
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-catalyzed methylthiolation of chloroarenes and diverse aryl electrophiles†
Sae Toyoda,
Keiichiro Iizumi and
Junichiro Yamaguchi *
*
Department of Applied Chemistry, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: junyamaguchi@waseda.jp
First published on 22nd May 2025
Abstract
In this study, we report the first development of metal catalyzed methylthiolation of chloroarenes and diverse aromatic electrophiles, addressing the persistent challenges of catalyst and intermediate deactivation in the functionalization of less reactive substrates. To overcome these issues, we designed a novel anion-shuttle-type methylthiolation agent, 4-((methylthio)methyl)morpholine, which enables the controlled in situ release of methylthiolate anions, thereby preventing catalyst poisoning and enhancing reactivity. This strategy allows efficient methylthiolation not only of chloroarenes but also of a broad range of aryl electrophiles, including bromoarenes, aryl triflates, aryl tosylates, aryl pivalates, aryl nitriles, and aryl carboxylic acids. The developed system exhibits excellent functional group compatibility, making it applicable to the derivatization of pharmaceuticals and natural products. Furthermore, detailed mechanistic investigations revealed key factors underlying the exceptional efficiency of this methylthiolation agent, providing new insights into C–S bond formation under practical conditions.
Introduction
Arenes bearing methylthio groups are prevalent structural motifs in pharmaceuticals, agrochemicals, and organic materials (Fig. 1A).1 Notable examples include the antipsychotic drug thioridazine and the natural aromatic sulfide lissoclibadin 1, which features a unique disulfide bridge.2,3 Additionally, compounds such as sulmazole and firocoxib are readily accessible through oxidation of their methylthio groups.4,5 The incorporation of methylthio units has also enabled functional modifications of BODIPY dyes via methylthio and sulfoxide functionalities.6 Given the widespread occurrence in functional molecules and their significant influence on molecular properties, the development of efficient methods for carbon–sulfur (C–S) bond formation has become a key focus in synthetic chemistry.7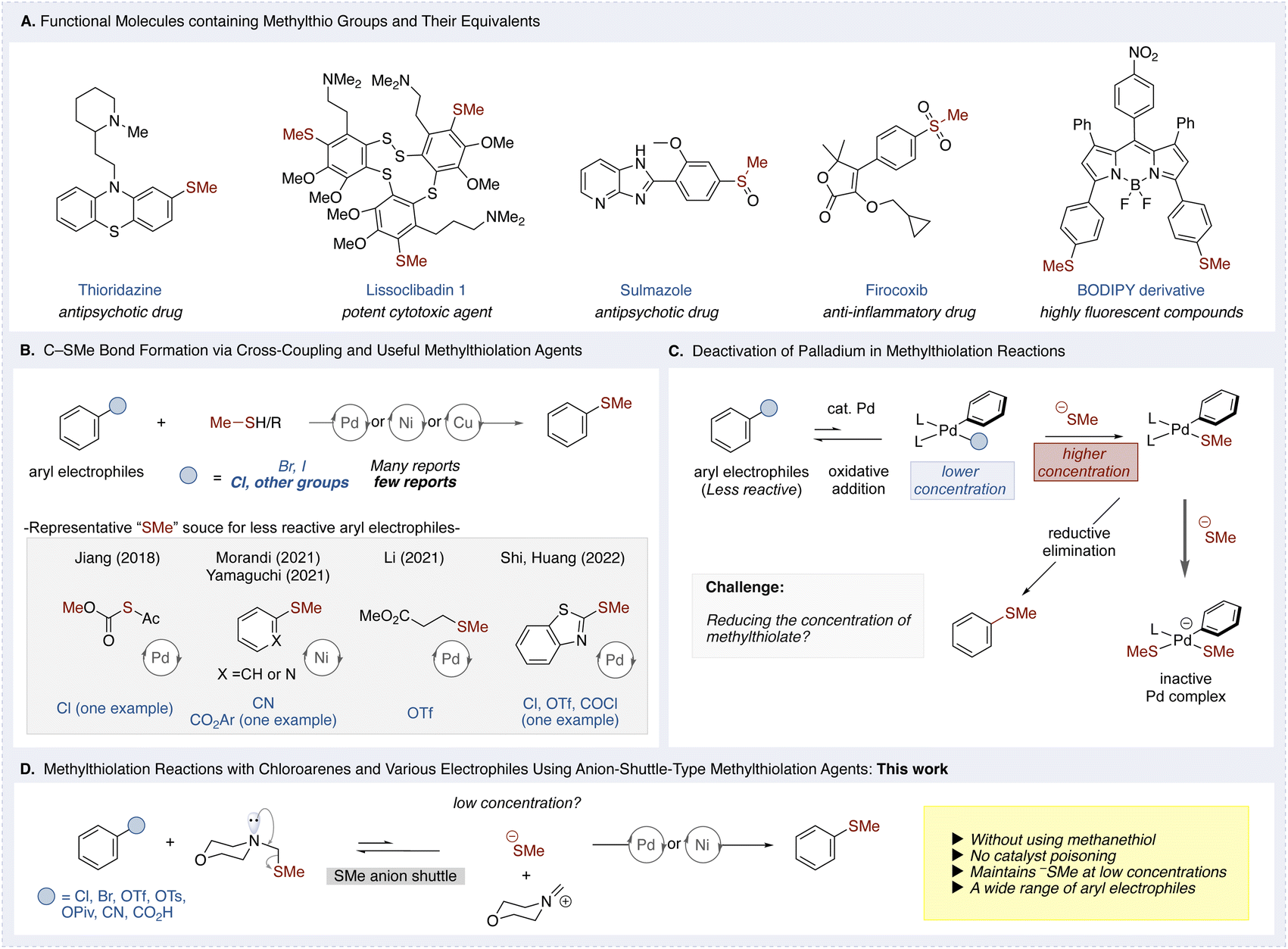 | ||
| Fig. 1 (A) Functional compounds containing methylthio groups and related structures. (B) Cross-coupling strategies for C–SMe bond formation with aryl electrophiles and common methylating agents. (C) Palladium catalyst deactivation in methylthiolation reactions. (D) Methylthiolation reactions with chloroarenes and various electrophiles using an anion-shuttle-type methylthiolation agent (this work). | ||
