
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic RNA ligands as activators of type I toxin–antitoxin systems: a novel antimicrobial strategy targeting Helicobacter pylori†
Céline
Martin
a,
Marc
Panosetti
 a,
Eleonora
Tesini
a,
Stéphane
Azoulay
a,
Anthony
Bugaut
b,
Véronique
Sinou
c,
Nadia
Patino
a,
Audrey
Di Giorgio
a,
Fabien
Darfeuille
*b and
Maria
Duca
*a
a,
Eleonora
Tesini
a,
Stéphane
Azoulay
a,
Anthony
Bugaut
b,
Véronique
Sinou
c,
Nadia
Patino
a,
Audrey
Di Giorgio
a,
Fabien
Darfeuille
*b and
Maria
Duca
*a
aUniversité Côte d’Azur, CNRS, Institute of Chemistry of Nice (ICN), 28 Avenue Valrose, 06100 Nice, France. E-mail: maria.duca@univ-cotedazur.fr
bUniversity of Bordeaux, INSERM U1212, CNRS UMR5320, ARNA Laboratory, F-33000, Bordeaux, France. E-mail: fabien.darfeuille@inserm.fr
cAix Marseille Université, INSERM, SSA, MCT, 13385 Marseille, France
First published on 23rd May 2025
Abstract
Targeting RNAs with synthetic small molecules represents a privileged avenue for the discovery of new therapeutic approaches and offers the possibility to identify original targets escaping the classical rules of druggability and resistance. In the context of multidrug resistance to antibiotics, an urgent need for new antimicrobial compounds is emerging; however both academia and industry are mostly working on known extensively explored targets susceptible to inducing resistance again. In this work, we present a new potential target for antibiotics represented by a Helicobacter pylori type I toxin–antitoxin system where RNA–RNA interactions are responsible for silencing the synthesis of a toxin that is lethal to bacteria and that is activated only under particular conditions. We report the design and synthesis of new RNA binders to inhibit these RNA–RNA interactions and to artificially activate toxin production and kill bacteria. After screening these compounds using several complementary assays, we identified a selective inhibitor of the targeted RNA–RNA interaction showing specific antibiotic activity against H. pylori. This represents an unprecedented antimicrobial strategy based on the use of compounds that are not toxic by themselves but activate the production of an endogenous toxin produced by the bacteria themselves. Finally, this work allowed us to explore new compounds to inhibit RNA–RNA interactions, which also represents an underexplored field of RNA targeting.
Introduction
Infectious diseases constitute one of the leading causes of human deaths throughout the world.1 Despite the development of numerous antimicrobial compounds during the last century, multidrug resistance is now a major threat worldwide.2 Overcoming this challenge calls for the discovery of new antibiotics and new targets that are less susceptible to inducing resistance.Toxin–antitoxin (TA) systems are bacterial small genetic elements composed of a toxin gene and its cognate antitoxin, both coding for the corresponding toxin and antitoxin products.3 Toxins of all known TA systems are proteins, whereas antitoxins are either proteins or small regulatory RNAs that neutralize toxicity during normal growth conditions.4 When bacteria are subject to environmental stress, such as temperature fluctuations, oxidative challenges, nutrient deprivation or antibiotic treatment, toxicity can be triggered by rapidly depleting the antitoxin pool. Eventually, toxins may target a wide range of cellular processes and structures, mainly acting as inhibitors of translation and leading to bacterial growth inhibition and/or bacterial cell death. In the context of antibiotic treatment, bacteria activate toxin–antitoxin systems in order to (i) slow their growth so