
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyridine-N-oxide catalyzed asymmetric N-acylative desymmetrization of sulfonimidamides†
Cui-Mei
Guo
a,
Fang-Yuan
Zhang
a,
Yin
Tian
 *b,
Ming-Sheng
Xie
*a and
Hai-Ming
Guo
*a
*b,
Ming-Sheng
Xie
*a and
Hai-Ming
Guo
*a
aState Key Laboratory of Antiviral Drugs, Pingyuan Laboratory, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China. E-mail: xiemingsheng@htu.edu.cn; ghm@htu.edu.cn
bState Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China. E-mail: ytian227@outlook.com
First published on 8th April 2025
Abstract
A highly efficient enantioselective N-acylative desymmetrization of sulfonimidamides with chloroformates was reported using chiral 4-arylpyridine-N-oxide as the catalyst, affording N-acylative sulfonimidamides with sulfur(VI)-stereocenters in high yields and excellent enantioselectivities. Experiments and DFT calculations support an acyl transfer mechanism, and the nucleophilic substitution of sulfonimidamide by the O-acyloxypyridinium cation intermediate is the enantio-determining step of the reaction. The reaction features variability for acyloxy groups and compatibility with moisture.
Introduction
Sulfonimidamides are bioisosteres of sulfonamides, in which the sulfur(VI)-bonded oxygen atom is replaced by a nitrogen atom (Fig. 1). This modification brings significant changes, because the achiral sulfur(VI) center becomes a stereogenic S(VI) center, and chemical space on S(VI) expands to three dimensions. Meanwhile, the structure of the substituent group on the newly introduced nitrogen atom is tunable, allowing for adjusting physicochemical properties.1,2 When sulfonamide LY181984,3–5 which exhibits good antitumor activity, becomes its bioisostere sulfonimidamide, the activity of (−)-isomer I significantly increases, while the activity of (+)-isomer II obviously decreases, indicating that the absolute configuration of the S(VI) center for sulfonimidamide has a vital impact on bioactivity. Representative bioactive molecules are carboxypeptidase A (CP-A) inhibitor,6 BACE 1 inhibitor,7 and NLRP3 antagonist8 (Fig. 1). Thus, developing an efficient method to construct chiral sulfonimidamides is highly desirable. | ||
| Fig. 1 Representative sulfonimidamides possessing bioactivity. | ||
For the construction of optically active sulfonimidamides, the traditional approach mainly relied on a chiral substrate strategy, through the nucleophilic substitution reaction of chiral sulfonimidoyl halides with different amines.9–14 In 2021, Willis and co-workers reported that para-methoxybenzyl (PMB) protected N,N′-sulfonimidamides are prone to rapid tautomerization under a variety of conditions, such as in toluene, upon treatment with KOH, or upon addition of trifluoroacetic acid, creating opportunities for asymmetric catalysis. Using bis-quaternized cinchona alkaloid Cat. 1 as the phase-transfer catalyst, Willis and co-workers developed the first catalytic enantioselective desymmetrization of sulfonimidamides via an asymmetric alkylation reaction, generating enantiomerically enriched sulfonimidamides in excellent yields and enantioselectivities (Scheme 1a).15 In 2024, Laconsay, Di Maso, Shaw, and co-workers reported a desymmetrization of cyclic sulfonimidamides by palladium-catalyzed asymmetric allylation using a Trost-type ligand.16 In addition to catalytic asymmetric alkylation and allylation reactions, the reported strategies mainly focused on constructing enantioenriched N-acylated sulfonimidamides.
 | ||
| Scheme 1 Asymmetric synthesis of sulfonimidamides using different strategies and catalysts. | ||
As for nitrogen nucleophiles, direct catalytic enantioselective N-acylation reaction is challenging,17–22 mainly due to the strong nucleophilicity of amines leading to strong background reactions. In 2023, Lim, Toste, Sigman, and co-workers reported a palladium-catalyzed enantioselective aryl-carbonylation of sulfonimidamides with aryl and heteroaryl iodides, affording diverse enantioenriched mono-acylated sulfonimidamides in excellent yields and enantioselectivities (Scheme 1b).23 With unprotected sulfonimidamides as the reactants, a small amount of undesired bis-acylation products was generated. In 2024, Lim, Miller, Sigman, and co-workers developed the desymmetrization of unprotected sulfonimidamides via asymmetric acylation with a cinchona-phosphinate catalyst Cat. 2, delivering enantioenriched mono-acylation sulfonimidamides in excellent results with no observed bis-acylation products (Scheme 1c).24 In the cinchona-phosphinate catalyst, the quinuclidine nitrogen and the phosphinate oxygen served as hydrogen-bond acceptors to anchor the sulfonimidamide substrate, promoting the nucleophilic addition of sulfonimidamide to 2,2,2-trifluoroethyl trifluoroacetate for the formation of a tetrahedral intermediate. Obviously, the proposed mechanism is different from the widely accepted acyl transfer mechanism.25–29 The electrophile was limited to 2,2,2-trifluoroethyl trifluoroacetate, resulting in the introduction of only trifluoroacetyl groups into the products, which restricted the structural diversity of the products. Meanwhile, trace moisture may hydrolyze 2,2,2-trifluoroethyl trifluoroacetate to generate trifluoroacetic acid, leading to a decrease in conversion and enantioselectivity. Thus, the catalytic system was sensitive to moisture. Considering that N-acyl sulfonimidamides are potential carboxylic acid bioisosteres with tunable properties,30 developing a new and effective asymmetric acylative desymmetrization of sulfonimidamides for the construction of sulfur(VI)-stereocenters remains a worthy endeavor.
