
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the toolbox for supramolecular chemistry: probing host–guest interactions and binding with in situ FTIR spectroscopy†
Shiva
Moaven
 a,
Douglas A.
Vander Griend
b,
Darren W.
Johnson
*a and
Michael D.
Pluth
*a
a,
Douglas A.
Vander Griend
b,
Darren W.
Johnson
*a and
Michael D.
Pluth
*a
aDepartment of Chemistry and Biochemistry, Materials Science Institute, 1253 University of Oregon, Eugene, OR 97403, USA. E-mail: dwj@uoregon.edu; pluth@uoregon.edu
bDepartment of Chemistry and Biochemistry, Calvin University, Grand Rapids, MI 49546, USA
First published on 6th May 2025
Abstract
Association constant (Ka) measurements provide fundamental information on host–guest interactions in supramolecular chemistry and other areas of science. Here we report the use of in situ FTIR spectroscopy to measure the Ka values across three classes of host–guest complexes that involve hydrogen bonding and halogen bonding. This approach can be performed with minimal sample preparation, does not require deuterated solvents, can measure association based on changes in host or guest vibrations, and benefits from a much shorter timescale than NMR spectroscopy. Due to its fast timescale, FTIR spectroscopy also provides details on host/guest conformational changes, such as the presence of unsymmetrical host conformations that are not in the ideal binding conformation until treatment with a suitable guest. These changes would not be observable by standard time-averaged NMR titration measurements. Using this approach, we demonstrated the capabilities and challenges of this technique to investigate host–guest interactions of three anion receptors that use hydrogen or halogen bonding with both mono- and polyatomic anions. In addition to directly observing how host–guest interactions impact bonding within the individual molecules, we also demonstrate that global fitting of the FTIR spectra is an effective and robust approach to measure Ka values of these host–guest complexes. We anticipate that this method will provide a new and useful approach to investigating the dynamics and specific interactions across broad areas of science.
Introduction
Supramolecular chemistry plays important roles across many research areas including biomedicine, sensing, drug delivery, catalysis, and other disciplines.1–7 For systems in which host–guest interactions govern the final properties of the system, understanding the individual supramolecular interactions between specific components, including the strength of the association constant (Ka), provides a quantitative measure of host–guest complexation. Such binding interactions are commonly measured by well-established methods, such as NMR, UV-vis, and fluorescence spectroscopy. Although perhaps less widely used, isothermal titration calorimetry (ITC) can also provide binding information, including direct information on other thermodynamic parameters such as enthalpy (ΔH) and entropy (ΔS) of the host–guest complexation.8–10One common challenge in investigating host–guest interactions is choosing a method that pairs with the common analytical approaches for measuring binding affinities. For example, the need for specific NMR-active nuclei for NMR titrations, and chromophores or fluorescent species are generally required for UV-vis and fluorescence measurements, although dye-displacement approaches have also been used to circumvent this need.9 Among these techniques, NMR spectroscopy often provides more details on the structural changes of the host and guest molecules during guest binding resulting from chemical shifts due to hydrogen bonding, π-stacking, or other changes in host or guest conformation. Unfortunately, NMR spectroscopy, which is commonly used in the majority of supramolecular host–guest measurements, provides only a time-averaged snapshot of structures in solution. This timescale complicates interpretations when guest exchange or dynamic conformational changes occur faster than the NMR timescale.11 By contrast, optoelectronic spectroscopic techniques, including UV-vis and fluorescence spectroscopy, provide binding information with a much faster timescale corresponding to electronic transitions, but require the presence of suitable chromo/fluorophores that are sufficiently responsive to host–guest interactions, although the global fitting of spectroscopic data is sensitive to nearly any change in electronic structure. In addition, these spectral changes often only occur in larger conjugated systems, rather than individual bonds or isolated small molecule/ion interaction motifs, which lowers both the generality and the structural information available from these techniques for investigating host–guest chemistry. Similarly, although fluorimetry can be used to improve sensitivity over UV-vis measurements, it reports on the excited state host–guest structure, which can result in additional complications if host- or host–guest exciplexes are formed.
In contrast to the above spectroscopic methods, infrared (IR) spectroscopy provides both individual bond level information and a fast timescale that should provide powerful insights into changes in structure, electronics, and other interactions upon host–guest binding. The fast timescale corresponding to individual bond vibrations provides another advantage that allows for the dynamics of complex processes to be monitored.12,13 As an example of this impact, biophysicists have used this technique to understand the dynamics of biomolecules and study the different states of those molecules.14–16 In addition, in situ FTIR spectroscopy has been used more broadly to investigate functional group changes for kinetic and mechanism measurements in organic, inorganic, and organometallic chemistry.17–21
FTIR has been used previously to investigate host–guest interactions in both solid and solution states. Most prior solution-state examples used individually prepared samples with different host–guest ratios to measure Ka values. These studies have typically focused on direct observation of N–H or O–H vibrational changes associated with guest binding.22–25 We are unaware, however, of prior examples using in situ FTIR to measure real-time changes in host–guest dynamics during the course of titrations to measure binding affinities. Using in situ approaches significantly reduces sample preparation requirements and allows for continuous monitoring of host–guest changes during the course of a titration, thereby bridging the gap between IR measurements and common titration techniques. Motivated by this opportunity, we demonstrate here the utility of this technique to investigate host–guest interactions and measure Ka values. We highlight this approach in three different types of anion binding receptors including those using hydrogen bond (C–H and N–H) and halogen bond (N–Br) interactions. We also highlight the benefits of this approach to investigate how binding impacts the bonding interactions in polyatomic anionic guests.