that the antibiotic treatment becomes ineffective (persistence) or (ii) kill sensitive cells to favor the survival of the resistant population.3 Eight different types of TA systems have been identified depending on the antitoxin nature.4 Type I TA (T1TA) systems are composed of a non-coding RNA (ncRNA) that acts as the antitoxin and inhibits toxin synthesis by base pairing with the messenger RNA (mRNA) coding for the toxin.5 Toxin inactivation by the antitoxin involves RNA–RNA interactions, starting with kissing loops and leading to an extended duplex that inhibits toxin mRNA translation (Fig. 1 top). When a stress condition induces the degradation of the antitoxin ncRNA, the toxin mRNA is translated and induces bacterial death (Fig. 1 bottom). T1TA systems are thus involved in the irreversible killing of bacterial cells.6 In this work, we decided to focus on a particular T1TA system called AapA/IsoA in the major human gastric pathogen Helicobacter pylori where AapA is the toxin protein and IsoA is the antitoxin RNA (Fig. 1 and S1†).7,8H. pylori is a Gram-negative bacterium that thrives in the acidic environment of the stomach and affects about 50% of the world's population. While efficient antibiotic therapies exist, renewed interest in the discovery of innovative therapeutic strategies has emerged as H. pylori infection is recognized as the main and specific infectious cause of gastric cancer worldwide.9 Similarly to other type I TA systems, the toxin is a small membrane protein whose production is inhibited by a small antisense RNA (antitoxin) that binds to the 5′ untranslated region of the mRNA.10 Importantly, the toxicity of these systems is controlled at the level of gene expression. They are involved in the irreversible killing of bacterial cells and their artificial activation by a small-molecule drug would induce death of the entire bacterial population.7 It was also previously shown that its chromosome encodes several homologs (up to 9 copies) of this TA pair that functions as a true killing system in H. pylori. Indeed, blocking the interaction of the toxin with the antitoxin would induce the synthesis of the toxin and bacterial cell death.
 | ||
| Fig. 1 Type I TA (T1TA) systems and the proposed strategy to artificially activate toxin production. Illustration of toxin mRNA silencing by antitoxin ncRNA (top) and stress conditions inducing antitoxin degradation, leading to toxin production, growth arrest and cell death (bottom). An RNA binder preventing toxin–antitoxin interaction could act as an antimicrobial by activating toxin production (bottom). | ||
Here, we thus explored small-molecule RNA binders as artificial activators of T1TA systems upon inhibition of toxin–antitoxin RNA/RNA interactions, thereby enabling translation of the toxin mRNA into a functional toxin that ultimately induces bacterial cell death (Fig. 1 bottom). To this aim, we synthesized a new series of RNA binders using multicomponent reactions, screened these compounds for their ability to inhibit the targeted RNA–RNA interaction, studied the affinity and selectivity for the targets and finally tested the compounds for their antibiotic activity. The use of various complementary assays allowed us to gather information about the newly synthesized compounds, to study their effect on the targeted RNA structures and to identify a promising hit able to inhibit IsoA/AapA interaction both in vitro and in vivo. This could establish a novel antimicrobial target with an innovative mechanism, minimizing resistance since the toxic agent is the species-specific toxin encoded at multiple chromosomal loci, rather than the synthetic RNA ligand.