Chiral pyridine-N-oxides,31–36 with oxygen atoms as nucleophilic sites, have been developed as efficient nucleophilic organocatalysts since the pioneering study by Spivey and co-workers in 2017, in which an atropisomeric 4-dimethylaminopyridine (DMAP)-N-oxide was applied to the kinetic resolution of 2-substituted indolines by N-sulfonylation.37 Afterwards, our group developed L-prolinamide derived chiral DMAP-N-oxides and 4-aryl-pyridine-N-oxides (ArPNO) as acyl transfer catalysts, and applied them in asymmetric C-acylation, O-acylation, and sulfinylation reactions.38–44 As far as we know, chiral pyridine-N-oxide has never been used in the asymmetric N-acylation reaction. For the asymmetric N-acylation desymmetrization of sulfonimidamides with chloroformates, stereocontrol is challenging due to their strong background reaction.24 Therefore, we assume that an asymmetric acyl transfer reaction using an L-prolinamide derived chiral pyridine-N-oxide as a nucleophilic catalyst could be used to address the aforementioned challenge, where pyridine-N-oxide attacks the chloroformate to give the O-acyloxypyridinium cation, and N–H in the prolinamide moiety forms an H-bond interaction with sulfonimidamide, resulting in a synergistic effect (Scheme 1d). Herein, with chloroformates as the acylation reagent, we report a highly enantioselective N-acylation desymmetrization of sulfonimidamides using chiral ArPNO as the bifunctional catalyst, allowing efficient construction of sulfur(VI)-stereocenters in excellent yields and enantioselectivities. The reaction follows an acyl transfer mechanism, with variability for acyloxy groups and compatibility with moisture.
Results and discussion
Initially, the reaction of sulfonimidamide 1a and methyl chloroformate 2a was selected as the exemplary reaction (Table 1). In the absence of catalyst, the N-acylation reaction could occur and gave N-acylation product 3aa in 19% yield at −20 °C for 48 h, indicating that the background reaction was relatively strong (entry 1). When L-prolinamide derived 3-substituted 4-pyrrolidinopyridine (PPY)-N-oxide C1 was used as the catalyst, desired sulfonimidamide 3aa was obtained in 30% yield and 26% ee (entry 2). Then, the position of the L-prolinamide moiety and the electronegativity of the substituent at the C4 position for the pyridine-N-oxides (C2–C4) were evaluated (entries 3–5), and 2-substituted 4-arylpyridine-N-oxide (ArPNO) C4 could afford product 3aa with better results in 40% yield and 74% ee (entry 5). Changing the solvent from DCM to CHCl3 improved the enantioselectivity of sulfonimidamide 3aa with 82% ee (entry 6). Afterwards, different bases were added (entries 7–9), and Cs2CO3 could deliver product 3aa with better results in 94% yield and 84% ee (entry 8). Upon lowering the reaction temperature to −50 °C and prolonging the reaction time to 96 h, the enantioselectivity of 3aa reached 92% ee, although the yield decreased to 51% (entry 10). When 4 Å MS was used as the additive, the N-acylative desymmetrization reaction proceeded well, affording the corresponding sulfonimidamide 3aa in 90% yield and 92% ee (entry 11). When 5 μL of H2O was added, the yield and enantioselectivity of the reaction were maintained (entry 12). Even with the addition of 15 μL of H2O, a similar good result could still be obtained (entry 13), indicating that this reaction is compatible with moisture. When 25 μL of H2O was added, the yield of product 3aa was reduced from 89% to 75% (entry 13 vs. 14). However, upon further addition of 4 Å MS to 50 mg, the yield of product 3aa increased from 75% to 88% (entry 14 vs. 15). The experimental results suggested that the major function of 4 Å MS is to sequester the water of the catalyst system.|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Base | Yieldb (%) | eec (%) |
| a Unless otherwise noted, reaction conditions were as follows: 1a (0.05 mmol), 2a (0.075 mmol), Cat. (5 mol%), base (1.0 equiv.) in solvent (0.5 mL) at −20 °C for 48 h. b The yields were determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard. c The ee values were determined by chiral HPLC analysis. d At −50 °C for 96 h. e 4 Å MS (25 mg) was added. PMB = p-methoxybenzyl. f 5 μL H2O was added. g 15 μL H2O was added. h 25 μL H2O was added. i 4 Å MS (50 mg) was added. | |||||
| 1 | — | DCM | — | 19 | 0 |
| 2 | C1 | DCM | — | 30 | 26 |
| 3 | C2 | DCM | — | 25 | 50 |
| 4 | C3 | DCM | — | 43 | −17 |
| 5 | C4 | DCM | — | 40 | 74 |
| 6 | C4 | CHCl3 | — | 51 | 82 |
| 7 | C4 | CHCl3 | KOH | 71 | 55 |
| 8 | C4 | CHCl3 | Cs2CO3 | 94 | 84 |
| 9 | C4 | CHCl3 | Et3N | 38 | 79 |
| 10d | C4 | CHCl3 | Cs2CO3 | 51 | 92 |
| 11d,e | C4 | CHCl3 | Cs2CO3 | 90 | 92 |
| 12d,e,f | C4 | CHCl3 | Cs2CO3 | 90 | 92 |
| 13d,e,g | C4 | CHCl3 | Cs2CO3 | 89 | 92 |
| 14d,e,h | C4 | CHCl3 | Cs2CO3 | 75 | 92 |
| 15d,h,i | C4 | CHCl3 | Cs2CO3 | 88 | 92 |

Under the optimized reaction conditions (Table 1, entry 11), the scope of sulfonimidamides was explored (Scheme 2). For the exemplary reaction, the desired N-acylative sulfonimidamide 3aa was isolated in 90% yield and 92% ee. For sulfonimidamides 1b–f bearing electron-donating or electron-withdrawing substituents at the para-position of the aryl moieties, the adducts 3ba–3fa were obtained in 78–93% yields and 90–96% ee. 3-Bromophenyl derived sulfonimidamide 1h exhibited better enantioselectivity than 3-methylphenyl derived sulfonimidamide 1g. 2-Bromophenyl derived sulfonimidamide 1i afforded product 3ia in good yield, albeit with low enantioselectivity. In the case of 2-thienyl derived sulfonimidamide 1j, the N-acylative desymmetrization reaction proceeded well to deliver adduct 3ja. When aliphatic benzyl and methyl derived sulfonimidamides 1k and 1l were used, the corresponding adducts 3ka and 3la were obtained in 82–85% yields and 95–97% ee. Complex sulfonimidamide 1m was also a suitable reactant, affording valdecoxib analogue 3ma in good results with 91% ee.