In this context, transition-metal-catalyzed C–S cross-coupling reactions have advanced significantly since the pioneering work of Migita and co-workers, who first reported palladium-catalyzed alkylthiolation and arylthiolation of haloarenes.8 Subsequent studies have expanded this strategy to various (pseudo)haloarenes, enabling the synthesis of diverse sulfur-containing arenes.9 However, methylthiolation of aryl electrophiles, particularly less reactive ones, remains a major challenge due to the volatility and toxicity of methanethiol, as well as catalyst poisoning by thiolate species. To circumvent these issues, organic methylthiolation agents have been developed as alternatives to methanethiol. Nonetheless, most existing methylthiolation methods have been limited to highly reactive bromoarenes and iodoarenes, while chloroarenes and other less reactive aryl electrophiles remain challenging substrates (Fig. 1B).10 To the best of our knowledge, the Jiang group has primarily focused on bromoarenes, reporting only a single example involving a chloroarene.11 Morandi and our group have demonstrated sulfide transfer reactions involving aryl nitriles12 and in one case, esters13 using nickel catalysis, Li and co-workers reported the methylthiolation of aryl triflates via a retro-Michael reaction that generates the methylthiolate in situ.14 Additionally, Shi and Huang's group reported one example of methylthiolation for chloroarenes, aryl triflates, and acid chlorides, although yields for the chloroarenes were notably low.15 These results underscore the continued difficulty of achieving efficient methylthiolation of chloroarenes and other less reactive aryl electrophiles, and highlight the need for a more general, broadly applicable strategy.
This limitation is particularly pronounced in palladium-catalyzed systems, where oxidative addition to less reactive electrophiles proceeds slowly and with lower efficiency. Under these conditions, excess thiolate nucleophiles, such as methylthiolates, can coordinate to the palladium center, leading to deactivation of the Ar–Pd–SR intermediate (Fig. 1C).16 As a result, most current methylthiolation protocols remain limited to highly reactive substrates like iodoarenes and bromoarenes, where the rapid formation and higher concentration of oxidative addition complexes help mitigate catalyst deactivation. Therefore, there is a pressing need to develop methylthiolation agents and methodologies that can efficiently functionalize less reactive aryl electrophiles, such as chloroarenes, under practical conditions.
To address these challenges, we designed a novel methylthiolation agent capable of regulating methylthiolate release (Fig. 1D). By leveraging the nucleophile-releasing properties of tertiary amines at the α-position, we developed a system in which the methylthiolates generated could nucleophilically coordinate to the metal catalyst without causing deactivation. This system operates via a dynamic equilibrium between the methylthiolate and an iminium ion, effectively minimizing the concentration of free methylthiolate available to interact undesirably with the metal catalyst or its intermediates.17 Herein, we report the first systematic study of the metal-catalyzed methylthiolation of chloroarenes and diverse aryl electrophiles using this anion-shuttle-type reagent.
