
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow elastically flexible can molecular crystals be? – a new record†
Zhengzheng
Zhou
 ab,
Vikram Chandrashekhar
Joshi
b,
Yiwang
Guo
b,
Tianyi
Xiang
b,
Zijian
Wang
b and
Changquan Calvin
Sun
*b
ab,
Vikram Chandrashekhar
Joshi
b,
Yiwang
Guo
b,
Tianyi
Xiang
b,
Zijian
Wang
b and
Changquan Calvin
Sun
*b
aNMPA Key Laboratory for Safety Evaluation of Cosmetics, Guangdong Provincial Key Laboratory of Tropical Disease Research, Department of Hygiene Inspection & Quarantine Science, School of Public Health, Southern Medical University, Guangzhou, Guangdong 510515, China
bPharmaceutical Materials Science and Engineering Laboratory, Department of Pharmaceutics, College of Pharmacy, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: sunx0053@umn.edu
First published on 21st March 2025
Abstract
The elastic strain limit, which quantifies the elastic flexibility of a material, is critical for technological applications of functional materials in a number of fields. Although the elastic flexibility of molecular crystals has been recognized, the extent of elastic flexibility of such materials remains to be defined. Here, we report a molecular crystal, i.e., form I polymorph of celecoxib (CEL), exhibiting exceptional elastic flexibility with an elastic strain of at least 8.70%. The record high elastic strain is accompanied by low Young's modulus (E = 3.18 ± 1.01 GPa) and hardness (H = 39.8 ± 15.6 MPa), as determined by single crystal nanoindentation, along with the high plasticity of the bulk powder observed in in-die Heckel analysis.
Extensive efforts have been devoted to designing and synthesizing mechanically flexible molecular crystals,1–13 because of their potential applications in a wide range of technological areas, including pharmaceuticals,14,15 sensors,16,17 electronics,18,19 optical waveguides,20,21 and wearable devices.22,23 It was projected that the flexible electronics market would reach a value of 70 billion US dollars by 2026.24 An elastically deformed crystal spontaneously regains its original shape once the applied stress is removed. However, irreversible deformation, either plastic deformation or brittle fracture, takes place once a certain elastic strain is exceeded. Thus, the maximum elastic strain determines the elastic flexibility of a single crystal, which ultimately determines the maximum flexibility of the device employing such crystals. Hence, knowledge of the elastic limit of an elastic crystal is critically important for its successful application in various areas.25
Elastic strain can be calculated from specimen thickness and the radius of curvature using Euler–Bernoulli beam-bending theory.26,27 The maximum elastic strain of known elastically flexible organic crystals typically ranges from 1% to 3%, with only a handful of cases reaching 6%.28–33 The most commonly adopted three-point bending method, performed using forceps and a needle for studying the elastic flexibility of molecular crystals, requires large crystals (typically mm in length) and physical contact with a stiff tool, which likely introduce defects and cause premature failure before reaching the theoretical elastic limit of the crystal.34,35 Here, using a contact-free bending method performed during melt crystallization, we show that an elastic strain of at least 8.70% could be attained for the form I polymorph of a nonsteroidal anti-inflammatory drug (NSAID), celecoxib (CEL), setting a new record of elastic strain for molecular crystals.
CEL has been widely used to treat acute pain since 1998.36 To date, four polymorphs of CEL, forms I, II, III, and IV, have been discovered.37 Form III is the polymorph present in commercial capsule products due to its thermodynamic stability under ambient conditions. Despite the extensive research performed on CEL, only the form III single crystal structure has been reported in the crystal structure database (CSD), likely because of the difficulty of growing sufficiently large single crystals of other polymorphs of CEL using the solution crystallization process. Form I CEL can crystallize from an amorphous film when stored overnight at 80 °C (Fig. 1a).38 However, the form I crystals thus obtained were too small with low quality for structure elucidation by single crystal X-ray diffraction (SCXRD) analysis. Hence, we attempted the melt crystallization method to grow large form I CEL single crystals for structure elucidation by SCXRD.