Results and discussion
C–D/H⋯X− interactions in imidazolium hosts
A key requirement of using IR to measure binding events is for these interactions to occur in a region of the IR spectrum void of other major competing vibrations. One particularly good region is from 1800–2500 cm−1, which is often referred to as the “transparent window” due to the limited number of vibrations that appear in this spectral region. Relevant to host–guest chemistry, this region provides an ideal window to investigate vibrational changes in carbon–deuterium (C–D), cyano (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), alkyne (CC), thiocyanate (SCN), and azide (N3) stretches.14 Based on these parameters and the growth of C–H based receptors for anions, we reasoned that hosts with C–D bonds involved in anion binding could be monitored directly using in situ FTIR.
N), alkyne (CC), thiocyanate (SCN), and azide (N3) stretches.14 Based on these parameters and the growth of C–H based receptors for anions, we reasoned that hosts with C–D bonds involved in anion binding could be monitored directly using in situ FTIR.
To start with a model proof-of-concept system, we chose to investigate the imidazolium host D-IPr·PF6 (Fig. 1a) based on the simple binding environment and previously demonstrated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host:guest binding with anions.26 To better isolate the hydrogen bonding interactions in the system, we selectively exchanged the imidazolium C–H to a C–D bond (vC–D = 2314 cm−1). Although the molar absorptivity of the C–D stretch is low (<10 M−1 cm−1), these vibrations are directly involved in anion binding and should show large shifts when interacting with guests.27
:1 host:guest binding with anions.26 To better isolate the hydrogen bonding interactions in the system, we selectively exchanged the imidazolium C–H to a C–D bond (vC–D = 2314 cm−1). Although the molar absorptivity of the C–D stretch is low (<10 M−1 cm−1), these vibrations are directly involved in anion binding and should show large shifts when interacting with guests.27
 | ||
| Fig. 1 (a) In situ FTIR spectra of D-IPr·PF6 treated with different TBAX (X = Cl−, Br−, and I−) salts in acetone. The new C–D stretch for each host–guest complex is highlighted and color coded. In situ FTIR titration spectra of TBACl and D-IPr·PF6 were recorded in anhydrous acetone (48.3 mM) at 296 K. Panels (b) and (d) show FTIR spectra and changes in absorbance of vC–D upon titration of TBACl. Panels (c) and (e) show FTIR spectra and changes in absorbance vN–CD upon titration of TBACl. | ||
We titrated D-IPr·PF6 with tetra-n-butylammonium (TBA) salts of Cl−, Br−, and I− in anhydrous acetone and monitored the changes by in situ FTIR spectroscopy (Fig. 1a). Upon addition of 6 equiv. of Cl−, we observed a shift of the C–D stretch from 2314 cm−1 to 2130 cm−1, which matched the 184 cm−1 redshift in the solid-state samples between the two salts (Fig. S40†), suggesting similar interactions in solution and in the solid state. We also observed similar shifts in C–D stretches upon treatment with Br− and I− centered at 2168 cm−1 and 2200 cm−1, respectively. The smaller redshifts for complexes with Br− (146 cm−1) and I− (114 cm−1) are attributed to the weaker interactions between the host and the heavier halides. These shifts in C–D stretches also highlight the potential for differentiating between the halide anions by monitoring changes in the stretching frequency of a C–D bond.