Results and discussion
Methylthiolation agent 1 was synthesized in large scale (100 mmol) in high yield (72%) from inexpensive, commercially available morpholine and chloromethyl methyl sulfide (Fig. 2A). The product is a colorless liquid that requires no column chromatography for purification and can be easily purified by distillation. This approach eliminates the need for methanethiol or its salts, thereby avoiding issues related to volatility and odor. Using Pd catalysis with methylthiolation agent 1, the methylthiolation of haloarenes was explored (Fig. 2B). Treatment of 1-bromonaphthalene (Br-2a) with 1 (1.5 equiv.) under Pd(OAc)2 (5.0 mol%) and xantphos (10 mol%) catalysis, along with Cs2CO3 (2.0 equiv.) in toluene at 120 °C for 12 h, afforded the desired methylthionaphthalene (3a) in excellent yield (96%, detailed optimization provided in the ESI†). This result confirmed that 1 is an effective methylthiolation agent under these conditions. In contrast, the reaction of ortho-methyl-substituted bromonaphthalene Br-2b under similar conditions gave only trace amounts of the product. However, the addition of zinc improved the yield to 91%. | ||
| Fig. 2 (A) Large-scale synthesis of methylthiolation agent 1 from morpholine and chloromethyl methyl sulfide. (B) Methylthiolation of haloarenes using Pd catalysis with agent 1. (C) Optimization of methylthiolation conditions across various aryl electrophiles. Xantphos: 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene. dppf: 1,1′-bis(diphenylphosphino)ferrocene dcypt: 3,4-bis(dicyclohexylphosphino)thiophene. dcype: 1,2-bis(dicyclohexylphosphino)ethane. | ||
Given that efficient reactivity observed with bromoarenes, we next investigated the more challenging chloroarenes, which have been rarely explored for methylthiolation. Without zinc, the reaction of Cl-2a exhibited minimal progress (2% yield), whereas the addition of zinc significantly improved the yield to 53%. Although zinc may function as a reductant for Pd(II), the lack of reactivity observed with stronger reductants such as Mg and Mn suggests that zinc's role is not solely as a simple reductant. It was further hypothesized that Zn2+, generated by the reduction of Pd(II), might facilitate the reaction. However, the addition of Zn2+ salts had no significant effect. The precise role of zinc in this reaction remains unclear and will be discussed in detail later (in Fig. 4B).
With these findings, the methylthiolation of not only chloroarenes but also a diverse range of aryl electrophiles using the novel methylthiolation agent 1 was further investigated. The optimized conditions were found to be effective for bromoarenes, aryl triflates, tosylates, pivalates, aryl nitriles, and aryl carboxylic acids, demonstrating the broad applicability of this catalytic system to various electrophiles with different reactivity profiles (Fig. 2C). For bromoarenes, the conditions outlined in Fig. 2B were found to be optimal. For chloroarenes and aryl triflates, ligands with large bite angle proved effective, with dppf providing high yields.18 Ni catalysis demonstrated high efficiency for tosylates and mesylates, affording the desired methylthiolated product 3c in excellent yields (caution: scaling up reactions in diethyl ether at high temperatures should be avoided unless appropriate equipment is used). Similarly, Ni catalysis with the dcypt ligand enabled efficient activation of C–O bonds in pivalates, producing methylthiolated product 3c in a good yield.13 All of the Ni catalysis described herein employed Ni(cod)2. Although Ni(II) salts were also tested, the reactions proceeded with reduced efficiency for triflates and pivalates, while no reaction occurred for tosylates and mesylates (for details, see the ESI†). In contrast, Pd catalysis facilitated the methylthiolation of aryl nitriles with good yields,19 while Ni catalysis enabled methylthiolation of aryl carboxylic acids, albeit with lower yields.20 Notably, this study represents the first example of Pd-catalyzed sulfidation of aryl nitriles.
Based on the reaction conditions described in Fig. 2C, the substrate scope of aryl electrophiles was further explored (Fig. 3). Initially, we examined naphthalene, anthracene, and polycyclic aromatic hydrocarbon (PAH) electrophiles. Under optimized conditions, Cl-2a afforded the desired product 3a in an excellent yield of 96%. The triflate derivative also gave the product in high yield, while tosylate and mesylate derivatives provided the product 3a in moderate yields but confirmed the reaction's applicability. During the investigation of commercially available aryl electrophiles, we primarily used bromoarenes due to their widespread availability. The reaction also proceeded efficiently with anthracene (3d and 3e), phenanthrene (3f), and pyrene (3g). Notably, when cyanopyrene was used as a substrate, a decyanative methylthiolation reaction occurred, affording 3g in 43% yield.