Results and discussion
Very few examples of RNA binders capable of affecting RNA/RNA interactions have been reported suggesting that identifying such compounds is challenging.11–14 To access efficient inhibitors, we performed a de novo design of binders by combining different RNA binding domains to confer both affinity and selectivity towards the target. This approach was already successful in our previous studies for the targeting of oncogenic miRNA precursors or viral RNAs.15–21 For the generation of these new RNA ligands, we chose to apply the 4-component Ugi reaction that relies on the condensation of carboxylic acid (red moiety), amine (blue moiety), aldehyde (green moiety) and isocyanide compounds (black moiety) to form a α-aminoacyl amide compounds (Fig. 2A). This platform is interesting for RNA targeting thanks to its ability to conjugate different interacting domains in one step under sustainable conditions.22 First, we decided to use naphthalene to initiate the target RNA recognition thanks to π–π and π–cation interactions or intercalation (Fig. 2B). Thus, naphthalene carbaldehyde was chosen as aldehyde component in the Ugi reaction (shown in green in Fig. 2C). Second, we decided to employ the neutral tert-butylisocyanide for all compounds. Third, we employed amino acids as carboxylic acids, since their lateral chains contain RNA recognition motifs known to be involved in high affinity RNA/peptides or protein complexes via various kinds of interactions, particularly with unpaired nucleobases (shown in red in Fig. 2B and C). We chose in particular lysine and histidine, that are basic amino acids known to form strong interactions with RNA both in RNA-binding proteins and small molecule ligands.15,23 Phenylalanine and alanine were chosen as aromatic and aliphatic amino acids, respectively, to assess whether the side chain of this moiety is important in the interaction. Lastly, as amine components, we chose various amino-substituted heteroaromatic moieties capable of interacting with paired and unpaired nucleobases (shown in blue in Fig. 2B and C) through specific hydrogen bonding interactions. The O-alkylated naphthyridine scaffold was introduced (compounds 1a–1c) since it is known to interact with unpaired G bases.24 We also selected an N-alkylated naphthyridine scaffold (compounds 2a–2d) which could interact with paired nucleobases via H-bond formation or by intercalation.12 Also, we chose a phenylthiazole moiety, previously used in the design of RNA binders as it can form specific hydrogen bond interactions with paired AU bases.20 This moiety was incorporated either via a glycine linker (compounds 3a–3d) or directly using the aromatic amine present on the phenyl ring (compound 4a) to assess if the flexibility of the substituents affects activity. Furthermore, we introduced the 2-(4-aminophenyl)benzothiazole moiety, with or without a glycine spacer (compounds 5a and 6a) to assess if an extended aromatic substituent could enhance interaction and inhibition activity. Finally, the nucleobase thymine (as in 7a and 7c), an imidazole (as in 8c) and an unsubstituted phenyl substituent (as in 9c) were introduced for comparison. Noteworthily, even though it remains difficult to predict which physico–chemical properties will enable bacterial cell membrane penetration, the compounds were designed to be positively charged and to bear amino groups, features that have been recognized as favorable for accumulation of compounds in bacteria.25 | ||
| Fig. 2 RNA ligands synthesized using the Ugi 4-component reaction. (A) General reaction scheme of the Ugi reaction. (B) General structure of the RNA binding domains and their potential interactions with RNA bases. (C) Starting from 1-naphthaldehyde (green moiety) upon addition of lysine, tryptophan, alanine or phenylalanine (red moieties) and various heteroaromatic amines (blue moieties) compounds, 1a–7a, 1b–3b, 1c–3c, 7c–9c and 2d–3d were obtained in two steps. | ||
Ugi reactions were individually performed in methanol, yielding desired Boc-protected compounds 1a′–7a′, 1b′–3b′, 1c′–3c′, 7c′–9c′ and 2d′–3d′ (Schemes S1 and S2†) in 33–90% yields including two diastereoisomers. A final deprotection step using TFA in CH2Cl2 led to the desired derivatives 1a–7a, 1b–3b, 1c–3c, 7c–9c and 2d–3d in quantitative yields (Fig. 2C).
After preparation and characterization of all compounds, we assessed whether they could inhibit the targeted RNA–RNA interaction. Therefore, we developed a new anisotropy-based assay employing one of the kissing loops involved in IsoA1/AapA1 (one out of six loci) complex formation (red and blue regions in Fig. 3A and S1†). Both IsoA1 and AapA1 stem-loops were labeled at the 5′ position with a fluorophore (fluorescein) and incubated with the corresponding unlabeled AapA1 or IsoA1 RNAs leading to comparable results. When the stem-loops are structured and evaluated separately, the anisotropy is low, while when the extended duplex is formed, the anisotropy level increases (Fig. 3B). Formation of the duplex when the two pre-structured stem loops are mixed at 37 °C is extremely fast, with anisotropy levels increasing within 15 minutes after addition (black line in Fig. 3C) compared to the AapA1 stem-loop alone or the pre-formed duplex (red and green line, respectively, in Fig. 3C). Formation of the duplex was also confirmed by PAGE (Fig. S2†), showing that incubation at 37 °C for 2 hours was effective in inducing the complete conversion from the individual stem-loops to the extended duplex. All compounds were tested by fluorescence anisotropy and only compounds 1a–c, sharing the naphthyridine scaffold, inhibit the extended duplex formation with IC50 values of 2.35, 1.79 and 6.57 μM, respectively. As a control, we employed neomycin, an aminoglycoside antibiotic known as a promiscuous RNA binder as its interactions with its primary target, i.e. prokaryotic ribosomal RNA, are mainly electrostatic and non-specific.26 Neomycin was previously reported to affect the IsoA1/AapA1 interaction7 and showed an IC50 of 88.8 μM (Fig. 3D and S3†).