Having demonstrated that the C–D stretching frequency is sensitive to anion binding in D-IPr·PF6, we next wanted to determine whether these shifts could be used to measure binding affinities. We titrated D-IPr·PF6 with TBACl in anhydrous acetone and monitored the resultant changes in the FTIR spectrum (Fig. 1b). We observed a shift in vC–D from 2314 cm−1 to 2130 cm−1 as expected. We used SIVUU,28–32 which has been an effective and accurate approach for analyzing titration data from a variety of supramolecular host–guest systems,33–38 to analyze and globally fit the IR spectrum from 2350–2000 cm−1 to measure Ka values. Importantly, this approach allows for small changes throughout an entire spectral region to be included into the global fitting, rather than solely one peak of interest. In addition, this method allows for 95% confidence intervals (CIs) to be modelled through numerical bootstrapping, which further improves the robustness to these measurements. In general, these CIs are asymmetric with larger uncertainty on the exergonic side of the equilibrium due to the mathematics of equilibrium models. In addition, these uncertainties are larger than often-reported standard deviations because they take into account variances in the global fitting, rather than just standard deviations from individually measured values. Performing this analysis for D-IPr·PF6 treated with TBACl provided a Ka of 13+2−4 (13, 95% CI [9, 15]) M−1 (Table S1, Fig. S7 and S8†), which puts this initial experiment in a medium binding regime ([H]Ka = 0.048 M × 13 M−1 = 0.6).39
In addition to changes in the C–D stretching frequency, we also observed significant changes in the imidazolium C–N vibration at 1518 cm−1, which is weakened by the increased participation of the C–D bond in guest binding. Upon addition of Cl−, we observed both an intensity decrease and a new red-shifted vibration at 1508 cm−1 (Fig. 1c). Fitting these features provided a Ka value of 35+5−3 M−1 (Table S1, Fig. S6 and S9†). In fitting the data from these experiments, we excluded points from the 1800–1650 cm−1 range due to the large absorbance from the solvent carbonyl group. To ensure that the features being fit were related to the imidazolium C–N groups, we repeated these experiments with H-IPr·PF6. In this system, we observed the C–N stretching of the free host is centered at 1540 cm−1, which redshifted to 1536 cm−1 upon titration with Cl− and resulted in a Ka value of 9+48−3 M−1 (Table S2, Fig. S10 and S12†).
To validate the in situ FTIR binding data, we also measured the binding affinities by 1H NMR spectroscopy. Titrations were performed on both D-IPr·PF6 and H-IPr·PF6 with Cl− and revealed Ka values of 210+150−120 M−1 and 210+110−60 M−1 (Tables S10, S12, Fig. S32 and S33†), respectively, and still in the medium binding regime since these titrations were carried out at 1.5 mM. We attribute the differences in Ka values from the NMR and FTIR titrations to the necessity of using highly concentrated (due to weak signals) samples in in situ FTIR titrations. Both host and guest salts are ionic and at 48 mM concentration, their activity coefficient can deviate from ideal behavior. As an example, the mean activity coefficient (γ±) of TBABr in acetone at 25 °C is 0.25 and 0.05 for 0.016 M and 0.30 M concentrations, respectively.40 These differences in activity suggest that higher ion-pair concentrations are present under the FTIR experimental conditions. To test this hypothesis, we performed additional 1H NMR titrations with 48.9 mM of H-IPr·PF6 to normalize the ionic strength and measured a Ka value of 39+35−10 M−1 (Table S14 and Fig. S34†), which was ∼6-fold lower than the value measured at 1.5 mM and better matched the Ka values measured by in situ FTIR, confirming that these values are affected by the ion-pairing of both host and guest complexes. To better simplify comparisons between NMR and FTIR measurement techniques, we next investigated uncharged host compounds to better evaluate this technique for measuring association constants.
N–H⋯X− interactions in tris(urea) tren-based hosts
Based on our observation in the D/H-IPr·PF6 system that changes in the C–N stretches, which are not directly interacting with the guest, could also be used to measure Ka values we expanded to urea-based receptors to determine whether the strongly IR-active carbonyl group could be used to monitor guest binding. The large dipole moment of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O results in significantly more intense stretches than the C–D/C–H bonds used to investigate binding in the D/H-IPr·PF6 system. We chose to focus on tren-based tris(urea) compounds, which have been extensively studied for anion binding and transport.41–46 These hosts bind different oxyanions, such as sulfates, phosphates, nitrite, nitrate, and carbonate, with most binding investigations being evaluated by 1H NMR and UV-vis spectroscopy. Based on the strong interaction of the N–H bonds of the urea with the guest upon anion binding, we expected that guest binding would result in significant changes to the urea CO stretch, which could be measured directly by in situ FTIR to determine Ka values.
O results in significantly more intense stretches than the C–D/C–H bonds used to investigate binding in the D/H-IPr·PF6 system. We chose to focus on tren-based tris(urea) compounds, which have been extensively studied for anion binding and transport.41–46 These hosts bind different oxyanions, such as sulfates, phosphates, nitrite, nitrate, and carbonate, with most binding investigations being evaluated by 1H NMR and UV-vis spectroscopy. Based on the strong interaction of the N–H bonds of the urea with the guest upon anion binding, we expected that guest binding would result in significant changes to the urea CO stretch, which could be measured directly by in situ FTIR to determine Ka values.