 | ||
| Fig. 3 Substrate scope. Reaction conditions: for bromoarenes; 2 (0.2 mmol), 1 (0.3 mmol), 5.0 mol% Pd(OAc)2, 10 mol% xantphos, Cs2CO3 (0.4 mmol), toluene, 120 °C for 12 h. For chloroarenes; 2 (0.2 mmol), 1 (0.3 mmol), 5.0 mol% Pd(OAc)2 10 mol% dppf, Zn (0.4 mmol), Cs2CO3 (0.4 mmol) toluene, 150 °C for 12 h. For aryl tosylates and mesylates: 2 (0.2 mmol), 1 (0.3 mmol), 5.0 mol% Ni(cod)2, 10 mol% dppf, Cs2CO3 (2.0 equiv.), Zn (2.0 equiv.), Et2O, 150 °C for 12 h. For aryl pivalates; 2 (0.2 mmol), 1 (0.3 mmol), 10 mol% Ni(cod)2, 20 mol% dcypt, Cs2CO3 (0.4 mmol), Zn (0.4 mmol), toluene, 150 °C for 12 h. For aryl cyanides, 2 (0.2 mmol), 1 (0.3 mmol), 5.0 mol% Pd(OAc)2, 10 mol% dcype, NaOtBu (0.4 mmol), Zn (0.4 mmol), toluene, 150 °C for 12 h. For aryl carboxylic acids; 2 (0.2 mmol), 1 (0.3 mmol), 10 mol% Ni(cod)2, 20 mol% dcypt, Piv2O (0.24 mmol), Zn (0.4 mmol), toluene, 150 °C for 12 h. a10 mol% Pd(OAc)2, 20 mol% dppf were used. bZinc was added. c0.4 mmol scale. dYield was determined by 1H NMR. e1 (0.6 mmol, 3.0 equiv.) was used. f24 h. | ||
Next, we explored benzene derivatives and functionalized aryl electrophiles. Substituted benzene derivatives afforded the corresponding methylthiolated products in good yields (3h and 3i). Expanding the substrate scope, we tested biphenyl derivatives, finding that bromo, chloro, and triflate derivatives reacted smoothly. However, tosylate and pivalate derivatives exhibited significantly lower yields of 3j compared to naphthalene derivatives. This decreasing yield is likely due to the higher aromaticity of biphenyl relative to naphthalene, which makes oxidative addition more challenging. Cyanated biphenyl derivatives provided 3j in moderate yields. Reactions involving biphenyl derivatives with meta- or para-substituted phenyl groups proceeded efficiently, affording the products in moderate yields (3k–3l). However, for ortho-disubstituted haloarenes, including both bromo and chloro derivatives, the reaction barely progressed, likely due to steric hindrance at the catalyst's active site. The methylthiolation reaction also exhibited broad functional group compatibility, successfully delivering the methylthiolated products without affecting ketone (3n), esters (3o, 3p and 3q), nitro group (3r), and alcohol (3s).
Next, we investigated heteroaromatic electrophiles. Chloro-substituted pyridine reacted efficiently under the optimized conditions, affording 3t in 65% yield. Owing to the high volatility of the product, the yield was determined by 1H NMR spectroscopy. The reaction also proceeded with other pyridines (3u), quinolines (3v and 3w), dibenzothiophene (3x), and thiophene (3y). However, the corresponding tosylate and mesylate derivatives remained unreactive under these conditions. The only exception was 2-cyanoquinoline, which afforded the methylthiolated product 3v in low yield (14%).
To further enhance the practical utility of this methylthiolation reaction, we applied it to substrates derived from natural products and pharmaceutical APIs with diverse functional groups. In these cases, tosylate and pivalate derivatives again showed low reactivity, so most reactions were conducted using chloro or triflate derivatives.
Aryl electrophiles containing flavone and coumarin skeletons underwent smooth methylthiolation, affording 3A and 3B. Similarly, substrates such as fenofibrate (3C), vanillin (3D), eugenol (3E), and phenothiazine derivative (3F)—which feature ester, ketone, aldehyde, olefin, and amine functionalities—were successfully methylthiolated without compromising the integrity of these functional groups. For estrone, the ketone moiety was reactive under standard conditions; however, when protected as an acetal, the triflate derivative afforded 3G in good yield. Additionally, converting the ketone into an enol triflate and reacting it with 3.0 equiv. of methylthiolation agent 1 enabled efficient dual methylthiolation, affording 3H in high yield. These results demonstrate that the developed conditions are applicable not only to aryl electrophiles but also to enol triflates,21 representing, to the best of our knowledge, the first example of methylthiolation of enol triflates. Finally, when ticlopidine was used as a substrate, reacting it with 3.0 equiv. of 1 for 12 h afforded the mono-methylthiolated product 3I. Extending the reaction time to 24 h selectively gave the di-methylthiolated product 3J. It should be noted that while Cs2CO3 was added in most reactions, certain substrates did not require its presence. Although it is presumed that the base primarily facilitates the exchange of the anionic species before the methylthiolate reacts with the metal-aryl complex following oxidative addition, its precise role remains unclear (for details on substrate-dependent variations, see the ESI†). In addition, although zinc was added in all reactions, there were substrates for which its presence had no significant effect, and the reaction proceeded even in its absence. Zinc appeared to be particularly effective for sterically hindered substrates or those for which oxidative addition is challenging (for details, see the ESI†).
In this methylthiolation reaction, three key mechanistic questions emerged: (1) does the methylthiolation agent effectively regulate the concentration of the methylthiolate, keeping it low? (2) Why does the addition of zinc significantly improve the reaction yield? (3) How is the methylthiolate generated from 1? To address these, we performed a series of mechanistic investigations. First, to investigate the concentration of the methylthiolate, to evaluate the effect of methylthiolate concentration, we performed control experiments (Fig. 4A). Under the optimized conditions using 1.5 equiv. of methylthiolation agent 1 and Cl-2a, product 3a was obtained in 96% yield. However, when 1.2 equiv. of sodium methanethiolate (NaSMe, 15% in water, 100 μL) was added, the yield of 3a dropped to 26%, with 64% the starting material Cl-2a recovered. To assess the role of water, we added 100 μL of water under otherwise identical conditions; in this case, the yield increased to 60%, indicating that the NaSMe itself—not water alone—was detrimental to the reaction. Furthermore, using 2.5 or 5.0 equiv. of agent 1 led to lower yields (74% and 53%, respectively) and substantial recovery of Cl-2a, highlighting that excessive thiolate negatively impacts catalysis. These findings clearly demonstrate that controlling the thiolate concentration is crucial for maintaining catalyst activity.
