c1 and c2). | ||
| Fig. 4 Comparison of hydration capability of QASs and Na+ in different electrolytes. (a) Water distribution around the QAS; (b) water distribution around Na+. | ||
Upon the introduction of Na+, only two water layers persist, likely due to the hydration of Na+ disrupting the organized water network surrounding TMA. This disruption is further evidenced by an increase in the number of oxygen atoms at N–O distances ranging from 3.5 Å to 8 Å, resulting in a flattened peak distribution. When TEA and Na+ coexist, there is an increase in oxygen atoms at distances of 4–4.5 Å and 6.5–7.5 Å, accompanied by a reduction in oxygen atoms within the 5–8 Å range. Comparable observations can be made at metal/water interfaces in the presence of both TPA and Na+. This behavior can be attributed to hydration competition between QASs and Na+, where they vie for the oxygen atoms of water molecules, thereby altering the orientation of these water molecules. This competition can also be noted by the decreased weight of Na–O at 2.6 Å after the introduction of TEA and TPA (Fig. 4(b)).
The hydrogen bond network oriented along the Z-direction has been thoroughly analyzed and is visually depicted in Fig. 4. Specifically, Fig. 4(a) reveals the presence of two distinct oxygen layers, centered at 7.9 Å and 10.4 Å respectively, in the absence of any cations. This arrangement underscores a well-structured hydrogen bond network at the interface. Upon the introduction of Na+ ions, the distribution of oxygen atoms remains relatively unchanged. However, the addition of QASs leads to a decrease in the peak intensity of the second water layer and shifts the first layer closer to the metal surface (Fig. S13†), thereby enhancing the hydrogen bond network. Conversely, in the presence of both QASs and Na+, the peak of the first layer shifts further away from the metal surface, suggesting a disruption in the hydrogen bond network.
The number of hydrogen bonds along the Z direction on Pt is depicted in Fig. 6(f), which additionally illustrates two layers of water devoid of any cations. Upon the introduction of Na+, a reduction in the hydrogen bond count is observed in the first layer, particularly in the case of double and triple hydrogen bonds. This decrease is attributed to the hydration process, which alters the orientation of water molecules and disrupts the hydrogen bond network. Similarly, the inclusion of TEA alone results in a decline in the hydrogen bond count of the first layer, yet the proportion of double or triple bonds remains relatively constant. The second layer experiences a significant decrease in hydrogen bond count due to the large size of TEA, which pushes away adjacent water molecules. Furthermore, the peak of the first layer shifts closer to the metal surface, providing an explanation for the enhanced HER activity observed in TEAOH.
However, in the presence of both TEA and Na+, the distance between the two water layers increases notably, hindering proton transport during hydrogen evolution. This conclusion is further supported by the average bond length data presented in Fig. 6(g). In pure water, the hydrogen bond length is uniformly distributed within the range of 1.8 to 2.0 Å and remains unchanged in the presence of TEA, suggesting a compact hydrogen bond network in the EDL and the robust interaction between metal sites and water molecules, when comparing the Cd/water interface to the Pt/water interface. In contrast, the introduction of Na+ alone leads to a slight increase in hydrogen bond length near the metal surface, consistent with the disruptive effect of Na+. Intriguingly, when both TEA and Na+ are present, the bond length at the interface increases significantly. Such a long length means the water is “free” rather than constrained in the hydrogen bond network, which explains the high peak wavenumber of the weak ν(O–H) shown in Fig. 3(e). This enlargement can be attributed to the competitive hydration between TEA and Na+, where both cations attract the oxygen atom, leaving the hydrogen atoms in the hydration layer facing each other and creating a larger water gap (Fig. 3(a)).