 | ||
| Scheme 2 Substrate scope of sulfonimidamidesa. aReaction conditions: 1 (0.1 mmol), 2a (0.15 mmol), C4 (5 mol%), Cs2CO3 (0.1 mmol) and 4 Å MS (50 mg) in CHCl3 (1 mL)at −50 °C for 96 h. Isolated yields are reported. The ee values were determined by chiral HPLC analysis. bAt r.t. for 48 h. | ||
Subsequently, the scope of chloroformates was evaluated (Scheme 3). Allyl chloroformate 2b and propargyl chloroformate 2c reacted smoothly in the N-acylative desymmetrization reactions, generating adducts 3ab and 3ac in 80–86% yields and 90–93% ee. Straight chain alkane derived chloroformates 2d–e could give adducts 3ad–3ae in 87–88% ee. In the case of branched alkene derived chloroformate 2f, the corresponding adduct 3af was generated with a slight decrease in enantioselectivity. As for cyclic alkane derived chloroformate 2g, the desired N-acylative product 3ag was obtained in moderate enantioselectivity. Benzyl chloroformate 2h was a suitable electrophile. When phenyl chloroformate 2i was employed, the desired adduct 3ai was afforded in 78% yield and 78% ee. As for aryl chloroformates 2j–m, the enantioselectivity of chloroformates with electron-rich aromatic groups (2j–k) was superior to that of electron-deficient aromatic groups (2l–m). With ent-C4 as the catalyst, complex chloroformates 2n and 2o, which were derived from (+)-menthol and cholesterol, were also suitable electrophiles, affording the corresponding N-acylative adducts 3an and 3ao in good results.
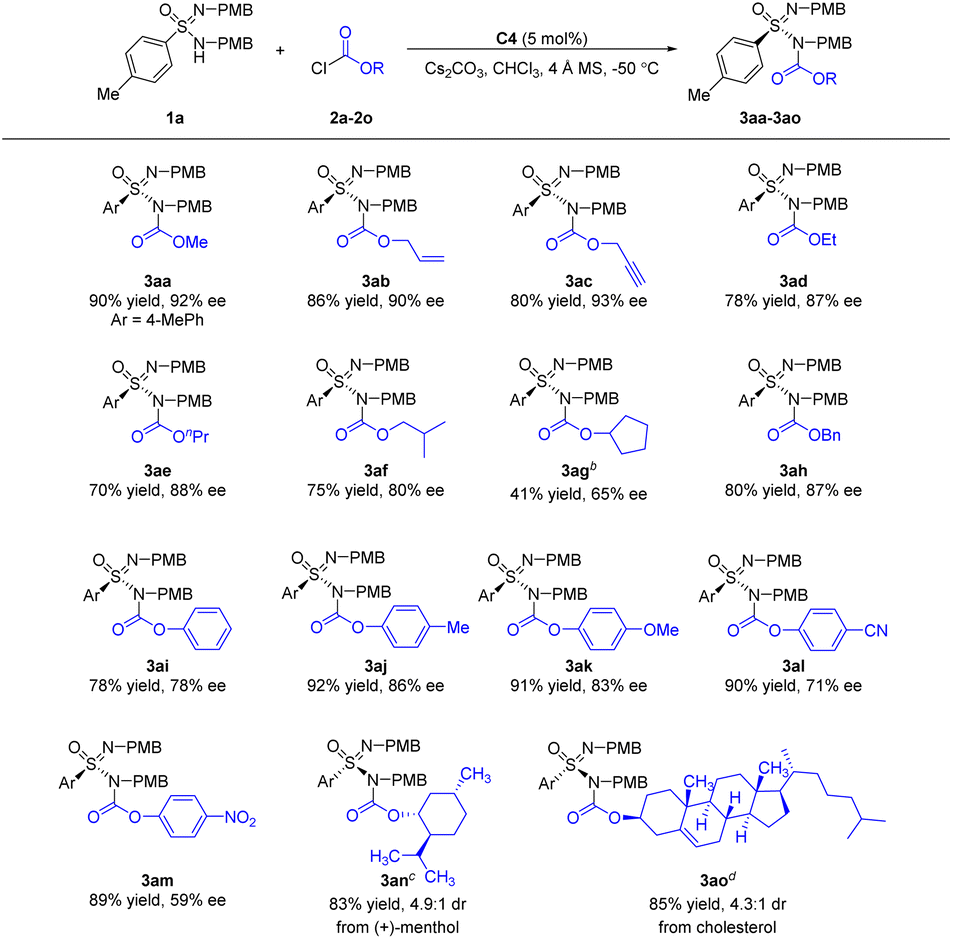 | ||
| Scheme 3 Substrate scope of chloroformatesa. aReaction conditions: 1a (0.1 mmol), 2 (0.15 mmol), C4 (5 mol%), Cs2CO3 (0.1 mmol) and 4 Å MS (50 mg) in CHCl3 (1 mL) at −50 °C for 96 h. Isolated yields are reported. The ee values were determined by chiral HPLC analysis. bAt −20 °C. cent-C4 (5 mol%) at r.t. for 72 h. dent-C4 (5 mol%) at 0 °C for 72 h. | ||
Different electrophiles including acid anhydrides, acyl chlorides, carbamic chlorides, and sulfonyl chlorides were also evaluated in the N-acylative desymmetrization reactions (Scheme 4). With acetic anhydride 2p as the electrophile, the corresponding adduct 3ap was obtained in low yield as a racemic mixture. The reactivity of acyl chloride was higher than that of the acid anhydride, and a racemic product was still formed. When methylcarbamic chloride 2r was used, the desired product 3ar was obtained in 53% yield and 0% ee. In the case of dimethylcarbamic chloride 2s, the reaction did not occur. The above comparative experiments among 2p, 2q, 2r, and 2s indicated that the oxygen atom in the O–Me moiety of methyl chloroformate 2a plays a vital role. Sulfonyl chloride 2t also afforded the corresponding adducts 3at without stereocontrol. As a result, the desymmetrization reactions have high structural requirements for electrophilic reagents, and chloroformates with variability for ester groups can achieve satisfactory stereocontrol.