 | ||
| Fig. 4 (A) Investigation of the role of methylthiolate concentration using sodium methanethiolate under optimized conditions. (B) Examination of zinc's role in the reaction. The red bars and numbers indicate the yield of the methylthiolated product 3, while the blue bars and numbers represent the recovery rate of the starting material. (C) Zinc as a reducing agent for disulfides. (D) Generation of methylthiolate. (E) The fate of the iminium ion. aThe product contains c.a. 10% of a regioisomeric methylthiolated compound. | ||
Next, we investigated the role of zinc in the reaction (Fig. 4B). Without zinc, Cl-2m remained largely unreacted (75% recovery), and only 5% of product 3m was formed. When 2.0 equiv. of zinc was added, the yield improved dramatically to 98%. Replacing Pd(OAc)2 with Pd(dba)2 (a Pd0 complex) resulted in only 45% yield, but adding zinc to this system raised the yield to 90%. Furthermore, we confirmed that using Mn or Mg did not promote the reaction (see the ESI†). These results indicate that zinc acts as a reductant to generate Pd(0) from Pd(II), but also provides an additional beneficial effect.
We hypothesized that trace disulfides, generated by palladium catalyst or intermediates, may deactivate the catalyst and zinc might restore activity by reducing these disulfides.22 To test this, we added 10 mol% dimethyl disulfide to the Pd(dba)2-catalyzed reaction. The reaction was completely inhibited, and the starting material was fully recovered—even in the presence of zinc. Using Pd(OAc)2 and zinc allowed the reaction to proceed, albeit with a reduced yield (33%). These observations suggest that dimethyl disulfide poisons Pd(0) catalysts, likely through strong coordination. Pd(II) is also inhibited, though to a lesser extent, possibly after its in situ reduction to Pd(0).
To further explore the zinc-disulfide hypothesis, we performed the reaction using dimethyl disulfide as a methylthiolation agent (Fig. 4C). Without zinc, no reaction occurred. However, with zinc (2.0 equiv.), 3a was obtained in 60% yield. For this reaction as well, it was confirmed that Mn and Mg had no effect and 3a was not obtained (see the ESI†). We then synthesized Zn(SMe)2 by heating dimethyl disulfide with zinc in DMSO at 150 °C, affording a white, odorless solid. Although Zn(SMe)2 was insoluble in toluene, it gave 3a in 80% yield under standard reaction conditions, with 12% starting material remaining. While Zn(SMe)2 was not more effective than agent 1, its thiolation reactivity suggests that it may act as a methylthiolating species. However, since dimethyl disulfide was not detected in the catalytic system, Zn(SMe)2 is unlikely to be the dominant active species.
We then examined the conditions for generating the methylthiolate from agent 1 (Fig. 4D). Since its formation is expected to produce an iminium ion, we used 4-methoxyphenol, a known iminium trap, to monitor this process. Heating agent 1 with 4-methoxyphenol at 150 °C afforded adduct 4 in 92% yield. The yield decreased to 45% and 29% at 120 °C and 80 °C, respectively, and was negligible at 50 °C or room temperature. Time-course monitoring confirmed that 4 was gradually formed over 4–6 hours. These results show that thermal generation of methylthiolate is efficient above 80 °C, with 150 °C being optimal.
Finally, we probed the behavior of the iminium ion (Fig. 4E). After methylthiolation of Cl-2a, addition of 4-methoxyphenol afforded 4 in 26% yield, confirming the generation of the cation. Interestingly, adding Cs2CO3 to the mixture of 4 and 4-methoxyphenol led to a similar yield, suggesting that the base may also trap the cation, although the exact mechanism remains unclear. In contrast, using 1,4-dimethoxybenzene did not yield any trapping product, indicating that the iminium ion does not react significantly with less nucleophilic aromatics such as chloroarenes under the reaction conditions (see the ESI†).
In summary of these mechanistic studies clarify that: (1) agent 1 maintains a low steady-state concentration of methylthiolate, which is essential to avoid catalyst deactivation. (2) Zinc acts both as a reductant and as a scavenger for inhibitory disulfides, enhancing overall efficiency. (3) Methylthiolate is thermally released from agent 1 via a reversible process that also generates an iminium ion.