3.4 Comparison of Cd/water and Pt/water interfaces in the hydrogen bond network
Our experimental findings also uncover distinct kinetic behaviors of QASs on Cd and Pt surfaces, particularly highlighting that the HER activity of Cd is more susceptible to QASs. Consequently, we conducted a comparative analysis of the hydrogen bond network at the Cd/water and Pt/water interfaces. As depicted in Fig. 5(e) and 6(e), beyond the presence of two discernible water layers, a minor peak emerges between the metal surface and the first water layer, attributed to the introduction of Na+ which not only diminishes the adsorbed water layer on Pt but also notably disrupts the second interfacial water layer. Intriguingly, the solitary addition of TEA leads to the formation of an additional hydrogen bond layer adjacent to the Pt surface, thereby enhancing the HER activity on Pt. This extra layer with a Pt–O distance of 2.0 Å corresponds to the surface adsorbed water molecule. In other electrolytes, though water can also be adsorbed on the Pt surface, it could not participate in the hydrogen bond network. Fig. 5(f) further illustrates that the combined addition of Na+ and TEA increases the hydrogen bond length at the interface, albeit with a moderate expansion in the gap. This observation elucidates the less influence of QASs on the HER activity of Pt (Fig. 6) compared to Cd in a NaOH solution. | ||
| Fig. 5 Hydrogen bond network at the electrochemical interface of Cd(101)/H2O in different electrolytes. (a–d) Snapshots of the interfacial structures after 10 ps AIMD simulation; (e) oxygen atom density profile along the z-axis (normal to the interface); (f) statistical analysis of single, double, and triple hydrogen bonds; (g) average hydrogen bond lengths. | ||
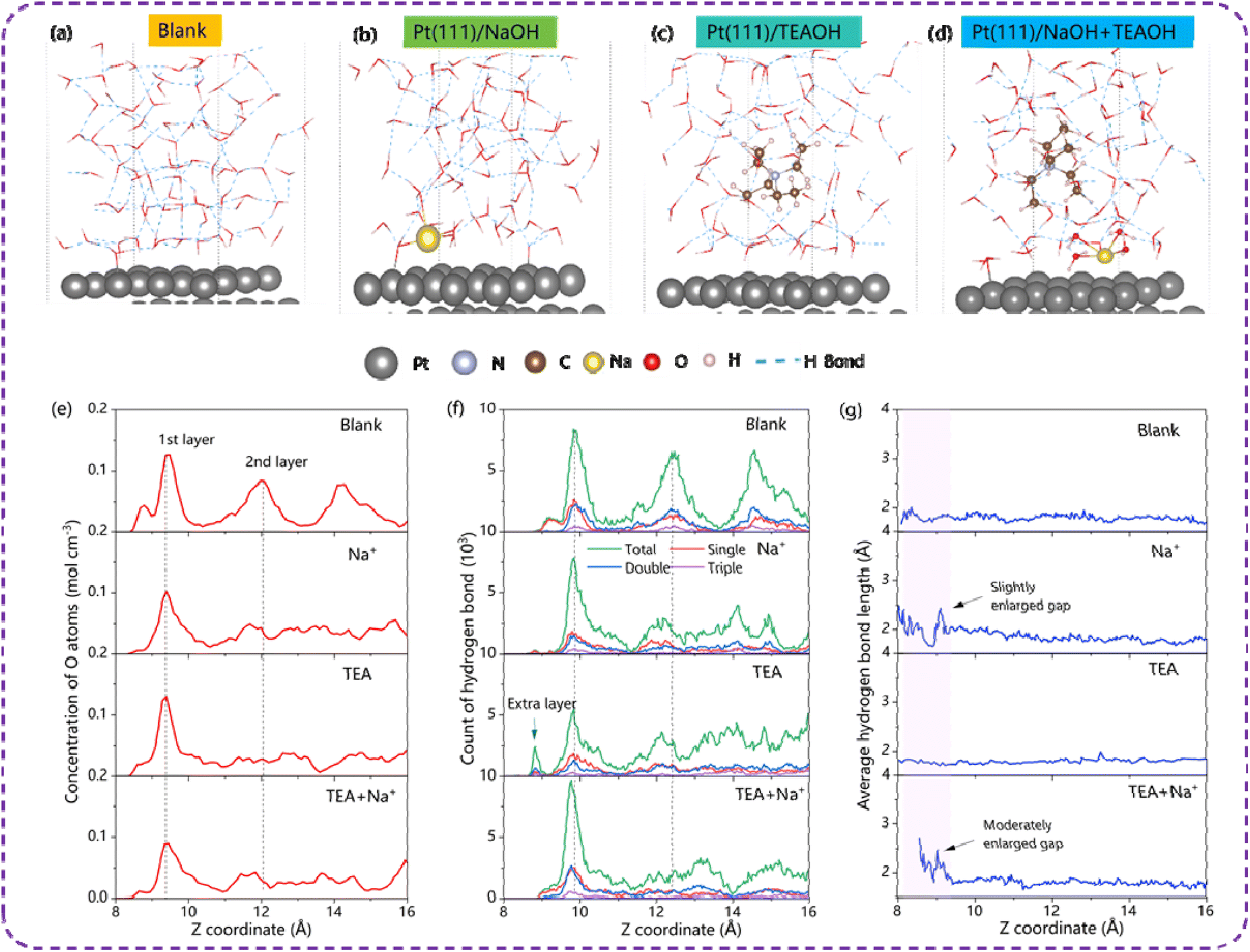 | ||
| Fig. 6 Hydrogen bond network at the electrochemical interface of Pt(111)/H2O in different electrolytes. (a–d) Snapshots of the interfacial structures after 10 ps AIMD simulation; (e) oxygen atom density profile along the z-axis (normal to the interface); (f) statistical analysis of single, double, and triple hydrogen bonds; (g) average hydrogen bond lengths. | ||
To eliminate the effect of potential, we doubled the number of Na+ ions at the Cd(101)/water interface, and found that the potential does not influence the changes in the hydrogen bond network (Fig. S21 and S22†). In other words, the difference between Na+ and Na+ + TEA is mainly due to the nature of TEA rather than the number of cations. We also replaced the Na+ with divalent cation Mg2+, and found that TEA can still lead to a gap in the hydrogen bond network, indicating a universal modulation of TEA on the metal cation containing interfacial structure (Fig. S23†).