To test this hypothesis directly, we prepared a p-CF3-tren-tris(urea) receptor (Fig. 2),42 which we expected would allow us to use lower concentrations than in the D/H-IPr·PF6 system due to the 20–30 times more intense CO stretch when compared to the C–D stretch.47 The CO stretching region of p-CF3-tren-tris(urea) spans from 1720–1640 cm−1, and Gaussian deconvolution of this non-symmetric region showed two distinct peaks centered at 1702 cm−1 and 1680 cm−1, with relative integrated intensities of 2.2 to 1.0, respectively (Fig. S13†). These peak ratios suggest that one urea is interacting intramolecularly with the carbonyl group of another urea group on the adjacent arm, causing a >20 cm−1 redshift in the hydrogen bonded CO stretching frequency. This observation further supports that these receptors prefer a nonsymmetrical conformation as shown in Fig. 2, which would otherwise be time-averaged into a single peak using NMR spectroscopy. This observation highlights the utility of in situ FTIR in better understanding the structural changes of flexible compounds such as the tren-tris(urea) anion receptors.
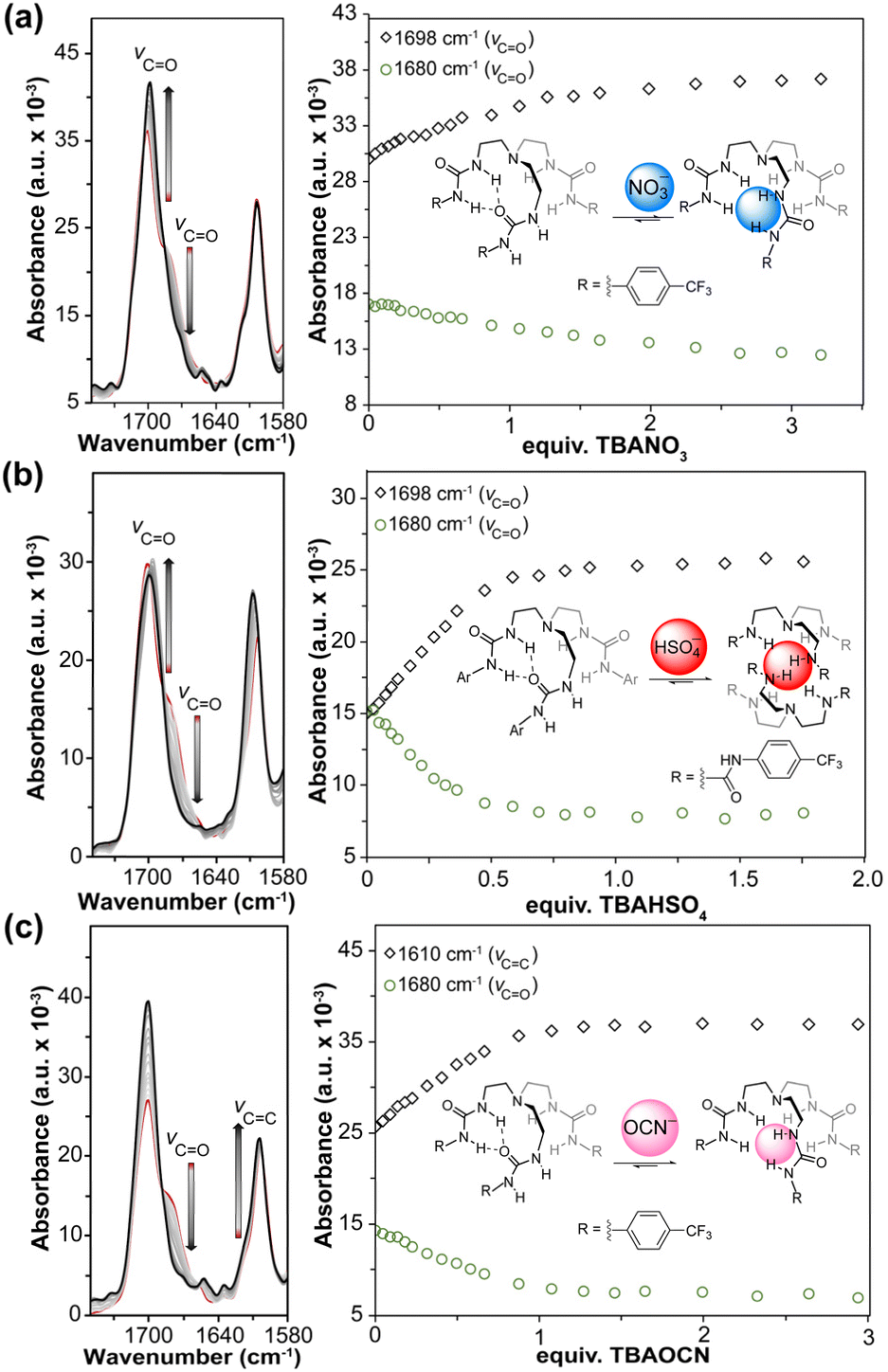 | ||
| Fig. 2 In situ FTIR titration spectra (left) and plot of absorbance changes (right) for p-CF3-tren-tris(urea) with (a) TBANO3, (b) TBAHSO4, and (c) TBAOCN in anhydrous 20% DMSO in MeCN at 298 K. | ||
We investigated three different oxyanions for in situ FTIR titrations, including HSO4−, NO3−, and OCN−. Prior studies have shown the high affinity of the tren-tris(urea) compounds for tetrahedral anions such as sulfates and phosphates, and lower affinity towards the trigonal nitrate anion. Based on these prior results, we selected TBAHSO4 and TBANO3 as strongly and weakly binding anions, respectively. We also chose to investigate TBAOCN because the vibrational frequencies of the OCN− anion can be monitored directly in the 1800–2500 cm−1 region of the IR spectrum, allowing for changes in OCN− bonding to be investigated directly upon binding to the host. Upon treatment of the host with each anion, we observed significant changes in the CO stretch of the receptor with a decrease at 1680 cm−1 and a concomitant increase of a new feature at 1702 cm−1 corresponding to formation of the host–guest complex. To measure the Ka values, we fit the entire 1750 – 1580 cm−1 region, which primarily includes changes in CO and CC stretching (Fig. 2).