 | ||
| Scheme 4 Substrate scope of electrophilesa. aReaction conditions: 1a (0.1 mmol), 2 (0.15 mmol), C4 (5 mol%), Cs2CO3 (0.1 mmol) and 4 Å MS (50.0 mg) in CHCl3 (1 mL) at −50 °C for 96 h. Isolated yields are reported. The ee values were determined by chiral HPLC analysis. | ||
To further evaluate the synthetic utility of this catalytic system, gram-scale synthesis of sulfonimidamide 3aa was performed. Using 5 mol% of catalyst C4, 3 mmol of ulfonimidamide 1a reacted with methyl chloroformate 2a for an extended time of 8 days, affording the corresponding N-acylative product 3aa in 1.18 g (84% yield) with 90% ee (Scheme 5a). Considering that N-acylative product 3ac has a terminal alkyne group, a Cu(I)-catalyzed click reaction with drug molecule zidovudine was carried out, affording the corresponding ligation product triazole 4ac in 89% yield and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (Scheme 5b). By treatment with Pd/C, the hydrogenation of sulfonimidamide 3aa could proceed to remove a PMB protecting group, giving sulfonimidamide 5aa in 65% yield with no loss of enantiomeric purity (Scheme 5c). The absolute configuration of the sulfonimidamide 5aa was determined to be the R configuration via single-crystal X-ray diffraction, which also proved that the absolute configuration of chiral sulfonimidamide 3aa was R-configuration.
:1 dr (Scheme 5b). By treatment with Pd/C, the hydrogenation of sulfonimidamide 3aa could proceed to remove a PMB protecting group, giving sulfonimidamide 5aa in 65% yield with no loss of enantiomeric purity (Scheme 5c). The absolute configuration of the sulfonimidamide 5aa was determined to be the R configuration via single-crystal X-ray diffraction, which also proved that the absolute configuration of chiral sulfonimidamide 3aa was R-configuration.
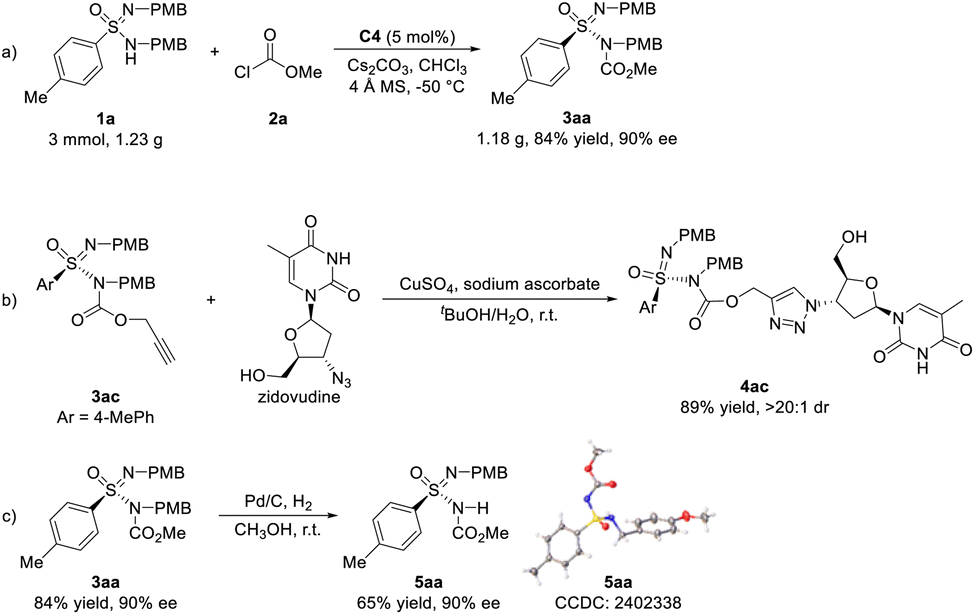 | ||
| Scheme 5 (a) Gram-scale synthesis of 3aa (b and c) further synthetic transformations. Reaction conditions: (a) 1a (3 mmol), 2a (4.5 mmol), C4 (5 mol%), Cs2CO3 (3 mmol) and 4 Å MS (1.5 g) in CHCl3 (30 mL) at −50 °C for 8 days. (b) 3ac (0.1 mmol), zidovudine (0.15 mmol), CuSO4 (20 mol%) and sodium ascorbate (60 mol%) in tBuOH/H2O (1:1, v/v, 1 mL) at r.t. for 12 h. (c) 3aa (0.1 mmol), Pd/C (0.3 mmol) and H2 (1 atm) in CH3OH (1 mL) at r.t. for 48 h. | ||
To probe the mechanism of the N-acylative desymmetrization reaction, the reaction of catalyst C4 and methyl chloroformate 2a was analyzed by HRMS, where the peak at m/z 614.3952 corresponded to the possible formation of O-acyloxypyridinium cation Int-A (Fig. 2a). Afterwards, the kinetic order of each reaction component was established by studying the initial rates of reaction (Fig. 2b). Catalyst C4 and sulfonimidamide 1a exhibited approximately first-order rate dependence, suggesting that they were involved in the rate-determining step and nucleophilic attack of sulfonimidamide 1a to O-acyloxypyridinium cation Int-A may be the rate-determining step of the reaction. In the case of methyl chloroformate 2a, the rate showed an approximately zero- order kinetic effect in the reaction, indicating that the formation of O-acyloxypyridinium cation Int-A between methyl chloroformate 2a and catalyst C4 was not involved in the rate-determining step. Furthermore, Cs2CO3 exhibited approximately first-order rate dependence, which suggested that the addition of Cs2CO3 was beneficial for binding HCl. In addition, examination of the 1H NMR spectrum of sulfonimidamide 1a in CDCl3 at room temperature showed that the two PMB groups appeared to be chemically and magnetically equivalent, which suggests that sulfonimidamide 1a is prone to rapid tautomerization in CDCl3 (see Fig. S1 in the ESI† for details).