As illustrated in Fig. S18 and S19,† we have computed the electronic local density of states for both the Cd (101) slice and the Pt (111) slice. Our calculations reveal that the electronic local density of states energy surrounding Cd is lower compared to that surrounding Pt. A higher work function suggests that electrons are less inclined to escape from the metal surface, which provides an explanation for the higher hydrogen evolution overpotential observed on the Cd electrode compared to the Pt electrode (Fig. 1a1 and a2).
Meanwhile, we also evaluated the electrostatic potential distribution of the Cd (101) crystal plane; the addition of the alkali metal cation Na+ increases the PZC potential at the double layer interface by 0.13 eV, while the organic cation TEA increases it by 0.29 eV. When both are present simultaneously, the PZC potential increases by 0.33 eV, indicating that the cations at the double layer interface can effectively regulate the electrode potential (Fig. S18†). This underscores the pivotal role of organic cations in modulating the electrostatic environment.
Furthermore, we evaluated the electrostatic potential distribution of the Pt (111) crystal plane in an alkaline environment containing TEA, as shown in Fig. S18(b).† Our results indicate that under alkaline conditions, the point of zero charge (PZC) is −1.62 eV, and under acidic conditions, the PZC is 0.57 eV, suggesting a negative double-layer capacitance at the Pt interface; the addition of Na+ ions can regulate the electrode potential at the double layer interface (Fig. S18e and h†). The calculation of the work function at the double layer interface and the zero-charge potential helps us understand the electrode potential of the system being studied, demonstrating that the addition of cations can effectively regulate the double layer capacitance, which is particularly important for charged systems.
By calculating the diffusion coefficient of QASs (Fig. S11†), we found that the diffusion coefficient gradually decreases as the ion size increases. Comparing the results in the presence and absence of Na+, we observed that the presence of Na+ slows down the movement of QAS molecules. To understand the distance between EDL water molecules, we statistically analysed the radial distribution functions of water molecules on different metal surfaces (Fig. S12†). The results strongly corroborate our proposed theory regarding the enhancement and weakening of the hydrogen bonding network (Fig. S15 and S20†).
4. Conclusion
In this study, we experimentally investigated the effect of the interplay between Na+ and QASs on the HER under alkaline conditions, specifically focusing on Cd and Pt cathodes. Our results demonstrated that, in the absence of Na+, QASs enhance HER activity compared to Na+ alone, with a more pronounced effect observed for QASs possessing longer side alkyl chains. However, in NaOH solutions, the incorporation of QASs, particularly those with extended side chains, led to a decrement in HER activity.Utilizing a combination of AIMD and the in situ SEIRAS-ATR test, we unveiled that Na+ weakens the hydrogen bond network at the interface, particularly within the water layer adjacent to the metal surface. Conversely, QASs strengthened this hydrogen bond network. The coexistence of Na+ and QASs disrupted the order of interfacial water and reduced its connectivity, resulting in a significant decline in HER activity.
A comparative analysis of the Cd/water and Pt/water interfaces revealed that the inhibitory effect of QASs in NaOH electrolyte is less pronounced on the HER activity of Pt. This is attributed to its lower work function, which facilitates stronger interactions with surface water molecules, thereby preventing the formation of gaps at the interface. Our findings underscore the significance of understanding and manipulating the interfacial water structure and the role of organic cations in optimizing electrochemical processes, particularly in promoting or inhibiting the hydrogen evolution reaction.
Data availability
All data are available in the main manuscript or the ESI.†Author contributions
Xun Huang and Zidong Wei conceived this project. Shilin Bo and Yang Xiang carried out the experiments. Shilin Bo, Li Li and Qiong Xiang performed the simulations. All the authors contributed to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFA1504200) and National Natural Science Foundation of China (22178033 and 22090030).References
- Y. Men, Y. Tan, P. Li, Y. Jiang, L. Li, X. Su, X. Men, X. Sun, S. Chen and W. Luo, Angew. Chem., 2024, 63, e202411341 CrossRef CAS PubMed
.