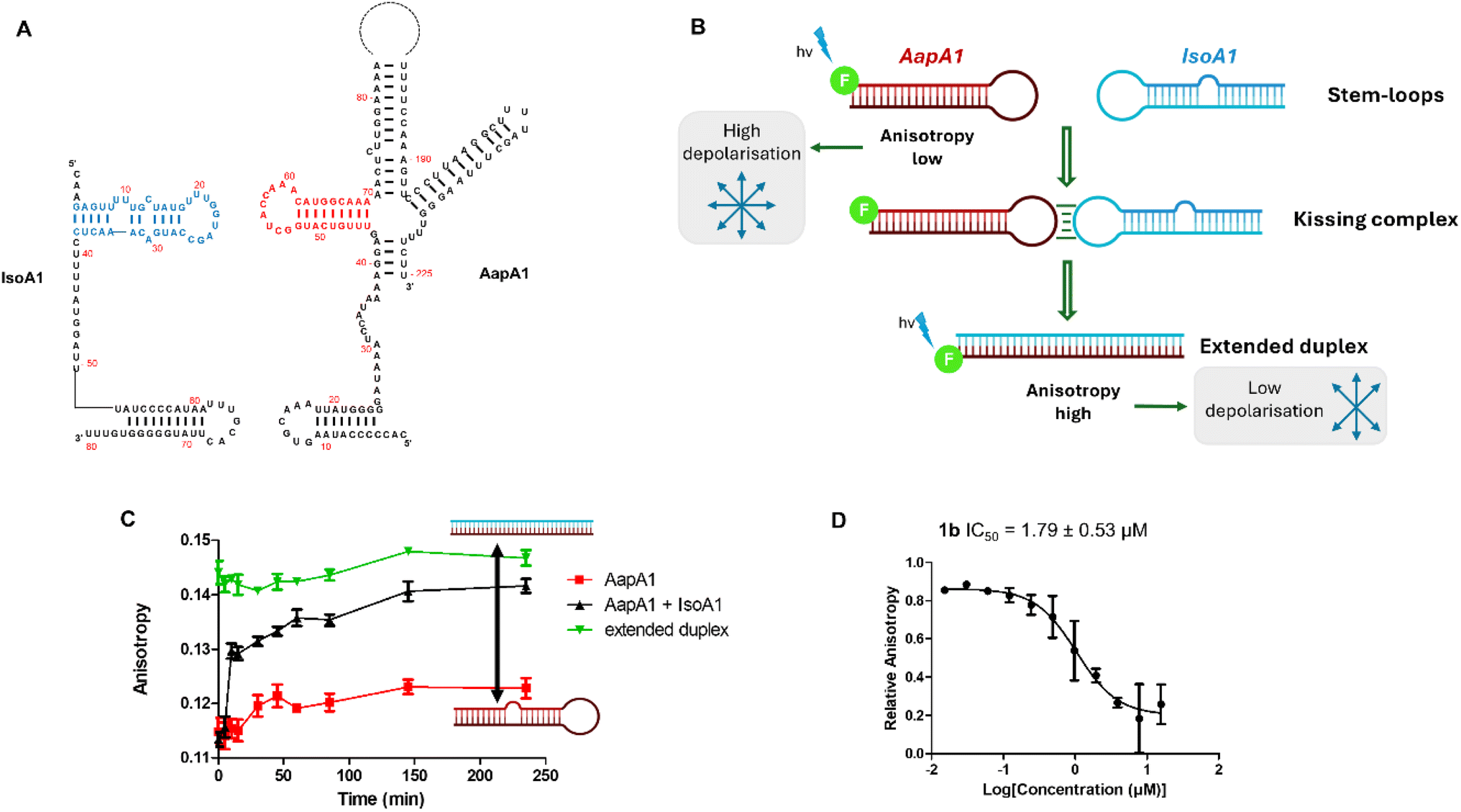 | ||
| Fig. 3 Fluorescence anisotropy assay to assess inhibition activity. (A) Primary and secondary structures of IsoA1 (1–80) and AapA1 (1–225) RNAs. (B) Principle of the anisotropy-based assay. (C) Kinetics of duplex formation as observed using fluorescence anisotropy. (D) Inhibition curve measured by fluorescence anisotropy on compound 1b showing the best IC50 value. | ||
These inhibitory activities were confirmed using PAGE analysis of IsoA1/AapA1 interaction in the presence of increasing concentrations of compounds 1a–c and of neomycin supporting the relative trend observed between the four compounds and further suggesting that compound 1b is the most efficient inhibitor (Fig. 4 and S4†).
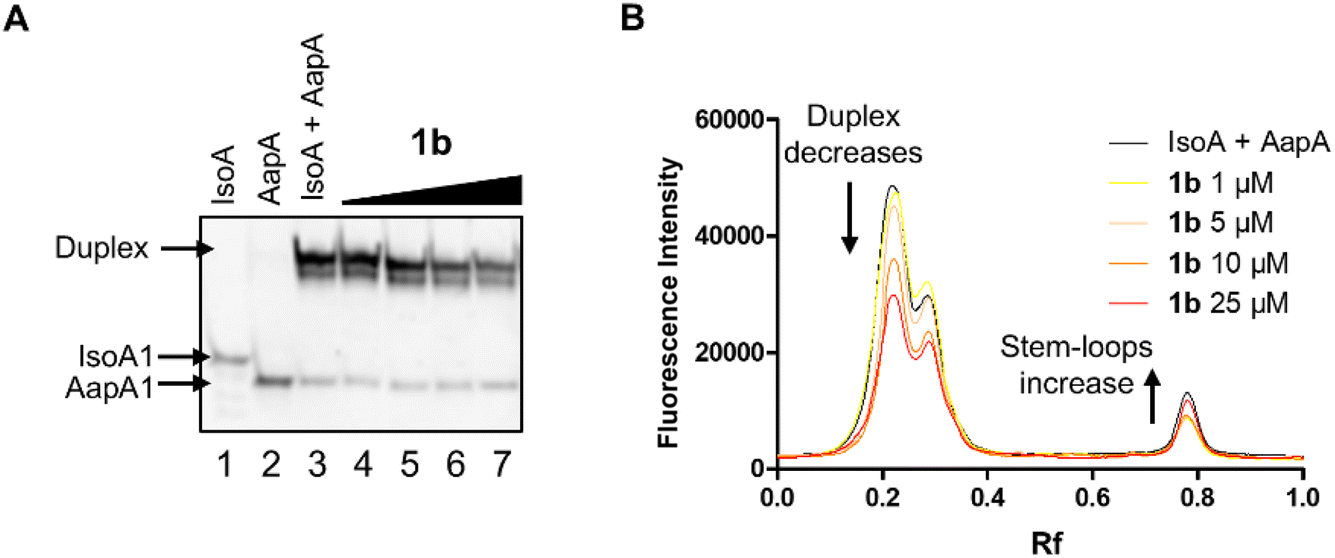 | ||
| Fig. 4 PAGE analysis of the inhibition of the formation of the extended duplex between AapA1 and IsoA1 stem-loop RNAs in the presence of compound 1b. (A) Non-denaturing gel where lane 1 is IsoA1 RNA, lane 2 is AapA1 RNA, lane 3 is the mixture of the two RNAs and lanes 4–7 are the mixture of the two RNAs in the presence of increasing concentrations (from 1 to 25 μM) of compound 1b. (B) Quantification of the non-denaturing gel illustrating precisely the decrease in the duplex amount and increase in stem loops bands. | ||
To assess if the inhibition activity is correlated with the affinity for AapA1 or IsoA1 stem-loop fragments, we measured dissociation constants (KD) for all compounds using a previously developed fluorescence-based assay.20 The obtained results (Table S1 and Fig. S5†) show that the best inhibitors 1a, 1b and 1c, characterized by the naphthyridine moiety, are submicromolar binders of AapA1 (with KD values of 0.301, 0.428 and 