 | ||
| Fig. 2 (a) HRMS experiment shows the formation of intermediate Int-A. (b) Kinetic orders of C4, 1a, Cs2CO3 and 2a. | ||
To gain insight into the reaction mechanism catalyzed by chiral 4-aryl-pyridine-N-oxide C4, density functional theory (DFT) is utilized to explore and analyze the reaction process. As described in Fig. 3, the mechanism consists of a two-step process: nucleophilic attack of catalyst C4 on methyl chloroformate 2a to form the O-acyloxypyridinium cation (step 1), and nucleophilic substitution of sulfonimidamide 1a by the O-acyloxypyridinium cation (step 2). In step 1, methyl chloroformate 2a and free catalyst C4 approach each other to form a reactant complex RC. With the oxygen atom as the nucleophilic site, the nucleophilic attack of catalyst C4 on methyl chloroformate 2a occurred along the Si face of methyl chloroformate 2avia transition state TS1 with the energy barrier of 5.3 kcal mol−1. Simultaneously, the chloride anion generated via cleavage of the C–Cl bond of 2a was trapped and stabilized by the H-bond from amide N–H on C4, and intermediate IM1 was then formed.
 | ||
| Fig. 3 DFT-computed relative energy profiles (kcal mol−1) of C4 nucleophilic attack along the Si-face of the substrate 2a plane at the M06-2X/6-31G(d,p)/SMD(trichloromethane) level. R1 = 4-MePh. | ||
With the addition of sulfonimidamide 1a, the H-bond between the chloride anion and N–H of sulfonimidamide 1a was formed, which generated the intermediate IM2. Then, nucleophilic substitution of sulfonimidamide 1a by the O-acyloxypyridinium cation occurred via transition state TS2 with the energy barrier of 6.6 kcal mol−1. Simultaneously, the C–O single bond of the O-acyloxypyridinium cation was cleaved, and the electron of N–H in 1a was transferred to S–N to form S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. Finally, the product complex PC was generated, which then released the catalyst C4 and (R)-3aa·HCl. With the help of Cs2CO3, (R)-3aa was finally generated from (R)-3aa·HCl.
N. Finally, the product complex PC was generated, which then released the catalyst C4 and (R)-3aa·HCl. With the help of Cs2CO3, (R)-3aa was finally generated from (R)-3aa·HCl.
The geometry information of the enantio-determining transition states and free energy were further analyzed to understand the stereoselectivity of the reaction (Fig. 4). As described in Fig. 4a and b, the relative free energy of (Si,R)-TS2 was 4.9 kcal mol−1 (ΔΔG‡) lower than that of (Si,S)-TS2, which indicated that (R)-3aa was the dominant adduct and displayed good agreement with the experimental results (Table 1, entry 11). In the case of (Si,S)-TS2, the observed increase of the energy barrier was mainly caused by the obvious steric repulsion between the 2,6-iPr2Ph group and R1 group of sulfonimidamide, which also resulted in the increase of the H-bond length between the chloride anion and N–H of sulfonimidamide. When catalyst C4 attacked methyl chloroformate 2a from the Re face (Fig. 4c and d), the relative free energies of transition states (Re,R)-TS2 and (Re,S)-TS2 were much higher than those of (Si,R)-TS2 and (Si,S)-TS2, resulting in the Si face attack as the dominant attack pathway. Compared with (Si,R)-TS2 and (Re,R)-TS2 (Fig. 4a and c), the increase of the energy barrier was mainly caused by the lack of π–π interaction between the pyridinium cation of C4 and CO of the O-acyloxypyridinium cation in (Re,R)-TS2, which is displayed in ESI Fig. S2.†
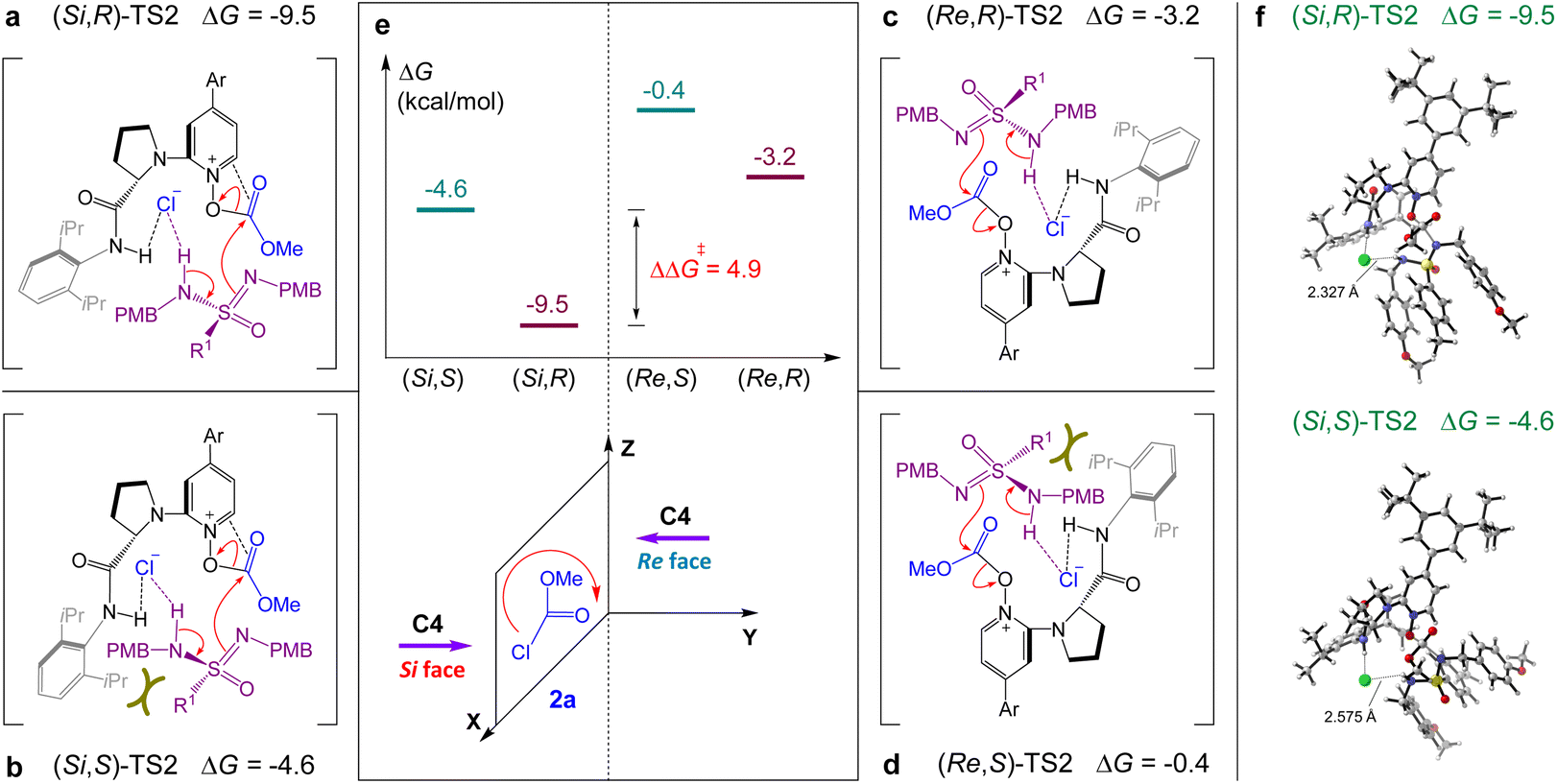 | ||