Based on the results, we propose the reaction mechanism shown in Fig. 5A. First, Pd(II) is reduced to Pd(0) species A by zinc. Subsequently, oxidative addition of Pd(0) to the aryl electrophile 2 forms complex B. Methylthiolation agent 1 undergoes thermal decomposition to generate the methylthiolate and an iminium ion in equilibrium. The methylthiolate undergoes nucleophilic metallation (ligand exchange) with complex B, and in certain substrates, Cs2CO3 facilitates this exchange process, forming Pd–SMe complex C. If the concentration of the methylthiolate becomes too high, deactivation occurs via formation of the anionic complex D.16 Complex C that avoids conversion to complex D undergoes direct reductive elimination, affording the desired methylthiolated product 3 and regenerating Pd(0) to complete the catalytic cycle.
 | ||
| Fig. 5 (A) Proposed mechanism. (B) Oxidation of methylthio groups. (C) Application to other thiosulfide reagents. | ||
For less reactive aryl electrophiles, oxidative addition is slower, allowing free methylthiolate to coordinate directly with Pd(0), forming an inactive Pd(SMe)2 species.22 Although reductive elimination from this species can regenerate Pd(0) and dimethyl disulfide, the reaction is reversible and proceeds slowly. However, the dimethyl disulfide generated can be reduced by zinc, thereby shifting the equilibrium toward productive catalysis and enabling smooth turnover of the reaction.
Additionally, we explored simple derivatizations of the obtained methylthiolated products (Fig. 5B).23 When 1.0 equiv. of mCPBA was added to 3c, the product was efficiently converted to sulfoxide 5 in good yield. Increasing the amount of mCPBA to 2.0 equiv. afforded the sulfone 6 in high yield.
Finally, as an extension of the methylthiolation agent's application, we synthesized reagents in which the methylthio group was replaced with other alkyl or phenyl groups. These modified reagents were then evaluated for thioalkylation and thiophenylation of chloroarene Cl-2c. For alkyl-substituted reagents, the reaction proceeded efficiently, affording 7. Even with a bulky adamantyl group, the transformation occurred smoothly to give 8. Similarly, thiophenylation was successful, affording 9 in 45% yield.
Conclusions
In this study, we developed a highly efficient methylthiolation method for chloroarenes and diverse aryl electrophiles using a novel methylthiolation agent synthesized from inexpensive, commercially available reagent. This agent facilitates methylthiolation without the use of volatile and odor-intensive methanethiol. Given the scarcity of successful methylthiolation examples for chloroarenes, we first established the applicability of our method to bromoarenes and then expanded its scope to the more challenging chloroarenes. Furthermore, the method proved effective for other less reactive aryl electrophiles, achieving efficient C–S bond formation despite their inherently lower reactivity. Zinc was found to serve dual functions: facilitating the reduction of Pd(II) to Pd(0) and scavenging trace disulfides, thereby enhancing the reaction efficiency. Mechanistic investigations revealed that methylthiolates are generated through a thermally driven equilibrium, which is crucial for the reaction's success. Furthermore, the method demonstrated excellent functional group compatibility, accommodating complex structures found in pharmaceuticals and natural products, underscoring its versatility and synthetic utility.This first systematic study on the methylthiolation of chloroarenes and other less reactive aryl electrophiles provides a robust and practical protocol for methylthiolation with broad substrate scope and functional group tolerance, significantly expanding the toolbox for aryl functionalization and offering promising applications in the synthesis of sulfur-containing aromatic compounds.24
Data availability
The data supporting this article have been included as part of the ESI.† Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Author contributions
J. Y. supervised the entire project. The reaction was initially discovered by K. I., and all experimental work was carried out by S. T. The manuscript was prepared by J. Y., based on feedback and suggestions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21H05213 (Digi-TOS) (to J. Y.). This work was partly supported by JST CREST Grant Number JPMJCR24T3 (to J. Y.) We thank Iwatani Corporation for providing Cs2CO3 as a gift. The Materials Characterization Central Laboratory in Waseda University is acknowledged for the support of HRMS measurement.Notes and references
- (a) T. Kimoto, K. Tanaka, M. Kawahata, K. Yamaguchi, S. Otsubo, Y. Sakai, Y. Ono, A. Ohno and K. Kobayashi, J. Org. Chem., 2011, 76, 5018–5025 CrossRef CAS PubMed; (b) A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef PubMed; (c) R. Zhang, H. Ding, X. Pu, Z. Qian and Y. Xiao, Catalysts, 2020, 10, 1339 CrossRef CAS; (d) X. Li, X. Wang, Y. Li, J. Xiao and Y. Du, Org. Biomol. Chem., 2022, 20, 4471–4495 RSC.
- M. E. Cameron, J. M. Lawrence and J. G. Olrich, Br. J. Ophthalmol., 1972, 56, 131 CrossRef CAS PubMed.
- T. Tatsuta, M. Hosono, H. Rotinsulu, D. S. Wewengkang, D. A. Sumilat, M. Namikoshi and H. Yamazaki, J. Nat. Prod., 2017, 80, 499–502 CrossRef CAS PubMed.
- W. J. Parsons, V. Ramkumar and G. L. Stiles, Mol. Pharmacol., 1988, 33, 441–448 CrossRef CAS PubMed.