 | ||
| Fig. 1 Single crystal growth of CEL form I by the gradient-cooling melt crystallization method. Scale bars are 100 μm. (a) Amorphous CEL with form I seeds, (b) sample held at 169.5 °C till only a single form I CEL seed remaining, (c–h) sample held at gradually lowered temperatures, from 167 °C to 160 °C, to facilitate single crystal growth, (i) a single crystal covered with fine curled crystals, grown from remaining melt, after being held at 160 °C much longer than 2 hours. | ||
The melt crystallization technique, which is material-sparing (∼10 μg per experiment), has been used for preparing polymorphs that are thermodynamically unstable near ambient temperature, e.g., indomethacin, ROY, and piroxicam.39–43 The temperature suitable for single crystal growth is usually within the range of 0.97–0.99 times the melting temperature (Tm).44 However, in that temperature range, uncontrolled late growth of curled fine CEL form I crystals covered the surface of the initial single crystal before it grew sufficiently large for SCXRD. To overcome this problem, we developed a gradient-cooling melt crystallization method, which involves the following steps: (1) a small sample of amorphous CEL containing form I crystal seeds (Fig. 1a) was prepared by storing an amorphous CEL film overnight at 80 °C.38 This was heated on a glass slide at 10 °C min−1 to 169.5 °C, close to the end of the form I melting range observed from a DSC thermogram (Fig. S1†), on a hot stage, and held at this temperature until only one seed crystal remained (Fig. 1b). (2) The temperature was lowered to 167 °C and held for 20 min (Fig. 1c), followed by lowering the temperature to 165 °C, 163 °C, and 161 °C sequentially (held for 20 min at each temperature, Fig. 1d–f) and finally to 160 °C (held for 1–2 hours, Fig. 1g and h) to obtain a single crystal with a sufficient size. This cooling program was intended to maintain the growth of the single crystal while avoiding the generation of new crystal nuclei invariably encountered during single step cooling. However, even with this gradient program, the growth of an acicular crystal was very slow once the fast-growing ends of the crystal exceeded the boundary of the melt (Fig. 1h). With a long holding time, thin crystals would always form and cover the surface of the single crystal before all the liquid was consumed by single crystal growth (Fig. 1i). Directly isolating the single crystal from the melt would still cause crystallization of the residual melt into fine crystals. To overcome this problem, a drop of silicone oil was placed to overlap with the remaining melt at 160 °C, and then the single crystal was isolated from the remaining melt by pushing it into the oil with a needle. By following this procedure, high-quality acicular single crystals (1–2 mm long and 0.05–0.1 mm wide) suitable for crystal structure elucidation by SCXRD were successfully prepared (Fig. S2†).
CEL form I crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with Z′ = 3 (Table S1†), belonging to the rare class of structures with Z′ = 3 (total 6387 crystal structures, corresponding to 0.52% of all structures in the CSD), out of which only 30.5% are in the P space group.45 The unit cell parameters and global crystal packing structures of CEL forms I differ from those of form III (Fig. S3 and Table S1†). There are three crystallographically unique molecules in the asymmetric unit (labelled with different colors in Fig. 2a), which exhibit significantly different conformations owing to the rotation of 4-methylphenyl (dihedral angles of 25.80°, 29.71°, and 61.12°) and benzenesulfonamide (dihedral angles of 59.18°, 60.80°, and 33.83°) rings about the trifluoromethyl-1H-pyrazole ring (Fig. S4a–c†). The three molecules in the asymmetric unit stack along the a-axis (Fig. S4d†) to form molecular columns, which are fortified through several types of weak intermolecular interactions, such as C–H⋯F (3.266 Å and 138.30°, Table S2†) and π⋯π stacking (centroid-to-centroid distance = 5.030 Å, Fig. 2f). The plane defined by the 4-methylphenyl and trifluoromethyl-1H-pyrazole rings in CEL form III is not observed in the form I structure,46 where the dihedral angles are all greater than 25° in form I but less than 18° in form III. The (001) plane is identified as a slip plane (Fig. 2b) with an uneven surface contour (Fig. 2c). The three conformationally different CEL molecules form six unique types of hydrogen-bonded tetramers (Fig. S5†), which can be classified into two categories based on their compositions: (a) an asymmetric unit plus one CEL molecule from a stacking asymmetric unit and (b) a pair of CEL molecules from the asymmetric unit with the same pair from a stacking asymmetric unit (Fig. S6†). In contrast, CEL form III only forms one type of dimer in the crystal structure. The different types of tetramers are interconnected through a number of hydrogen bonds (Table S2†). A one-dimensional (1D) chain along the a-axis (Fig. S7a†) is formed by stacking tetramers, fortified via weak π⋯π (4.953 Å, centroid-to centroid), C–H⋯π (3.347 Å), and N–H⋯O (d/θ: 2.961 Å/170.57° and 2.858 Å/160.83°) interactions. Adjacent 1D columns connect via π⋯π (5.030 Å, centroid-to-centroid) interactions between two aromatic rings to form a 2D layer of CEL molecules (Fig. S7b†).