0.870 μM, respectively) while their KD values for IsoA1 RNA are two to three times higher. Compounds 2a, 4a and 6a, sharing lysine, also exhibit submicromolar affinities for AapA1, suggesting that this residue is highly favorable for binding. However, none of these latter compounds could inhibit AapA1/IsoA1 duplex formation suggesting that the naphthyridine scaffold and its mode of binding are crucial for directing the compounds toward a binding site that encompasses essential interactions between the two RNAs. Compounds bearing thymine (7a and 7c) or simple aromatic substituents (8c–9c) were the weakest binders demonstrating that, irrespective of the nature of the amino acid, thymine and monocyclic aromatic moieties are not well suited to interact. Regarding 3a–d and 5a, these were found to be partially degraded into the corresponding diketopiperazines (2,5-DKP) and thus their KD values could not be determined. Noteworthily, all binders showed very good selectivity in competition experiments as illustrated by K′D and K′′D values (Table S1†) obtained in the presence of a large excess of tRNA and DNA, respectively.
Given that compound 1b is the best inhibitor of AapA1/IsoA1 duplex formation and one of the best binders for these RNAs, we decided to assess if its inhibition activity is maintained when using full-length IsoA1 and AapA1 RNAs (Fig. S1†). As demonstrated by PAGE analysis of the AapA1/IsoA1 complex (Fig. 5A and S6†), compound 1b reduces extended duplex formation in a dose-dependent manner, while 1a and neomycin were not able to maintain a detectable inhibition under these conditions (Fig. S6†).
 | ||