Prior 1H NMR and UV-vis titration studies with the p-CF3-tren-tris(urea) host and its analogs showed minimal to no binding with NO3− indicating weak interactions with this anion and/or minimal spectral changes allowing for binding measurements.41,42,48 Repeating these measurements using in situ FTIR titrations of TBANO3 with the host, revealed change in the CO stretch upon guest addition (Fig. 2a). Additionally, increasing the NO3− concentration decreased the feature at 1680 cm−1, which is consistent with disruption of the intramolecular interaction between the two arms of the receptor to favor tripodal anion binding. After reaching equilibrium, the feature corresponding to intramolecular H-bonding is still visible, indicating that the weak interaction between the host and guest does not fully outcompete this intramolecular stabilization. Data fitting of four titration measurements resulted in a Ka value of 74+46−34 M−1 (Table S3, Fig. S14–S16†), which was substantiated by 1H NMR titration data that provided a Ka value of 52+3−3 M−1 (Table S16 and Fig. S35†) under similar conditions (∼14 mM, 25 °C). Both titration experiments showed comparable binding affinities and correlated well with the previously reported results in the literature.
Similar to the NO3− titration, the addition of TBAHSO4 to the host resulted in a decrease in the CO feature at 1680 cm−1. These changes plateaued after 0.5 equiv. of HSO4− were added, which is consistent with 2:1 host:guest binding (Fig. 2b). We also observed a new intense feature at 1610 cm−1 corresponding to the CC stretches of the p-CF3-phenyl rings. Titration data further supported the 2:1 host:guest model, providing a Ka value of 540000+77000000−340000 M−1 (Table S4, Fig. S17–S19†) for this receptor and HSO4−, which is similar to the reported values in the literature.28 In addition, we further validated this binding using 1H NMR titrations under similar conditions, which provided a Ka value of 490000+460000−100000 M−1 (Table S18 and Fig. S36†), confirming the utility of in situ FTIR for measuring both strongly and weakly interacting complexes. This model also suggests that the significant changes in the CC stretch (compared to NO3− and OCN−) of the p-CF3-phenyl rings can result from π–π interactions when the anion is encapsulated between two host molecules. We note that only with global fitting of entire spectra can one hope to zero in on a binding constant in such a strong binding regime, which also contributes to the extremely high upper confidence boundary in this binding regime.30
Using the same receptors, we also wanted to determine whether in situ FTIR could be used to measure changes in guest stretching upon binding to a host. By using the strong OCN− stretching between 2100–2200 cm−1, we monitored both conventional (guest into host solution) and reversed (host into guest solution) titrations to better track changes in both the host CO and guest stretches. During the conventional titrations, we observed intense changes in the stretching frequencies of the CO groups (Fig. 2c), which were fit to a 1:2 (host:guest) model with Ka(1:1) = 2700+11000−1900 M−1 and Ka(1:2) = 37+220−33 M−1 (Table S5, Fig. S20–S22†). A broad feature at 2148 cm−1 is present at lower OCN− concentrations (Fig. S20†), and a second feature at 2140 cm−1 grows in after reaching equilibrium. These two features correspond to bound and free OCN−, respectively. The change in vibrational frequencies between the free and bound anions is due to the change in the charge localization on the oxygen atom and the resultant increase in CN bond order.