space group with Z′ = 3 (Table S1†), belonging to the rare class of structures with Z′ = 3 (total 6387 crystal structures, corresponding to 0.52% of all structures in the CSD), out of which only 30.5% are in the P space group.45 The unit cell parameters and global crystal packing structures of CEL forms I differ from those of form III (Fig. S3 and Table S1†). There are three crystallographically unique molecules in the asymmetric unit (labelled with different colors in Fig. 2a), which exhibit significantly different conformations owing to the rotation of 4-methylphenyl (dihedral angles of 25.80°, 29.71°, and 61.12°) and benzenesulfonamide (dihedral angles of 59.18°, 60.80°, and 33.83°) rings about the trifluoromethyl-1H-pyrazole ring (Fig. S4a–c†). The three molecules in the asymmetric unit stack along the a-axis (Fig. S4d†) to form molecular columns, which are fortified through several types of weak intermolecular interactions, such as C–H⋯F (3.266 Å and 138.30°, Table S2†) and π⋯π stacking (centroid-to-centroid distance = 5.030 Å, Fig. 2f). The plane defined by the 4-methylphenyl and trifluoromethyl-1H-pyrazole rings in CEL form III is not observed in the form I structure,46 where the dihedral angles are all greater than 25° in form I but less than 18° in form III. The (001) plane is identified as a slip plane (Fig. 2b) with an uneven surface contour (Fig. 2c). The three conformationally different CEL molecules form six unique types of hydrogen-bonded tetramers (Fig. S5†), which can be classified into two categories based on their compositions: (a) an asymmetric unit plus one CEL molecule from a stacking asymmetric unit and (b) a pair of CEL molecules from the asymmetric unit with the same pair from a stacking asymmetric unit (Fig. S6†). In contrast, CEL form III only forms one type of dimer in the crystal structure. The different types of tetramers are interconnected through a number of hydrogen bonds (Table S2†). A one-dimensional (1D) chain along the a-axis (Fig. S7a†) is formed by stacking tetramers, fortified via weak π⋯π (4.953 Å, centroid-to centroid), C–H⋯π (3.347 Å), and N–H⋯O (d/θ: 2.961 Å/170.57° and 2.858 Å/160.83°) interactions. Adjacent 1D columns connect via π⋯π (5.030 Å, centroid-to-centroid) interactions between two aromatic rings to form a 2D layer of CEL molecules (Fig. S7b†).
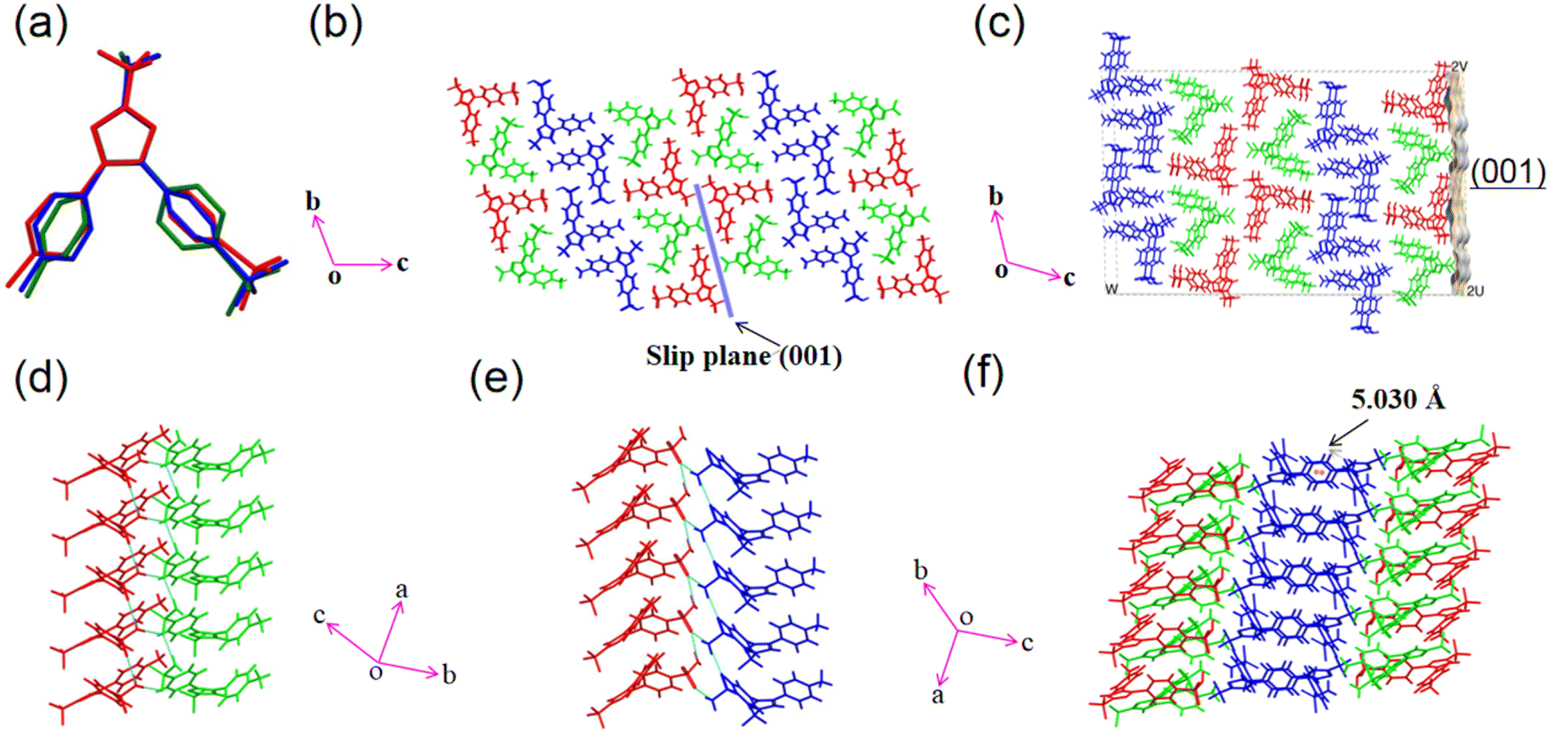 | ||
| Fig. 2 Crystal structure analysis of CEL form I: (a) the overlay of three molecules in the asymmetric unit showing conformational differences; (b) the molecular arrangement viewed along the a-axis; (c) calculated surface contour of the (001) slip plane; (d) hydrogen bonding involving one pair of CEL molecules; (e) hydrogen bonding involving the other pair of molecules; (f) packing of three molecules. | ||