| Fig. 5 Effect of 1b on the extended duplex amount and stability. (A) Quantification of PAGE analysis of full-length IsoA1 and AapA1 RNA sequences with increasing 1b concentrations. The red arrows indicate a decrease in the duplex structure and an increase in AapA1 stem-loop. (B) DSF analysis of a mixture of pre-structured IsoA1 (80 nucleotides) and AapA1 (225 nucleotides). The green arrow indicates the transition stabilized by compound 1b. | ||
To explore the interaction mechanism of compound 1b, we performed differential scanning fluorimetry (DSF) experiments in which variations in the melting temperatures (Tm) of each structure are evaluated. When individually structured full-length AapA1 and IsoA1 RNAs were mixed they showed multiple transitions between 30 and 40 °C together with a major one at 55.0 °C and one at 79.0 °C (Fig. 5B, dark green line). Based on the individual DSF profiles (Fig. S7†), these two peaks correspond to the melting of the stem-loop structures and the extended duplex, respectively. In the presence of compound 1b, a ΔTm stabilization of +4.5 °C was observed for the peak at 55.0 °C that shifted to 59.5 °C while the extended duplex stability remained unaffected. This suggests stabilization of the stem-loop structures that inhibits the formation of the extended duplex, thus inducing the inhibition activity. Neomycin exhibits a very different behavior since it stabilizes all observed structures including the duplex (Fig. S7†). These results were also confirmed using the truncated IsoA1 and AapA1 sequences used for anisotropy and affinity experiments (Fig. S8–S10†). In this context, neomycin strongly stabilizes both the duplex (+7.0 °C) and the separate stem loops (+6 °C to +9 °C) while compound 1b stabilizes the stem-loop structures without affecting the duplex as observed for the full-length sequences.