Reversing the order of the titration, the single feature at 2140 cm−1 corresponding to free OCN− was observed, which shifted to 2148 cm−1 upon host addition (Fig. 3). We measured the Ka values from the reverse titrations to be 2500+6200−1000 M−1 and 170+720−96 M−1 (Table S6, Fig. S23–S25†) for the 1:1 and 1:2 (guest:host) binding modes, respectively, which match the values measured by 1H NMR spectroscopy of 2200+6800−1500 M−1 and 10+20−7 M−1 (Table S20 and Fig. S37†). These experiments not only show the utility of the in situ FTIR for measuring the association constants but also help understand different structural changes in the receptors and the polyatomic anions.
 | ||
| Fig. 3 (a) Binding scheme and (b) in situ FTIR titration of p-CF3-tren-tris(urea) (12.7 mM) into TBAOCN in anhydrous 20% DMSO in MeCN at 298 K. | ||
N–Br⋯X− interactions in halogen bond donor
Building from these results, we also wanted to demonstrate the utility of the in situ FTIR approach to investigate less conventional non-covalent interactions, such as halogen bond (XB) systems. As a simple test system, we chose N-bromosuccinimide (NBS) due to the presence of the two carbonyl groups near the N–Br group, which should provide strong CO stretching signals. The highly polar N–Br bond should bind to halides, which is supported by prior solid-state structures of NBS interacting with Cl− and Br− in 2:1 and 1:1 ratios.49
Upon treatment of NBS with TBACl and TBABr in MeCN, we observed significant changes in the CO stretching between 1700 and 1750 cm−1 (Fig. 4). By increasing the concentration of the TBAX salts, the signal intensity of the free NBS CO groups centered at 1726 cm−1 started to diminish and a new feature centered at 1696 cm−1 emerged. This pattern is similar to what was observed for the imidazolium hosts and indicates an interaction between NBS and anion. When interacting with anions via halogen bonding, the bond order in the NBS CO groups decrease, which subsequently results in a red-shifted feature. Using a global fit of this data, we determined that NBS forms a 2:1 complex with both anions with Ka values of 26000+7900−14000 M−1 and 13000+18000−8200 M−1 for Cl− and Br−, respectively (Table S7, Fig. S26–S28, Table S8, Fig. S29–S31†). NBS binding to Br− also results in a color change to form a light-yellow solution. This change in absorbance was used to also measure the Ka value by UV-vis spectroscopy, which revealed a Ka value of ∼11000 M−1, although the uncertainty in this measurement was larger than the value making it statistically insignificant (Table S21†). These results further highlight the utility of in situ FTIR to obtain the Ka values for less-common classes of non-covalent interactions such as XB donors and demonstrates its potential to be used for other types of non-covalent interactions such as chalcogen and pnictogen bonding.
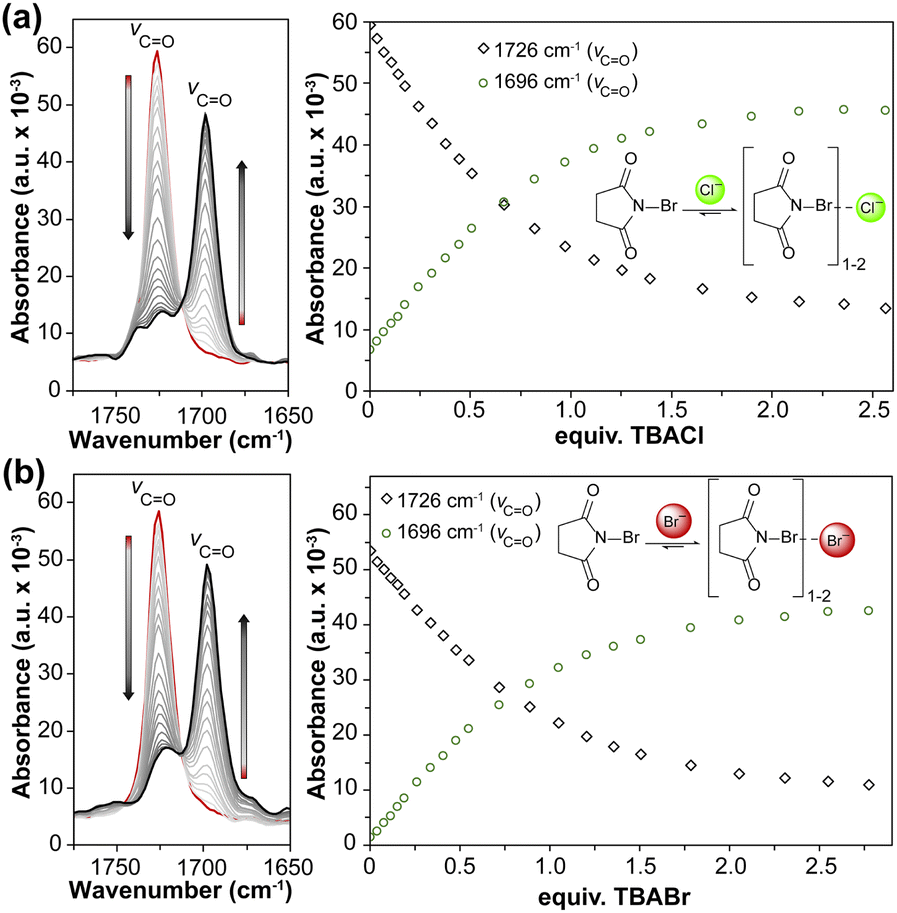 | ||
| Fig. 4 In situ FTIR titration spectra (left) and plot of absorbance changes (right) for NBS titrated with (a) TBACl and (b) TBABr in MeCN at 298 K. | ||
Conclusions
In this study we demonstrated the applicability of in situ FTIR spectroscopy for measuring Ka values of different host guest systems that predominantly interact through hydrogen or halogen bonding. This approach can be used in combination with other common methods to obtain additional or new information on host–guest interactions across different host and guest types. We showed the potential of this technique to differentiate between halides by studying changes in the C–D stretches in hydrogen bond donating receptors. We also found that CO and CN stretches near hydrogen and halogen bond donors were sufficiently sensitive to assess binding. This technique also provides detailed information on the conformational and electronic changes in both host and guest compounds, which is often difficult to obtain through