- A. Rangel-Nava, J. M. Ramírez-Uribe, S. Recillas-Morales, J. A. Ibancovichi-Camarillo, A. Venebra-Muñoz and P. Sánchez-Aparicio, J. Equine Vet. Sci., 2019, 77, 36–42 CrossRef PubMed.
- X.-D. Jiang, X. Liu, T. Fang and C. Sun, Dyes Pigments, 2017, 146, 438–444 CrossRef CAS.
- (a) J. Li, S. Yang, W. Wu and H. Jiang, Org. Chem. Front., 2020, 7, 1395–1417 RSC; (b) N. Sundaravelu, S. Sangeetha and G. Sekar, Org. Biomol. Chem., 2021, 19, 1459–1482 RSC; (c) F. Abedinifar, S. Bahadorikhalili, B. Larijani, M. Mahdavi and F. Verpoort, Appl. Organomet. Chem., 2022, 36, e6482 CrossRef CAS; (d) V. J. Geiger, R. M. Oechsner, P. H. Gehrtz and I. Fleischer, Synthesis, 2022, 54, 5139–5167 CrossRef CAS; (e) S. Huang, M. Wang and X. Jiang, Chem. Soc. Rev., 2022, 51, 8351–8377 RSC; (f) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed; (g) E. Rufino-Felipe, H. Valdés and D. Morales-Morales, Eur. J. Org Chem., 2022, e202200654 CrossRef CAS; (h) I. H. Lindenmaier, R. C. Richter and I. Fleischer, Org. Chem. Front., 2024, 11, 2485–2493 RSC.
- (a) T. Migita, T. Shimizu, Y. Asami, J. Shiobara, K. Kato and M. Kosugi, Bull. Chem. Soc. Jpn., 1980, 53, 1385–1389 CrossRef CAS; (b) M. Kosugi, T. Shimizu and T. Migita, Chem. Lett., 1978, 13–14 CrossRef CAS; (c) T. Migita, T. Shimizu, Y. Asami, J. Shiobara, Y. Kato and M. Kosugi, Bull. Chem. Soc. Jpn., 2006, 53, 1385–1389 CrossRef.
- Representative C–S bond formations from (pseudo)haloarenes, see, (a) T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587–4590 CrossRef CAS PubMed; (b) M. A. Fernández-Rodríguez, Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180–2181 CrossRef PubMed; (c) K. D. Jones, D. J. Power, D. Bierer, K. M. Gericke and S. G. Stewart, Org. Lett., 2018, 20, 208–211 CrossRef CAS PubMed; (d) T. Scattolin, E. Senol, G. Yin, Q. Guo and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 12425–12429 CrossRef CAS PubMed; (e) J. Xu, R. Y. Liu, C. S. Yeung and S. L. Buchwald, ACS Catal., 2019, 9, 6461–6466 CrossRef CAS PubMed; (f) M. T. Martín, M. Marín, C. Maya, A. Prieto and M. C. Nicasio, Chem.–Eur. J., 2021, 27, 12320–12326 CrossRef PubMed; (g) R. M. Oechsner, J. P. Wagner and I. Fleischer, ACS Catal., 2022, 12, 2233–2243 CrossRef CAS; (h) R. M. Oechsner, I. H. Lindenmaier and I. Fleischer, Org. Lett., 2023, 25, 1655–1660 CrossRef CAS PubMed.
- (a) N. Taniguchi, J. Org. Chem., 2004, 69, 6904–6906 CrossRef CAS PubMed; (b) V. Gómez-Benítez, O. Baldovino-Pantaleón, C. Herrera-Álvarez, R. A. Toscano and D. Morales-Morales, Tetrahedron Lett., 2006, 47, 5059–5062 CrossRef; (c) O. Baldovino-Pantaleón, S. Hernández-Ortega and D. Morales-Morales, Adv. Synth. Catal., 2006, 348, 236–242 CrossRef; (d) F. Luo, C. Pan, L. Li, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 5304–5306 RSC; (e) P. J. A. Joseph, S. Priyadarshini, M. L. Kantam and B. Sreedhar, Tetrahedron, 2013, 69, 8276–8283 CrossRef CAS; (f) K. Ghosh, S. Ranjit and D. Mal, Tetrahedron Lett., 2015, 56, 5199–5202 CrossRef CAS; (g) V. Gómez-Benítez, H. Valdés, S. Hernández-Ortega, J. M. German-Acacio and D. Morales-Morales, Polyhedron, 2018, 143, 144–148 CrossRef; (h) J. Heidebrecht, C. Gendy, B. S. Gelfand and R. Roesler, Polyhedron, 2018, 143, 138–143 CrossRef CAS; (i) Y. Wang, X. Wu and M. Yang, Synlett, 2020, 31, 1226–1230 CrossRef CAS; (j) G. B. Li and X.-F. Wu, Chem. Sci., 2020, 11, 2187–2192 RSC; (k) B. A. Hopkins, B. Zavesky and D. White, J. Org. Chem., 2022, 87, 7547–7550 CrossRef CAS PubMed; (l) B. Xu, S. Ling, S. Zheng, X. Feng, H. Liu, Y. Dong, X. Li, B. Hong and F.-G. Sun, Org. Lett., 2025, 27, 1620–1625 CrossRef CAS PubMed.