The dominant faces of the form I CEL crystal are (001)/(00−1), with the side faces running along the long axis of the crystal being (010)/(0−10) and the capping ends being (100)/(−100) (Fig. S8†). Interestingly, some of the CEL form I single crystals bent when growing from a melt at 160–167 °C (Fig. 3a). These crystals straightened up when the viscosity of the melt was sufficiently decreased as the temperature was raised to 167–168 °C (Fig. 3c, Video S1†), indicating the elastic nature of the deformation. The elastic strains of different bent crystals ranged from 0.54 to 8.7% (Fig. S9, S10 and Table S3†), calculated from the crystal thickness and diameter of the curvature arc based on Euler–Bernoulli beam theory. Thus, the maximum elastic strain of CEL form I is at least 8.7%, which is significantly higher than those of all reported elastic molecular crystals, including CEL form III (3.56%).47 Form I crystals underwent brittle fracture when the (010) face was bent during crystal mounting for SCXRD (Fig. S2†). Thus, similar to CEL form III, form I CEL is 1D elastic.
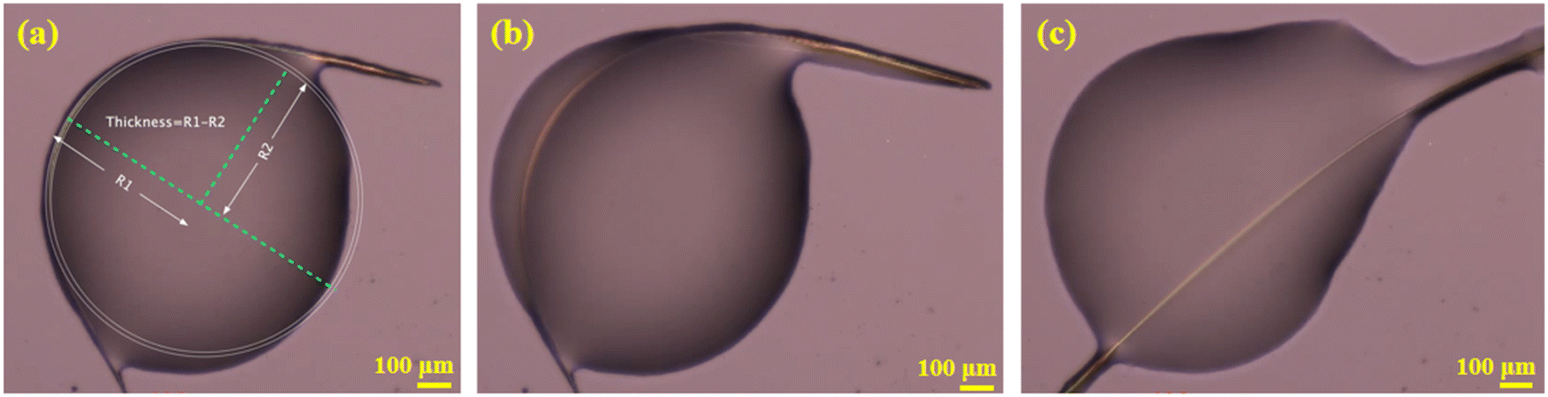 | ||
| Fig. 3 CEL form I: (a) a bent crystal (two circles were drawn to fit the outer and inner side of the bent region, from which the crystal thickness was calculated); (b) elastic recovery in the process; (c) the crystal with shape recovered. | ||
The Young's modulus (E = 3.18 ± 1.01 GPa) and hardness (H = 39.8 ± 15.6 MPa) of the elastically bendable (001)/(00−1) crystal face determined by nanoindentation (Fig. 4 and S11†) are much lower than those of CEL form III (E = 16.27 ± 0.43 GPa and H = 450 ± 20 MPa, (001)/(00−1) crystal face).46 The experimentally measured room temperature true density of form I (1.4758 g cm−3) is ∼3.28% lower than that of form III (1.526 g cm−3).47 Thus, the greater elastic flexibility of CEL form I is accompanied by both lower stiffness and lower density. Moreover, the form I CEL bulk powder also exhibits greater plasticity, as shown by its lower in-die mean yield pressure (Py,i = 41.7 ± 3.0 MPa) than that of form III powder (Py,i = 74.6 ± 3.6 MPa) (Table S4 and Fig. S13, S14†). The lower stiffness of form I, confirmed by both single crystal nanoindentation and bulk powder Heckel analysis, is consistent with its higher elastic flexibility than form III.
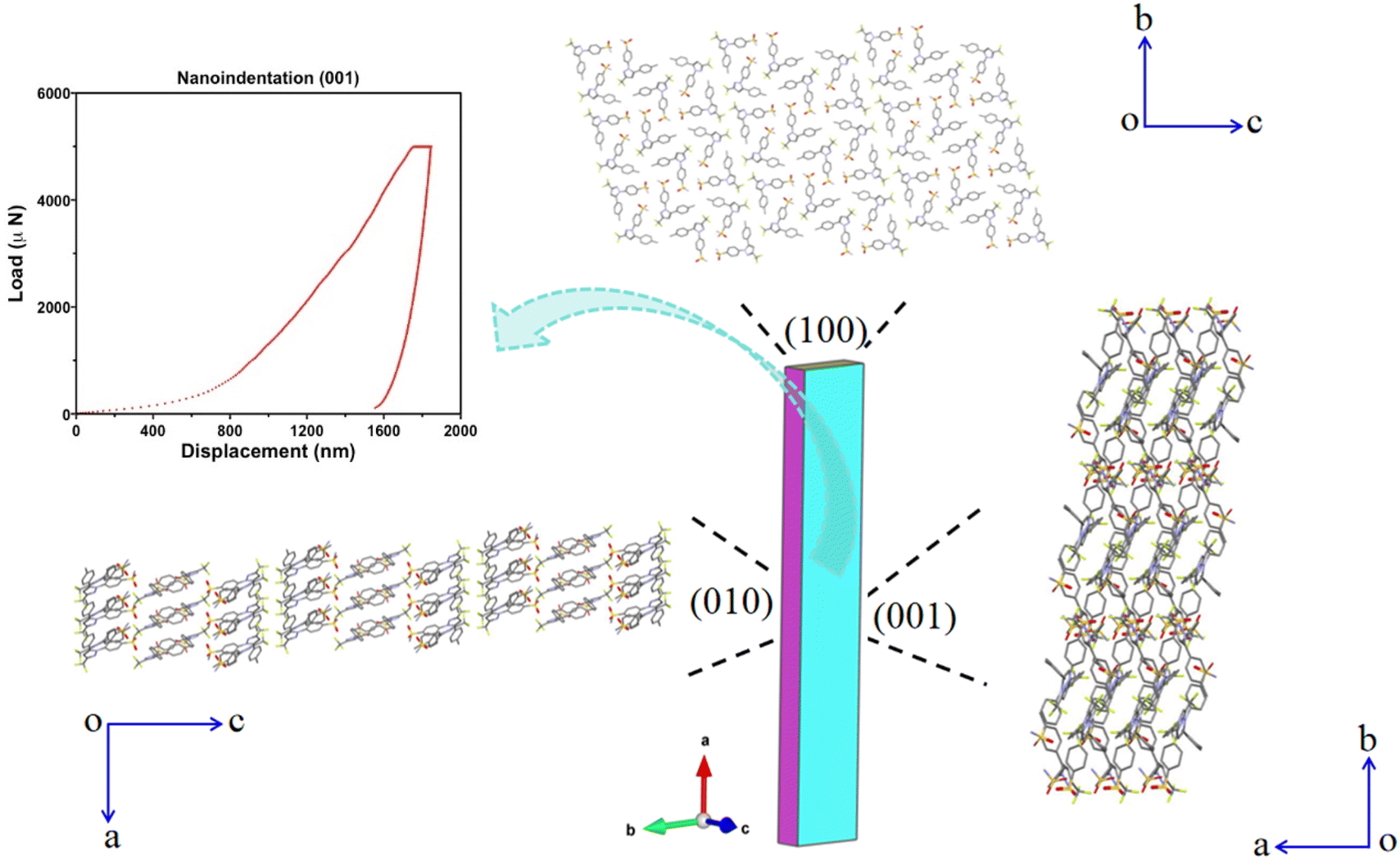 | ||
| Fig. 4 Packing diagrams of CEL form I viewed along the (100), (001) and (010) crystal faces (with hydrogens omitted for clarity), accompanied by a nanoindentation load-displacement curve for the (001) crystal face. | ||