-
P. Sabatier, La catalyse en chimie organique, ed., C. Béranger, 1920, vol. 3 Search PubMed
-
J. M. Bockris, M. Devanathan and K. Müller, in Electrochemistry, Elsevier, 1965, pp. 832–863 Search PubMed
- A. Narten and S. Lindenbaum, J. Chem. Phys., 1969, 51, 1108–1114 CrossRef CAS
- R. Futrelle and D. McGinty, Chem. Phys. Lett., 1971, 12, 285–287 CrossRef CAS
- M. Baizer, Chem. Ind., 1979, 7, 435 Search PubMed
- A. Li, X. Tang, R. Cao, D. Song, F. Wang, H. Yan, H. Chen and Z. Wei, Adv. Mater., 2024, 2401818 CrossRef CAS PubMed
- Y. Zhou, W. Yuan, M. Li, Z. Xie, X. Song, Y. Yang, J. Wang, L. Li, W. Ding and W.-F. Lin, Nat. Energy, 2024, 1–13 Search PubMed
- Z. Wei, X. Huang, H. Duan, M. Shao, R. Li, J. Zhang, C. Li and X. Duan, Chin. J. Catal., 2024, 58, 1–6 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- M. Flór, D. M. Wilkins, M. de la Puente, D. Laage, G. Cassone, A. Hassanali and S. Roke, Science, 2024, eads4369 CrossRef PubMed
- P. Li, Y. Jiao, J. Huang and S. Chen, JACS Au, 2023, 3, 2640–2659 CrossRef CAS PubMed
- M. Tang, C. Wang, Z. Zheng, X. Wang, F. Zhu and S. Chen, Energy Environ. Sci., 2024, 17, 9479–9486 RSC
- H. Zhang, S. Wu, X. Huang, L. Li, Q. Liao and Z. Wei, Chem. Eng. J., 2022, 428, 131534 CrossRef CAS
- Q. Xiang, S. Jiang, J. Liu, X. Chen, L. Li and Z. Wei, ACS Appl. Mater. Interfaces, 2025, 17(5), 7619–7628 CrossRef CAS PubMed
- Y. Qin, C. Xia, T. Wu, J. Zhang, G. Gao, B. Y. Xia, M. L. Coote, S. Ding and Y. Su, J. Am. Chem. Soc., 2024, 146(47), 32539–32549 CrossRef CAS PubMed
- Z. Mi, T. Wang, L. Xiao, G. Wang and L. Zhuang, J. Am. Chem. Soc., 2024, 146(25), 17377–17383 CrossRef CAS PubMed
- Y. Zhou, W. Yuan, M. Li, Z. Xie, X. Song, Y. Yang, J. Wang, L. Li, W. Ding and W.-F. Lin, Nat. Energy, 2024, 9, 1297–1309 CrossRef CAS
- S. Zheng, X. Yang, Z.-Z. Shi, H. Ding, F. Pan and J.-F. Li, J. Am. Chem. Soc., 2024, 146, 26965–26974 CrossRef CAS PubMed
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. Van Der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900–911 CrossRef CAS
- Y. Tian, B. Huang, Y. Song, Y. Zhang, D. Guan, J. Hong, D. Cao, E. Wang, L. Xu, Y. Shao-Horn and Y. Jiang, Nat. Commun., 2024, 15, 7834, DOI:10.1038/s41467-024-52131-w
- P. Li, Y.-L. Jiang, Y. Men, Y.-Z. Jiao and S. Chen, Nat. Commun., 2025, 16, 1844, DOI:10.1038/s41467-025-56966-9
- L. Zeng, D. Dong, J. Lu, K. He, X. Liu, J. Wang, Z. Wei, M. Gresil, J. Ratcliffe and Z. Li, Adv. Funct. Mater., 2025, 2424662 CrossRef
- B. Tang, Y. Fang, S. Zhu, Q. Bai, X. Li, L. Wei, Z. Li and C. Zhu, Chem. Sci., 2024, 15, 7111–7120, 10.1039/d3sc06904d
- W. Wu, S. Yang, H. Qian, L. Zhang, L. Peng, L. Li, B. Liu and Z. Wei, Chin. J. Catal., 2024, 66, 1–19 CrossRef CAS
- F. Chen, L. Guo, D. Long, S. Luo, Y. Song, M. Wang, L. Li, S. Chen and Z. Wei, J. Am. Chem. Soc., 2024, 146, 30388–30396 CrossRef CAS PubMed
- Y. Song, Y. Liu, D. Long, X. Tao, S. Luo, Y. Yang, H. Chen, M. Wang, S. Chen and Z. Wei, Adv. Funct. Mater., 2024, 34, 2410305 CrossRef CAS
- Z. Zeng, S. Wu, X. Huang and Z. Wei, Electrochemical Oxidation of Furfural on NiMoP/NF: Boosting Current Density with Enhanced Adsorption of Oxygenates, Small, 2023, 2305462 Search PubMed