- M. Wang, Z. Qiao, J. Zhao and X. Jiang, Org. Lett., 2018, 20, 6193–6197 CrossRef CAS PubMed.
- T. Delcaillau, P. Boehm and B. Morandi, J. Am. Chem. Soc., 2021, 143, 3723–3728 CrossRef CAS PubMed.
- R. Isshiki, M. B. Kurosawa, K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 10333–10340 CrossRef CAS PubMed.
- D. Pan, S. Xu, Q. Tian and Y. Li, Eur. J. Org Chem., 2021, 2021, 4616–4619 CrossRef CAS.
- X. Liang, K. Wen, Q. Shi, B. Zhang, S. Pei, Q. Lin, B. Ma, S. Wang, M. Zhang, X. Li, Z. Wang and H. Huang, Chem.–Eur. J., 2022, 28, e202200869 CrossRef CAS PubMed.
- M. A. Fernández-Rodríguez, Q. Shen and J. F. Hartwig, Chem.–Eur. J., 2006, 12, 7782–7796 CrossRef PubMed.
- A cyanating agent our group and others previously reported, which operates via a similar mechanism to release cyanide anions, see, (a) R. Takise, K. Itami and J. Yamaguchi, Org. Lett., 2016, 18, 4428–4431 CrossRef CAS PubMed; (b) K. Iizumi, M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, Synlett, 2021, 32, 1555–1559 CrossRef CAS; (c) K. Iizumi, H. Tanaka, K. Muto and J. Yamaguchi, Org. Lett., 2024, 26, 3977–3981 CrossRef CAS PubMed; (d) S. Kotani, M. Sakamoto, K. Osakama and M. Nakajima, Eur. J. Org Chem., 2015, 6606–6609 CrossRef CAS; (e) M. S. Ahmad, Z. Shafiq and K. Meguellati, Synthesis, 2022, 54, 3077–3084 CrossRef CAS; (f) L. Wang, Y. Wang, J. Shen, Q. Chen and M.-Y. He, Org. Biomol. Chem., 2018, 16, 4816–4820 RSC; (g) T. Zhang, J. Qiao, H. Song, F. Xu, X. Liu, C. Xu, J. Ma, H. Liu, Z. Sun and W. Chu, N. J. Chem., 2019, 43, 9084–9089 RSC.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS PubMed.
- P. Boehm, P. Müller, P. Finkelstein, M. A. Rivero-Crespo, M.-O. Ebert, N. Trapp and B. Morandi, J. Am. Chem. Soc., 2022, 144, 13096–13108 CrossRef CAS PubMed.
- (a) H. Ji, H. Cao, G. Wang, F. Xing, M. Szostak and C. Liu, Org. Chem. Front., 2023, 10, 4275–4281 RSC; (b) C. E. Brigham, C. A. Malapit, N. Lalloo and M. S. Sanford, ACS Catal., 2020, 10, 8315–8320 CrossRef CAS PubMed.
- (a) Y. Imazaki, E. Shirakawa and T. Hayashi, Tetrahedron, 2011, 67, 10212–10215 CrossRef CAS; (b) A. B. Dürr, G. Yin, I. Kalvet, F. Napoly and F. Schoenebeck, Chem. Sci., 2015, 7, 1076–1081 RSC; (c) F. Zhang, Y. Wang, Y. Wang and Y. Pan, Org. Lett., 2021, 23, 7524–7528 CrossRef CAS PubMed; (d) B. Xu, H. Xiang, Y. Tan, Z. Li, S. Li, X.-Y. Ye and Y. Ye, J. Org. Chem., 2023, 88, 4592–4605 CrossRef CAS PubMed; (e) G. Du, Y. Zhao, P. Zhu, S. Ling, B. Xu, H. Liu, X. Li and F.-G. Sun, Org. Chem. Front., 2023, 10, 6185–6191 RSC.
- (a) N. Taniguchi, J. Org. Chem., 2004, 69, 6904–6906 CrossRef CAS PubMed; (b) L. Bettanin, S. Saba, F. Z. Galetto, G. A. Mike, J. Rafique and A. L. Braga, Tetrahedron Lett., 2017, 58, 4713–4716 CrossRef CAS; (c) K. D. Jones, D. J. Power, D. Bierer, K. M. Gericke and S. G. Stewart, Org. Lett., 2018, 20, 208–211 CrossRef CAS PubMed.
- G. F. Pasha, S. Asghari, M. Tajbakhsh and M. Mohseni, Res. Chem. Intermed., 2017, 43, 7291–7306 CrossRef CAS.
- B. Mouhsine, M. Norlöff, J. Ghouilem, A. Sallustrau, F. Taran and D. Audisio, J. Am. Chem. Soc., 2024, 146, 8343–8351 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental procedures, spectroscopic data for compounds including 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d5sc01428j |
| This journal is © The Royal Society of Chemistry 2025 |