In order to better understand the lower stiffness and higher elastic flexibility of CEL form I, we performed an integrated energy framework and topology analysis.48 CEL molecular columns, running along the a-axis and strengthened by numerous C–H⋯O, N–H⋯O, and C–H⋯F interactions, are stabilized by π⋯π and C–H⋯π inter-columnar interactions, resulting in a highly anisotropic energy framework. The energy frameworks of CEL form I and III differ significantly, where it appears discontinuous in form I (Fig. S15†), but continuous in form III (Fig. S16†). The (001) slip plane based on visual analysis of the crystal structure (Fig. 2b), i.e., on the basis of flat molecular layers with only van der Waals type inter-layer interactions, is confirmed by the integrated energy framework and full interaction map (FIM, Fig. S17†). The (001) slip plane in CEL form III exhibits a rather different FIM than that in form I, corresponding to their different crystal packing patterns and intermolecular interactions. In addition, the attachment energy for the slip plane (001) in form I (−3.165 kJ mol−1) is significantly weaker than that in form III (−28.077 kJ mol−1), while the lattice energy of form I is also weaker than that of form III (Table S5†), all of which is consistent with the lower density, lower stiffness, higher plasticity, and higher elastic flexibility of form I. The existence of multiple long-acting dispersive interactions between (001) slip planes, which is also the elastically bending crystal face, and the rough topology of the slip planes that hinders the onset of plastic slip during bending explain the significantly higher elastic flexibility of form I than form III.49 Among the four known mechanisms for the elastic flexibility of molecular crystals,50 the reversible molecular rotation model8 best explains the exceptional elasticity of CEL form I crystals. Its structure lacks key features typically associated with other mechanisms, such as interlocked stacking layers,51 fibril lamellae,52 and layered nanorods.53
In summary, using the gradient-cooling melt crystallization method, we successfully obtained a high-quality single crystal of CEL form I and solved its structure 27 years after its first discovery. The correspondence between the record-high elastic flexibility and low crystal stiffness, if proven robust with more systems, suggests the possible use of low stiffness as one of the criteria for screening elastically flexible molecular crystals that otherwise could have been overlooked because of the difficulty of forming large acicular crystals required for performing the classical three point bending test.