- J. Huang, P. Li and S. Chen, J. Phys. Chem. C, 2019, 123, 17325–17334 CrossRef CAS
- X. Luo, Y. Wang, B. Wu, Y. Wang, C. Li, M. Shao, B. Liu and Z. Wei, J. Phys. Chem. Lett., 2024, 15, 10435–10441 CrossRef CAS PubMed
- S. Wu, H. Zhang, X. Huang, Q. Liao and Z. Wei, Ind. Eng. Chem. Res., 2021, 60, 8324–8330 CrossRef CAS
- K. Zhao, H. Yu, H. Xiong, Q. Lu, Y. Q. Gao and B. Xu, Chem. Sci., 2023, 14, 11076–11087 RSC
- D. Danly, J. Electrochem. Soc., 1984, 131, 435C CrossRef
- F. Karimi, S. Ashrafizadeh and F. Mohammadi, Chem. Eng. J., 2012, 183, 402–407 CrossRef CAS
- J. Li, X. Li, C. M. Gunathunge and M. M. Waegele, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9220–9229 CrossRef CAS PubMed
- Y. Wang, J. Zhang, J. Zhao, Y. Wei, S. Chen, H. Zhao, Y. Su, S. Ding and C. Xiao, ACS Catal., 2024, 14, 3457–3465, DOI:10.1021/acscatal.3c05880
- W. Zhang, W. Ge, Y. Qi, X. Sheng, H. Jiang and C. Li, Angew. Chem., Int. Ed., 2024, 63, e202407121 CrossRef CAS PubMed
- C. Deacon-Price, L. Changeur, C. S. Santana and A. C. Garcia, ACS Catal., 2024, 14, 12928–12939 CrossRef CAS PubMed
- D. Sheng, X. Liu, Z. Yang, M. Zhang, Y. Li, P. Ren, X. Yan, Z. X. Shen and D. Chao, Adv. Funct. Mater., 2024, 2402014 CrossRef CAS
- F. Fiegenbaum, E. M. Martini, M. O. de Souza, M. R. Becker and R. F. de Souza, J. Power Sources, 2013, 243, 822–825, DOI:10.1016/j.jpowsour.2013.06.077
- M. Qiu, P. Sun, K. Han, Z. Pang, J. Du, J. Li, J. Chen, Z. L. Wang and W. Mai, Nat. Commun., 2023, 14, 601 CrossRef CAS PubMed
- A. Döner, Int. J. Hydrogen Energy, 2018, 43, 22797–22806 CrossRef
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory and O. Voznyy, Nat. Energy, 2019, 4, 107–114 CrossRef CAS
- J. Fernández-Vidal and M. T. M. Koper, ACS Catal., 2024, 14, 8130–8137, DOI:10.1021/acscatal.4c01765
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48, 13115 CrossRef CAS PubMed
- J. Carrasquilla and R. G. Melko, Nat. Phys., 2017, 13, 431–434 Search PubMed
- Z. Li, J. R. Kermode and A. De Vita, Phys. Rev. Lett., 2015, 114, 096405 CrossRef PubMed
- A. P. Bartók, J. Kermode, N. Bernstein and G. Csányi, Phys. Rev. X, 2018, 8, 041048 Search PubMed
- V. L. Deringer and G. Csányi, Phys. Rev. B, 2017, 95, 094203 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed
- Y. Crespo and A. Hassanali, J. Chem. Phys., 2016, 144, 074304 CrossRef PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Sakong, K. Forster-Tonigold and A. Groß, J. Chem. Phys., 2016, 144, 194701 CrossRef PubMed
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS
- M. Dunwell, Y. Yan and B. Xu, Curr. Opin. Chem. Eng., 2018, 20, 151–158 CrossRef
- P. J. Rheinländer, J. Herranz, J. Durst and H. A. Gasteiger, J. Electrochem. Soc., 2014, 161, F1448–F1457 CrossRef
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem.
Soc., 2010, 157, B1529–B1536 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational details; visualizations of the structural models. See DOI: https://doi.org/10.1039/d5sc01034a |
| This journal is © The Royal Society of Chemistry 2025 |