Based on these results, we next assessed the in vitro activity of all compounds against the targeted bacteria H. pylori in comparison with other bacteria that do not express the same TA system, such as S. aureus, E. coli, K. pneumoniae, K. aerogenes, A. baumanii and P. aeruginosa. At first, MIC-based susceptibility assay showed that compounds 6a and 1b were very effective at inhibiting H. pylori growth with MIC values of 6.25 μM (Table S2†). When antibacterial activity was evaluated against other bacteria (Table S3†), we observed that 6a arrested the growth of E. coli and S. aureus with significant potency suggesting that this compound exerts its antibiotic effect with a non-specific mechanism, while 1b exhibited selectivity for H. pylori supporting a specific mechanism of action targeting the inhibition of the IsoA/AapA T1TA system. Determination of the minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) for compound 1b and 6a resulted in MBC/MIC ratios ≤ 4, consistent with a bactericidal mode of action (Table S2†).27 While in the case of 6a, this suggests a promising but non-specific mechanism of action, these results further support the proposed mechanism for the specific antibiotic effect of compound 1b.
Conclusion
In this work, we designed, synthesized and evaluated a new series of RNA binders using a large set of biochemical experiments to validate the inhibition of IsoA/AapA interaction, affinity for the target RNAs, the effect on the structures of the targets and eventually the antibiotic activity. Overall, compound 1b is among the best ligands (KD of 428 nM), the best inhibitor in all in vitro assays (IC50 1.79 μM) and the only selective compound against H. pylori, suggesting that its in vivo activity relies on the artificial activation of the TA system. While it is not possible to quantify the expression of the toxin because of bacterial death in the presence of the compounds, it would be suitable to develop further in vitro assays in the near future to test the direct effect of such compounds on toxin expression. Confirmation of the bactericidal effect induced by the compounds will also be performed. The fact that compound 1b is selective for the targeted T1TA system and for H. pylori while compound 6a shows antibiotic activity on multiple strains, suggests that while the mechanism of action of 1b is selective, compound 6a acts on a different target. This latter target could be the prokaryotic ribosomal RNA as is the case for neomycin. Given the results obtained with compound 1b, we propose type I TA systems as promising and innovative antimicrobial targets. Artificial activation of the IsoA/AapA T1TA system using synthetic small molecules leverages an endogenous toxin expressed by multiple loci in the bacterial genome, reducing mutation-based resistance risks. Noteworthily, inhibition of RNA–RNA interactions by small molecules remains underexplored and this work stands among the rare examples of such proposed compounds.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
C. M., M. P. were responsible for the synthesis of all compounds and their biochemical evaluation. E. T. was responsible for DSF assays. S. A. contributed to the design and performance of the biochemical experiments. A. B. and V. S. performed experiments in bacteria. N. P. contributed to the analysis of the obtained results and the writing. A. D. G. contributed to the design of the compounds and oversight the entire project. F. D. and M. D. designed the project and all the experiments and performed the writing of the article.Conflicts of interest
No conflict of interest to declare.Acknowledgements
This work and the PhD fellowship to C. M. were funded by ANR grant ANR-17-CE18-0009-01. The authors thank J. M. Guigonis (CEA TIRO) for the HRMS analysis.References
- W. R. Miller and C. A. Arias, Nat. Rev. Microbiol., 2024, 22, 598–616 CrossRef CAS PubMed.
- Antimicrobial Resistance Collaborators, Lancet, 2022, 399, 629–655 CrossRef PubMed.
- D. Jurenas, N. Fraikin, F. Goormaghtigh and L. Van Melderen, Nat. Rev. Microbiol., 2022, 20, 335–350 CrossRef CAS PubMed.
- J. Qiu, Y. Zhai, M. Wei, C. Zheng and X. Jiao, Microbiol. Res., 2022, 264, 127159 CrossRef CAS PubMed.
- B. A. Berghoff and E. G. H. Wagner, Curr. Genet., 2017, 63, 1011–1016 CrossRef CAS PubMed.