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Zhengzheng Zhou: formal analysis, investigation, writing – original draft, visualization, and project administration; Vikram Chandrashekhar Joshi: single crystal X-ray diffraction, nanoindentation data collection, and writing – review & editing; Yiwang Guo: nanoindentation and mean yield pressure (Py) data collection; Tianyi Xiang: single crystal X-ray diffraction data collection; Zijian Wang: scanning electron microscope data collection; Changquan Calvin Sun: conceptualization, resources, writing – review & editing, supervision, project administration, and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that will influence this work.Acknowledgements
Z. Z. was supported by the “International Training Program for Outstanding Young Scientific Research Talents in Universities of Guangdong Province” (G819002020) and Young Scientists Promotion Funding of the Natural Science Foundation of Guangdong Province (No. 2023A1515030128). C. C. S. thanks the National Science Foundation for support through the Industry University Collaborative Research Center (IUCRC) grant IIP-2137264, Center for Integrated Materials Science and Engineering for Pharmaceutical Products (CIMSEPP). Nanoindentation was conducted at the Characterization Facility, University of Minnesota, which is a member of the NSF-funded Materials Research Facilities Network (https://www.mrfn.org) via the MRSEC program. Single crystal X-ray diffraction and face indexing were performed on a Bruker-AXS D8 Venture diffractometer, which was purchased through a grant from NSF/MRI (#1229400) and a grant-in-aid from the University of Minnesota. We thank Ms. Bonita VanHeel and Dr Alex Fok of the Minnesota Dental Research Center for Biomaterials and Biomechanics, MDRCBB, for assistance in nanoindentation. We also thank Mr. Alex Lovstedt and Dr Victor G. Young Jr of the Department of Chemistry, University of Minnesota, for helpful discussions related to face indexing and structure solution of the single crystal.References
- W. M. Awad, D. W. Davies, D. Kitagawa, J. M. Halabi, M. B. Al-Handawi, I. Tahir, F. Tong, G. Campillo-Alvarado, A. G. Shtukenberg, T. Alkhidir, Y. Hagiwara, M. Almehairbi, L. Lan, S. Hasebe, D. P. Karothu, S. Mohamed, H. Koshima, S. Kobatake, Y. Diao, R. Chandrasekar, H. Zhang, C. C. Sun, C. Bardeen, R. O. Al-Kaysi, B. Kahr and P. Naumov, Chem. Soc. Rev., 2023, 52, 3098–3169 CAS
.
- C. M. Reddy, G. Rama Krishna and S. Ghosh, CrystEngComm, 2010, 12, 2296–2314 Search PubMed