| Fig. 4 DFT-computed relative free energies (ΔG, kcal mol−1) of enantio-determining transition states (direction, configuration)-TS2 along C4-catalyzed pathways at the M06-2X/6-31G(d,p)/SMD(trichloromethane) level. Ar = 3,5-tBu2Ph, R1 = 4-MePh. | ||
Conclusions
In summary, we have developed an asymmetric N-acylative desymmetrization of sulfonimidamides with chloroformates for the construction of sulfur(VI)-stereocenters. With 5 mol% ArPNO as the catalyst, diverse N-acylative sulfonimidamides were obtained in high yields (up to 93% yield) and excellent enantioselectivities (up to 97% ee). In the presence of ArPNO, the mechanism of the reaction is an acyl transfer mechanism, which is different from Lim, Miller, and Sigman's work. Using chloroformates as the electrophiles, the structure of the ester moiety is variable, which provides an opportunity for post modification of the N-acylative products. Furthermore, the catalyzed reaction is compatible with moisture. Control experiments and DFT calculations revealed that the nucleophilic substitution of sulfonimidamide by the O-acyloxypyridinium cation intermediate is the enantio-determining step of the reaction. The high enantioselectivity of the reaction is governed by steric factors, and the H-bonding interactions between the N–H of the amide moiety of the catalyst, chloride ion, and N–H of sulfonimidamide play a vital role.Data availability
The exploratory investigation results, experimental procedures, computational data, and characterization data are available.Author contributions
Methodology, M.-S. X. and H.-M. G.; investigation, C.-M. G. and F.-Y. Z.; computational studies, Y. T.; writing – original draft, M.-S. X., C.-M. G. and Y. T.; writing – review & editing, C.-M. G., M.-S. X., Y. T., and H.-M. G; supervision, M.-S. X., Y. T., and H.-M. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from NSFC (U22A20378 and 22422103) and the Program for Innovative Research Team in Science and Technology in University of Henan Province (23IRTSTHN003). We are also thankful for the financial support from Henan Key Laboratory of Organic Functional Molecules and Drug Innovation, and NMPA Key Laboratory for Research and Evaluation of Innovative Drug.Notes and references
- P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Sulfonimidamides in Medicinal and Agricultural Chemistry, Angew. Chem., Int. Ed., 2017, 129, 4160–4170 Search PubMed.
- X. Zou, B. Shen, G.-l. Li, Q. Liang, Y. Ouyang, B. Yang, P. Yu and B. Gao, Strain-promoted S-arylation and alkenylation of sulfinamides using arynes and cyclic alkynes, Sci. China Chem., 2024, 67, 928–935 CrossRef CAS.
- J. E. Toth, G. B. Grindey, W. J. Ehlhardt, J. E. Ray, G. B. Boder, J. R. Bewley, K. K. Klingerman, S. B. Gates, S. M. Rinzel, R. M. Schultz, L. C. Weir and J. F. Worzalla, Sulfonimidamide Analogs of Oncolytic Sulfonylureas, J. Med. Chem., 1997, 40, 1018–1025 CrossRef CAS PubMed.
- B. L. Neubauer, R. L. Merriman, K. L. Best, R. L. Goode, M. F. Sarosdy, L. R. Tanzer and J. J. Howbert, Inhibition of PAIII Rat Prostatic Adenocarcinoma Growth and Metastasis by a New Diarylsulfonylurea Antitumor Agent, LY181984, J. Urol., 1992, 147, 500–504 CrossRef CAS PubMed.
- D. J. Morré and T. Reust, A Circulating Form of NADH Oxidase Activity Responsive to the Antitumor Sulfonylurea N-4-(methylphenylsulfonyl)-N′-(4-chlorophenyl)urea(LY181984) Specific to Sera from Cancer Patients, J. Bioenerg. Biomembr., 1997, 29, 281–289 CrossRef PubMed.
- B. E. Cathers and J. V. Schloss, The sulfonimidamide as a novel transition state analog for aspartic acid and metallo proteases, Bioorg. Med. Chem. Lett., 1999, 9, 1527–1532 CrossRef CAS PubMed.