other commonly used spectroscopies focused on host–guest interactions. We also showed that this method can also be used to study less conventional non-covalent interactions such as halogen bond donors. Finally, this study demonstrates the compatibility of this technique for Ka measurements of neutral compounds and its potential to be used as a complementary method with NMR or UV-vis titration techniques for Ka measurements and dynamic studies on flexible host or guest compounds. The higher host/guest concentrations needed for in situ FTIR investigations when compared to other spectroscopic techniques, may make this approach challenging for very strongly binding systems. However, it also offers a significant advantage for studying systems that form weaker interactions, allowing for significant titration data to be acquired and fit in binding regimes that are often difficult to measure with other conventional titration methods. For systems in which strong IR absorbances are present in the host or guest (particularly if common NMR nuclei are absent or chemical shifts are obscured from fast exchange or overlapping signals), we anticipate that in situ FTIR can be used as a valuable approach to gain additional information on host/guest bonding as well as intermolecular interactions. The commercial availability of ready-to-use systems may further advance the inclusion of these systems as useful tools for measuring different host–guest interactions.
Data availability
Experimental data, general methods, spectroscopic data, fitting parameters and data are all available in the ESI.†Author contributions
S. M. completed the experimental work, S. M. and D. A. V. G. analyzed and modeled the data. S. M., D. A. V. G., D. W. J., and M. D. P. designed experiments, organized project data, analyzed data, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF (CHE-2107602) to MDP.Notes and references
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- M. Sayed and H. Pal, Phys. Chem. Chem. Phys., 2021, 23, 26085–26107 RSC.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS.
- F. H. Huang and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 5879–5880 RSC.
- Z. Q. Q. Feng, T. F. Zhang, H. M. Wang and B. Xu, Chem. Soc. Rev., 2017, 46, 6470–6479 RSC.
- L. You, D. J. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS.
- K. Hirose, Determination of Binding Constants, in Analytical Methods in Supramolecular Chemistry, John Wiley & Sons, Ltd, 2006, pp 17–54 Search PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- E. G. Sheetz, D. Van Craen and A. H. Flood, Anion Recognition and Binding Constant Determination, in Anion-Binding Catalysis, John Wiley & Sons, Ltd, 2022; pp pp 79–109 Search PubMed.
- I. R. Kleckner and M. P. Foster, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 942–968 CrossRef CAS PubMed.
- P. R. Griffiths, Introduction to the Theory and Instrumentation for Vibrational Spectroscopy, in Handbook of Vibrational Spectroscopy, John Wiley & Sons, Ltd, 2010 Search PubMed.
- I. D. Campbell, Biophysical Techniques, Oxford University Press, 2012 Search PubMed.
- R. Adhikary, J. Zimmermann and F. E. Romesberg, Chem. Rev., 2017, 117, 1927–1969 CrossRef CAS PubMed.
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 Search PubMed.
- R. B. Dyer, F. Gai and W. H. Woodruff, Acc. Chem. Res., 1998, 31, 709–716 CrossRef CAS.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, J. Phys. Chem., 1985, 89, 588–591 CrossRef CAS.
- N. Y. Topsoe, Science, 1994, 265, 1217–1219 CrossRef CAS.
- Y. Chae, S. Min, E. Park, C. Lim, C. H. Cheon, K. Jeong, K. Kwak and M. Cho, Anal. Chem., 2021, 93, 2106–2113 CrossRef CAS PubMed.
- B. Wei, J. C. Sharland, D. G. Blackmond, D. G. Musaev and H. M. L. Davies, ACS Catal., 2022, 12, 13400–13410 CrossRef CAS.
- H. F. Mao, H. M. Xing, M. M. Jin, J. B. Liu, Y. L. Yao and Y. Zhao, Anal. Methods, 2022, 14, 2833–2840 RSC.