- M. K. Mishra, U. Ramamurty and G. R. Desiraju, Solid State Mater. Sci., 2016, 20, 361–370 Search PubMed
- S. Saha and G. R. Desiraju, Chem. Commun., 2016, 52, 7676–7679 Search PubMed
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2017, 139, 1975–1983 CAS
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CAS
- S. Li and D. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 22703–22710 CAS
- A. Worthy, A. Grosjean, M. C. Pfrunder, Y. Xu, C. Yan, G. Edwards, J. K. Clegg and J. C. McMurtrie, Nat. Chem., 2018, 10, 65–69 CrossRef CAS PubMed
- M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem.–Eur. J., 2019, 25, 5105–5112 CrossRef CAS PubMed
- T. Seki, N. Hoshino, Y. Suzuki and S. Hayashi, CrystEngComm, 2021, 23, 5686–5696 Search PubMed
- J. Li, J. Li, H. Liu, L. Zhang, Y. Lu and Z. Zhou, Chin. Chem. Lett., 2022, 33, 4069–4073 CrossRef CAS
- I. S. Divya, S. Kandasamy, S. Hasebe, T. Sasaki, H. Koshima, K. Woźniak and S. Varughese, Chem. Sci., 2022, 13, 8989 RSC
- R. Samanta, S. Das, S. Mondal, T. Alkhidir, S. Mohamed, S. P. Senanayak and C. M. Reddy, Chem. Sci., 2023, 14, 1363–1371 RSC
- C. C. Sun, J. Pharm. Sci., 2009, 98, 1671–1678 CrossRef CAS PubMed
- S. Hu and C. C. Sun, Chem. Mater., 2019, 31, 3818–3822 Search PubMed
- Q. Di, L. Li, X. Miao, L. Lan, X. Yu, B. Liu, Y. Yi, P. Naumov and H. Zhang, Nat. Commun., 2022, 13, 5280 Search PubMed
- R. Zhang, Q. Wang and X. Zheng, J. Mater. Chem. C, 2018, 6, 3182 Search PubMed
- X. Yang, L. Lan, X. Pan, X. Liu, Y. Song, X. Yang, Q. Dong, L. Li, P. Naumov and H. Zhang, Nat. Commun., 2022, 13, 7874 CrossRef CAS PubMed
- K. Chen, J. Wang, W. Wu, H. Shan, H. Zhao, N. Wang, T. Wang, X. Huang and H. Hao, Dyes Pigm., 2023, 219, 111536 Search PubMed
- H. Liu, Z. Lu, B. Tang, C. Qu, Z. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 12944–12950 CrossRef CAS PubMed
- L. Lan, L. Li, P. Naumov and H. Zhang, Chem. Mater., 2023, 35, 7363–7385 CrossRef CAS
- A. J. Thompson, A. I. C. Orué, A. J. Nair, J. R. Price, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021, 50, 11725–11740 CAS
- S. Takamizawa and Y. Miyamoto, Angew. Chem., Int. Ed., 2014, 53, 6970–6973 CAS
- X. Guo, X. Yong, S. Ogier, T. N. Ng, M. Caironi, A. Perinot, L. Li, J. Zhao, W. Tang, R. A. Sporea, A. Nejim, J. Carrabina, P. Cain and F. Yan, IEEE Trans. Electron Devices, 2017, 64, 1906–1921 CAS
- C. C. Sun and C. M. Reddy, CrystEngComm, 2021, 23, 5683–5685 CAS
-
L. D. Landau and E. M. Lifshitz, Theory of Elasticity, Pergamon Press, 1959 Search PubMed
- J. Peng and G. J. Snyder, Science, 2019, 366, 690–691 CAS
- A. G. Shtukenberg, Y. O. Punin, E. Gunn and B. Kahr, Chem. Rev., 2012, 112, 1805–1838 CAS