- G. Wolfgang, H. Wolfgang, K. Andreas, P. Jens-Uwe, O. S. Ulrike, S. Christian, W. Roger and W. Thomas, 5-Aryl-1-imino-1-oxo-[1,2,4]thiadiazines, WO2015091595A1, 2015.
- K. Jason, R. William, S. H. Martin, D.-M. Shen and V. Shankar, Compounds and compositions for treating conditions associated with NLRP activity, WO2020102100, 2020.
- C. S. Richards-Taylor, C. Martínez-Lamenca, J. E. Leenaerts, A. A. Trabanco and D. Oehlrich, The Synthesis of Trifluoromethyl-sulfonimidamides from Sulfinamides, J. Org. Chem., 2017, 82, 9898–9904 CrossRef CAS PubMed.
- S. Greed, E. L. Briggs, F. I. M. Idiris, A. J. P. White, U. Lücking and J. A. Bull, Synthesis of Highly Enantioenriched Sulfonimidoyl Fluorides and Sulfonimidamides by Stereospecific Sulfur–Fluorine Exchange (SuFEx) Reaction, Chem.–Eur. J., 2020, 26, 12533–12538 CrossRef CAS PubMed.
- G.-f. Yang, Y. Yuan, Y. Tian, S.-q. Zhang, X. Cui, B. Xia, G.-x. Li and Z. Tang, Synthesis of Chiral Sulfonimidoyl Chloride via Desymmetrizing Enantioselective Hydrolysis, J. Am. Chem. Soc., 2023, 145, 5439–5446 CrossRef CAS PubMed.
- H.-s. Huang, Y. Yuan, W. Wang, S.-q. Zhang, X.-k. Nie, W.-t. Yang, X. Cui, Z. Tang and G.-x. Li, Enantioselective Synthesis of Chiral Sulfonimidoyl Fluorides Facilitates Stereospecific SuFEx Click Chemistry, Angew. Chem., Int. Ed., 2024, 63, e202415873 Search PubMed.
- M. Liao, Y. Liu, H. Long, Q. Xiong, X. Lv, Z. Luo, X. Wu and Y. R. Chi, Enantioselective sulfinylation of alcohols and amines by condensation with sulfinates, Chem, 2024, 10, 1541–1552 CAS.
- B. Li, J. Hu, M. Liao, Q. Xiong, Y. Zhang, Y. R. Chi, X. Zhang and X. Wu, Catalyst Control over S(IV)-stereogenicity via Carbene-derived Sulfinyl Azolium Intermediates, J. Am. Chem. Soc., 2024, 146, 25350–25360 Search PubMed.
- M. J. Tilby, D. F. Dewez, A. Hall, C. Martínez Lamenca and M. C. Willis, Exploiting Configurational Lability in Aza-Sulfur Compounds for the Organocatalytic Enantioselective Synthesis of Sulfonimidamides, Angew. Chem., Int. Ed., 2021, 60, 25680–25687 CrossRef CAS PubMed.
- D. A. Gutierrez, G. Toth-Williams, C. J. Laconsay, M. Yasuda, J. C. Fettinger, M. J. Di Maso and J. T. Shaw, Desymmetrization of Cyclic Sulfonimidamides by Asymmetric Allylation, Angew. Chem., Int. Ed., 2024, 63, e202407114 Search PubMed.
- S. Arai, S. Bellemin-Laponnaz and G. C. Fu, Kinetic Resolution of Amines by a Nonenzymatic Acylation Catalyst, Angew. Chem., Int. Ed., 2001, 40, 234–236 CrossRef CAS PubMed.
- C. K. De and D. Seidel, Catalytic Enantioselective Desymmetrization of meso-Diamines: A Dual Small-Molecule Catalysis Approach, J. Am. Chem. Soc., 2011, 133, 14538–14541 CrossRef CAS PubMed.
- M. Binanzer, S.-Y. Hsieh and J. W. Bode, Catalytic Kinetic Resolution of Cyclic Secondary Amines, J. Am. Chem. Soc., 2011, 133, 19698–19701 CrossRef CAS PubMed.
- W. Lin, Q. Zhao, Y. Li, M. Pan, C. Yang, G.-H. Yang and X. Li, Asymmetric synthesis of N–N axially chiral compounds via organocatalytic atroposelective N-acylation, Chem. Sci., 2022, 13, 141–148 RSC.
- K. Balanna, S. Barik, S. Barik, S. Shee, N. Manoj, R. G. Gonnade and A. T. Biju, N-Heterocyclic Carbene-Catalyzed Atroposelective Synthesis of N–N Axially Chiral 3-Amino Quinazolinones, ACS Catal., 2023, 13, 8752–8759 CrossRef CAS.
- C. Song, C. Pang, Y. Deng, H. Cai, X. Gan and Y. R. Chi, Catalytic N-Acylation for Access to N–N Atropisomeric N-Aminoindoles: Choice of Acylation Reagents and Mechanistic Insights, ACS Catal., 2024, 14, 6926–6935 CrossRef CAS.
- L. van Dijk, B. C. Haas, N.-K. Lim, K. Clagg, J. J. Dotson, S. M. Treacy, K. A. Piechowicz, V. A. Roytman, H. Zhang, F. D. Toste, S. J. Miller, F. Gosselin and M. S. Sigman, Data Science-Enabled Palladium-Catalyzed Enantioselective Aryl-Carbonylation of Sulfonimidamides, J. Am. Chem. Soc., 2023, 145, 20959–20967 Search PubMed.
- B. C. Haas, N.-K. Lim, J. Jermaks, E. Gaster, M. C. Guo, T. C. Malig, J. Werth, H. Zhang, F. D. Toste, F. Gosselin, S. J. Miller and M. S. Sigman, Enantioselective Sulfonimidamide Acylation via a Cinchona Alkaloid-Catalyzed Desymmetrization: Scope, Data Science, and Mechanistic Investigation, J. Am. Chem. Soc., 2024, 146, 8536–8546 CrossRef CAS PubMed.