- J. W. M. Nissink, H. Boerrigter, W. Verboom, D. N. Reinhoudt and J. H. van der Maas, J. Chem. Soc., Perkin Trans. 2, 1998, 1671–1676 RSC.
- J. W. M. Nissink, H. Boerrigter, W. Verboom, D. N. Reinhoudt and J. H. van der Maas, J. Chem. Soc., Perkin Trans. 2, 1998, 2541–2546 RSC.
- J. W. M. Nissink, H. Boerrigter, W. Verboom, D. N. Reinhoudt and J. H. van der Maas, J. Chem. Soc., Perkin Trans. 2, 1998, 2623–2630 RSC.
- J. M. Widom, R. J. Philippe and M. E. Hobbs, J. Am. Chem. Soc., 1957, 79, 1383–1386 CrossRef CAS.
- M. H. Dunn, N. Konstandaras, M. L. Cole and J. B. Harper, J. Org. Chem., 2017, 82, 7324–7331 CrossRef CAS.
- M. C. Thielges, D. A. Case and F. E. Romesberg, J. Am. Chem. Soc., 2008, 130, 6597–6603 CrossRef CAS.
- D. A. D. Vander, M. J. Griend, M. Greely, Y. Kim, D. Buist and C. Ulry, 2021, http://sivvu.org.
- D. A. V. Griend, D. K. Bediako, M. J. DeVries, N. A. DeJong and L. P. Heeringa, Inorg. Chem., 2008, 47, 656–662 CrossRef.
- N. P. Kazmierczak, J. A. Chew, A. R. Michmerhuizen, S. E. Kim, Z. D. Drees, A. Rylaarsdam, T. Thong, L. Van Laar and D. A. V. Griend, J. Chemom., 2019, 33, e3119 CrossRef.
- N. P. Kazmierczak and D. A. Vander Griend, J. Chemom., 2019, 33, e3183 CrossRef CAS.
- N. P. Kazmierczak, J. A. Chew and D. A. Vander Griend, Anal. Chem. Acta, 2022, 1227, 339834 CrossRef CAS.
- Y. J. Li, D. A. V. Griend and A. H. Flood, Supramol. Chem., 2009, 21, 111–117 CrossRef CAS.
- S. Lee, C. H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed.
- E. M. Fatila, E. B. Twum, J. A. Karty and A. H. Flood, Chem.–Eur. J., 2017, 23, 10652–10662 CrossRef CAS.
- S. I. Etkind, D. A. Vander Griend and T. M. Swager, J. Org. Chem., 2020, 85, 10050–10061 CrossRef CAS.
- E. G. Sheetz, D. Van Craen and A. H. Flood, Anion Recognition and Binding Constant Determination, in Anion-Binding Catalysis, John Wiley & Sons, Ltd, 2022, pp. 79–109 Search PubMed.
- N. Bhattacharjee, X. F. Gao, A. Nathani, J. R. Dobscha, M. Pink, T. Ito and A. H. Flood, Chem.–Eur. J., 2023, 29, e202302339 CrossRef CAS PubMed.
- K. Hirose, Quantitative Analysis of Binding Properties, in Analytical Methods in Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012, pp 27–66 Search PubMed.
- J. Barthel, R. Neueder, H. Poepke and H. Wittmann, J. Solution Chem., 1999, 28, 1277–1287 CrossRef CAS.
- D. A. Jose, D. K. Kumar, B. Ganguly and A. Das, Inorg. Chem., 2007, 46, 5817–5819 CrossRef CAS PubMed.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- A. Pramanik, D. R. Powell, B. M. Wong and M. A. Hossain, Inorg. Chem., 2012, 51, 4274–4284 CrossRef CAS PubMed.
- R. Dutta, S. Chakraborty, P. Bose and P. Ghosh, Eur. J. Inorg. Chem., 2014, 4134–4143 CrossRef CAS.
- R. Chutia, S. K. Dey and G. Das, Cyst. Growth Design, 2015, 15, 4993–5001 CrossRef CAS.
- W. Zuo, C. D. Jia, H. Z. Zhang, Y. X. Zhao, X. J. Yang and B. Wu, Chem. Sci., 2019, 10, 2483–2488 RSC.
- I. M. Pazos, A. Ghosh, M. J. Tucker and F. Gai, Angew. Chem., Int. Ed., 2014, 53, 6080–6084 CrossRef CAS.
- A. Pramanik, B. Thompson, T. Hayes, K. Tucker, D. R. Powell, P. V. Bonnesen, E. D. Ellis, K. S. Lee, H. T. Yua and M. A. Hossain, Org. Biomol. Chem., 2011, 9, 4444–4447 RSC.
- M. H. Ghassemzadeh, K. Dehnicke and D. Fenske, Z. Naturforsch, 1994, 49, 593–601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01329a |
| This journal is © The Royal Society of Chemistry 2025 |