- S. Ghosh, M. K. Mishra, S. B. Kadambi, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2015, 54, 2674–2678 CAS
- P. Commins, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2019, 58, 10052–10060 CAS
- H. Liu, K. Ye, Z. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2019, 58, 19081–19086 Search PubMed
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 CrossRef CAS PubMed
- H. Liu, H. C. S. Chan, L. Zhang, Y. Lu, J. Li, L. Li and Z. Zhou, Chin. Chem. Lett., 2023, 34, 108057 CrossRef CAS
- C. Wang and C. C. Sun, Int. J. Pharm., 2022, 629, 122409 CrossRef CAS
- Z. Zhuo, J. Lin, J. Li, S. Wu, W. Hu and J. Gong, Chem. Eng. J., 2022, 450, 138333 CrossRef CAS
-
L. J. Ferro and P. S. Miyake, US Pat., 0087640A1, 2004 Search PubMed
- G. W. Lu, M. Hawley, M. Smith, B. M. Geiger and W. Pfund, J. Pharm. Sci., 2006, 95, 305–317 CrossRef CAS PubMed
- K. Wang and C. C. Sun, Cryst. Growth Des., 2019, 19, 3592–3600 CAS
- C. Yao, L. A. Guzei, Y. Jin, S. Ruan, G. Sun, Y. Gui, L. Wang and L. Yu, Cryst. Growth Des., 2020, 20, 7874–7881 Search PubMed
- X. Li, X. Qu, H. Rong, S. Huang, J. Nyman, L. Yu and M. Lu, Cryst. Growth Des., 2020, 20, 7093–7097 Search PubMed
- J. Zhang, Q. Shi, M. Guo, Z. Liu and T. Cai, Mol. Pharmaceutics, 2020, 17, 2064–2071 CAS
- H. Liu, H. H. Y. Tong and Z. Zhou, J. Therm. Anal. Calorim., 2022, 147, 12947–12963 CAS
- M. Lightowler, S. Li, X. Qu, X. Zou, M. Lu and H. Xu, Angew. Chem., Int. Ed., 2022, 61, e202114985 Search PubMed
- X. Qu, X. Li, H. Rong, L. Yu and M. Lu, Chem. Commun., 2020, 56, 9950–9953 Search PubMed
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 Search PubMed
- K. Wang, M. K. Mishra and C. C. Sun, Chem. Mater., 2019, 31, 1794–1799 CAS
- M. Goldenberg, G. Vreeman, D. J. Sun, M. Moffit, M. Li, M. Zernik, S. Ahuja, Y. Kim, D. Semin and C. C. Sun, Int. J. Pharm., 2023, 635, 122694 CAS
- C. Wang and C. C. Sun, Cryst. Growth Des., 2018, 18, 1909–1916 CAS
- M. K. Mishra and C. C. Sun, Cryst. Growth Des., 2020, 20, 4764–4769 CAS
- Z. Wang, W. Han, R. Shi, X. Han, Y. Zheng, J. Xu and X. H. Bu, JACS Au, 2024, 4, 279–300 CAS
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319–10323 CAS
- S. Hayashi and T. Koizumi, Angew. Chem., Int. Ed., 2016, 55, 2701–2704 CAS
- C. Wei, L. Bai, X. An, M. Xu, W. Liu, W. Zhang, M. Singh, K. Shen, Y. Han, L. Sun, J. Lin, Q. Zhao, Y. Zhang, Y. Yang, M. Yu, Y. Li, N. Sun, Y. Han, L. Xie, C. Ou, B. Sun, X. Ding, C. Xu, Z. An, R. Chen, H. Ling, W. Li, J. Wang and W. Huang, Chem, 2022, 8, 1427–1441 CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2343182. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01260k |
| This journal is © The Royal Society of Chemistry 2025 |