- S. Bonabal and F. Darfeuille, Biochimie, 2024, 217, 95–105 CrossRef CAS PubMed.
- H. Arnion, D. N. Korkut, S. Masachis Gelo, S. Chabas, J. Reignier, I. Iost and F. Darfeuille, Nucleic Acids Res., 2017, 45, 4782–4795 CAS.
- C. M. Sharma, S. Hoffmann, F. Darfeuille, J. Reignier, S. Findeiss, A. Sittka, S. Chabas, K. Reiche, J. Hackermuller, R. Reinhardt, P. F. Stadler and J. Vogel, Nature, 2010, 464, 250–255 CrossRef CAS PubMed.
- A. Dart, Nat. Rev. Cancer, 2024, 24, 651 CrossRef CAS PubMed.
- S. Masachis and F. Darfeuille, Microbiol. Spectrum, 2018, 6 DOI:10.1128/microbiolspec.rwr-0030-2018.
- S. Bernacchi, S. Freisz, C. Maechling, B. Spiess, R. Marquet, P. Dumas and E. Ennifar, Nucleic Acids Res., 2007, 35, 7128–7139 CrossRef CAS PubMed.
- C. Dohno and K. Nakatani, ChemBioChem, 2019, 20, 2903–2910 CrossRef CAS PubMed.
- S. Kovachka, M. Panosetti, B. Grimaldi, S. Azoulay, A. Di Giorgio and M. Duca, Nat. Rev. Chem., 2024, 8, 120–135 CrossRef CAS PubMed.
- C. Schwergold, G. Depecker, C. Di Giorgio, N. Patino, F. Jossinet, B. Ehresmann, R. Terreux, D. Cabrol-Brass and R. Condom, Tetrahedron, 2002, 58, 5675–5687 CrossRef CAS.
- J. P. Joly, G. Mata, P. Eldin, L. Briant, F. Fontaine-Vive, M. Duca and R. Benhida, Chemistry, 2014, 20, 2071–2079 CrossRef CAS PubMed.
- C. Martin, M. Bonnet, N. Patino, S. Azoulay, A. Di Giorgio and M. Duca, ChemPlusChem, 2022, 87, e202200250 CrossRef CAS PubMed.
- C. Maucort, M. Bonnet, J. C. Ortuno, G. Tucker, E. Quissac, M. Verreault, S. Azoulay, C. Di Giorgio, A. Di Giorgio and M. Duca, J. Med. Chem., 2023, 66, 10639–10657 CrossRef CAS PubMed.
- C. Maucort, D. D. Vo, S. Aouad, C. Charrat, S. Azoulay, A. Di Giorgio and M. Duca, ACS Med. Chem. Lett., 2021, 12, 899–906 CrossRef CAS PubMed.
- D. D. Vo, C. Becquart, T. P. A. Tran, A. Di Giorgio, F. Darfeuille, C. Staedel and M. Duca, Org. Biomol. Chem., 2018, 16, 6262–6274 RSC.
- D. D. Vo, C. Staedel, L. Zehnacker, R. Benhida, F. Darfeuille and M. Duca, ACS Chem. Biol., 2014, 9, 711–721 CrossRef CAS PubMed.
- D. D. Vo, T. P. Tran, C. Staedel, R. Benhida, F. Darfeuille, A. Di Giorgio and M. Duca, Chem.–Eur. J., 2016, 22, 5350–5362 CrossRef CAS PubMed.
- A. Domling and I. I. Ugi, Angew Chem. Int. Ed. Engl., 2000, 39, 3168–3210 CrossRef CAS PubMed.
- M. Corley, M. C. Burns and G. W. Yeo, Mol. Cell, 2020, 78, 9–29 CrossRef CAS PubMed.
- A. Murata, T. Otabe, J. Zhang and K. Nakatani, ACS Chem. Biol., 2016, 11, 2790–2796 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- G. A. Pankey and L. D. Sabath, Clin. Infect. Dis., 2004, 38, 864–870 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01412c |
| This journal is © The Royal Society of Chemistry 2025 |