- R. P. Wurz, Chiral Dialkylaminopyridine Catalysts in Asymmetric Synthesis, Chem. Rev., 2007, 107, 5570–5595 CrossRef CAS PubMed.
- C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Isothiourea-Catalyzed Enantioselective Carboxy Group Transfer, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed.
- C. McLaughlin and A. D. Smith, Generation and Reactivity of C(1)-Ammonium Enolates by Using Isothiourea Catalysis, Chem.–Eur. J., 2021, 27, 1533–1555 CrossRef CAS PubMed.
- M. Wang, Z. Zhang and W. Zhang, Design, Synthesis, and Application of Chiral Bicyclic Imidazole Catalysts, Acc. Chem. Res., 2022, 55, 2708–2727 CrossRef CAS PubMed.
- M. Wang, Z. Zhang and W. Zhang, Lewis base catalyzedasymmetric C-acylation, Sci. Sin.: Chim., 2023, 53, 388–401 Search PubMed.
- S. R. Borhade, R. Svensson, P. Brandt, P. Artursson, P. I. Arvidsson and A. Sandström, Preclinical Characterization of Acyl Sulfonimidamides: Potential Carboxylic Acid Bioisosteres with Tunable Properties, ChemMedChem, 2015, 10, 455–460 CrossRef CAS PubMed.
- G. C. Fu, Enantioselective Nucleophilic Catalysis with “Planar-Chiral” Hetero cycles, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS PubMed.
- G. C. Fu, Asymmetric Catalysis with “Planar-Chiral” Derivatives of 4-(Dimethylamino) pyridine, Acc. Chem. Res., 2004, 37, 542–547 CrossRef CAS PubMed.
- A. V. Malkov and P. Kocovsky, Chiral N-Oxides in Asymmetric Catalysis, Eur. J. Org Chem., 2007, 2007, 29–36 CrossRef.
- X. H. Liu, L. L. Lin and X. M. Feng, Chiral N,N′-Dioxides: New Ligands and Organo catalysts for Catalytic Asymmetric Reactions, Acc. Chem. Res., 2011, 44, 574–587 Search PubMed.
- X. H. Liu, L. L. Lin and X. M. Feng, Chiral N,N′-dioxide Ligands: Synthesis, Coordination Chemistry and Asymmetric Catalysis, Org. Chem. Front., 2014, 1, 298–302 RSC.
- X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Asymmetric Cycloaddition and Cyclization Reactions Catalyzed by Chiral N,N′-Dioxide−Metal Complexes, Acc. Chem. Res., 2017, 50, 2621–2631 Search PubMed.
- J. I. Murray, N. J. Flodén, A. Bauer, N. D. Fessner, D. L. Dunklemann, O. Bob-Egbe, H. S. Rzepa, T. Bürgi, J. Richardson and A. C. Spivey, Kinetic Resolution of 2-Substituted Indolines by N-Sulfonylation using an Atropisomeric 4-DMAP-N-oxide Organocatalyst, Angew. Chem., Int. Ed., 2017, 56, 5760–5764 CrossRef CAS PubMed.
- M.-S. Xie, Y.-F. Zhang, M. Shan, X.-X. Wu, G.-R. Qu and H.-M. Guo, Chiral DMAP-N-oxides as Acyl Transfer Catalysts: Design, Synthesis, and Application in Asymmetric Steglich Rearrangement, Angew. Chem., Int. Ed., 2019, 58, 2839–2843 CrossRef CAS PubMed.
- M.-S. Xie, B. Huang, N. Li, Y. Tian, X.-X. Wu, Y. Deng, G.-R. Qu and H.-M. Guo, Rational Design of 2-Substituted DMAP-N-oxides as Acyl Transfer Catalysts: Dynamic Kinetic Resolution of Azlactones, J. Am. Chem. Soc., 2020, 142, 19226–19238 CrossRef CAS PubMed.
- M. Shan, T. Liang, Y.-F. Zhang, M.-S. Xie, G.-R. Qu and H.-M. Guo, Enantioselective Rearrangement of Indolyl Carbonates Catalyzed by Chiral DMAP-N-oxides, Org. Chem. Front., 2019, 6, 3874–3878 RSC.
- M.-S. Xie, N. Li, Y. Tian, X.-X. Wu, Y. Deng, G.-R. Qu and H.-M. Guo, Dynamic Kinetic Resolution of Carboxylic Esters Catalyzed by Chiral PPY N-Oxides: Synthesis of Nonsteroidal Anti-Inflammatory Drugs and Mechanistic Insights, ACS Catal., 2021, 11, 8183–8196 CrossRef CAS.
- M.-S. Xie, M. Shan, N. Li, Y.-G. Chen, X.-B. Wang, X. Cheng, Y. Tian, X.-X. Wu, Y. Deng, G.-R. Qu and H.-M. Guo, Chiral 4-Aryl-pyridine-N-oxide Nucleophilic Catalysts: Design, Synthesis, and Application in Acylative Dynamic Kinetic Resolution, ACS Catal., 2022, 12, 877–891 CrossRef CAS.
- T. Wei, H.-L. Wang, Y. Tian, M.-S. Xie and H.-M. Guo, Enantioselective construction of stereogenic-at-sulfur(IV) centres via catalytic acyl transfer sulfinylation, Nat. Chem., 2024, 16, 1301–1311 Search PubMed.
- D. Liu, Z.-C. Shu, Z. Zhang, Z.-T. Wang, L. Wang, M.-S. Xie, H.-M. Guo, L.-Q. Lu and W.-J. Xiao, Asymmetric Cascade Cyclization of Enynamides with Photogenerated Ketenes via Relay Gold and N-Oxide Catalysis, ACS Catal., 2024, 14, 1741–1749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental procedures and characterization data of the synthesized compounds. CCDC 2402338. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01270h |
| This journal is © The Royal Society of Chemistry 2025 |